

SUMMARY
The current paradigm indicates that naive T cells are primed in secondary lymphoid organs. Here, we present evidence that intranasal administration of peptide antigens appended to nanofibers primes naive CD8+ T cells in the lung independently and prior to priming in the draining mediastinal lymph node (MLN). Notably, comparable accumulation and transcriptomic responses of CD8+ T cells in lung and MLN are observed in both Batf3KO and wild-type (WT) mice, indicating that, while cDC1 dendritic cells (DCs) are the major subset for cross-presentation, cDC2 DCs alone are capable of cross-priming CD8+ T cells both in the lung and draining MLN. Transcription analyses reveal distinct transcriptional responses in lung cDC1 and cDC2 to intranasal nanofiber immunization. However, both DC subsets acquire shared transcriptional responses upon migration into the lymph node, thus uncovering a stepwise activation process of cDC1 and cDC2 toward their ability to cross-prime effector and functional memory CD8+ T cell responses.
Graphical Abstract
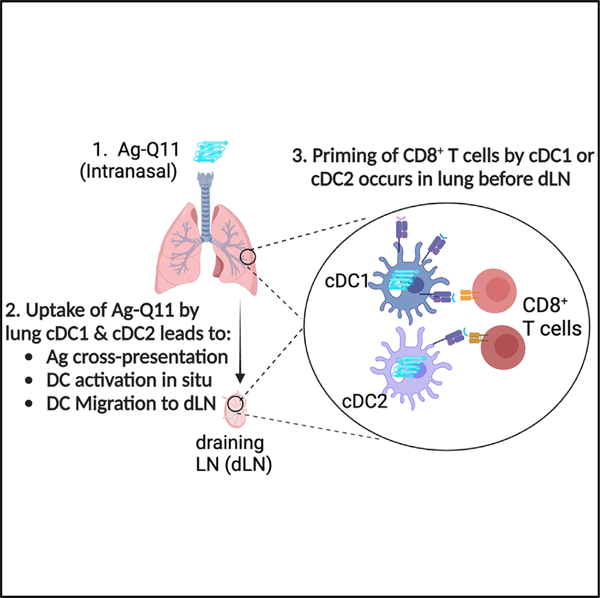
In brief
Si et al. show that intranasal nanofiber immunization cross-primes naive CD8+ T cells in the lung before lymph nodes. In the absence of cDC1, cDC2 are sufficient for cross-priming. While lung cDC1 and cDC2 have distinct transcriptional responses, they converge into shared transcriptional responses in the lymph node.
INTRODUCTION
T cell responses in the lung have been extensively investigated during the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic.1–6 It is widely acknowledged that respiratory infections lead to the priming of T cell responses in the mediastinal lymph node (MLN) by lung professional antigen-presenting cells (APCs), which capture antigens at the site of infection in the lung then migrate to the draining lymphoid tissues to activate T cells.7–11 However, recent studies have challenged this paradigm, suggesting that virus-specific CD8+ T cells can be primed in the spleen after respiratory influenza virus infection.12 Jenkins et al. also demonstrated a lung-dendritic cell trafficking pathway connecting the lungs with the spleen, indicating CD8+ T cells primed in the spleen and have an enhanced ability to differentiate into long-lived memory cells compared to MLN primed counterparts after influenza infection.13 Intriguingly, Lund et al. reported delayed but functional influenza-specific CD8+ T cell responses in LTα−/− mice that entirely lack lymph nodes (LNs) and Peyer’s patches and have a disrupted splenic architecture.14 These studies suggest that LNs and spleen may not be absolutely required for the generation of effective CD8+ T cell responses in the lung.
The respiratory tract presents a unique immunological challenge as it is constantly exposed to diverse pathogens, environmental particulates, and harmless antigens. Achieving a comprehensive understanding of how lung immunity maintains a delicate balance between pathogen defense and tissue inflammation remains an ongoing challenge. To date, much of our knowledge concerning lung immunity has been derived from studying respiratory infections15,16 or utilizing sub-unit vaccines that contain antigens formulated with adjuvants.17,18 However, in those scenarios, immune and inflammatory responses are simultaneously elicited, making it difficult to distinguish the signals that are necessary for eliciting immunity from the bystander pro-inflammatory signals that contribute to vaccine reactogenicity and infection pathogenicity but are unnecessary for eliciting adaptive protective immunity. As a result, the elucidation of the dynamic process of CD8+ T cell responses with minimal local or systemic inflammation proves to be a challenging yet crucial task, particularly when designing safe and effective respiratory tract-targeted vaccines.
Biomaterials have emerged as effective tools to elicit adaptive immune responses, are being designed for immunomodulatory interventions and immunotherapies, and are especially well suited for dissecting T and B cell responses.19,20 We previously reported that peptide antigens linked to self-assembling QQKFQFQFEQQ (Q11) peptide nanofibers are minimally inflammatory but are able to elicit robust T cell and antibody responses when delivered either subcutaneously or intranasally.21–23 In the present study, we use self-assembling peptide nanofibers to study how long-term CD8+ T cell responses are elicited when delivered intranasally. These nanofibers are generated by linking the major histocompatibility complex (MHC) class I-restricted epitope OVA257–264 (SIINFEKL) to Q11 peptide (termed as SIIN-Q11) and allowing these peptides to self-assemble into β sheet nanofibers that display SIINFEKL epitopes. We show that intranasally delivered SIIN-Q11 nanofibers were able to cross-prime naive CD8+ T cells and induce functional memory responses without requiring help from CD4+ T cells. Both lung CD103+ cDC1 and CD11b+ cDC2 cross-presented CD8+ T cell epitopes, albeit at different efficiencies, and Batf3-independent cDC2 were sufficient to prime and induce CD8+ T cell responses. CD8+ T cell responses were elicited in the lung before the draining MLN. Finally, transcriptomic analysis defined distinctive transcriptional responses induced by SIIN-Q11 in lung cDC1 and cDC2 that matured into a shared transcriptional response in non-antigen-presenting cDC1 and cDC2 from the MLN that was associated with exogenous antigen processing and presentation. In contrast, SIINFEKL-presenting cDC1 from the MLN downregulated genes in the MHC II presentation pathway, and, instead, upregulated genes involved in MHC I presentation. These observations expand our understanding of how immunity can be elicited in the respiratory tract with minimal inflammation.
RESULTS
Antigens appended to Q11 peptide nanofibers generate protective systemic and tissue-resident CD8+ T cell responses in the lung without requiring CD4+ T cell help
We previously showed that intranasally delivered Q11 nanofibers displaying CD8+ epitopes, but without exogenous adjuvant or CD4+ epitopes, generated lung-resident memory antigen-specific CD8+ T cell responses with minimal inflammation in the lung.23 To define the mechanisms by which Q11 nanofibers elicit memory CD8+ T cell responses, we appended the OVA257–264 (SIINFEKL) epitope to Q11 to generate SIIN-Q11 nanofibers that were used to intranasally immunize mice that lacked CD4+ T cells (CD4KO) or WT control (Figures 1A and 1B). The accumulation of endogenous SIINFEKL-specific CD8+ T cells in CD4KO mice following intranasal SIIN-Q11, and challenged on day 28 with intranasal influenza virus expressing SIINFEKL (PR8-OVA124), were quantified. Significant accumulation of SIINFEKL-specific CD8+ T cells on day 10 post SIIN-Q11 was observed in both WT and CD4KO mice and also upon challenge with intranasal PR8-OVA1 virus (day 8 post infection; Figures 1C–1E). These data indicate that endogenous memory CD8+ T cells can be elicited by SIIN-Q11 without the need for CD4+ T cell help and that the challenge with PR-OVA1 infection 28 days later induced the accumulation CD8+ T cells in the lung as a result of the recruitment of circulating and/or expansion of lung-resident memory CD8+ T cells.
Figure 1. Intranasal Q11 vaccines elicit endogenous memory CD8+ T cells capable of rapid response to viral infection.
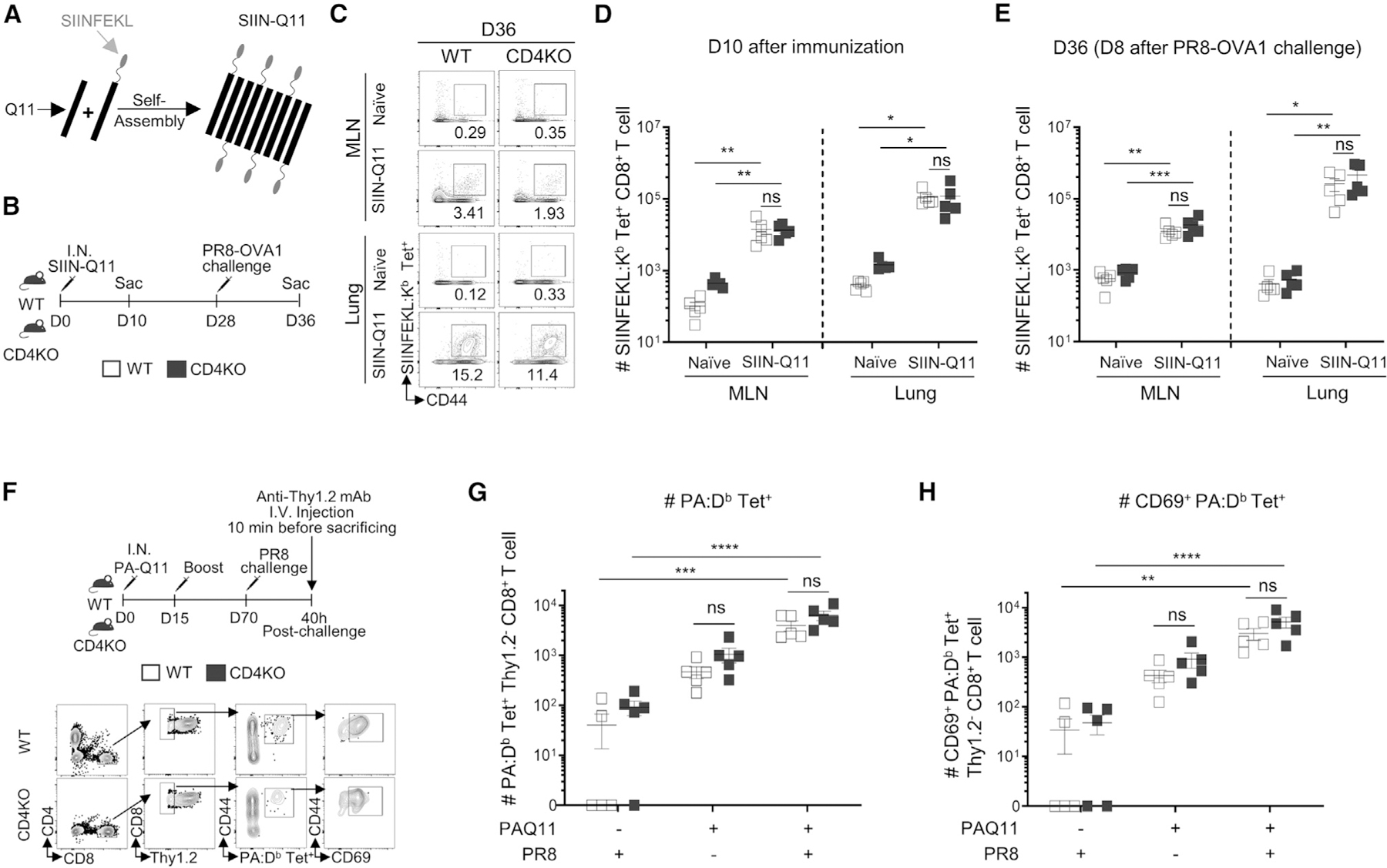
(A) Schematic of SIIN-Q11 fibers displaying SIINFEKL epitopes.
(B) Experimental design for (C)–(E). WT or CD4KO mice were immunized intranasally (i.n.) with SIIN-Q11 on day 0 and challenged with PR8 OVA1 on day 28. Lungs and mediastinal draining MLNs were collected on day 10 or 36 for evaluation of primary or secondary responses, respectively. Antigen-specific memory/effector CD8+ T cells were identified as CD8+ CD44+ SIINFEKL:H-2Kb tetramer-positive cells.
(C) Representative flow cytometry plots showing secondary antigen-specific CD8+ T cell responses in lung and draining MLN of WT vs. CD4KO mice.
(D and E) Number of tetramer+ CD8+ T cells on day 10 (D) and day 36 (E) of WT vs. CD4KO mice.
(F) Experimental design (top) for (G)–(H). WT or CD4KO mice were immunized i.n. with PA-Q11 on day 0 and boosted on day 15. Eight weeks after boost, mice were challenged with a sub-lethal dose of PR8. Lung cells were labeled by intravenous anti-Thy1.2 mAb 10 min before sacrifice at 40 h post challenge. Response of tissue-resident memory (TRM) PA-specific upon PR8 infection were analyzed with PA:Db tetramers and by flow cytometry.
(G and H) Gating strategy for the identification of TRM PA-specific CD8+ T cells (bottom). Numbers of (G) total and (H) CD69+ lung PA-specific CD8+ T cells in WT vs. CD4KO mice. Data shown are means ± SEM from two independent experiments. ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant by two-way ANOVA (D, E, G, H).
Our studies tracking OT-1 and endogenous CD8+ responses to the model antigen SIINFEKL leaves open the possibility that our findings are relevant only to the ovalbumin-derived epitope. To address this concern, we generated a Q11 nanofiber that displayed a class I-restricted epitope from influenza acid polymerase, PA224–233 (PA-Q11) and tracked the endogenous PA-specific CD8+ response using PA:Db tetramers.23 Mice were challenged and boosted with PA-Q11 on days 0 and 15 and then infected with PR8 on day 70 (Figure 1F). Extravascular CD8+ T cells from the lung were identified by intravenous administration of PE/Cyanine7-conjugated anti-Thy1.2 at 10 min before sacrifice and gating on anti-Thy1.2− CD8+ T cells. We observed a significant increase in PA:Db tetramer-binding CD8+ T cells in the lungs of PA-Q11-vaccinated compared to non-vaccinated mice at 40 h after PR8 infection and that the majority of these cells expressed CD69, which promotes lung tissue residence. This rapid increase was driven in part by the high numbers of PA-specific tissue-resident memory (TRM) cells in the lung even before PR8 challenge (Figures 1F–1H).25 Importantly, the magnitudes of the lung-resident CD8+ T cell response were comparable in WT or CD4KO mice. Taken together, these studies show that Q11 nanofibers displaying only class I epitopes were able to elicit persistent endogenous memory CD8+ responses that rapidly expand in the lung upon challenge with influenza infection, even in the absence of CD4+ T cell help. Thus, SIIN-Q11, which is composed of only an antigenic peptide appended to a self-assembling peptide scaffold, is an ideal tool for investigating how CD8+ T cell responses in the lung can be induced in the absence of local inflammation.23
Naive CD8+ OT-1 cells were primed in the lung in situ prior to draining MLN
CD8+ T cells play critical protective roles in clearing respiratory viral infections, and the kinetics of these responses that lead to the clearance of virus determines the speed at which a healthy state is restored and host survival.15 To track CD8+ T cell priming in the lung, we adoptively transferred naive CD8+ OVA-specific OT-1 cells (specific for OVA257–264 [SIINFEKL] presented by Kb) 1 day before intranasal immunization with SIIN-Q11 nanofibers (Figure 2A). We quantified OT-1 cells from the lung and draining MLN because we and others have demonstrated that intranasal Q11 or infection result in T cell priming in the MLN and not the cervical LN, the major draining LN from nasal-associated lymphoid tissues.21,26–30 By day 3 post SIIN-Q11, the number of OT-1 cells had significantly increased in the lung, whereas the increase in the MLN was not yet significant (Figures 2B–2D). By day 5 post immunization, OT-1 numbers were significantly increased both in the lung and MLN, and, by day 7 post immunization, ~1.5× more interferon (IFN) γ-producing OT-1 cells accumulated in the lung compared to MLN. Importantly, modest increases in endogenous IFNγ- and interleukin (IL)-17-producing CD8+ T cells accompanied the increase in OT-1 cells in the lung (Figure S5). These observations raise the possibility that CD8+ T cells may be independently primed in the lung and with more rapid kinetics than priming in the draining MLN.
Figure 2. SIIN-Q11 i.n. immunization results in situ priming and proliferation of naive CD8+ T cells in the lung before draining MLN.
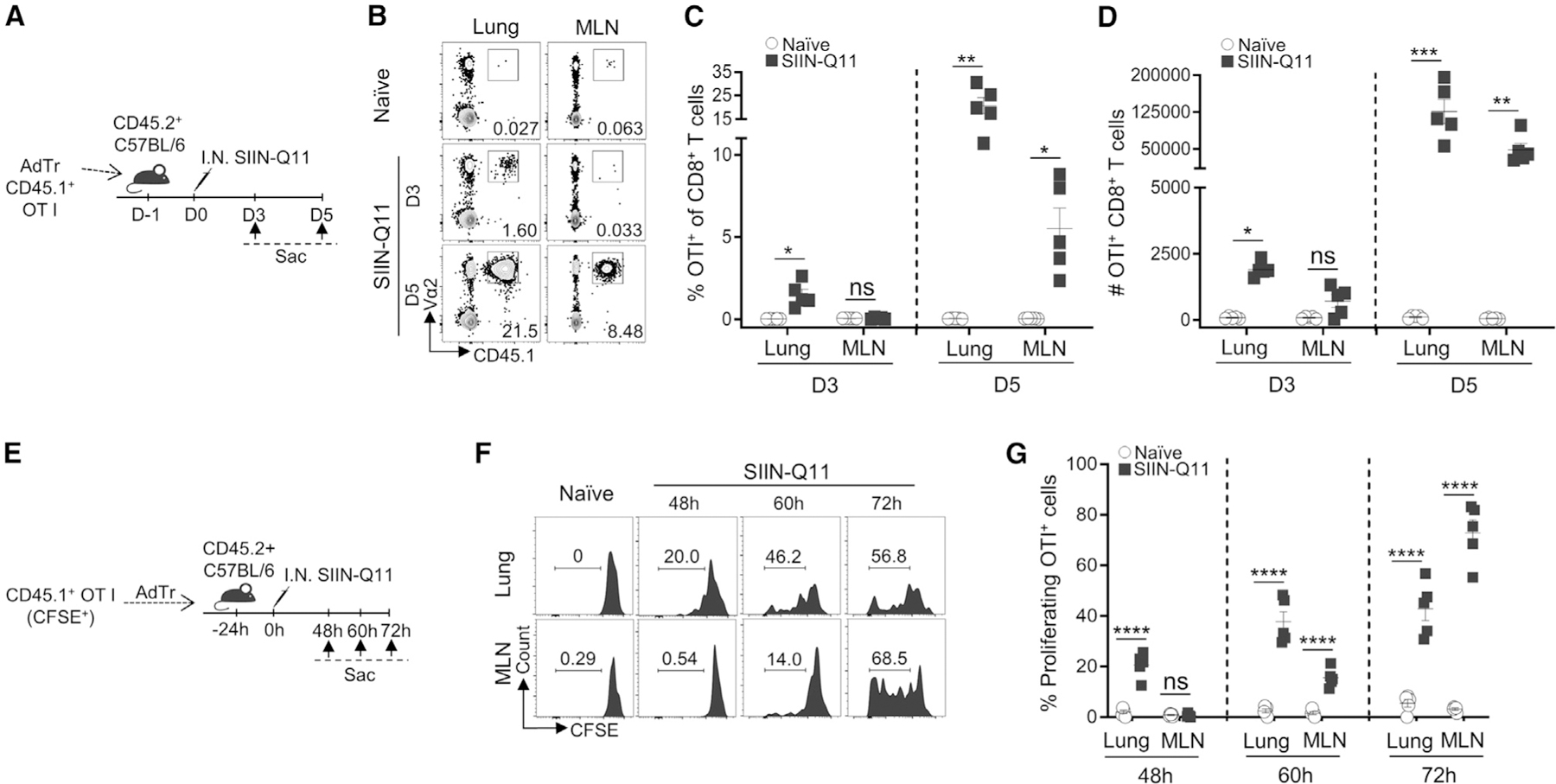
(A) Experimental design for (B)–(D). OT-1 T cells were purified from OT-1 TCR-Tg CD45.1+ mice and adoptively transferred to CD45.2+ C57BL/6 mice (50,000/mouse) at 24 h before immunization. Mice were sacrificed on day 3 (D3) or day 5 (D5) post immunization. OT-1 T cells were identified as CD45.1+Va2+CD8+ T cells. OT-1 cell accumulation in the lung vs. MLN was analyzed.
(B) Representative flow cytometry plots identifying OT-1 cells in lung (left) and MLN (right).
(C and D) (C) Percentage of OT-1 T cells of CD8+ T cells, and total number (D) in lung and draining MLN on day 3 vs. day 5 after immunization.
(E) Experimental design for (F) and (G). CD45.1+ OT-1 cells were labeled with CFSE prior to adoptive transfer to CD45.2+ C57BL/6 mice (50,000/mouse) at 24 h before immunization. Mice were sacrificed and lungs and MLNs were harvested at the indicated time points after immunization. Proliferation of adoptively transferred OT-1 cells were analyzed by flow cytometry for CFSE dilution.
(F) Representative flow cytometry plots showing CFSE dilution of OT-1 T cells in the lung (top) and MLN (bottom) at different time points.
(G) Percentage CFSE-diluted cells of OT-1 cells at 48, 60, and 72 h after immunization. Each dot represents one mouse. Data shown are means ± SEM from two independent experiments (C, D, G). ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant by two-way ANOVA (C, D, G).
To test this possibility, carboxyfluorescein succinimidyl ester (CFSE)-labeled OT-1 cells were transferred into naive mice on day −1 and challenged with SIIN-Q11 on day 0 (Figures 2E–2G). OT-1 T cells in the lung were observed to proliferate before those in the MLN, with ~20% of lung OT-1 cells having diluted CFSE vs. 0% in the MLN at 48 h post SIIN-Q11. At 60 h post immunization, 30%–48% vs. 11%–21% of OT-1 cells in the lung and MLN had diluted CFSE, whereas, at 72 h, 31%–57% vs. 55%–83% of OT-1 cells had proliferated. At all three time points tested, OT-1 cells with the entire spectrum of CFSE dilution were observed in both locations, confirming that priming of OT-1 cells had indeed occurred independently in the lung and MLN. These observations contrast with the paradigm that naive CD8+ T cells are primed in the MLN before migrating to the site of immunization or infection.31
Lung cDC1 and cDC2 acquire and present MHC class I epitope SIINFEKL after intranasal SIIN-Q11 immunization
The ability of intranasal SIIN-Q11 to prime CD8+ T cells in the lung predicted the presence of cells presenting SIINFEKL:Kb complexes in the lung. We used the SIINFEKL:Kb-specific reporter T cell hybridoma, B3Z, to confirm that lung cDCs were presenting SIINFEKL and not CD45− lung-derived cells (Figure 3A). We next used the 25.D1.16 monoclonal antibody (mAb) that recognized SIINFEKL:Kb complexes to show that low numbers of both lung cDC1 and cDC2 were displaying SIINFEKL:Kb complexes (Figures 3B–3D and S1).32 Because expression of antigen alone is not sufficient to induce T cell responses and DC activation is necessary, we next compared the expression of co-stimulatory molecules, CD80, CD86, and CD40, on lung cDC1 and cDC2 2 days after intranasal SIIN-Q11 delivery (Figures 3E–3G). Both lung DC subsets upregulated CD80 and CD40 after intranasal SIIN-Q11 compared to naive controls, whereas CD86 was significantly upregulated only in cDC2. Additionally, intranasal Q11 without appended SIINFEKL was also able to modestly upregulate co-stimulatory molecules, although less than SIIN-Q11, suggesting that interaction with CD8+ T cells further promoted DC activation.
Figure 3. Lung cDC2 and cDC1 DCs present MHC class I epitope SIINFEKL and are activated after intranasal SIIN-Q11.
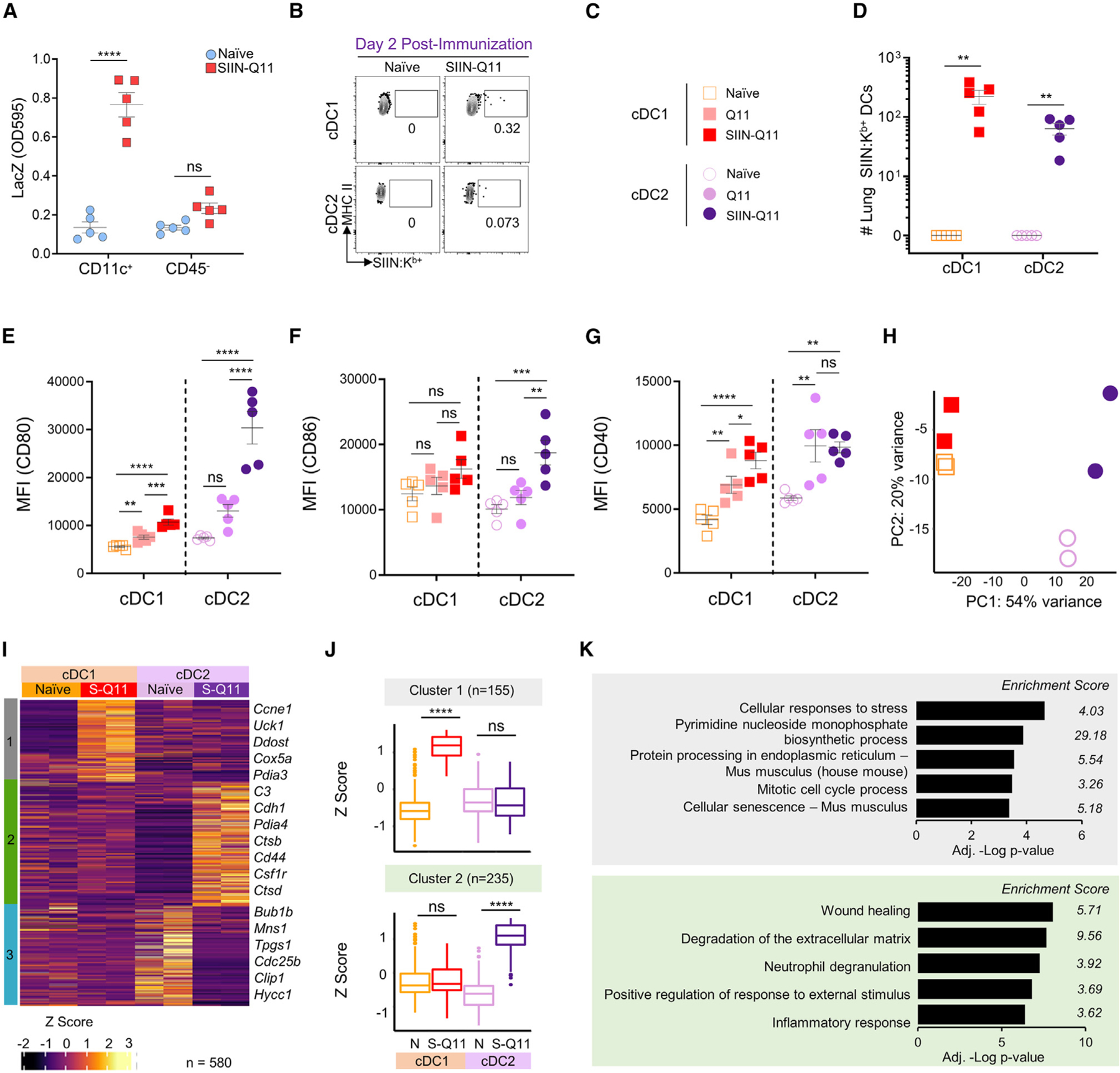
(A) Ability of CD11c+ DCs and CD45− cells enriched from the lungs of naive or SIIN-Q11 immunized mice to stimulate B3Z reporter cells. B3Z cells recognize and respond to SIINFEKL:Kb complexes with the upregulation of LacZ.
(B)Representative flow cytometry plots of lung SIINFEKL:Kb presenting cDC1 and cDC2 DCs at 2 days post intranasal SIIN-Q11 immunization. cDCs were identified as CD45+CD49b−TER119−CD19−CD3−SiglecF−Ly6G−CD11c+MHCIIhiCD64−cells, and SIINFEKL:H-2Kb-positive cDCs were detected using 25-D1.16 mAb.
(C) Groups for (D)–(H).
(D) Number of lung SIINFEKL:Kb presenting cDC1 and cDC2 DCs at day 2 post intranasal SIIN-Q11 immunization.
(E–G) CD80 (E), CD86 (F), and CD40 (G) mean fluorescence intensity (MFI) on lung cDC1 and cDC2 at 2 days post intranasal nanofibers immunization. Each dot represents the number of SIINFEKL:Kb-presenting lung DCs from one mouse. Data are means ± SEM from two independent experiments (A and D–G). ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant by unpaired two-tailed t test.
(H) PCA of RNA-seq from the four DC groups.
(I) Heatmap of DEGs and k-means clustering of DEGs based on pairwise comparison of four DC groups using DESeq2 (n = 580; adjusted p <0.01). The color key for Z score, and the three main clusters (S-Q11:SIIN-Q11).
(J) Boxplots of the Z scores of cluster 1 and 2 DEGs according to experimental groups. Significant differences in Z scores between the indicated DC groups were assessed by one-way ANOVA. ****p < 0.0001; ns, not significant.
(K) Bar charts of Metascape enrichment analysis of DEGs from clusters 1 (top) and 2 (bottom).
We next tested whether the transcriptional responses of lung cDC1 and cDC2 to intranasal SIIN-Q11 were comparable. We flow sorted lung cDC1 and cDC2 DCs at 18 h after intranasal SIIN-Q11, compared to lung DCs from naive mice, and subjected the cells to RNA sequencing (RNA-seq). Principal-component analysis (PCA) plot revealed that lung cDC1 and cDC2 were transcriptionally distinct at baseline (Figure 3H). Consistent with the upregulation of co-stimulatory molecules, SIIN-Q11 induced a more modest transcriptional change in cDC1 compared to cDC2, which exhibited extensive changes. To test whether differentially expressed genes (DEGs) induced by SIIN-Q11 were shared between lung cDC1 and cDC2, we performed pairwise comparison between all four DC subsets using DeSeq2, followed by k-means clustering analysis (Figures 3I–3K and S6). Notably, there was no clusters of shared up- or downregulated DEGs induced by SIIN-Q11 in cDC1 and cDC2. Cluster 1 DEGs were upregulated by SIIN-Q11 in cDC1 and were classified by Metascape enrichment analysis to be involved in cellular response to stress, pyrimidine monophosphate biosynthesis, protein processing in endoplasmic reticulum (ER), and cell cycle (Figures 3J, 3K, and S6C). Cluster 2 represented DEGs upregulated by SIIN-Q11 in cDC1 and were enriched for genes involved in wound healing, degradation of extracellular matrix, neutrophil degranulation, and inflammatory response (Figures 3J, 3K and S6D). Cluster 3 represented genes constitutively upregulated in cDC2 compared to cDC1 (Figures S6A and S6B). Thus, consistent with their distinct baseline transcriptional profile, lung cDC1 and cDC2 exhibited distinct transcriptional responses to intranasal SIIN-Q11, suggesting that cDC2 were not activating a cDC1 program, a possibility suggested by Bosteels et al.33
SIINFEKL-presenting cDC1 and cDC2 in the draining mediastinal MLN are migratory lung DCs
Because CD8+ T cells were also primed in the MLN, we compared the expression of co-stimulatory molecules, CD80, CD86, and CD40, on MLN cDC1 and cDC2 at 2 days after intranasal SIIN-Q11. Both DC subsets upregulated CD80, CD86, and CD40 after intranasal SIIN-Q11 compared to naive controls (Figures 4A–4D). Additionally, intranasal Q11 without appended SIINFEKL also induced a modest upregulation of CD80 and CD86 compared to SIIN-Q11, whereas CD40 was comparably upregulated. These observations were similar to those observed in lung DCs and raise the possibility that SIIN-Q11 was inducing the migration of lung DCs to the MLN.
Figure 4. SIINFEKL-presenting cDC1 and cDC2 DCs in draining MLN are migratory lung DCs.
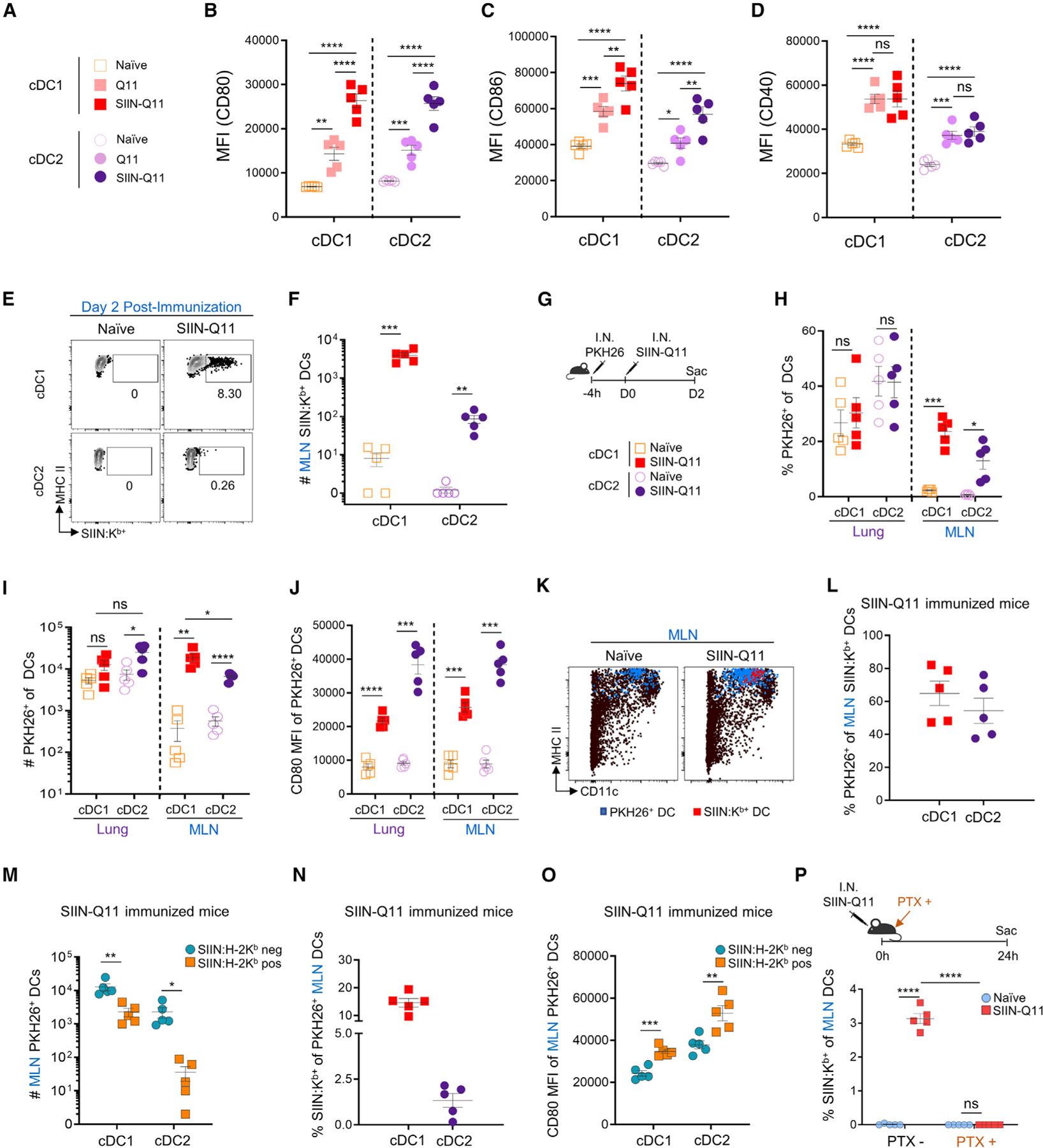
(A) Groups for (B)–(D).
(B–D) CD80 (B), CD86 (C), and CD40 (D) MFI on MLN cDC1 and cDC2 at 2 days post intranasal nanofibers immunization.
(E) Representative flow cytometry plots of cDC1 and cDC2 in draining MLN displaying SIINFEKL:H-2Kb on day 2 post-SIIN-Q11 immunization.
(F) Number of SIINFEKL:H-2Kb-positive cDC1 and cDC2 in draining MLN on day 2 after immunization.
(G) Experiment schema for tracking DC migration from lungs to MLN. Lung DCs were labeled with PKH26 4 h before SIIN-Q11 intranasal immunization (top). PKH26+ cDCs were harvested from the lung and draining MLN at day 2 after immunization. Symbols indicate experimental groups for (F), (H), (I), (J), (L) and (N) (bottom).
(H) Percentage PKH26+ cells of cDC1 and cDC2 DCs in lung and draining MLN 2 days after immunization. PKH26+ cells in draining MLN represent migrants from lung.
(I and J) (I) Number and (J) CD80 mean channel fluorescence (MFI) on PKH26+ cDC1 and cDC2 DCs from the MLN 2 days after SIIN-Q11.
(K) Representative flow cytometry plots displaying PKH26+ (blue dots) or SIINFEKL:H-2Kb-presenting (red dots) cDCs in draining MLN.
(L) Percentage PKH26+ of SIINFEKL:H-2Kb-presenting cDCs in MLN 2 days after SIIN-Q11.
(M–O) (M) Total number, (N) percentage, and (O) CD80 MFI of PKH26+ SIINFEKL:H-2Kb-presenting vs. non-presenting cDC1 and cDC2 DCs in MLN of SIIN-Q11-immunized mice.
(P) (Top) Experimental approach for inhibition of lung DC migration with pertussis toxin (PTX) treatment. C57BL/6 mice were treated with PTX immediately after SIIN-Q11 intranasal immunization, sacrificed at 24 h after immunization, and SIINFEKL-presenting cells in MLN were analyzed. (bottom) Percentage SIINFEKL:H-2Kb-presenting cDC1 and cDC2 DCs in draining MLN with or without PTX treatment. Data shown are means ± SEM from two independent experiments (B–D, F , H–J, L–P). ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant by unpaired, two-tailed t test (B–D, F , H–J, L–P).
We used the 25.D1.16 mAb to identify cells in the draining MLN displaying SIINFEKL:Kb complexes. MHC class II-expressing macrophages, monocyte-derived DCs, and B cells did not display SIINFEKL:Kb, whereas ~4,000/mouse cDC1 and ~100/mouse cDC2 from the draining MLNs were 25.D1.16 positive, consistent with cDC1 being more efficient at cross-presentation (Figures 4E and 4F).7 We next tested whether SIINFEKL-presenting DCs in the MLN were migrants from the lung, or MLN-resident DCs that captured SIIN-Q11 nanofibers draining into the MLN, or both. Intranasal PKH26 resulted in the labeling of ~17%–58% of lung DCs (Figures 4G and 4H).21,34 SIIN-Q11 induced a significant increase the percentage and total number of PKH26+ DCs in the MLN, with significantly more cDC1 reaching the MLN compared to cCD2 (Figure 4I). Finally, PKH26+ DCs reaching the MLN following SINN-Q11 immunization expressed significantly higher levels of CD80 compared to unimmunized controls (Figure 4J).
We next examined the DCs presenting SIINFEKL:Kb complexes in the MLN. Consistent with the incomplete labeling of lung DCs and migration of DCs after intranasal SIIN-Q11, 50%–65% of MLN DCs presenting SIINFEKL:Kb were positive for PKH26 (Figures 4K and 4L). Furthermore, ~10-fold higher percentage of cDC1 were presenting SIINFEKL compared to cDC2 after intranasal SIIN-Q11 (Figures 4M and 4N), and both DC subsets presenting antigen expressed significantly higher CD80 compared to their counterparts that did not (Figure 4O). Finally, we confirmed that the majority of antigen-presenting DCs in the MLN were migrants from the lung, as pertussis toxin treatment at the time of SIIN-Q11 immunization abrogated the accumulation of SIIN-FEKL:Kb-positive DCs in the draining MLN (Figure 4P).35
Intranasal SIIN-Q11 immunization induces distinct transcription signatures in antigen-presenting vs. non-antigen-presenting cDC1 from the MLN
To gain insights into the transcriptional profiles of antigen-presenting MLN cDC1 induced by SIIN-Q11, we conducted an RNA-seq analysis on flow-sorted cDC1 isolated from the MLN at 48 h after intranasal SIIN-Q11 administration compared to non-immunized controls. This time point was the earliest post SIIN-Q11 that we were able to isolate sufficient numbers of SIINFEKL-presenting (25.D1.16-positive) cDC1 for analysis. The number of antigen-presenting cDC2 remained insufficient for transcriptional analysis. Pairwise comparisons were performed followed by k-means clustering analysis to group the DEGs induced by SIIN-Q11 in antigen-presenting and non-presenting cDC1 in the MLN (Figure 5A). Cluster 1 represented genes downregulated by SIIN-Q11 in non-presenting MLN cDC1 and even more downregulated in antigen-presenting cDC1s (Figure 5B). Cluster 2 were genes upregulated in non-presenting cDC1 but downregulated in antigen-presenting cDC1 (Figure 5C), whereas cluster 3 represented genes upregulated in antigen-presenting cDC1 but downregulated in the non-presenting cDC1 (Figure 5D). Cluster 1 genes were classified as adaptive immune response and cell adhesion genes (Figures 5E and 5F). Downregulated genes in presenting cDC1s were enriched for MHC II antigen processing and presentation, cytokine production, and cell activation (Figures 5G and 5H). In contrast, genes upregulated in antigen-presenting cDC1 were involved in MHC I presentation (H2-K1, B2m, hspa8, hsp90), response to tumor necrosis factor (TNF), adaptive immune responses, and lymphocyte migration (Figures 5I and 5J).
Figure 5. Intranasal SIIN-Q11 induces distinct transcriptional profiles in antigen-presenting vs. non-presenting MLN cDC1 DCs.
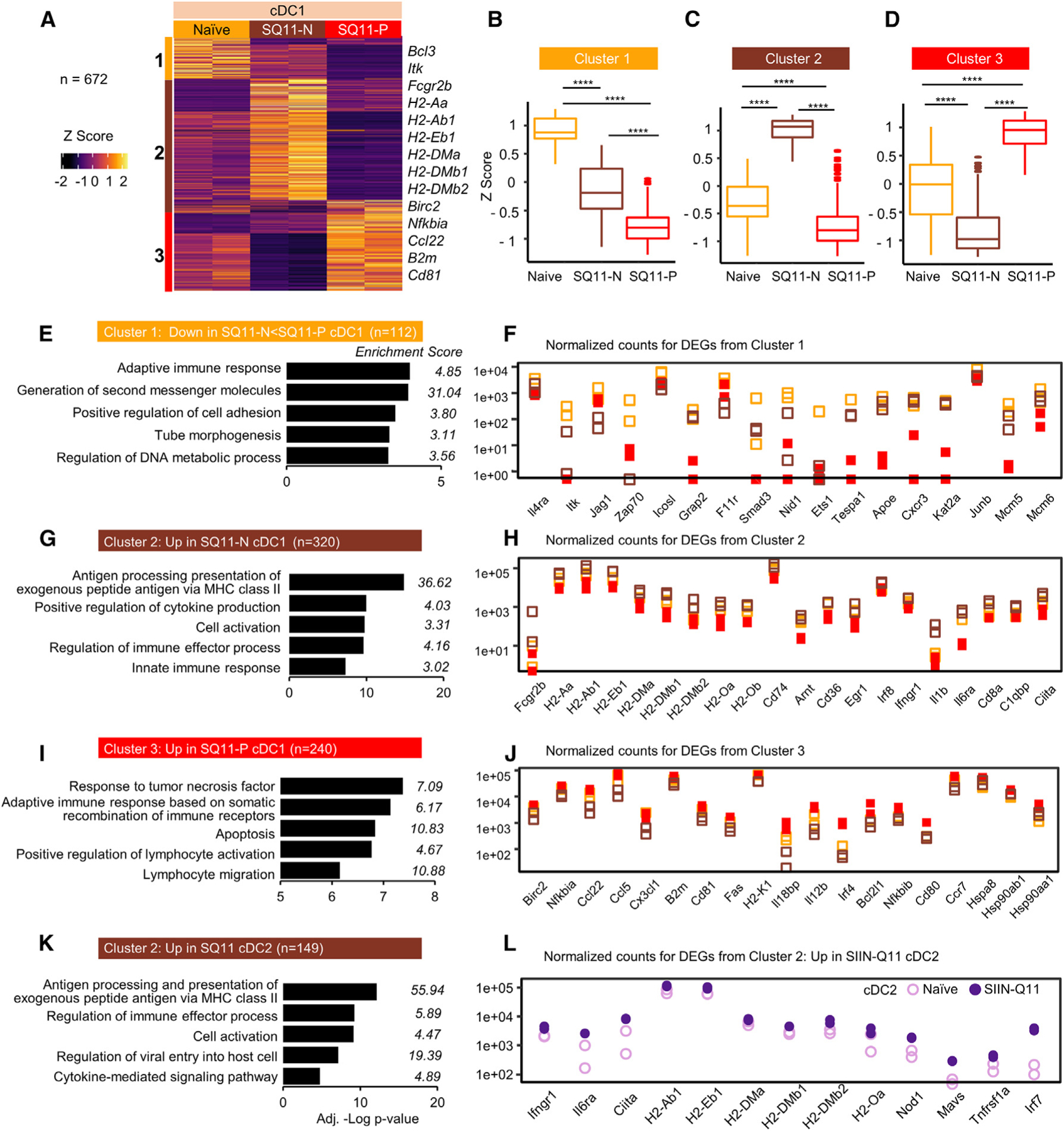
Flow-sorted SIINFEKL-presenting (SQ11-P) and non-presenting (SQ11-N) cDC1 DCs were isolated from the mediastinal MLN of SIIN-Q11 (48 h post immunization); controls were cDC1 DCs from naive mice.
(A) Heatmap and k-means clustering of DEGs based on pairwise comparison of indicated cDC1 samples (n = 672; adjusted p <0.01). Color key for Z scores, and the three k-means clusters.
(B–D) Boxplots of the Z scores of clusters 1–3 DEGs according to experimental groups. Significant differences in Z scores between the indicated DC groups were assessed by one-way ANOVA. ****p < 0.0001; ns, not significant.
(E–J) (E, G, and I) Bar charts of Metascape enrichment analysis and (F, H, and J) normalized gene counts of representative DEGs from of the DEGs from clusters 1, 2, and 3.
(K and L) Bar charts of Metascape enrichment analysis, and normalized gene counts of cluster 2 genes that were also upregulated in MLN cDC2 by SIIN-Q11.
We next queried if these clusters of genes were similarly induced in MLN cDC2 by SIIN-Q11, hypothesizing that, since ~99% of MLN cDC2 were non-presenting, they would be transcriptionally most similar to non-presenting MLN cDC1. Indeed, MLN cDC2 were transcriptionally distinct at baseline from MLN cDC1, and only 9% (10 of 112) of DEGs from cluster 1 were downregulated in both MLN non-presenting cDC1 and cDC2 following intranasal SIIN-Q11 (Figures S7A–S7C). In contrast, ~47% of cluster 2 genes (149 of 320) were upregulated in non-presenting cDC1 and cDC2 in SIIN-Q11-treated mice; strikingly, these genes were enriched for antigen processing and presentation via MHC class II (Figures 5K, 5L, and S7D). Finally, MLN cDC2 upregulated 12% (28 of 240) of DEGs upregulated by antigen-presenting cDC1; these included Hspa8, Hsp90ab1, Hsph1, and CD80 (Figure S7E). Thus, the minimally inflammatory response of the nanofibers allowed the evolution of the transcriptional response in distinct subsets of DCs to be precisely defined as they migrated from the lung to draining MLN and matured from antigen non-presenting to presenting cells.
cDC2 are sufficient for cross-priming naive OT-1 T cells after intranasal SIIN-Q11 immunization
The observation of significantly increased numbers and activated transcriptional profiles of cDC2 from the lung and MLN displaying SIINFEKL:Kb following intranasal SIIN-Q11 (Figures 3 and 4) raised the possibility that cDC2 might contribute to OT-1 priming. However, the low numbers of SIINFEKL-presenting cDC2 compared to SIINFEKL-presenting cDC1, and current literature indicating that cDC1 are necessary for the cross-presentation of exogenous antigens, prompted us to more rigorously test the sufficiency of cDC2.36,37 We interrogated OT-1 responses in Batf3KO, which are deficient in cDC1, and on days 3–5 post intranasal SIIN-Q11 since these were the earliest days that increased OT-1 numbers could be detected in the lung (day 3) or MLN (day 4) (Figures 6A and 6B). Comparable numbers of OT-1 cells recovered from SIIN-Q11 immunized WT vs. Batf3KO mice at all three time points (Figures 6C–6E). Moreover, accumulation of OT-1 cells was observed earlier in the lung than in draining MLN in both Batf3KO and WT mice after SIIN-Q11 immunization. These observations support the conclusion that cDC1 are not necessary and that cDC2 are sufficient to prime OT-1 responses following intranasal SIIN-Q11.
Figure 6. CD11b+ DCs in Baf3KO mice are sufficient for cross-priming naive OT-1 T cells after intranasal SIIN-Q11.
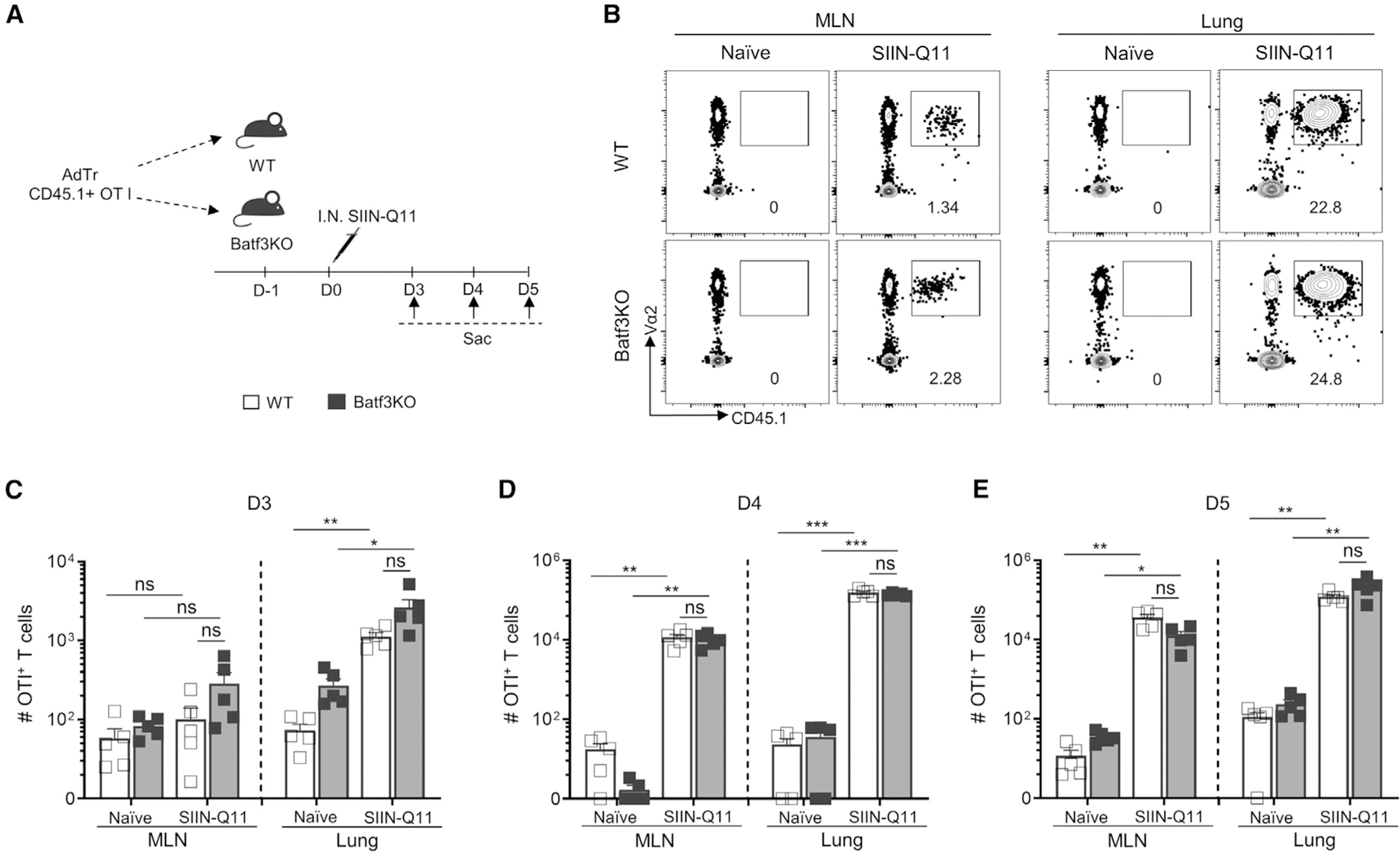
(A) Schema of experiment. CD45.1+ OT-1 T cells (50,000 cells/mouse) were adoptively transferred into WT or Batf3KO mice at 1 day before SIIN-Q11 immunization and sacrificed at indicated days post immunization. The total number of OT-1 T cells in lung and draining MLN of WT vs. Batf3KO mice was determined.
(B) Representative flow cytometry plots showing OT-1 T cells in lung and draining MLN of WT vs. Batf3KO or control mice at day 4 after immunization.
(C–E) Number of OT-1 T cells in lung and draining MLN of WT vs. Batf3KO or control mice at day 3 (C), day 4 (D), day 5 (E) after SIIN-Q11 immunization. Data shown are means ± SEM from two independent experiments. ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant by unpaired, two-tailed t test (C–E).
To more rigorously test this conclusion, whole-genome transcriptome analysis was performed to compare the quality of OT-1 activation in WT or Batf3KO mice; OT-1 cells were isolated from the lung and MLN on day 6 post intranasal SIIN-Q11 immunization (Figure 7A). PCA plots revealed that lung OT-1 cells clustered separately from MLN OT-1 cells; more importantly, there was no significant transcriptional difference between lung OT-1 cells from WT vs. Batf3KO mice (Figures 7B and 7C). Heatmap of DEGs between the five groups of OT-1 cells followed by k-means clustering identified three main clusters (Figures 7D–7F, S8, and S9). Cluster 1 identified DEGs more strongly upregulated in OT-1 cells from lung vs. MLN in SIIN-Q11 WT and Batf3KO mice; DEGs were enriched in response to virus and lymphocyte activation pathways. Cluster 2 were DEGs downregulated in lung and MLN OT-1 cells in SIIN-Q11 WT and Batf3KO mice; DEGs included lymphocyte activation and JAK-STAT signaling pathways. Cluster 3 DEGs were upregulated in lung and MLN OT-1 cells and were enriched for genes involved in leukocyte activation and cell adhesion.
Figure 7. Intranasal SIIN-Q11 induces comparable activation of OT-1 cells in Batf3KO and WT mice.
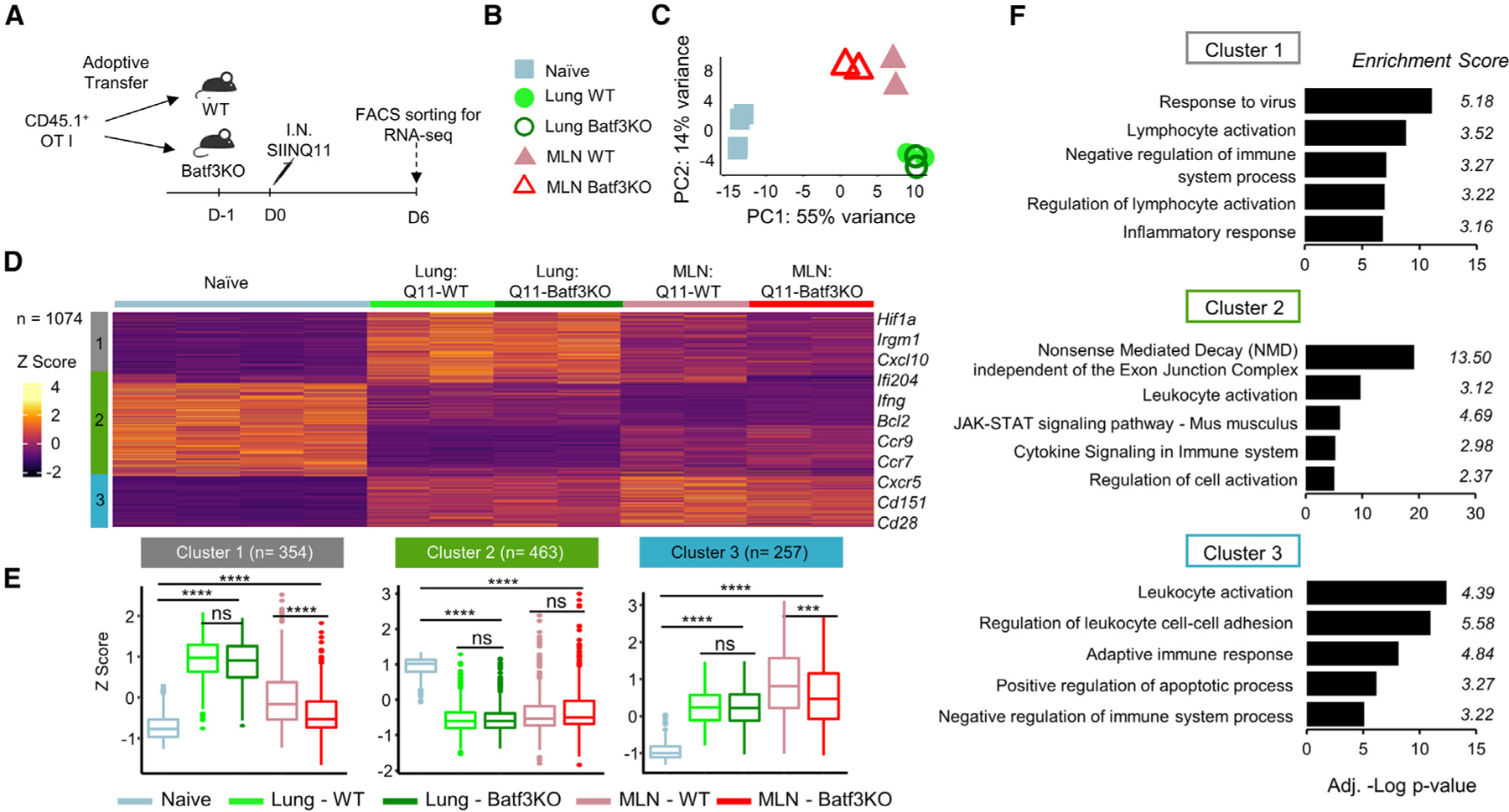
(A) Experimental schema for harvesting OT-1 cells from the lungs and draining MLN of Batf3KO and WT mice.
(B) Groups for (C).
(C) PCA plot of RNA-seq data.
(D) Heatmap and k-means clustering of DEGs based on pairwise comparison of the five OT-1 samples (n = 1,074; p < 0.01). Color key for Z score and the three main gene clusters.
(E) Boxplots of the Z scores of clusters 1–3 DEGs in the OT-1 experimental groups. Significant differences in Z scores between the indicated OT-1 groups were assessed by one-way ANOVA. ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant.
(F) Bar charts of Metascape pathway enrichment analysis of DEGs from clusters 1–3.
DEGs in clusters 1 and 3 were statistically significantly different in the MLN OT-1 cells from WT vs. Batf3KO mice. To identify these genes, a volcano plot performed to compare the global transcriptomes identified a total of 96 DEGs; 65 (67.6%) genes were upregulated in MLN OT-1 cells from WT compared to Batf3KO mice, and these DEGs were enriched for cell division and adaptive immune system (Figure S10). Collectively, our data demonstrated that cDC2 alone are sufficient for cross-priming naive OT-1 T cells, and the accumulation and overall transcriptomic response of lung OT-1 cells to intranasal SIIN-Q11 were comparable between WT and Batf3KO mice, whereas a slightly more optimal priming was observed in MLN OT1 cells from WT mice.
DISCUSSION
The inability of current vaccines to provide long-term protection against respiratory viral infections highlights the need for improving and diversifying vaccine options. Experimental nasal vaccines have recently received particular attention, with preclinical studies confirming their superiority at eliciting immunity against SARS-CoV-2 by facilitating protective immunity in the lung.38–40 Nasal-associated lymphoid tissue (NALT) is located in the nasal cavity and may be involved in generation of CD8+ T cell responses following intranasal vaccination. However, studies have shown that NALTs are stimulated by intranasal vaccination but fail to support naive T cell priming following both vaccination with live attenuated influenza virus (LAIV) and infection with a pathogenic influenza strain.30 We previously showed that the draining LN following intranasal administration of Q11 nanofibers bearing MHCII-restricted epitope (Ea) resulted in significant CD4+ T cell responses in the lung and MLN but minimal responses in the NALT.21 Those studies also documented the appearance of antigen-presenting DCs in the lung one day after Q11 delivery and in the MLN 2 days after intranasal Q11 displaying MHC II-restricted antigens, consistent with lung DCs migrating directly to the MLN. In the present study, we not only observed significant accumulation of SIINFEKL-specific CD8+ T cells (OTI cells) in the lung earlier than in the MLN but also detected OTI cells proliferation in the lung at 48 h post intranasal OVA-Q11 delivery, followed by detection in the MLN 12 h later. These data demonstrate that T cell priming occurs in the lung in situ prior to draining LNs.
While MLN cDC1 are the major subset cross-presenting SIINFEKL after intranasal SIIN-Q11 immunization, low numbers of cDC1 in the lung, as well as cDC2 in the lung and MLN, were capable of presenting SIINFEKL:Kb. Comparable accumulation of OT-1 cells in Batf3KO vs. WT mice in the lung and MLN as early as day 3–5 after SIIN-Q11 administration confirmed that cDC1 was not the only subset capable of cross-priming CD8+ T cells and that cDC2 were also capable and sufficient. Furthermore, global transcriptional analysis determined that the quality of lung OT-1 activation in the presence vs. absence of cDC2 was comparable, and only modest transcriptional differences toward reduced priming of MLN OT-1 cells were detected in Batf3KO mice. Whether these differences result in functionally distinct effector or memory CD8+ T cells requires further investigation. Furthermore, cDC2 exhibiting an IFN-stimulated transcription profile has been reported to acquire MHC I complexes by cross-decoration41–43; however, this is unlikely to be an explanation for our observations with Batf3KO mice since the main source of SIINFEKL:Kb complexes following intranasal SIIN-Q11 would have to be from cells capable of cross-presentation, namely cDC1 (Figure S4). Based on these observations, we speculate that redundancy in the types of APCs mediating CD8+ T cell priming may be a unique feature of the respiratory mucosa being a major site for pathogen invasion and thus requiring vigilant immune surveillance. Indeed, lung DCs have been reported to be capable of cross-presentation and stimulating CD8+ T cells, depending on their mode of activation.44,45
A unique feature of the immunogenicity of nanofibers is that it predominantly activates the DCs that had internalized the nanofiber, with minimal effects on DCs that did not.21,46 This novel feature of Ag-Q11 allows us to more precisely define the transcriptomes in DCs associated with immunogenicity compared to classical adjuvants or infections that are broadly inflammatory.47 We showed that the transcriptional responses induced in each lung cDC subset to intranasal SIIN-Q11 were distinct, consistent with disparate baseline transcriptome. Genes involved in metabolic and cell cycle processes were upregulated in lung cDC1, while those involved in migration and inflammatory responses were upregulated in cDC2. Consistent with the priming of CD8+ T cells in the lung, MHC I (H2-D1) and genes encoding the peptide chaperones of MHC I antigen presentation, namely hsp90aa1, hsp90ab1, and hspa8(Hsp70), were upregulated.48–50 These observations together with the increased expression of co-stimulatory molecules, CD80, CD86, and CD40 are consistent with the ability of lung DCs to prime CD8+ T cells in situ following intranasal SIIN-Q11.
In contrast, to the distinct transcriptomes elicited by SIIN-Q11 in lung cDC1 and cDC2, as well as the distinct baseline differences in transcriptomes between MLN cDC1 and cDC2, non-presenting MLN cDC1 and cDC2 upregulated a shared set of 149 DEGs following intranasal SIIN-Q11. Of note, upregulated DEGs were involved in MHC II complex assembly and presentation, namely Ciita, H2-Ab1, H2-Eb1, H2-DMa, H2-DMb1, H2-DMb2, H2-Oa, H-2Ob, and CD74, as well as in cytokine responsiveness, Ifngr1 and Il6ra. Furthermore, we defined the transcriptomes of antigen-presenting vs. non-presenting cDC1 in the MLN. The most striking observation was the downregulation in antigen-presenting cDC1 of genes involved in MHC II complex assembly and upregulation of genes involved in MHC I antigen presentation (H-2K1, B2m, hsp90aa1, hsp90ab1, and hspa8).48–52 These observations indicate that the maturation of cDC1 toward CD8+ T cell priming is associated with a broad downregulation of MHC II and upregulation of MHC I presentation. Further investigation is required to determine if these differences reflect differential kinetics in cDC1 activation driven by the processing of SIIN-Q11 and presentation of SIINFEKL peptide or in response to additional signals provided by cognate interaction with CD8+ T cells. Support for the latter possibility comes from observations that lung and MLN DCs expressed higher levels of CD80, CD86, and/or CD40 when stimulated with SIIN-Q11 compared to Q11. It also remains to be tested whether the downregulation of MHC II processing is avoided in DCs that are co-presenting CD4+ and CD8+ T cell epitopes. Finally, antigen-presenting cDC1 upregulated transcripts for chemokine ligands and receptors, Ccl22 Ccl5, Ccr7, and Cx3cl1, which guide DC migration from the lung to MLN or are involved in recruiting CXCR1-expressing CD8+ T cells.27,53,54 These data provide new insights into the activation of cDC1 poised for antigen cross-presentation beyond increased expression of co-stimulatory molecules, CD80, CD86, and CD40.
Respiratory infections are advantaged by multiple modes of transmission through direct or indirect contact (fomite), droplets and aerosols, and a large area of infectivity,55–57 and both humoral and cellular immunity are required to clear infection and limit morbidity and mortality.58 In this study, we show that antigen-displaying nanofibers raise long-lived memory CD8+ T cell responses by inducing the in situ priming of CD8+ T cells in the lung and subsequently in the MLN. We speculate that lung-primed T cells may contribute to the accumulation of endogenous CD8+ T cells with the capacity for IFNγ and IL-17 production in the lung at day 7 post SIIN-Q11. Both cDC1 and cDC2 were capable and sufficient of cross-priming CD8+ T cells. While distinct transcriptional responses are induced in lung cDC1 and cDC2 by intranasally delivered nanofibers, these DCs acquired shared transcriptional responses upon migration into the LN, with the antigen-presenting cDC1 uniquely downregulating MHC class II and upregulating class I genes, consistent with a specialization toward cognate CD8+ T cell priming. In conclusion, our study reveals novel insights into the dynamics of CD8+ T cell priming in the lung and the contribution of different DC subsets. The ability of antigen-displaying nanofibers to elicit long-lived memory CD8+ T cell responses through in situ priming in the lung has significant implications for the development of respiratory vaccines. These findings may be beneficial for understanding how respiratory infections can be overcome and facilitating a faster return to health. Further research in this area has the potential to advance the field of vaccine development and enhance our understanding of respiratory immune responses.
Limitations of the study
As the respiratory tract is constantly exposed to a wide array of pathogens and environmental factors, redundancy in the types of APCs cross-priming CD8+ T cells may be unique to the lung. We speculate that naive CD8+ T cells priming in the lung prior to LN may be advantageous for eliciting a more rapid and localized immunity to respiratory infections. However, the precise mechanisms mediating naive CD8+ T cell cross-priming in the lung versus the LN, and whether cDC1 and cDC2 in each of the locations differentially activate CD8+ T cells, remain to be clarified. Finally, additional studies are needed to comprehensively test whether these processes identified in this study apply to all inhaled antigens or are unique to antigens appended to nanofibers.
STAR✰METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Anita S Chong (achong@surgery.bsd.uchicago.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data is available in the main text or the supplementary materials. The RNA-seq data has been uploaded to the GEO repository (GEO: GSE216097, GSE216098, GSE216099). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
C57BL/6 mice were purchased from Harlan-Envigo Laboratories. Batf3KO mice (B6.129S(C)-Batf3tm1Kmm/J) and CD4KO mice (B6.129S2-Cd4tm1Mak/J) were purchased from Jackson Laboratories. OT-1 TCR-Tg Rag1 knockout CD45.1 mice were a gift from the Huang Lab at the University of Chicago. Both male and female mice were used at 6–8 weeks old. All mice were housed under specific pathogen-free conditions in the animal facilities at the University of Chicago. All procedures performed were approved by the Institutional Animal Care and Use Committees of the University of Chicago.
METHOD DETAILS
Peptides and nanofiber preparation
Q11 (Ac-QQKFQFQFEQQ-CONH2) and SIIN-Q11 (Ac-SIINFEKL-GGAAY-QQKFQFQFEQQ-CONH2) were synthesized by the Collier Lab, using standard Fmoc solid-phase chemistry, purified by high-performance liquid chromatography (HPLC) and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS), and lypohilized as previously reported.59 To prepare the nanofibers, lyophilized peptides were weighed and intermixed as dry powders by vortexing for 30 min. Ultrapure water was added to form 8 mM total peptide solutions. After overnight incubation at 4°C, the peptide solution was diluted to 2 mM in 1× phosphate-buffered saline (PBS) by adding ultrapure water (Invitrogen) and sterile 10× PBS (Fisher Scientific) and then incubated at room temperature for 3 h.
Intranasal administration
For intranasal immunizations, mice were anesthetized intraperitoneally with ketamine (Par Pharmaceutical, Inc) and xylazine (Lloyd Laboratories), and 40 μL of vaccine formulations (2 mM SIIN-Q11) was slowly administered to one nostril using a micropipette.23 For influenza virus challenge, anesthetized mice were challenged with a low dose of PR8 or PR8-OVA1 viruses, which were kindly provided by David J. Topham, University of Rochester, by i.n. administration of 40 μL virus suspension diluted in PBS.24,46
Adoptive transfer of OT-1 T cells
Spleen and MLNs were collected from OT-1 TCR-Tg Rag1 knockout CD45.1 mice. OT-1 T cells were isolated using a CD8+ T cell Isolation kit (Miltenyi Biotec) according to the manufacturer’s protocol. The purity of OT-1 T cells was confirmed to be >90%, which was evaluated by flow cytometry using anti-Vα2 TCR (BD Biosciences) and anti-CD45.1 (BD Biosciences). 50,000 OT-1 T cells in 200 μL PBS were injected into the tail vein of each recipient mouse 1 day before immunization. In some experiments, OT-1 T cells were labeled with CFSE (Invitrogen) prior to the adoptive transfer. Mice were sacrificed at indicated time points, single-cell suspensions were obtained from lung and MLNs for flow cytometry.
In vivo labeling of lung DCs
Lung DCs were labeled in vivo by intranasal delivery of 40 μL of 1:100 diluted PKH26 dye (Sigma-Aldrich) at 4 h before SIIN-Q11 intranasal immunization.
Pertussis toxin (PTX) treatment
Lyophilized pertussis toxin (PTX) from Bordetella pertussis (Sigma-Aldrich) was reconstituted to a concentration of 10 μg/mL. Mice were treated with intranasal injection of 40 μL and intravenous injection of 200 μL of PTX. Lungs and MLNs were collected at indicated time for FACS analysis.
Detection of SIINFEKL presentation by B3Z T cells
8-week-old female C57BL/6 mice were intranasally immunized with SIIN-Q11. Single-cell suspensions were obtained from lungs after perfusion on day 2 post-immunization. CD11c+ and CD45− cells were isolated using CD11c MicroBeads UltraPure or CD45 MicroBeads respectively, according to the manufacturer’s instructions (Miltenyi Biotec). Subsequently, 1×105 CD11c+ or CD45− cells were co-cultured with 5 × 105 B3Z T cellT-cell hybridoma, which specifically recognizes OVA257–264 (SIINFEKL) in the context of H-2Kb and produce β-galactosidase upon recognition of SIINFEKL presented in MHC-I. B3Z was a kind gift from Dr. H. Schreiber (University of Chicago) with permission from Dr. N. Shastri (University of California, Berkeley, CA).60 Following an 18 h incubation at 37°C, cells were lysed in a buffer containing 9 mM MgCl2, 0.13% Nonidet P-40, 0.15 mM Chlorophenol red-β-D-galactopyranoside.61 After incubation for 4 h at 37°C, absorbances at 595 nm and 615 nm were measured.
Cell preparation and flow cytometry
Mice were sacrificed at the indicated time after immunization. In some experiments, mice were injected intravenously with 3 μg PE/Cyanine7-conjugated anti-Thy1.2 antibody (Thermo Fisher) 10 min before sacrifice to distinguish circulating T cells from tissue-localized T cells. Lungs were collected and perfused with 20 mL DPBS through the right ventricle. Each lung was cut into small pieces with scissors followed by digestion using Collagenase (Sigma-Aldrich) and Deoxyribonuclease I (Worthington Biochemicals) for 1h in 10mL DMEM with 5% FBS with slow tilt rotation. After enzymatic digestion, the tissue was further dissociated by repeated pipetting of digested lung tissue adequately and filtered through a 100-μm filter strainer to obtain single-cell suspensions. Lymph node capsule was disrupted and digested as described above. Red blood cells (RBCs) were removed with ACK lysis buffer followed by washing with PBS twice. Cells were then counted and stained with the Zombie NIR Fixable Viability Kit (BioLegend), and blocked with TruStain FcX PLUS (anti-mouse CD16/32) (BioLegend) to exclude dead cells and to prevent unspecific binding. For cell surface staining, cells were then stained with fluorophore-conjugated antibodies listed in Table S1 at 4°C for 30 min.
To detect IFN-γ, IL-4 and IL-17A production, cells were cultured in the presence of PMA (50 ng/mL; Sigma-Aldrich), ionomycin (750 ng/mL; Sigma-Aldrich), and brefeldin A (5 μg/mL; Sigma-Aldrich) for 3 h. After surface staining with the corresponding cocktail of antibodies, cells were fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) for 30 min at room temperature, followed by two washes with staining buffer (PBS +2% fetal calf serum). Cells were then washed twice with Permeabilization Wash Buffer and incubated with anti-IFN-γ, anti-IL-4, and anti-IL-17A overnight at 4°C. Flow cytometry was performed on Cytek Aurora, BD LSRFortessa X-20, or BD LSRII and analyzed with FlowJo software (BD Biosciences).
Cell sorting and RNA-Seq
Sorting was performed on BD FACSAria III (BD Biosciences) (100 mm nozzle) at the University of Chicago flow cytometry core. Target subsets were sorted as follows: CD11b+ DCs (CD49b−TER119−CD19−CD3− SiglecF−CD11c+MHCIIhiCD64−CD11b+CD103−); CD103+SIINFEKL non-presenting DCs (CD49b−TER119 −CD19−CD3−SiglecF−CD11c+MHCIIhiCD64−CD11b−CD103+SIIN: H-2Kb−); CD103+SIINFEKL presenting DCs (CD49b−TER119−CD19−CD3−SiglecF−CD11c+MHCIIhiCD64−CD11b−CD103+SIIN: H-2Kb+); OT-1 cells (CD49b−TER119−CD19−CD3+CD4−CD8+CD45.1+Vα2+ T cells). Live cells were gated and sorted into 96-well plates (Thomas Scientific) (100 cells per well) containing 4 μL of the catching solution at 4°C. Sequencing libraries were generated using Nextera XT DNA Library Prep Kit and Nextera XT Index Kit (Illumina) according to the SmartSeq2 protocol (Detailed information see the reference)62. Sequencing and initial raw data analyses were processed by Novogene Corporation Inc. FASTQ files were aligned using STAR 2.5.2a against the mm10 mouse reference genome. The aligned files were processed using PORT gene-based normalization (https://github.com/itmat/Normalization). Differential gene expression was performed using DESeq2 package (Andres et al., Genome Biology 2010; 11(10): R106) in R to identify transcripts that were significantly (adjusted p value of <0.05) differentially expressed between experimental groups, and ordered by k-means clustering. Metascape pathway enrichment analysis for each cluster was conducted comparing the top 100 genes that were differentially expressed (Table S2).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as means ± SEM. Statistical analyses were performed using GraphPad Prism software. Differences between groups were analyzed using one-way, two-way ANOVA, or Student’s t test as indicated in the figure legend.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rat Anti-Mouse CD49b (BV421); Clone DX5 | BD Biosciences | Cat# 563063; RRID: AB_2737983 |
| Rat Anti-Mouse TER-119 (BV421); Clone: TER-119 | BD Biosciences | Cat# 563998; RRID:AB_2738534 |
| Rat Anti-Mouse CD19 (BV421); Clone 1D3 | BD Biosciences | Cat# 562701; RRID: AB_2737731 |
| Rat Anti-Mouse CD88 (BV711); Clone 20/70 | BD Biosciences | Cat# 743773; RRID: AB_2741741 |
| Rat Anti-Mouse Siglec-F (BV650); Clone E50-2440 | BD Biosciences | Cat# 740557; RRID: AB_2740258 |
| Rat Anti-Mouse Siglec-F (PE); Clone E50-2440 | BD Biosciences | Cat# 562068; RRID: AB_394341 |
| Rat Anti-Mouse CD44 (APC-R700); Clone IM7 | BD Biosciences | Cat# 565480; RRID: AB_2739259 |
| Rat Anti-Mouse Vα2 TCR (PE); Clone B20.1 | BD Biosciences | Cat# 553289; RRID: AB_394760 |
| Rat Anti-Mouse IL-4 (PerCP-Cy™5.5); Clone 11B11 | BD Biosciences | Cat# 560700; RRID: AB_1727549 |
| Rat Anti-Mouse IFN-γ (APC); Clone XMG1.2 | BD Biosciences | Cat# 554413; RRID: AB_398551 |
| Rat Anti-Mouse IL-17A (BV510); Clone TC11-18H10 | BD Biosciences | Cat# 564168; RRID: AB_2738639 |
| Rat Anti-Mouse CD103 (APC-R700); Clone M290 | BD Biosciences | Cat# 565529; RRID: AB_2739282 |
| Rat Anti-Mouse Siglec-F (BV711); Clone E50-2440 | BD Biosciences | Cat# 740764; RRID: AB_2740427 |
| Ly-6G Monoclonal (PerCP-eFluor 710); Clone 1A8-Ly6g | eBioscience | Cat# 46-9668-82; RRID: AB_2573893 |
| anti-mouse CD45 (PE/Cyanine7); Clone 30-F11 | BioLegend | Cat# 103113; RRID: AB_312978 |
| anti-mouse CD45.1 (APC/Cyanine7); Clone A20 | BioLegend | Cat# 110715; RRID: AB_313504 |
| anti-mouse CD64 (APC); Clone X54-5/7.1 | BioLegend | Cat# 139305; RRID: AB_11219205 |
| anti-mouse CD11c (PE/Cyanine7); Clone N418 | BioLegend | Cat# 117317; RRID: AB_493569 |
| anti-mouse/human CD11b (BV605); Clone M1/70 | BioLegend | Cat# 101237; RRID: AB_11126744 |
| anti-mouse H-2Kb bound to SIINFEKL (APC); Clone 25-D1.16 | BioLegend | Cat# 141605; RRID: AB_11219402 |
| anti-mouse/human CD44 (PerCP/Cyanine5.5); Clone IM7 | BioLegend | Cat# 103031; RRID: AB_2076206 |
| anti-mouse I-A/I-E (BV510); Clone M5/114.15.2 | BioLegend | Cat# 107635; RRID: AB_2561397 |
| anti-mouse CD80 (PE/Dazzle 594); Clone 16-10A1 | BioLegend | Cat# 104737; RRID: AB_2564174 |
| anti-mouse CD4 (PE/Cyanine7); Clone RM4-5 | BioLegend | Cat# 100527; RRID: AB_312728 |
| anti-mouse CD8a (BV510); Clone 53-6.7 | BioLegend | Cat# 100751; RRID: AB_2561389 |
| anti-mouse CD3ε (BV421); Clone 145-2C11 | BioLegend | Cat# 100335; RRID: AB_10898314 |
| anti-mouse CD3ε (PE/Dazzle 594); Clone 145-2C11 | BioLegend | Cat# 100347; RRID: AB_2564028 |
| anti-Mouse CD90.2 (Thy-1.2) (PE-Cyanine7); Clone 53-2.1 | eBioscience | Cat# 25-0902-82; RRID: AB_469642 |
| TruStain FcX™ PLUS (anti-mouse CD16/32); Clone S17011E | BioLegend | Cat# 156603; RRID: AB_2783137 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Influenza A/Puerto Rico/8/1934 H1N1 (PR8) | Topham Lab, University of Rochester | N/A |
| PR8-OVA1 | Topham Lab, University of Rochester | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Q11 (Ac-QQKFQFQFEQQ-CONH2) | Collier Lab, Duke University | N/A |
| SIIN-Q11 (Ac-SIINFEKL-GGAAY-QQKFQFQFEQQ-CONH2) | Collier Lab, Duke University | N/A |
| UltraPure DNase/RNase-Free Distilled Water | Invitrogen | Cat# 10977015 |
| PBS (10X) | Fisher Scientific | Cat# BP399-500 |
| ACK lysis buffer | Quality Biological | Cat# 118-156-101 |
| MgCl2 (1 M) | Invitrogen | Cat# AM9530G |
| Nonidet-P40 | UNITED STATES BIOLOGICAL INC | Cat# N3500-100mL |
| CPRG Chlorophenol red-β-D-galactopyranoside | Roche Diagnostics | Cat# 10884308001 |
| PMA | Sigma-Aldrich | Cat# P1585-1MG |
| Ionomycin | Sigma-Aldrich | Cat# I3909-1ML |
| Brefeldin A | Sigma-Aldrich | Cat# B6542-5MG |
| PKH26 Red Fluorescent Cell Linker Mini Kit | Sigma-Aldrich | Cat# MINI26-1KT |
| Pertussis toxin from Bordetella pertussis | Sigma-Aldrich | Cat# P7208-50UG |
| Collagenase | Sigma-Aldrich Cat# | C5138-1G-PW |
| DNase I (Deoxyribonuclease I) | WORTHINGTON BIOCHEMICAL CORP | CAT# LS002138 |
| ODN 1826 (TYPE B) ENDOTO 1 MG | Fisher Scientific | Cat# 50596710 |
| Ketamine | Par Pharmaceutical, Inc | NDC:42023-113-10 |
| Xylazine | Lloyd Laboratories | NADA #139-236 |
| Random Access 96 well Skirted PCR Plate | Thomas Scientific | Cat# 4ti-0960/RA |
| DPBS | Genesee Scientific | Cat #: 25-508 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Zombie NIR™ Fixable Viability Kit | BioLegend | Cat# 423105 |
| UltraComp eBeads™ Compensation Beads | Invitrogen | Cat# 501129040 |
| CellTrace CFSE Cell Proliferation Kit | Invitrogen | Cat# C34554 |
| Foxp3/Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523 |
| LS Columns | Miltenyi Biotec | Cat# 130-042-401 |
| CD11c MicroBeads UltraPure, mouse | Miltenyi Biotec | Cat# 130-125-835 |
| CD45 MicroBeads, mouse | Miltenyi Biotec | Cat# 130-052-301 |
| CD8a+ T cell Isolation Kit, mouse | Miltenyi Biotec | Cat# 130-104-075 |
| Nextera XT DNA Library Prep Kit | Illumina | Cat# FC-131-1024 |
| Nextera XT Index Kit (24 indexes, 96 samples) | Illumina | Cat# FC-131-1001 |
| HiSeq PE150 lane sequencing | Novogene Corporation Inc | N/A |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| B3Z T cellT-cell hybridomas | Hans Lab, The University of Chicago | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6 | Harlan - Envigo | 71236(6) HSD |
| Mouse: Batf3−/− (B6.129S(C)-Batf3tm1Kmm/J) | Jackson Laboratory | Stock# 013755 |
| Mouse: CD4KO (B6.129S2-Cd4tm1Mak/J) | Jackson Laboratory | Stock# 002663 |
| Mouse: OT-1 TCR-Tg Rag1 knockout CD45.1 | Huang Lab, The University of Chicago | N/A |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq data | This paper | GEO: GSE216097, GSE216098, GSE216099 |
| Software and algorithms | ||
|
| ||
| FlowJo v10.5.3 | FlowJo, LLC | https://www.flowjo.com/ |
| Prism 8 | GraphPad | https://www.graphpad.com/ |
Highlights.
Intranasal nanofiber immunization cross-primes naive CD8+ T cells in the lung before LN
cDC2 cross-prime CD8+ T cells in Batf3KO mice with comparable transcriptomes to WT
Lung cDC1 and cDC2 show distinct transcription profiles post intranasal immunization
cDC1 and cDC2 acquire shared antigen processing transcriptional response in the LN
ACKNOWLEDGMENTS
We thank Flow Cytometry Core (UCFlow) and Animal Resources Center (ARC) at the University of Chicago for instruments and services, the NIH Tetramer Core Facility for the synthesis of class I tetramers, David J. Topham (University of Rochester) for the gift of PR8 and PR8-OVA1 viruses, Jun Huang (University of Chicago) for OT-1 TCR-Tg mice, H. Schreiber (University of Chicago), and N. Shastri (University of California, Berkeley, CA) for the B3Z cell line. This study was funded in part by grants from the NIH (NIAID: 75N93019C00041-0-9999-1 NIBIB 5R01EB009701; NIAID 5R01AI118182).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113299.
DECLARATION OF INTERESTS
J.H.C. is listed as an inventor on US Patent no. 9,241,987, which is associated with the technology described. The patent covers the design and use of supramolecular peptide-polymer conjugates. J.H.C. is an inventor on a patent related to this work filed by USPTO (US Patent 10,596,238, published on 24 March 2020).
REFERENCES
- 1.Kedzierska K, and Thomas PG (2022). Count on us: T cells in SARS-CoV-2 infection and vaccination. Cell Rep. Med 3, 100562. 10.1016/j.xcrm.2022.100562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, Rawlings SA, Sutherland A, Premkumar L, Jadi RS, et al. (2020). Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 181, 1489–1501.e15. 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertoletti A, Le Bert N, Qui M, and Tan AT (2021). SARS-CoV-2-specific T cells in infection and vaccination. Cell. Mol. Immunol 18, 2307– 2312. 10.1038/s41423-021-00743-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhuang Z, Lai X, Sun J, Chen Z, Zhang Z, Dai J, Liu D, Li Y, Li F, Wang Y, et al. (2021). Mapping and role of T cell response in SARS-CoV-2-infected mice. J. Exp. Med 218, e20202187. 10.1084/jem.20202187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng Y, Felce SL, Dong D, Penkava F, Mentzer AJ, Yao X, Liu G, Yin Z, Chen J-L, Lu Y, et al. (2022). An immunodominant NP105-113-B*07:02 cytotoxic T cell response controls viral replication and is associated with less severe COVID-19 disease. Nat. Immunol 23, 50–61. 10.1038/s41590-021-01084-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bange EM, Han NA, Wileyto P, Kim JY, Gouma S, Robinson J, Greenplate AR, Hwee MA, Porterfield F, Owoyemi O, et al. (2021). CD8+ T cells contribute to survival in patients with COVID-19 and hematologic cancer. Nat. Med 27, 1280–1289. 10.1038/s41591-021-01386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenbarth SC (2019). Dendritic cell subsets in T cell programming: location dictates function. Nat. Rev. Immunol 19, 89–103. 10.1038/s41577-018-0088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim TS, Gorski SA, Hahn S, Murphy KM, and Braciale TJ (2014). Distinct Dendritic Cell Subsets Dictate the Fate Decision between Effector and Memory CD8+ T Cell Differentiation by a CD24-Dependent Mechanism. Immunity 40, 400–413. 10.1016/j.immuni.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, and Carbone FR (2006). Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 25, 153–162. 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 10.Gurevich I, Feferman T, Milo I, Tal O, Golani O, Drexler I, and Shakhar G (2017). Active dissemination of cellular antigens by DCs facilitates CD8+ T-cell priming in lymph nodes. Eur. J. Immunol 47, 1802–1818. 10.1002/eji.201747042. [DOI] [PubMed] [Google Scholar]
- 11.Mettelman RC, Allen EK, and Thomas PG (2022). Mucosal immune responses to infection and vaccination in the respiratory tract. Immunity 55, 749–780. 10.1016/j.immuni.2022.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner DL, Bickham KL, Farber DL, and Lefranç ois, L. (2013). Splenic Priming of Virus-Specific CD8 T Cells following Influenza Virus Infection. J. Virol 87, 4496–4506. 10.1128/JVI.03413-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jenkins MM, Bachus H, Botta D, Schultz MD, Rosenberg AF, Leόn B, and Ballesteros-Tato A (2021). Lung dendritic cells migrate to the spleen to prime long-lived TCF1hi memory CD8+ T cell precursors after influenza infection. Sci. Immunol 6, eabg6895. 10.1126/sciimmunol.abg6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lund FE, Partida-Sánchez S, Lee BO, Kusser KL, Hartson L, Hogan RJ, Woodland DL, and Randall TD (2002). Lymphotoxin-a-Deficient Mice Make Delayed, But Effective, T and B Cell Responses to Influenza. J. Immunol 169, 5236–5243. 10.4049/jimmunol.169.9.5236. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt ME, and Varga SM (2018). The CD8 T Cell Response to Respiratory Virus Infections. Front. Immunol 9, 678. 10.3389/fimmu.2018.00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiu C, and Openshaw PJ (2015). Antiviral B cell and T cell immunity in the lungs. Nat. Immunol 16, 18–26. 10.1038/ni.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zens KD, Chen JK, and Farber DL (2016). Vaccine-generated lung tissue–resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 1, e85832. 10.1172/jci.insight.85832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng MZM, and Wakim LM (2022). Tissue resident memory T cells in the respiratory tract. Mucosal Immunol 15, 379–388. 10.1038/s41385-021-00461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irvine DJ, Swartz MA, and Szeto GL (2013). Engineering synthetic vaccines using cues from natural immunity. Nat. Mater 12, 978–990. 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Backlund C, Jalili-Firoozinezhad S, Kim B, and Irvine DJ (2023). Biomaterials-Mediated Engineering of the Immune System. Annu. Rev. Immunol 41, 153–179. 10.1146/annurev-immunol-101721-040259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Si Y, Tian Q, Zhao F, Kelly SH, Shores LS, Camacho DF, Sperling AI, Andrade MS, Collier JH, and Chong AS (2020). Adjuvant-free nanofiber vaccine induces in situ lung dendritic cell activation and TH17 responses. Sci. Adv 6, eaba0995. 10.1126/sciadv.aba0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Si Y, Wen Y, Chen J, Pompano RR, Han H, Collier J, and Chong AS (2018). MyD88 in antigen-presenting cells is not required for CD4+ T-cell responses during peptide nanofiber vaccination. Medchemcomm 9, 138–148. 10.1039/C7MD00367F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Si Y, Wen Y, Kelly SH, Chong AS, and Collier JH (2018). Intranasal delivery of adjuvant-free peptide nanofibers elicits resident CD8+ T cell responses. J. Contr. Release 282, 120–130. 10.1016/j.jconrel.2018.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Topham DJ, Castrucci MR, Wingo FS, Belz GT, and Doherty PC (2001). The role of antigen in the localization of naive, acutely activated, and memory CD8(+) T cells to the lung during influenza pneumonia. J. Immunol 167, 6983–6990. 10.4049/jimmunol.167.12.6983. [DOI] [PubMed] [Google Scholar]
- 25.Walsh DA, Borges da Silva H, Beura LK, Peng C, Hamilton SE, Masopust D, and Jameson SC (2019). The Functional Requirement for CD69 in Establishment of Resident Memory CD8+ T Cells Varies with Tissue Location. J. Immunol 203, 946–955. 10.4049/jimmunol.1900052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amezcua Vesely MC, Pallis P, Bielecki P, Low JS, Zhao J, Harman CC, Kroehling L, Jackson R, Bailis W, Licona-Limó n, P., et al. (2019). Effector TH17 Cells Give Rise to Long-Lived TRM Cells that Are Essential for an Immediate Response against Bacterial Infection. Cell 178, 1176– 1188.e15. 10.1016/j.cell.2019.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rawat K, Tewari A, Li X, Mara AB, King WT, Gibbings SL, Nnam CF, Kolling FW, Lambrecht BN, and Jakubzick CV (2023). CCL5-producing migratory dendritic cells guide CCR5+ monocytes into the draining lymph nodes. J. Exp. Med 220, e20222129. 10.1084/jem.20222129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anthony SM, Van Braeckel-Budimir N, Moioffer SJ, van de Wall S, Shan Q, Vijay R, Sompallae R, Hartwig SM, Jensen IJ, Varga SM, et al. (2021). Protective function and durability of mouse lymph node-resident memory CD8+ T cells. Elife 10, e68662. 10.7554/eLife.68662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eriksson M, Nylé n, S., and Grö nvik, K.O. (2022). T cell kinetics reveal expansion of distinct lung T cell subsets in acute versus in resolved influenza virus infection. Front. Immunol 13, 949299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pizzolla A, Wang Z, Groom JR, Kedzierska K, Brooks AG, Reading PC, and Wakim LM (2017). Nasal-associated lymphoid tissues (NALTs) support the recall but not priming of influenza virus-specific cytotoxic T cells. Proc. Natl. Acad. Sci. USA 114, 5225–5230. 10.1073/pnas.1620194114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paik DH, and Farber DL (2021). Influenza infection fortifies local lymph nodes to promote lung-resident heterosubtypic immunity. J. Exp. Med 218, e20200218. 10.1084/jem.20200218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Porgador A, Yewdell JW, Deng Y, Bennink JR, and Germain RN (1997). Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity 6, 715–726. 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- 33.Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, De Prijck S, Bosteels V, Vandamme N, Martens L, et al. (2020). Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 52, 1039–1056.e9. 10.1016/j.immuni.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakano H, Burgents JE, Nakano K, Whitehead GS, Cheong C, Bortner CD, and Cook DN (2013). Migratory properties of pulmonary dendritic cells are determined by their developmental lineage. Mucosal Immunol 6, 678–691. 10.1038/mi.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klimova N, Holubova J, Streparola G, Tomala J, Brazdilova L, Stanek O, Bumba L, and Sebo P (2022). Pertussis toxin suppresses dendritic cell-mediated delivery of B. pertussis into lung-draining lymph nodes. PLoS Pathog 18, e1010577. 10.1371/journal.ppat.1010577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joffre OP, Segura E, Savina A, and Amigorena S (2012). Cross-presentation by dendritic cells. Nat. Rev. Immunol 12, 557–569. 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 37.Heath WR, and Carbone FR (2009). Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol 10, 1237–1244. 10.1038/ni.1822. [DOI] [PubMed] [Google Scholar]
- 38.Hassan AO, Shrihari S, Gorman MJ, Ying B, Yuan D, Raju S, Chen RE, Dmitriev IP, Kashentseva E, Adams LJ, et al. (2021). An intranasal vaccine durably protects against SARS-CoV-2 variants in mice. Cell Rep 36, 109452. 10.1016/j.celrep.2021.109452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Americo JL, Cotter CA, Earl PL, Liu R, and Moss B (2022). Intranasal inoculation of an MVA-based vaccine induces IgA and protects the respiratory tract of hACE2 mice from SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 119, e2202069119. 10.1073/pnas.2202069119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bošnjak B, Odak I, Barros-Martins J, Sandrock I, Hammerschmidt SI, Permanyer M, Patzer GE, Greorgiev H, Gutierrez Jauregui R, Tscherne A, et al. (2021). Intranasal Delivery of MVA Vector Vaccine Induces Effective Pulmonary Immunity Against SARS-CoV-2 in Rodents. Front. Immunol 12, 772240. 10.3389/fimmu.2021.772240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Das Mohapatra A, Tirrell I, Bénéchet AP, Pattnayak S, Khanna KM, and Srivastava PK (2020). Cross-dressing of CD8α+ Dendritic Cells with Antigens from Live Mouse Tumor Cells Is a Major Mechanism of Cross-priming. Cancer Immunol. Res 8, 1287–1299. 10.1158/2326-6066.CIR-20-0248. [DOI] [PubMed] [Google Scholar]
- 42.Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, Bhutkar A, Wittrup KD, and Spranger S (2022). Type I interferon activates MHC class I-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ T cell immunity. Immunity 55, 308–323.e9. 10.1016/j.immuni.2021.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacNabb BW, Tumuluru S, Chen X, Godfrey J, Kasal DN, Yu J, Jongsma MLM, Spaapen RM, Kline DE, and Kline J (2022). Dendritic cells can prime anti-tumor CD8+ T cell responses through major histocompatibility complex cross-dressing. Immunity 55, 982–997.e8. 10.1016/j.immuni.2022.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desch AN, Gibbings SL, Clambey ET, Janssen WJ, Slansky JE, Kedl RM, Henson PM, and Jakubzick C (2014). Dendritic cell subsets require cis-activation for cytotoxic CD8 T-cell induction. Nat. Commun 5, 4674. 10.1038/ncomms5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ballesteros-Tato A, Leόn B, Lund FE, and Randall TD (2010). Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8+ T cell responses to influenza. Nat. Immunol 11, 216–224. 10.1038/ni.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J, Pompano RR, Santiago FW, Maillat L, Sciammas R, Sun T, Han H, Topham DJ, Chong AS, and Collier JH (2013). The use of self-adjuvanting nanofiber vaccines to elicit high-affinity B cell responses to peptide antigens without inflammation. Biomaterials 34, 8776–8785. 10.1016/j.biomaterials.2013.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petrovsky N (2015). Comparative Safety of Vaccine Adjuvants: A Summary of Current Evidence and Future Needs. Drug Saf 38, 1059–1074. 10.1007/s40264-015-0350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castelli C, Ciupitu AM, Rini F, Rivoltini L, Mazzocchi A, Kiessling R, and Parmiani G (2001). Human heat shock protein 70 peptide complexes specifically activate antimelanoma T cells. Cancer Res 61, 222–227. [PubMed] [Google Scholar]
- 49.Srivastava P (2002). Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol 2, 185–194. 10.1038/nri749. [DOI] [PubMed] [Google Scholar]
- 50.Murshid A, Gong J, and Calderwood SK (2012). The Role of Heat Shock Proteins in Antigen Cross Presentation. Front. Immunol 3, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Accolla RS, Ramia E, Tedeschi A, and Forlani G (2019). CIITA-Driven MHC Class II Expressing Tumor Cells as Antigen Presenting Cell Performers: Toward the Construction of an Optimal Anti-tumor Vaccine. Front. Immunol 10, 1806. 10.3389/fimmu.2019.01806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neefjes J, Jongsma MLM, Paul P, and Bakke O (2011). Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol 11, 823–836. 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 53.Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, and Schall TJ (1997). A new class of membrane-bound chemokine with a CX3C motif. Nature 385, 640–644. 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 54.Combadiere C, Salzwedel K, Smith ED, Tiffany HL, Berger EA, and Murphy PM (1998). Identification of CX 3CR1: A CHEMOTACTIC RECEPTOR FOR THE HUMAN CX 3C CHEMOKINE FRACTALKINE AND A FUSION CORECEPTOR FOR HIV-1 *. J. Biol. Chem 273, 23799–23804. 10.1074/jbc.273.37.23799. [DOI] [PubMed] [Google Scholar]
- 55.Richard M, and Fouchier RAM (2016). Influenza A virus transmission via respiratory aerosols or droplets as it relates to pandemic potential. FEMS Microbiol. Rev 40, 68–85. 10.1093/femsre/fuv039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Killingley B, and Nguyen-Van-Tam J (2013). Routes of influenza transmission. Influenza Other Respir. Viruses 7, 42–51. 10.1111/irv.12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kutter JS, Spronken MI, Fraaij PL, Fouchier RA, and Herfst S (2018). Transmission routes of respiratory viruses among humans. Curr. Opin. Virol 28, 142–151. 10.1016/j.coviro.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leung NHL (2021). Transmissibility and transmission of respiratory viruses. Nat. Rev. Microbiol 19, 528–545. 10.1038/s41579-021-00535-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rudra JS, Tian YF, Jung JP, and Collier JH (2010). A self-assembling peptide acting as an immune adjuvant. Proc. Natl. Acad. Sci. USA 107, 622–627. 10.1073/pnas.0912124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karttunen J, Sanderson S, and Shastri N (1992). Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc. Natl. Acad. Sci. USA 89, 6020–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schliehe C, Redaelli C, Engelhardt S, Fehlings M, Mueller M, van Rooijen N, Thiry M, Hildner K, Weller H, and Groettrup M (2011). CD8 — Dendritic Cells and Macrophages Cross-Present Poly(D,L-lactate-co -glycolate) Acid Microsphere-Encapsulated Antigen In Vivo. J. ICE 187, 2112–2121. 10.4049/jimmunol.1002084. [DOI] [PubMed] [Google Scholar]
- 62.Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181. 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the main text or the supplementary materials. The RNA-seq data has been uploaded to the GEO repository (GEO: GSE216097, GSE216098, GSE216099). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.