

Abstract
The lack of targeted therapies for traumatic brain injury (TBI) remains a compelling clinical unmet need. Although knowledge of the pathophysiologic cascades involved in TBI has expanded rapidly, the development of novel pharmacological therapies has remained largely stagnant. Difficulties in creating animal models that recapitulate the different facets of clinical TBI pathology and flaws in the design of clinical trials have contributed to the ongoing failures in neuroprotective drug development. Furthermore, multiple pathophysiological mechanisms initiated early after TBI that progress in the subacute and chronic setting may limit the potential of traditional approaches that target a specific cellular pathway for acute therapeutic intervention. We describe a reverse translational approach that focuses on translating endogenous mechanisms known to influence outcomes after TBI to develop druggable targets. In particular, numerous clinical observations have demonstrated an association between apolipoprotein E (apoE) polymorphism and functional recovery after brain injury. ApoE has been shown to mitigate the response to acute brain injury by exerting immunomodulatory properties that reduce secondary tissue injury as well as protecting neurons from excitotoxicity. CN-105 represents an apoE mimetic peptide that can effectively penetrate the CNS compartment and retains the neuroprotective properties of the intact protein.
Keywords: Traumatic brain injury, Neuroinflammation, Neurodegeneration, Drug development, Neuroprotection
Introduction
Traumatic brain injury (TBI) is an acquired condition that results from an external mechanical force applied to the skull and underlying intracranial contents resulting in temporary or permanent damage to affected brain tissue or function [1]. Globally, an estimated 64–74 million people sustain a TBI every year, the majority of which are classified as mild TBI, also commonly referred to as concussion [2, 3]. TBIs are associated with numerous mechanisms including sports-related injuries, falls, motor vehicle accidents, and assaults. Additionally, factors such as location, socioeconomic status, and biological sex can impact TBI incidence and outcome [4]. Between 2001 and 2012 in the USA, an estimated 3.42 million sports- and recreation-related TBIs were evaluated in US emergency departments [5]. According to the US Centers for Disease Control and Prevention, there were approximately 223,135 TBI-related hospitalizations in 2019, and 64,362 TBI-related deaths in 2020 [6]. For certain populations, additional mechanisms of brain injury exist, such as military personnel who may be exposed to blast and direct energy sources [7–9]. Between 2001 and 2021, more than 1.6 million military personnel deployed to Iraq or Afghanistan in support of the Global War on Terror (GWOT), with an estimated 5 to 35% of them having suffered a concussion [7]. As of August 2022, the Traumatic Brain Injury Center of Excellence (TBICoE) reports 463,392 TBIs across the Department of Defense since 2000, 82% of which are mild in severity [10]. Despite improvements in the recognition and documentation of concussions, it is widely believed that the incidence is drastically underestimated, with as few as 1 out of every 9 concussions being captured by current data collection methods [1, 5]. Practically, TBI can result in significant long-term cognitive and physical sequelae that have a significant impact on the individual’s personal and professional life, as well as the society and economy. Annual costs, both direct and indirect, have previously been calculated to total more than $60 billion annually in the USA [11]. As a result, there is significant interest in improving diagnostic accuracy and therapeutic options for TBI.
TBI Therapeutic Landscape: Unmet Needs
A combination of unknown incidence, poorly understood pathophysiology, historically unrecognized or misattributed signs and symptoms, and the paucity of options to aid in the diagnosis and treatment of TBI have led some to refer to it as a “silent epidemic” [12]. Between the increased attention to the long-term effects of head injury in sports and the prevalence of blast TBI in the GWOT over the last 20 years, there has been steadily increasing awareness of the effects and cost of TBI among the civilian and military populations [7, 13]. Indeed, significant progress has been made in many domains including public health initiatives that have led to seatbelt legislation, improved workplace and safety regulations, improved helmet design for sports and the military, effective screening tools and protocolized approaches to assessment and care, and return to activity recommendations for sports. The establishment of specialized neurocritical care units and the focus on specialized TBI rehabilitation have also contributed to improved outcomes, particularly in patients with moderate and severe TBI [7, 14]. Despite these advances, an important area where progress remains elusive is the development of effective neuroprotective agents.
Although decades of research have yielded pharmacological interventions with promising preclinical results, none have demonstrated benefit in improving long-term outcomes once they reached Phase 3 clinical trials. This 100% failure rate suggests a problem with study design at multiple levels. More specifically, criticisms of TBI trials can be divided into problems with preclinical testing or clinical trial design and testing [15]. Problematic issues in the preclinical setting include inadequate attention to the temporal profile of specific pathobiological responses that contribute to secondary injury [16]. For example, both the mechanical stretch and secondary ischemia associated with the primary concussive force lead to overactivation of glutamate receptors, resulting in neuronal excitotoxic injury [17]. The temporal window for therapeutic intervention to mitigate this injury cascade is brief compared to the neuroinflammatory response, which is associated with diffuse glial activation, leading to secondary tissue injury and cerebral edema, and may progress over a period of days [18–20].
In TBI studies, it is important to recognize the biological differences between human and rodent brain structure and function, and that laboratory methods often fail to replicate the heterogenous injury patterns that occur with real-world TBI [21–26]. The known anatomic differences, such as the absence of gyri and a small amount of white matter in the rodent brain, as well as geometric variations in the neuraxis of animal brains, limit the ability to produce injury patterns comparable to those in humans. Furthermore, many animal models focus on short-term functional outcomes and cannot reproduce emotional or language deficits that can occur in human TBI [14, 27]. Additional limitations associated with preclinical testing often include inadequate sample size [28] and inadequate blinding, randomization, reporting bias, attrition, and selective analysis [29–31]. Translational issues such as inadequate understanding of the pharmacokinetics, dosages, and differences between human and experimental animals may also limit the potential translatability of promising preclinical findings. Moreover, preclinical studies may not adequately address efficacy across, sex, age, and medical co-morbidities, which are needed to better assess the populations of patients who are most likely to benefit from therapies [14, 15, 32].
Methods for producing direct head trauma in animals generally produce homogenous injury patterns that are well-controlled by design [27]. The controlled cortical impact (CCI) method, for example, is one of the most widely utilized approaches because it can create consistent injuries associated with durable functional deficits [33, 34]. CCI is used post-craniotomy to produce mild to severe injuries via direct cortical deformation using a 3 mm or 5 mm impactor with a velocity of 3–6 m/s resulting in a displacement of the cortex of 0.5–1.5 mm [35]. Spatial learning and memory deficits, recognition, limbic and frontal dysfunction, and a variety of motor impairments can be reliably produced with CCI in rodents. Histologically, CCI can reproduce primary and secondary neuronal death, hippocampal and subventricular zone cell proliferation, blood–brain barrier disruption, and oxidative stress and neuroinflammation [33, 34]. While easy to use, reproducible, and capable of producing a range of injury severity, CCI can produce variable and sometimes unintended injuries. Recent data has shown that lateral movements of the piston delivering the cortical impact can increase the number of secondary impacts. While the animal skull is fixed in a stereotaxic frame to limit the potential for secondary impacts, this immobility risks increased tissue damage and injury severity [34]. Furthermore, CCI can lead to dural lacerations that create an open head injury, the type of impactor tip can affect the rate of neocortical degeneration, and the mild to moderate forces produced by this method may not induce brainstem injury necessary for severe TBI models [33, 35].
Importantly, although models that employ a targeted impact directly to brain tissue may create a standardized and homogenous injury, they are limited in their ability to recapitulate the biomechanical forces associated with clinical closed head injury [35], which are associated with complex pathology including parenchymal, subarachnoid, subdural and epidural hemorrhage, cortical contusion, and sheer injury. Although all lissencephalic rodent models of TBI imperfectly recapitulate the clinically relevant biomechanics seen in human closed head injury, we favor pneumatic impact models in which a controlled force and displacement are delivered stereotactically to the intact skull. These models allow for diffuse glial activation and injury to selectively vulnerable remote hippocampal and cortical populations and produce durable neurocognitive deficits that are modifiable by therapeutic intervention [29, 36, 37].
Limitations in clinical trial design for patients with closed head injuries have also played an important role in the failure of neuroprotective therapy development. These include the use of single-center randomized clinical trials (RCTs) [38], follow-up bias, inadequate or unclear random sequence generation or blinding with subjective endpoints [39], poor translation of doses from preclinical studies to clinical trials [40], poor patient selection [41], misclassification of outcomes reducing effect size (i.e., Glasgow Outcome Scale score) [42], and inadvertent bias due to external pressures [31]. Improvements in study design such as better patient selection criteria (such as stratification by serum biomarkers as well as age, sex, radiographic findings, and type of injury), adaptive designs that eliminate locked-in assignments in favor of changes in group assignments as the trial progresses, the inclusion of multiple quantitative outcome measures, optimization of dose and route administration, and a stronger understanding of drug mechanisms and metabolic characteristics may ultimately lead to a greater track record of success [32].
Reverse Translation: from Genetic Association to Therapy
Traditional drug development programs often utilize a reductionistic approach, whereby a specific cellular pathway believed to influence disease pathology is targeted with an optimized pharmacologic intervention. However, despite our increased understanding of the molecular and cellular mechanisms associated with secondary neuronal injury in the setting of acute CNS injury, no clinically effective neuroprotectant has been identified or developed. It is likely that this failure of translation is due, in part, to the pleiotropic effects of pathways involved in inflammation, oxidative stress, and excitotoxicity, as well as compensatory cellular mechanisms. An alternative to this traditional approach of “bench to bedside” is reverse translation. This “bedside to bench” approach focuses on the translation of an endogenous protein known to influence outcomes into a druggable target (Fig. 1). For example, functional outcome after traumatic brain injury is widely variable, and this has prompted the study of genetic influences on recovery [43]. Although several genetic polymorphisms have been demonstrated to influence outcomes, one of the leading candidates for this process of reverse translation is the apoE polymorphism, which encodes the human apolipoprotein E2, E3, and E4 protein isoforms. Although these common human isoforms only differ by single cysteine to arginine substitutions (apoE3: Cys 112 Arg 158; apoE4: Arg 112 Arg 158; apoE2: Cys 112 Cys 158), the cysteine to arginine substitution at position 112 is believed to cause domain interactions which result in structural alterations of apoE4 that influence its metabolism and functional activity [44, 45].
Fig. 1.

Translation vs. reverse translation. The conventional bench to bedside approach for development of therapeutic strategies, known a translation, involves basic science research in the lab, leading to drug development, and culminating in human clinical trials. Alternatively, a bedside to bench approach, known as reverse translation, takes clinical observations and utilizes them to inform the development of new therapies in the laboratory. Considering the slow progress in identifying an effective therapeutic for TBI using traditional methods thus far, reverse translation may augment traditional approaches by helping to identify therapeutic targets and influence trial design
Although initially identified in the context of cholesterol metabolism, in the 1990s apoE polymorphism was found to have a robust effect on the development of late-onset sporadic and familial Alzheimer’s disease [46]. In particular, the presence of one apoE4 allele resulted in an approximately fourfold increase in the development of AD, whereas homozygosity for apoE4 was associated with an approximately tenfold increase. Based on the identification of apoE4 as a genetic risk factor for AD, initial studies focused on disease-specific pathologies, such as isoform-specific effects on Aβ metabolism and deposition [47] and interactions with the microtubule-associated protein tau [48]. Interestingly, several studies have also demonstrated that the presence of apoE4 adversely affects outcomes following acute brain injury [49, 50]. Similarly, clinical and preclinical evidence suggests that apoE4 may be associated with impaired cognitive outcomes after mild TBI [51] and the development of cognitive dysfunction and chronic traumatic encephalopathy in older patients after repetitive head injuries [52–55], although the latter association remains somewhat controversial [56, 57].
As glial activation and neuroinflammatory responses play an important role in both acute brain injury responses and the development of neurodegenerative disease, one unifying hypothesis that would accommodate a role for apoE polymorphism in modifying outcomes across this spectrum of acute and chronic neurological disease is an isoform-specific effect on neuroinflammatory pathways. In fact, initial observations from more than 25 years ago have demonstrated a role for apoE in modifying glial activation and neuroinflammatory responses [58, 59]. Moreover, several preclinical and clinical studies have demonstrated isoform-specific effects on inflammation, with the apoE4 protein isoform associated with increased neuroinflammatory responses relative to apoE3 [60–64]. These isoform-specific effects on neuroinflammatory pathways may also extend to the blood–brain barrier permeability, which plays a particularly important role in the development of cerebral edema following traumatic brain injury [65]. A more complete understanding of the mechanism(s) of the isoform-specific effects by which apoE modifies neuroinflammatory mechanisms has important therapeutic implications and remains incompletely defined. ApoE binds a family of low-density lipoprotein (LDL) receptors, and observations suggest that interactions with the low-density lipoprotein receptor-related protein (LRP1) may initiate a signaling cascade [66–70] that are critical in mediating its immunomodulatory properties via mitogen-activated protein kinase (MAP kinase) pathways [71–73], and stabilization of the blood–brain barrier through the cyclophilin A-matrix metallopeptidase-9 pathway [74].
Despite the numerous clinical observations suggesting that the apoE isoform modifies functional outcomes after acute brain injury, clinical observational studies are often limited by the heterogeneity of injury and small population sizes. In this regard, the use of standardized injury paradigms in targeted replacement mice expressing the human apoE3 and apoE4 isoforms allows an important tool to validate the effect of apoE polymorphisms in the setting of acute brain injury, as well as to allow for the study of cellular mechanisms by which these effects are mediated.
Early preclinical studies have consistently demonstrated that apoE plays an important function in modulating systemic and CNS inflammatory responses [75–80]. ApoE4 mice have enhanced inflammatory responses as compared to their apoE3 counterparts in a variety of experimental paradigms. For example, after peripheral injection of bacterial endotoxin (lipopolysaccharide), there was enhanced evidence of peripheral and CNS pro-inflammatory responses, suggesting a dynamic interplay between peripheral and CNS inflammatory mediators [80]. Moreover, the presence of the apoE4 allele was associated with enhanced neuroinflammation and worsened outcomes in preclinical models of traumatic brain injury [81] and standardized models designed to mimic many of the pathological features of TBI, including subarachnoid hemorrhage [82] intraparenchymal hemorrhage [83] and cerebral ischemia [84, 85]. In total, these preclinical and clinical observations support the contention that apoE modifies acute brain injury responses in an isoform-specific fashion, in part via modulation of maladaptive neuroinflammatory responses [60].
Preclinical Development of CN-105, a Candidate apoE Mimetic Peptide
Based on the evidence that apoE3 plays an adaptive role following acute brain injury, a plausible therapeutic strategy would be to augment the beneficial effects of the endogenous protein. However, due to its size, peripherally administered apoE does not readily cross the blood–brain barrier, and in fact, the apoE produced primarily by astrocytes within the CNS compartment represents a relatively discrete pool from that found in the periphery, where synthesis occurs primarily in the liver [86]. However, dimerized peptides derived from the apoE receptor binding region had been demonstrated to have effects on lymphocyte proliferation [87–89]. Thus, to harness the therapeutic potential of apoE, a series of smaller peptides were created from the residues 130–149 of the apoE receptor binding region to explore the hypothesis that a smaller peptide capable of penetrating the CNS compartment could retain the receptor interactions necessary to mediate the bioactivity of the intact holoprotein [62, 90]. Although there was a size threshold by which the peptides lost the helicity necessary for bioactivity [62], the therapeutic peptide COG1410 represented a 12-amino acid peptide in which the helical structure was stabilized by two aminoisobutyric (Aib) residues that retained the anti-inflammatory effects of the apoE holoprotein [91].
As noted above, TBI is associated with heterogenous pathology, and to facilitate the translation of new therapeutic interventions, it is helpful to dissect the underlying pathobiological responses. The temporal profile of secondary tissue injury due to neuroinflammation may extend over several days, making it an attractive target for pharmacological intervention. Moreover, secondary tissue injury associated with neuroinflammation is common to many of the mechanisms of injury associated with TBI pathology, including ischemia, contusion, subarachnoid, and parenchymal hemorrhage [92]. The intracellular cascade associated with glutamate excitotoxicity is another mechanism of neuronal cell death following trauma and ischemia. In this regard, it is notable that in addition to its effects on microglial activation and neuroinflammation, apoE mimetic peptides also reduced excitotoxic neuronal injury in the same manner as the intact apoE [93], a property which was hypothesized to also be mediated by LRP-1 interactions [94, 95]. Thus, apoE mimetic peptides have pleiotropic effects on glutamate excitotoxicity and inflammation, and intravenous administration of the apoE mimetic peptide reduced histological injury and had a durable effect on improving functional outcomes in murine models of closed head injury [96, 97], as well as models of cerebral ischemia [98], and parenchymal and subarachnoid hemorrhage [82, 99, 100]. These models recapitulate different aspects of clinical TBI pathology [92].
Although well tolerated and associated with functional improvement, a limitation of COG1410 was its relatively large size, low potency, and the cost associated with the incorporation of non-naturally occurring Aib residues. To enhance druggability, a newer generation of apoE mimetic peptides was developed by modeling the polar receptor binding face of the amphipathic helical receptor binding region of apoE involved in direct receptor interactions. This library of peptides was tested in assays of glial activation [91] and excitatory [94] neuroprotection to generate the candidate apoE mimetic pentapeptide CN-105 (Ac-VSRRR-NH2) (Fig. 2). To date, CN-105 has demonstrated histological and functional improvement in standardized preclinical models of closed head injury [101–103], as well as preclinical models that were designed to mimic the heterogenous pathology associated with TBI, including blast injury [73], subarachnoid hemorrhage [103], intraparenchymal hemorrhage [104], and cerebral ischemia [105, 106]. Based on these compelling preclinical data of neuroprotection across the different facets of clinical TBI pathology [92], CN-105 was selected for further clinical development. Initial Phase 1 trials were performed in Western [107] and Asian populations [108] populations. These phase 1 studies confirmed a strong safety profile after intravenous dosing, a terminal half-life of approximately 3.5 h, and no significant drug accumulation upon repeated dosing [107, 108].
Fig. 2.
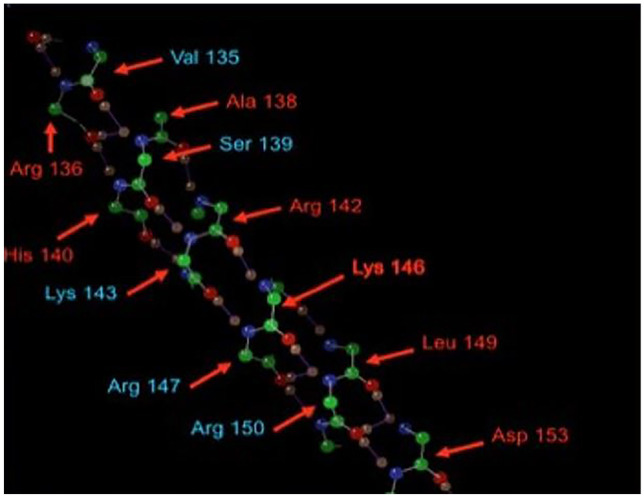
Figure demonstrating the amino acid structure of the receptor binding face of ApoE with the five peptide residues that comprise CN-105 highlighted in blue. Ala alanine, Arg arginine, Asp Aspartate, His histidine, Leu leucine, Lys lysine, Ser serine, and Val valine (used with permission: Guptill JT, Raja S, Ramey S, et al. Phase I randomized, double-blind, placebo-controlled study to determine the safety, tolerability, and pharmacokinetics of a single escalating dose and repeated doses of CN-105 in healthy adult subjects. J Clin Pharmacol. 2017;57(6):770–776)
Clinical Development of CN-105
Because CN-105 appears to affect maladaptive neuroinflammatory and excitotoxic pathways following acute brain injury, there is a spectrum of neurological indications for which it might be effective in addition to TBI. For example, as noted above, the preclinical efficacy of this approach has been demonstrated in preclinical models of stroke, blast injury, subarachnoid, and intraparenchymal hemorrhage, all of which are characterized by secondary tissue injury mediated by glial activation and neuroinflammation. Important criteria for the first in-disease state trial of CN-105 included relevance to TBI pathology and tractability of pilot trial design. Based on these considerations, intracranial hemorrhage was chosen as the initial therapeutic outlet, as parenchymal hemorrhage is a common component of TBI, and primary hypertensive intracranial hemorrhage (ICH) tends to have a more focal and homogenous pathology, which allows for the longitudinal radiographic monitoring of cerebral edema as a marker of target engagement.
The resulting trial, CN-105 in participants with acute supratentorial intracerebral hemorrhage (CATCH; NCT03168581) incorporated a dose of 1 mg/kg CN-105, administered intravenously at 6-h intervals for 72 h [109]. CATCH was designed as a multi-site, open-label study to establish molecular and radiographic markers of target engagement and surrogate markers of efficacy. Although primarily designed for safety and feasibility, to establish a preliminary signal of efficacy, CN-105-treated participants were compared 1:1 with participants closely matched for sex, race, and presentation ICH score that were drawn from the Ethnic/Racial Variations of Intracerebral Hemorrhage (ERICH) study, a cohort of 3000 patients with ICH prospectively assembled between 2010 and 2015 [110]. The results were promising, as CN-105-treated patients demonstrated improved 30-day functional outcomes (lower modified Rankin score) when compared to matched controls (odds ratio 2.69; 1.31–5.51 95% CI) [109]. Based on these encouraging results, we recently initiated a multicenter, randomized, double-blind, placebo-controlled clinical trial for patients with acute intracerebral hemorrhage in Singapore (S-CATCH; NCT03711903) [111].
In addition to these ongoing studies, the anti-inflammatory and neuroprotective effects of CN-105 are also currently being evaluated in the setting of postoperative cognitive dysfunction (POCD). Delirium and cognitive dysfunction following surgery represents a significant public health issue, especially in patients over the age of 60, and is associated with decreased quality of life and 1-year mortality [112]. Although the mechanisms underlying POCD are likely multifactorial, increasing evidence implicates maladaptive neuroinflammation leading to blood–brain barrier breakdown and secondary tissue injury [113]. To study the effects of CN-105 in this setting, the modulating ApoE signaling to reduce brain inflammation, delirium, and postoperative cognitive dysfunction (MARBLE; NCT 03802396) study was designed as a single-center, randomized, tiered, dose-escalation study evaluating perioperative neurocognitive disorders in patients undergoing prolonged non-cardiac surgery [114]. Of note, this study is also designed to provide information on the effect of CN-105 on cerebrospinal fluid markers of injury and inflammation and their correlation with neurocognition, which may be of relevance in evaluating the delayed effects of TBI.
CN-105 as a Potential Therapy in TBI
Given the societal impact of traumatic brain injury, the development of a pharmacological intervention to improve functional outcome has remained an intense focus of research. However, as noted above, despite hundreds of clinical trials the identification of successful therapeutic strategies has remained elusive. One major obstacle to the development of an effective pharmacological intervention is the heterogeneity of tissue pathology associated with TBI, which may include elements of subdural, epidural, intraparenchymal, and subarachnoid hemorrhage as well as cerebral ischemia, tissue contusion and diffuse axonal injury. Thus, it is reasonable that preclinical candidate therapies be chosen based on the demonstration of histological and functional improvement in preclinical models of these different facets of TBI pathology. Improvement in multiple preclinical models has generated much interest in apoE mimetic peptides for TBI and has been one of the major criteria for the selection of CN-105 (Table 1) [90, 96, 97, 101, 102, 115–120].
Table 1.
Preclinical studies evaluating apoE mimetic peptides in TBI
| Injury | Species | Peptide/dosing | Histological/biochemical outcome measures | Functional outcome measures | References |
|---|---|---|---|---|---|
| TBI (closed head injury) | Mouse | ApoE (133–149) high dose (406 1 µg/kg), low dose (203 µg/kg) or scrambled control IV 30 min post-injury | Reduction in oxidative stress (aconitase), neuronal degeneration, and TNFα mRNA | Improved vestibulomotor function and memory in peptide treated mice | Lynch et al. 2005 [90] |
| TBI (closed head injury) | Mouse | COG1410 0.6 mg/kg IV 120 min post-injury × 1 dose | Reduction in degenerating neurons, microgliosis | Improved vestibulomotor function and memory | Laskowitz et al. 2007 [96] |
| TBI (cortical contusion) | Rat | COG1410 0.8 mg/kg IV, 0.4 mg/kg IV 30 min post-injury | Smaller lesion volume and reduction in astrocytosis with 0.8 mg/kg dose | Improved motor outcomes with 0.8 mg/kg dose | Hoane et al. 2007 [115] |
| TBI (closed head injury) | Mouse | ApoE(133–149) 1 mg/kg IV × 1 vs. 100 µL isotonic saline 30 min post-injury | Reduction in degenerating neurons, microgliosis, TNFα, and Aβ | Improved vestibulomotor function | Wang et al. 2007 [116] |
| TBI (cortical contusion) | Rat | COG1410 0.8 mg/kg IV at 30 min and 24 h post-injury | Reduction in degenerating neurons | Improved sensorimotor function and working memory | Hoane et al. 2009 [97] |
| TBI (closed head injury) | Mouse | COG1410 1 mg/kg IV after scalp sutured close post-injury followed by 1 mg/kg IV daily until day prior to sacrifice | Suppressed activation of MMP-9, reduced breakdown of blood–brain barrier, reduced TBI lesion volume, and vasogenic edema | Decreased functional deficits compared with saline-treated TBI animals | Cao et al. 2016 [117] |
| TBI (closed head injury) | Mouse | CN-105 various doses, routes and timepoints administered prior to injury | Reduced hippocampal microgliosis in treatment group | Improved vestibulomotor function | Van Wyck et al. 2022 [101] |
| TBI (closed head injury) | Mouse | COG1410 1 mg/kg IV daily | Reduced traumatic axonal injury in pericontusional | N/A | Jiang and Brody 2012 [118] |
The heterogeneity of patients presenting with TBI also has implications for patient enrollment in clinical trials. Traditionally, clinical trials have relied on the presentation Glasgow Coma Scale (GCS) assessment of injury severity as a key enrollment criterion. However, in isolation, the presentation of GCS may be misleading. Thus, it would be reasonable for future trials to incorporate more quantitative radiographic and serum biomarkers of tissue injury to select patients with a degree of TBI severity that might mostly benefit from a particular therapy. Several protein biomarkers of neuronal injury and glial activation such as glial fibrillary acidic protein (GFAP), neurofilament light protein (NFL), phosphorylated tau protein (p-tau), and ubiquitin C-terminal hydrolase-L1 (UCHL-1), as well as other markers of inflammation have demonstrated promise in early clinical trials [121–123]. Of note, the recent development of point-of-care platforms that measure GFAP and UCHL-1 at bedside may increase the practicality of stratifying TBI severity prior to enrollment [124, 125]. Moreover, it is important to recognize that secondary injuries may commonly occur during the prehospital period where hypoxia, cerebral hypoperfusion, and expanding hematoma may exacerbate secondary tissue injury. Ideally, agents such as CN-105 should be selected based on safety profile which may ultimately facilitate administration in the prehospital setting.
Once appropriate neuroprotective candidates are defined for clinical translation, clinical trials are often difficult to directly compare due to widely varying trial designs. For example, the timing of intervention is a critical variable and should be based on presumptive mechanisms of action, as traditional neuroprotective agents designed to reduce initial excitotoxic injury might be expected to have a short therapeutic window as compared to interventions that mitigate secondary tissue injury by reducing maladaptive neuroinflammatory responses. Ideally, candidate therapies should be chosen with pleiotropic mechanisms of action or be used in combination to target different components of the injury cascade. In this regard, CN-105 was chosen based on preclinical evidence that it mitigated both glutamate-mediated excitotoxicity as well as neuroinflammatory responses.
In addition to the harmonization of trial design to integrate standardized radiographic and biochemical surrogates of tissue injury, the adoption of consensus endpoints that are functionally relevant and sensitive to intervention would increase the likelihood of trial success and allow more direct comparisons of efficacy between trials. Finally, it is important to recognize that trials in TBI are often extremely resource-intensive, and clinical development may be abandoned in an early trial that demonstrates promising but non-statistically significant results due to financial considerations. Similar proprietary and commercial concerns often make it difficult to design trials that test rational drug combinations targeting different components of the injury cascade.
Ultimately, one mechanism to address many of these limitations in TBI trials and accelerate the process of drug development may be the implementation of an adaptive platform design. A platform study is a randomized study design targeting a specific disease that is performed under a master protocol, which allows for the standardization of inclusion/exclusion criteria that incorporate consensus clinical, biomarker, and radiographic assessments, as well as validated clinical outcome assessments that are sensitive to therapeutic intervention. Platform designs also allow for statistical efficiencies which may optimize the use of resources. For example, the ability of multiple interventions to be evaluated simultaneously against a common control group and the addition of new interventions during the trial make this approach particularly attractive for early-phase clinical trials designed to identify promising candidate therapies for further development. Because adaptive platform trials take advantage of the stream of real-time information acquired during a trial, it also allows for statistical efficiencies with regard to conditional powering, which make it less likely that a potentially promising intervention is abandoned due to lack of statistical significance. This approach is being adopted by the TRACK-TBI network, which will be testing multiple promising TBI therapies in a multi-center, double-blind, placebo-controlled adaptive platform [126, 127].
Conclusions
Our understanding of TBI is rapidly evolving. With the development of novel methods for diagnosing TBI, including biomarker panels and advanced imaging, it is reasonable to expect that the incidence of TBI will continue to increase as we improve our diagnostic capabilities. This trend hastens the need for effective neuroprotectant strategies. As studies aiming to develop effective neuroprotectants continue, it will be necessary to consider and address shortcomings in existing trial designs and develop clearly defined and functionally relevant clinical outcomes when evaluating neuroprotectants. The disappointing results thus far of reductionist approaches may support increased efforts to identify endogenous proteins with pleiotropic effects that can affect TBI outcomes. Apolipoprotein E is one of the most studied genetic polymorphisms as it relates to outcomes following neurotrauma and has an adaptive role after TBI through the reduction of neuroinflammation, excitotoxicity, and secondary brain injury. Although the intact apoE lipoprotein is too large to cross the blood–brain barrier, apoE peptides such as CN-105 appear to effectively interact with receptors that can modulate neuroinflammatory and excitotoxic responses to brain injury. Furthermore, CN-105 is stable, demonstrates predictable pharmacokinetics, and has a favorable toxicity profile. A growing body of literature supports its efficacy in a wide variety of head injury subtypes, making it an attractive candidate for continued clinical evaluation in human trials of TBI.
Acknowledgements
The authors would like to thank Sarah Laskowitz for her assistance with the figure art.
Data Availability
All data supporting the findings of this study are available/cited within the paper. All pre-clinical data has been made public in the referenced works.
Declarations
Conflict of Interest
None.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Capizzi A, Woo J, Verduzco-Gutierrez M. Traumatic brain injury: an overview of epidemiology, pathophysiology, and medical management. Med Clin North Am. 2020;104(2):213–238. [DOI] [PubMed]
- 2.Dewan MC, Rattani A, Gupta S, et al. Estimating the global incidence of traumatic brain injury. J Neurosurg. 2018;2018(1):1–18. doi: 10.3171/2017.10.JNS17352. [DOI] [PubMed] [Google Scholar]
- 3.Janak JC, Pugh MJ, Ormal L. Epidemiology of TBI in: traumatic brain injury rehabilitation medicine. D.X. Cifu and B.C. Eapen (eds.). Elsevier: Philadelphia, PA. 2015:6–35.
- 4.Ponsford J. Factors contributing to outcome following traumatic brain injury. Neuro Rehabilitation. 2013;32(4):803–815. doi: 10.3233/NRE-130904. [DOI] [PubMed] [Google Scholar]
- 5.Pierpoint LA, Collins C. Epidemiology of sports-related concussion. Clin Sports Med. 2021;40(1):1–18. [DOI] [PubMed]
- 6.Centers for Disease Control and Prevention. National center for health statistics: mortality data on CDC WONDER. Available at: https://wonder.cdc.gov. Accessed December 16, 2022.
- 7.Lindberg MA, Moy-Martin EM, Marion DW. Military traumatic brain injury: the history, impact and future. J Neurotrauma. 2022;39(17–18):1133–1145. [DOI] [PMC free article] [PubMed]
- 8.Hubler G, Hoffman S, Andreadis TD, et al. Pulsed microwave energy transduction of acoustic phonon related brain injury. Front Neurol. 2020;11:753. doi: 10.3389/fneur.2020.00753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson R. Havana syndrome might be the result of energy pulses. Lancet. 2021;2021(19):396. doi: 10.1016/S0140-6736(20)32711-2. [DOI] [PubMed] [Google Scholar]
- 10.DoD worldwide numbers for TBI. In: TBI center of excellence [online]. 10 August 2022. Available at: https://health.mil/Military-Health-Topics/Centers-of-Excellence/Traumatic-Brain-Injury-Center-of-Excellence/DOD-TBI-Worldwide-Numbers. Accessed November 3, 2022.
- 11.Coronado VG, Haileyesus T, Cheng TA, et al. Trends in sports- and recreation-related traumatic brain injuries treated in US emergency departments: the National Electronic Injury Surveillance System-All Injury Program (NEISS-AIP) 2001–2012. J Head Trauma Rehabil. 2015;30(3):185–197. doi: 10.1097/HTR.0000000000000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lefkovits AN, Hicks AJ, Downing M, et al. Surviving the “silent epidemic”: a qualitative exploration of the long-term journey after traumatic brain injury. Neuropsych Rehabil. 2021;31(10):1582–1606. doi: 10.1080/09602011.2020.1787849. [DOI] [PubMed] [Google Scholar]
- 13.Wilson L, Stewart W, Dams-O’Connor K, et al. The chronic and evolving neurological consequences of traumatic brain injury. Lancet Neurol. 2017;16(10):813–825. [DOI] [PMC free article] [PubMed]
- 14.Khellaf A, Khan DZ, Helmy A. Recent advances in traumatic brain injury. J Neurol. 2019;266:2878–2889. [DOI] [PMC free article] [PubMed]
- 15.DeWitt DS, Hawkins BE, Dixon CE, et al. Pre-clinical testing of therapies for traumatic brain injury. J Neurotrauma. 2018;35:2737–2754. doi: 10.1089/neu.2018.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams JH, Graham DI. An introduction to neuropathology. 2. Edinburgh: Churchill Livingstone; 1994. [Google Scholar]
- 17.Guo H, Camargo LM, Yeboah F, et al. A NMDA-receptor calcium influx assay sensitive to stimulation by glutamate and glycine/D-serine. Sci Rep. 2017;7(1):11608. doi: 10.1038/s41598-017-11947-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giza C, Greco T, Prins ML. Concussion: pathophysiology and clinical translation. Handb Clin Neurol. 2018;158:51–61. doi: 10.1016/B978-0-444-63954-7.00006-9. [DOI] [PubMed] [Google Scholar]
- 19.Jallo J, Loftus C, editors. Pathophysiology of traumatic brain injury. In: Neurotrauma and critical care of the brain. 2nd ed. New York: Thieme. 2018.
- 20.Taber KH, Warden DL, Hurley RA. Blast-related traumatic brain injury: what is known? J Neuropsychiatry Clin Neurosci. 2006;18(2):141–145. doi: 10.1176/jnp.2006.18.2.141. [DOI] [PubMed] [Google Scholar]
- 21.Lu J, Gary KW, Neimeier JP, et al. Randomized controlled trials in adult traumatic brain injury. Brain Inj. 2012;16(13–14):1523–1548. doi: 10.3109/02699052.2012.722257. [DOI] [PubMed] [Google Scholar]
- 22.Dahdah MN, Barnes S, Buros A, et al. Variations in inpatient rehabilitation functional outcomes across centers in the traumatic brain injury model systems study and the influence of demographics and injury severity on patient outcomes. Arch Phys Med Rehabil. 2016;97(11):1821–1831. doi: 10.1016/j.apmr.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 23.Bramlett HM, Dietrich WD. Long-term consequences of traumatic brain injury: current status of potential mechanisms of injury and neurological outcomes. J Neurotrauma. 2015;32(23):1834–1848. doi: 10.1089/neu.2014.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Broglio SP, Lapointe A, O’Connor KL, et al. Head impact density: a model to explain the elusive concussion threshold. J Neurotrauma. 2017;34(19):2675–2683. doi: 10.1089/neu.2016.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tellier A, Marshall SC, Wilson KG, et al. The heterogeneity of mild traumatic brain injury: where do we stand? Brain Inj. 2009;23(11):879–887. doi: 10.1080/02699050903200555. [DOI] [PubMed] [Google Scholar]
- 26.Dikmen S, Machamer J, Temkin N. Mild traumatic brain injury: longitudinal study of cognition, functional status, and post-traumatic symptoms. J Neurotrauma. 2017;34(8):1524–1530. doi: 10.1089/neu.2016.4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marklund N, Hillered L. Animal modeling of traumatic brain injury in preclinical drug development: where do we go from here. Br J Pharmacol. 2011;164(4):1207–1229. doi: 10.1111/j.1476-5381.2010.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Button KS, Ionnidis JPA, Mokrysz C, et al. Power failure: why small sample size undermines the reliability of neuroscience. Nat Rev Neurosci. 2013;14:365–376. doi: 10.1038/nrn3475. [DOI] [PubMed] [Google Scholar]
- 29.Lynch JR, Pineda JA, Morgan D, et al. Apolipoprotein E affects the central nervous system response to injury and the development of cerebral edema. Ann Neurol. 2002;51(1):113–117. doi: 10.1002/ana.10098. [DOI] [PubMed] [Google Scholar]
- 30.Holman C, Piper SK, Grittner U, et al. Where have all the rodents gone? The effects of attrition in experimental research on cancer and stroke. PLoS Biol. 2016;14:e1002331. doi: 10.1371/journal.pbio.1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fanelli D, Costas R, Ioannidia PA. Meta-assessment of bias in science. PNAS. 2016;114:3714–3719. doi: 10.1073/pnas.1618569114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stein D. Embracing failure: what the phase III progesterone studies can teach about TBI clinical trials. Brain Inj. 2015;29(11):1259–1272. doi: 10.3109/02699052.2015.1065344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marklund N, Bakshi A, et al. Evaluation of pharmacological treatment strategies in traumatic brain injury. Curr Pharm Des. 2006;12(13):1645–1680. doi: 10.2174/138161206776843340. [DOI] [PubMed] [Google Scholar]
- 34.Sellappan P, Cote J, Kreth P, et al. Variability and uncertainty in the rodent controlled cortical impact model of traumatic brain injury. J Neurosci Methods. 2019;312:37–42. doi: 10.1016/j.jneumeth.2018.10.027. [DOI] [PubMed] [Google Scholar]
- 35.Zhang YP, Cai J, Shields LBE. Traumatic brain injury using mouse models. Transl Stroke Res. 2014;5:454–471. doi: 10.1007/s12975-014-0327-0. [DOI] [PubMed] [Google Scholar]
- 36.Pineda JA, Aono M, Sheng H, et al. Extracellular superoxide dismutase overexpression improves behavioral outcome from closed head injury in the mouse. J Neurotrauma. 2001;18(6):625–634. doi: 10.1089/089771501750291864. [DOI] [PubMed] [Google Scholar]
- 37.Dixon CE, Clifton GL, Lighthall James W, et al. A controlled cortical impact model of traumatic brain injury in the rat. J NeuroSci Methods. 1991;39(3):253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 38.Bafeta A, Dechartres A, Trinquart L, et al. Impact of single centre status on estimates of intervention effects in trials with continuous outcomes: meta-epidemiological study. BMJ. 2012;344:e813. doi: 10.1136/bmj.e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bragge P, Synnot A, Maas AI, et al. A state-of-the-science overview of randomized controlled trials evaluating acute management of moderate-to-severe traumatic brain injury. J Neurotrauma. 2016;33:1461–1478. doi: 10.1089/neu.2015.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maas AI, Murray GD, Roozenbeck B, et al. Advancing care for traumatic for traumatic brain injury: findings from the IMPACT studies and perspectives on future research. Lancet Neurol. 2013;12:1200–1210. doi: 10.1016/S1474-4422(13)70234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agoston DV, Risling M, Bellander BM. Bench-to-bedside and bedside back to bench; coordinating clinical and experimental traumatic brain injury studies. Front Neurol. 2012;3:1–5. doi: 10.3389/fneur.2012.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu J, Murray GD, Steveberg EW, et al. Effects of Glasgow Outcome Scale misclassification on traumatic brain injury clinical trials. J Neurotrauma. 2008;25:641–651. doi: 10.1089/neu.2007.0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeiler FA, McFadyen C, Newcombe VFJ, et al. Genetic influences on patient-oriented outcomes in traumatic brain injury: a living systematic review of non-apolipoprotein e single-nucleotide polymorphisms. J Neurotrauma. 2021;38:1107–1123. doi: 10.1089/neu.2017.5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weisgraber KH. Apolipoprotein E: structure-function relationships. Adv Protein Chem. 1994;45:249–302. doi: 10.1016/S0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- 45.Frieden C, Garai K. Concerning the structure of apoE. Protein Sci. 2013;22:1820–1825. doi: 10.1002/pro.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 47.Castellano JM, Kim J, Stewart FR, et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3(89):89ra57. [DOI] [PMC free article] [PubMed]
- 48.Fleming LM, Weisgraber KH, Strittmatter WJ, et al. Differential binding of apolipoprotein E isoforms to tau and other cytoskeletal proteins. Exp Neurol. 1996;138(2):252–260. doi: 10.1006/exnr.1996.0064. [DOI] [PubMed] [Google Scholar]
- 49.Zhou W, Xu D, Peng X, et al. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J Neurotrauma. 2008;25:279–290. doi: 10.1089/neu.2007.0489. [DOI] [PubMed] [Google Scholar]
- 50.Zeng S, Jiang JX, Xu MH, et al. Prognostic value of apolipoprotein E epsilon4 allele in patients with traumatic brain injury: a meta-analysis and meta-regression. Genet Test Mol Biomarkers. 2014;18:202–210. doi: 10.1089/gtmb.2013.0421. [DOI] [PubMed] [Google Scholar]
- 51.Deng H, Ordaz A, Upadhyayula PS, et al. Apolipoprotein E epsilon 4 genotype, mild traumatic brain injury, and the development of chronic traumatic encephalopathy. Med Sci. 2018;6(3):78. doi: 10.3390/medsci6030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Atherton K, Han X, Chung J, et al. Association of apoE genotypes and chronic traumatic encephalopathy. JAMA Neurol. 2022;79:787–796. doi: 10.1001/jamaneurol.2022.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jordan BD, Relkin NR, Ravdin LD, et al. Apolipoprotein E epsilon4 associated with chronic traumatic brain injury in boxing. JAMA. 1997;278(02):136–140. doi: 10.1001/jama.1997.03550020068040. [DOI] [PubMed] [Google Scholar]
- 54.Kutner KC, Erlanger DM, Tsai J, et al. Lower cognitive performance of older football players possessing apolipoprotein E epsilon 4. Neurosurgery. 2000;47(03):651–657, discussion 657–658. [DOI] [PubMed]
- 55.Stern RA, Daneshvar DH, Baugh CM, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology. 2013;81(13):1122–1129. doi: 10.1212/WNL.0b013e3182a55f7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136(Pt 1):43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bieniek KF, Ross OA, Cormier KA, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol. 2015;130(6):877–889. doi: 10.1007/s00401-015-1502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laskowitz DT, Goel S, Bennett E, Matthew WD. Apolipoprotein E suppresses glial secretion of TNF alpha. J Neuroimmunol. 1997;76(1–2):70–74. doi: 10.1016/S0165-5728(97)00021-0. [DOI] [PubMed] [Google Scholar]
- 59.Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388(6645):878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- 60.Parhizkar S, Holtzman DM. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin Imunol. 2022;59:101594. doi: 10.1016/j.smim.2022.101594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iannucci J, Sen A, Grammas P. Isoform-specific effects of apolipoprotein E on markers of inflammation and toxicity in brain glia and neuronal cells in vitro. Curr Issues Mol Biol. 2021;215–225. [DOI] [PMC free article] [PubMed]
- 62.Laskowitz DT, Thekde AD, Thekde SD, et al. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptide. Exp Neurol. 2001;167(1):74–85. doi: 10.1006/exnr.2001.7541. [DOI] [PubMed] [Google Scholar]
- 63.Moretti EW, Morris RW, Podgoreanu M, et al. APOE polymorphism is associated with risk of severe sepsis in surgical patient. Crit Care Med. 2005;33(11):2521–2526. doi: 10.1097/01.CCM.0000186368.96146.FB. [DOI] [PubMed] [Google Scholar]
- 64.Tao Q, TFA, Akhter-Khan S, , et al. Impact of C-reactive protein on cognition and Alzheimer disease biomarkers in homozygous apolipoprotein E ɛ4 carriers. Neurology. 2021;97(12):e1243–e1252. doi: 10.1212/WNL.0000000000012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCann MS, Washington PM, Rodriguez OC, et al. Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Mol Neurodegener. 2018;13(1):17. doi: 10.1186/s13024-018-0249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beisiegel U, Weber W, Ihrke G, et al. The ldl-receptor-related protein, lrp, is an apolipoprotein e-binding protein. Nature. 1989;341(6238):162–164. doi: 10.1038/341162a0. [DOI] [PubMed] [Google Scholar]
- 67.Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron. 1993;11:575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 68.Misra UK, Adlakha CL, Gawdi G, et al. Apolipoprotein E and mimetic peptide initiate a calcium-dependent signaling response in macrophages. J Leukoc Biol. 2001;70(4):667–683. doi: 10.1189/jlb.70.4.677. [DOI] [PubMed] [Google Scholar]
- 69.Croy JE, Brandon T, Komives EA. Two apolipoprotein E mimetic peptides, ApoE(130–149) and ApoE(141–155)2, bind to LRP1. Biochemistry. 2004;43(23):7328–7335. doi: 10.1021/bi036208p. [DOI] [PubMed] [Google Scholar]
- 70.Guttman M, Prieto JH, Handel TM, et al. Structure of the minimal interface between ApoE and LRP. J Mol Biol. 2010;398(2):306–319. doi: 10.1016/j.jmb.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang L, Liu CC, Zheng H, et al. LRP1 modulates the microglial immune response via regulation of JNK and NF-κB signaling pathways. J Neuroinflammation. 2016;13(1):304. doi: 10.1186/s12974-016-0772-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pocivavsek A, Mikhailenko I, Strickland DK, Rebeck GW. Microglial low-density lipoprotein receptor-related protein 1 modulates c-Jun N-terminal kinase activation. J Neuroimmunol. 2009;214(1–2):25–32. doi: 10.1016/j.jneuroim.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yu AW, Cutcliffe HC, Laskowitz DT, et al. Neuroprotective effect of an ApoE mimetic peptide in a gyrencephalic blast animal model. Presented at the 2018 Military Health System Research Symposium; August 29–23, 2018. Orlando.
- 74.Bell R, Winkler E, Singh I, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roselaar SE, Daugherty A. Apolipoprotein E-deficient mice have impaired innate immune responses to Listeria monocytogenes in vivo. J Lipid Res. 1998;39:1740–1743. doi: 10.1016/S0022-2275(20)32160-X. [DOI] [PubMed] [Google Scholar]
- 76.de Bont N, Netea MG, Demacker N, et al. Apolipoprotein E knock-out mice are highly susceptible to endotoxemia and Klebsiella pneumonia infection. J Lipid Res. 1999;40:680–685. doi: 10.1016/S0022-2275(20)32147-7. [DOI] [PubMed] [Google Scholar]
- 77.Laskowitz DT, Lee DM, Schmechel D, Staats HF. Altered immune responses in apolipoprotein E-deficient mice. J Lipid Res. 2000;41(4):613–620. doi: 10.1016/S0022-2275(20)32409-3. [DOI] [PubMed] [Google Scholar]
- 78.Lynch JR, Morgan D, Mance J, et al. Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J Neuroimmunol. 2001;114(1–2):107–113. doi: 10.1016/S0165-5728(00)00459-8. [DOI] [PubMed] [Google Scholar]
- 79.Van Oosten M, Rensen PC, Van Amersfoort ES, et al. Apolipoprotein E protects against bacterial lipopolysaccharide-induced lethality: a new therapeutic approach to treat gram-negative sepsis. J Biol Chem. 2001;276(12):8820–8824. doi: 10.1074/jbc.M009915200. [DOI] [PubMed] [Google Scholar]
- 80.Lynch JR, Tang W, Wang H, et al. APOE genotype and an apoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. 2003;278(49):48529–48533. doi: 10.1074/jbc.M306923200. [DOI] [PubMed] [Google Scholar]
- 81.Giarratana AO, Zheng C, Reddi S, et al. APOE4 genetic polymorphism results in impaired recovery in a repeated mild traumatic brain injury model and treatment with bryostatin-1 improves outcomes. Sci Rep. 2020;10(1):19919. doi: 10.1038/s41598-020-76849-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gao J, Wang H, Sheng H, et al. A novel apoE-derived therapeutic reduces vasospasm and improves outcome in a murine model of subarachnoid hemorrhage. Neurocrit Care. 2006;4(1):25–31. doi: 10.1385/NCC:4:1:025. [DOI] [PubMed] [Google Scholar]
- 83.James ML, Sullivan PM, Lascola CD, et al. Pharmacogenomic effects of apolipoprotein e on intracerebral hemorrhage. Stroke. 2009;40(2):632–639. doi: 10.1161/STROKEAHA.108.530402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sheng H, Laskowitz DT, Bennett E, et al. Apolipoprotein E isoform specific differences in outcome from focal ischemia in transgenic mice. J Cereb Blood Flow and Metab. 1998;18(4):361–366. doi: 10.1097/00004647-199804000-00003. [DOI] [PubMed] [Google Scholar]
- 85.Sheng H, Laskowitz DT, Mackensen GB, et al. Apolipoprotein E deficiency worsens outcome from global ischemia in the mouse. Stroke. 1999;30(5):1118–1124. doi: 10.1161/01.STR.30.5.1118. [DOI] [PubMed] [Google Scholar]
- 86.Linton MF, Gish R, Hubl ST, et al. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;88(1):270–281. doi: 10.1172/JCI115288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dyer CA, Smith RS, Curtiss LK. Only multimers of a synthetic peptide of human apolipoprotein e are biologically active. J Biol Chem. 1991;266(23):15009–15015. doi: 10.1016/S0021-9258(18)98578-9. [DOI] [PubMed] [Google Scholar]
- 88.Dyer CA, Cistola DP, Parry GC, Curtiss LK. Structural features of synthetic peptides of apolipoprotein E that bind the LDL receptor. J Lipid Res. 1995;36(1):80–88. doi: 10.1016/S0022-2275(20)39756-X. [DOI] [PubMed] [Google Scholar]
- 89.Clay MA, Anantharamaiah GM, Mistry MJ, et al. Localization of a domain in apolipoprotein E with both cytostatic and cytotoxic activity. Biochemistry. 1995;34:11142–11151. doi: 10.1021/bi00035a020. [DOI] [PubMed] [Google Scholar]
- 90.Lynch JR, Wang H, Mace B, et al. A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Exp Neurol. 2005;192(1):109–116. doi: 10.1016/j.expneurol.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 91.Laskowitz DT, Fillit H, Yeung M, et al. Apolipoprotein E-derived peptides reduce CNS inflammation: implications for therapy of neurological disease. Acta Neurol Scand Suppl. 2006;185:15–20. doi: 10.1111/j.1600-0404.2006.00680.x. [DOI] [PubMed] [Google Scholar]
- 92.Saatman KE, Duhaime A-C, Bullock R. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25(7):719–738. doi: 10.1089/neu.2008.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aono M, Bennett ER, Kim KS, et al. Protective effect of apolipoprotein E-mimetic peptides on N-methyl-d-aspartate excitotoxicity in primary rat neuronal-glial cultures. Neuroscience. 2003;116(2):437–445. doi: 10.1016/S0306-4522(02)00709-1. [DOI] [PubMed] [Google Scholar]
- 94.Sheng Z, Prorok M, Brown BE, Castellino FJ. N-methyl-d-aspartate receptor inhibition by an apolipoprotein E-derived peptide relies on low-density lipoprotein receptor-associated protein. Neuropharmacology. 2008;55(2):204–214. doi: 10.1016/j.neuropharm.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Qiu Z, Crutcher KA, Hyman BT, Rebeck GW. ApoE isoforms affect neuronal N-methyl-d-aspartate calcium responses and toxicity via receptor-mediated processes. Neuroscience. 2003;122(2):291–303. doi: 10.1016/j.neuroscience.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 96.Laskowitz DT, McKenna SE, Song P, et al. COG1410, a novel apolipoprotein E-based peptide, improves functional recovery in a murine model of traumatic brain injury. J Neurotrauma. 2007;24(7):1093–1107. doi: 10.1089/neu.2006.0192. [DOI] [PubMed] [Google Scholar]
- 97.Hoane MR, Kaufman N, Vitek MP, McKenna SE. COG1410 improves cognitive performance and reduces cortical neuronal loss in the traumatically injured brain. J Neurotrauma. 2009;26(1):121–129. doi: 10.1089/neu.2008.0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang H, Anderson LG, Lascola CD, et al. Apolipoprotein E mimetic peptides improve outcome after focal ischemia. Exp Neurol. 2013;241:67–74. doi: 10.1016/j.expneurol.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 99.Laskowitz DT, Lei B, Dawson HN, et al. The apoE-mimetic peptide, COG1410, improves functional recovery in a murine model of intracerebral hemorrhage. Neurocrit Care. 2012;16(2):316–326. doi: 10.1007/s12028-011-9641-5. [DOI] [PubMed] [Google Scholar]
- 100.Pang J, Chen Y, Kuai L, et al. Inhibition of blood-brain barrier disruption by an apolipoprotein e-mimetic peptide ameliorates early brain injury in experimental subarachnoid hemorrhage. Transl Stroke Res. 2017;8(3):257–272. doi: 10.1007/s12975-016-0507-1. [DOI] [PubMed] [Google Scholar]
- 101.Van Wyck D, Kolls BJ, Wang H, et al. Prophylactic treatment with CN-105 improves functional outcomes in a murine model of closed head injury. Exp Brain Res. 2022;240(9):2413–2423. doi: 10.1007/s00221-022-06417-4. [DOI] [PubMed] [Google Scholar]
- 102.Laskowitz DT, Wang H, Chen T, et al. Neuroprotective pentapeptide CN-105 is associated with reduced sterile inflammation and improved functional outcome in a traumatic brain injury murine model. Sci Rep. 2017;7:46461. doi: 10.1038/srep46461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu J, Zhou G, Kolls BJ, et al. The apolipoprotein E mimetic peptide CN-105 improves outcome in a murine model of SAH. Stroke and Vasc Neurol. 2018;3(4):222–230. doi: 10.1136/svn-2018-000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lei B, James ML, Liu J, et al. Erratum: neuroprotective pentapeptide CN-105 improves functional and histological outcomes in a murine model of intracerebral hemorrhage. Sci Rep. 2017;7:39580. doi: 10.1038/srep39580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tu TM, Kolls B, Soderblom EJ, et al. Apolipoprotein E mimetic peptide, CN-105, improves outcomes in ischemic stroke. Ann Clin Transl Neurol. 2017;9(4):246–265. doi: 10.1002/acn3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xue M, Li S, Xu M, et al. Antagonism of nicotinic acetylcholinergic receptors by CN-105, an apoE-mimetic peptide reduces stroke-induced excitotoxicity. Clin Transl Med. 2022;12(1):e677. doi: 10.1002/ctm2.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guptill JT, Raja S, Ramey S, et al. Phase I randomized, double-blind, placebo-controlled study to determine the safety, tolerability, and pharmacokinetics of a single escalating dose and repeated doses of CN-105 in healthy adult subjects. J Clin Pharmacol. 2017;57(6):770–776. doi: 10.1002/jcph.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li S, Wangqin R, Meng X, et al. Tolerability and pharmacokinetics of single escalating and repeated doses of CN-105 in healthy participants. Clin Ther. 2022;44(2):744–754. doi: 10.1016/j.clinthera.2022.03.006. [DOI] [PubMed] [Google Scholar]
- 109.James ML, Troy J, Nowacki, et al. CN-105 in participants with acute supratentorial intracerebral hemorrhage (CATCH) trial. Neurocrit Care. 2021;36(1):216–225. [DOI] [PubMed]
- 110.Woo D, Rosand J, Kidwell C, McCauley JL, et al. The ethnic/racial variations of intracerebral hemorrhage (erich) study protocol. Stroke. 2013;44:e120–125. doi: 10.1161/STROKEAHA.113.002332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000 Feb 29. Identifier NCT05826912, evaluation of CN-105 in subject with acute supratentorial intracerebral hemorrhage (S-CATCH). 2018 Oct 19 [cited 2023 Jul 03]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT03711903.
- 112.Berger M, Nadler JW, Browndyke J, et al. Postoperative cognitive dysfunction: minding the gaps in our knowledge of a common postoperative complication in the elderly. Anesthesiol Clin. 2015;33:517–555. doi: 10.1016/j.anclin.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hirsch J, Vacas S, Terrando N, et al. Perioperative cerebrospinal fluid and plasma inflammatory markers after orthopedic surgery. J Neuroinflammation. 2016;13(1):211. doi: 10.1186/s12974-016-0681-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Van Dusen KW, Eleswarpu S, Moretti EW, et al. The MARBLE study protocol: modulating apoE signaling to reduce brain inflammation, delirium, and postoperative cognitive dysfunction. J Alzheimers Dis. 2020;75(4):1319–1328. doi: 10.3233/JAD-191185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hoane MR, Pierce JL, Holland MA, et al. The novel apolipoprotein E-based peptide COG1410 improves sensorimotor performance and reduces injury magnitude following cortical contusion injury. J Neurotrauma. 2007;24(7):1108–1118. doi: 10.1089/neu.2006.0254. [DOI] [PubMed] [Google Scholar]
- 116.Wang H, Durham L, Dawson H, et al. An apolipoprotein E-based therapeutic improves outcome and reduces Alzheimer’s disease pathology following closed head injury: evidence of pharmacogenomic interaction. Neuroscience. 2007;144(4):1324–1333. doi: 10.1016/j.neuroscience.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 117.Cao F, Jiang Y, Wu Y, et al. Apolipoprotein E-mimetic COG1410 reduces acute vasogenic edema following traumatic brain injury. J Neurotrauma. 2016;33(2):175–182. doi: 10.1089/neu.2015.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jiang Y, Brody D. Administration of COG1410 reduces axonal amyloid precursor protein immunoreactivity and microglial activation after controlled cortical impact in mice. J Neurotrauma. 2012;13:2332–2341. doi: 10.1089/neu.2012.2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kaufman NA, Beare JE, Tan AA, et al. COG1410, an apolipoprotein E-based peptide, improves cognitive performance and reduces cortical loss following moderate fluid percussion injury in the rat. Behav Brain Res. 2010;214(2):395–401. doi: 10.1016/j.bbr.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Qin X, You H, Cao F, et al. Apolipoprotein E mimetic peptide increases cerebral glucose uptake by reducing blood-brain barrier disruption after controlled cortical impact in mice: an (18) F-fluorodeoxyglucose PET/CT study. J Neurotrauma. 2017;34(4):943–951. doi: 10.1089/neu.2016.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hier DB, Obafemi-Ajayi T, Thimgan MS, et al. Blood biomarkers for mild traumatic brain injury: a selective review of unresolved issues. Biomark Res. 2021;9(1):70. doi: 10.1186/s40364-021-00325-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mouhieddine TH, El Houjeiri L, Sabra M, et al. CNS trauma biomarkers and surrogate endpoints pipeline from bench to bedside: a translational perspective. In: Kobeissy FH, editor. Brain neurotrauma: molecular, neuropsychological, and rehabilitation aspects. Boca Rato: CRC Press/Taylor & Francis. 2015. [PubMed]
- 123.Korley FK, Jain S, Sun X, et al. Prognostic value of day-of-injury plasma GFAP and UCH-L1 concentrations for predicting functional recovery after traumatic brain injury in patients from the US TRACK-TBI cohort: an observational cohort study. Lancet Neurol. 2022;21(9):803–813. doi: 10.1016/S1474-4422(22)00256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bazarian JJ, Biberthaler P, Welch RD, et al. Serum GFAP and UCH-L1 for prediction of absence of intracranial injuries on head CT (ALERT-TBI): a multicentre observational study. Lancet Neurol. 2018;17(9):782–789. doi: 10.1016/S1474-4422(18)30231-X. [DOI] [PubMed] [Google Scholar]
- 125.Bazarian JJ, Welch RD, Caudle K, et al. Accuracy of rapid glial fibrillary acidic protein/ubiquitin carboxy-terminal hydrolase L1 test for the prediction of intracranial injuries on head computed tomography after mild traumatic brain injury. Acad Emerg Med. 2021;28(11):1308–1317. doi: 10.1111/acem.14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000 Feb 29. Identifier NCT05826912, Multi-arm multi-stage adaptive platform trial (APT) for the acute treatment of traumatic brain injury (APT-TBI-01); 2023 Apr 24 [cited 2023 Jul 03]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT05826912.
- 127.Katlowitz K, Gopinath S, Cruz Navarro J, Robertson C. HMG-CoA reductase inhibitors for traumatic brain injury. Neurotherapeutics. 2023 doi: 10.1007/s13311-023-01399-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data supporting the findings of this study are available/cited within the paper. All pre-clinical data has been made public in the referenced works.