

Summary
The central role of target antigen density on chimeric antigen receptor T cell potency highlights the need for accurate measurement of antigen levels on clinical tumor samples. Here, we present a protocol for quantifying antigen density for six cell-surface antigens on neuroblastoma cells metastatic to bone marrow. We describe steps for patient sample acquisition, flow cytometry panel development, instrument setup, and compensation and detail procedures for running clinical samples and data analysis.
For complete details on the use and execution of this protocol, please refer to Heitzeneder et al. (2022).1
Subject areas: Flow Cytometry, Cancer, Immunology
Graphical abstract
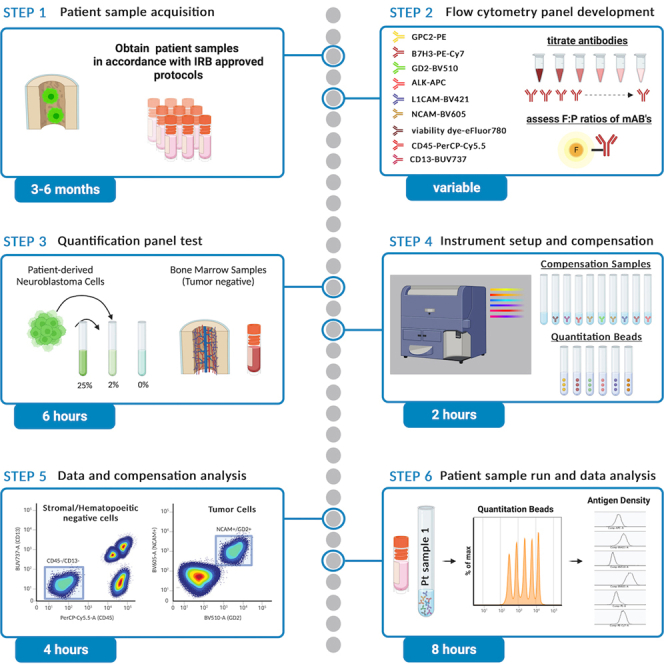
Highlights
-
•
Quantification of antigen density for multiple cell-surface tumor markers on clinical samples
-
•
Protocol for measurements on neuroblastoma-infiltrating bone marrow specimens
-
•
Flow cytometric assay using corresponding fluorescent calibration microbeads
-
•
Protocol can be modified for other antigens and tumor specimens
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
The central role of target antigen density on chimeric antigen receptor T cell potency highlights the need for accurate measurement of antigen levels on clinical tumor samples. Here, we present a protocol for quantifying antigen density for six cell-surface antigens on neuroblastoma cells metastatic to bone marrow. We describe steps for patient sample acquisition, flow cytometry panel development, instrument setup, and compensation and detail procedures for running clinical samples and data analysis.
Before you begin
Accurate measurement of quantitative CAR T cell target antigen density on clinical specimens is a critical correlative tool for CAR T cell potency, which is highly dependent on this metric.2 Insufficient reactivity against low-antigen density tumor cells has emerged as a crucial resistance mechanism, as evidenced in patients with refractory or recurrent acute B-cell lymphoblastic leukemia (B-ALL) post CD22 CAR T cell immune pressure.3
Note: We previously used quantitative measurements of CD19, CD20 and CD22 on lymph node fine-needle aspirate specimens of patients enrolled in clinical trials of CAR T therapies targeting CD19 (axicabtagene ciloleucel), and a novel bi-specific CD19/CD22 CAR T cell construct pre- and post-treatment to correlate patient responses.4 This work credentialed the measurement of CD19 molecules/cell on large B cell lymphoma (LBCL) clinical specimens as a critical tool to predict future disease progression associated with lower antigen levels post axi-cel treatment and allowed to define an antigen threshold associated with relapse. Most novel CAR T cell constructs are not tailored to detect antigen density thresholds present in clinical samples, which is a plausible cause for failure when tested in clinical trials. Hence, we harnessed the quantitative assessment of target antigen levels on clinical specimens to inform the rational design of novel CAR T cell receptors targeting Glypican-2 (GPC2).1
This protocol describes reagents and the specific steps for cell-surface antigen density quantification of six neuroblastoma markers including GPC2 on viable tumor cells metastatic to the bone marrow of patients. These antigens were chosen based on their potential or ongoing efforts as targets for novel CAR T cell therapy development.5 The protocol uses multi-color flow cytometry with well-defined antibody-fluorophore conjugate reagents and fluorescence quantitation beads for each of the six quantitation channels. The 9-color antibody panel is strategically designed to 1) identify the tumor cell population as the viable cell fraction negative for both the hematopoietic lineage marker CD45 and neuroblastoma-mimicking bone marrow stromal cells based on CD13 expression and 2) quantify antigen density (molecules per cell) of the six cell-surface tumor markers GPC2, B7-H3, GD2, ALK, L1CAM and NCAM to assess their potential as CAR T cell targets (Figure 1A).
Note: Measurement of antigen density on cells by flow cytometry is based on using saturating antibody to bind antigens, using antibody-fluorophore conjugates with known ratio of antibody to fluorophore (fluorophore-to-protein or F:P ratio), and carefully measuring the fluorescence intensity of each cell with a calibrated flow cytometer. Consequently, for every cell the fluorescence intensity can be related back to the number of antibodies bound per cell, and hence, because antibodies are saturating, to the number of antigens per cell. Calibration of the flow cytometer fluorescence scale is typically performed using microbeads with known numbers of fluorophore molecules to create a linear relationship between the fluorescence of a given flow cytometer and number of fluorescent molecules (Figure 1B). This approach has been used for single-color measurement of PE-labeled reagents for many years6,7,8,9,10 with calibration microbeads such as BD Quantibrite beads6 and more recently extended to multi-color measurements in the same sample with additional fluorescence quantitation beads.4
Figure 1.

Multicolor antigen density quantification panel using pre-calibrated beads alongside monoclonal antibodies of known fluorophore-to-protein ratios
(A) Entire 9-color antibody panel containing six antibodies specific for neuroblastoma cell surface markers used for antigen density quantitation and three additional colors for gating purposes.
(B) Fluorescent calibration beads contain several populations with known fluorophore molecules/bead and allow conversion of MFI into antigen density for antibody-fluorophore conjugates with known F:P ratio used at saturating concentrations.
(C) Plate set up for titration of antibodies used in the panel to assess saturating concentrations using the negative control cell line CHO-K1 and two neuroblastoma specimens (the cell line SMS-SAN and the patient-derived neuroblastoma cells ST16-4045). Serial dilutions range from no antibody to 0.1, 1, 5, 10, 20 and 30 μg/mL in a total of 100 μL FACS buffer containing fixable viability dye. Example plate setup for three antibodies (GPC2-PE, GD2-BV510, ALK-APC) is shown. A second plate with the same setup was run for B7H3-PE-Cy7, L1CAM-BV421, NCAM-BV605).
Here, we use BD Quantibrite beads in PE and custom quantitation beads in the fluorophores APC, PE-Cy7, BV421, BV510, and BV605, together with monoclonal antibodies labeled with matching fluorophores with defined F:P ratios, to perform the antigen density measurements. The protocol details the development and use of appropriate flow cytometry panels, specimen handling and sample preparation, and data acquisition and analysis and is applicable to other antigens and tumor types infiltrating the bone marrow if appropriate monoclonal antibodies with defined F:P ratios can be identified. The protocol can be extended to other specimen types with modified sample preparation if the population of interest can be definitively identified by flow cytometry.
Individuals performing these experiments must have received safety training (BSL2+) on the proper handling and disposal of human biological materials.
Institutional permissions
Patient tumor samples analyzed in this protocol were collected according to the guidelines of Stanford University Institutional Review Board approved protocols.
Obtaining patient samples
Timing: 3–6 months prior to experiment with patient specimens (depending on sample availability)
-
1.To obtain appropriate clinical samples for antigen density quantification, work with the respective Institutional Review Board (IRB) to receive bone-marrow samples containing viable tumor cells with intact cell-surface membranes under IRB-approved protocols.Note: We used tumor-infiltrated bone marrow samples obtained at the time of routine bone marrow aspiration from neuroblastoma patients, that were Ficoll-purified within 1–6 hours of specimen procurement.
-
a.Perform purification using Ficoll-Paque PLUS density gradient media according to manufacturer’s instructions.
-
b.After the final centrifugation step, resuspend samples in FBS and viably freeze after adding 2X cryomedia, consisting of 80% FBS and 20% DMSO. To ensure optimal cell preservation, transfer cryovials to a ‒80°C freezer in Nalgene Mr. Frosty containers, in which the recommended controlled cooling rate of 1°C/min is achieved. Move to liquid nitrogen storage within 24 h and keep there until use.Note: If feasible, samples can also be run fresh but were not extensively tested in our study.For further details about patient characteristics, please refer to Heitzeneder et al.1
-
a.
Flow cytometry panel development for antigen density quantitation
Note: If changes to the antibody panel described in this protocol are being made, antibody clones and fluorophore combinations should be selected carefully for optimum performance under saturating conditions. Unlike conventional flow cytometry where antibody titrations can be chosen to maximize stain index, saturating concentrations are critical for antigen density quantification. Further important considerations are the sensitivity of the flow cytometry instrument in different fluorescent channels, spillover between channels, available antibody clones, apparent brightness of available fluorophores, compensation controls, and the expected range of antigen density of the markers.
-
2.Develop the flow cytometry panel under careful considerations to ensure an adequate signal-to-noise stain index for defined populations.
-
a.Characterize the Flow Cytometer to-be-used with the quantitation beads to determine its sensitivity in each channel for quantitation.
-
b.Chose monoclonal antibody clones for antigens of interest and prioritize reagents such that the lowest expressed antigens are detected in the most sensitive fluorescent channels. Place highly expressed antigens in fluorescent channels with minimal spillover to those used for antigens expressed at low levels.
-
c.The final panel might be limited by available antibody conjugates. Test the panel comprehensively on representative preclinical samples, before moving to precious clinical samples.
-
a.
The quantitative flow cytometry panel developed for this assay is illustrated in Figure 1. Specific items related to optimization are detailed below.
Quantitative antibody panel setup with known fluorophore-to-protein ratios
-
3.Determine the fluorophore-to-protein (F:P) ratios for all monoclonal antibodies used in the quantitation panel.
-
a.Obtain F:P ratios on request from the manufacturer when possible. For antibodies used in this protocol, BioLegend provided manufacturer-determined information on F:P ratios for NCAM-BV605, B7-H3-PE-Cy7 and GD2-BV510.
-
b.Conjugate an unlabeled antibody to-be-used in the panel with PE using the Abcam PE conjugation kit Lightning-Link (or use a custom PE conjugation service from a reagent vendor). This kit results in an expected F:P ratio of 1:1 according to manufacturer’s instructions. In this protocol, the custom-made GPC2 antibody was conjugated to PE using this approach.
-
c.The 1:1 ratio does not generally apply to conjugation with other fluorophores. Estimate the F:P ratio for antibodies with unknown F:P ratios by comparison to the PE version (1:1 F:P) of the same clone at same titration with quantitation beads to determine number of antibodies bound per cell (ABC).
-
a.
Note: For instance, in this protocol, the ALK antibody was conjugated to both APC and PE using Abcam Conjugation Lightning-Link kits and the F:P ratio for the ALK-APC clone was calculated from comparison of cells stained with this antibody and the PE-conjugated version of the same clone (F:P = 1:1) at the same concentration using BD Quantibrite PE and APC quantitation bead measurements on a BD LSRFortessa X-20 flow cytometer. A similar approach was taken for L1CAM-BV421, for which a commercially available PE-conjugated version of the same clone with F:P of 1:1 was used. Table 1 shows calculations of the F:P ratios for the ALK-APC and L1CAM-BV421 antibody conjugates in the present study.
Table 1.
Example determination of F:P ratios for ALK-APC and L1CAM-BV421 antibody conjugates
| MFI values |
||||||
|---|---|---|---|---|---|---|
| Labeled antibody | BV421 | PE | APC | Fluorophore molecules/cell | F:P ratio | Antigen molecules/cell |
| ALK-APC | 595 | 4325 | 3.34b | 1296 | ||
| ALK-PE | 269 | 1296 | 1:1a | 1296 | ||
| L1CAM-BV421 | 864 | 8041 | 2.21b | 3632 | ||
| L1CAM-PE | 788 | 3632 | 1:1a | 3632 | ||
Determination of fluorophore-to-protein ratios of a given monoclonal antibody (ALK-APC and L1CAM-BV421) by staining neuroblastoma cells with these mABs and the same clone labeled in PE with 1:1 F:P ratios, respectively. Quantibrite-PE and quantitation beads in APC or BV421 allow calculation of F:P from fluorophore molecules/cell with equivalent antigen molecules/cell.
Known F:P ratio.
Calculated F:P ratio as detailed in Table 3 and according manuscript section.
Note: If the above approach is not feasible to determine F:P ratio, a conventional spectroscopy approach11 can be applied. For determination of ABC values using other fluorophores such as FITC, Rhodamine, Alexa Fluor dyes (with small molecular weights), the approach employed by Wang et al.8,12,13 should be carefully followed.
Antibody titrations
Prior to sample analysis, the optimal concentration of each antibody used in the panel needs to be determined. Antibodies used for antigen density quantitation should be used at the minimal saturating concentration; other antibodies can be titrated to maximize stain index. Cells used for the titrations should be carefully chosen from patient specimens, healthy donors, or a suitable model system that expresses the markers of interest. Neuroblastoma patient specimens are difficult to obtain in sufficient quantity for research studies, and availability of viably frozen samples is limited. Hence, in this protocol, we used CHO-K1 cells as negative controls, which do not specifically bind any of our antibodies of interest. As positive control specimens, we used the neuroblastoma cell line SMS-SAN known to express all antigens in this panel based on prior experiments and a patient-derived neuroblastoma cell line isolated from a patient’s bone marrow (ST16-BM4045), which we have generated. Cells were stained at increasing antibody concentrations and analyzed on a BD LSRFortessa X-20 flow cytometer in plate-mode (alternatively titration can be performed in tube mode). FACS buffer is prepared as outlined in the materials and equipment section and stored at 4°C for up to one month. Viability dye is used to ensure only live cells are analyzed.
-
4.Determine the optimal concentration of each conjugated monoclonal antibody utilized in the quantification panel to ensure usage at minimal saturating conditions.
-
a.Prepare antibody staining mixes.
-
i.Vortex and spin down antibodies and viability dye and place on ice protected from light.
-
ii.Prepare fixable viability dye staining mix by diluting 1:1000 in FACS buffer.
-
iii.Prepare serial dilutions of all antibodies to obtain concentrations ranging from 0.1 μg/mL to 30 μg/mL in a total of 100 μL fixable viability dye staining mix (Figure 1C).
-
i.
-
b.Prepare cultured positive and negative control cell lines.
-
i.Harvest cells and wash once with PBS, pellet by centrifugation at a temperature of 18°C–22°C for 5 min at 335 × g, resuspend in FACS buffer and count using an automated cell counter. For cells that form spheres, such as our patient-derived neuroblastoma cell line ST16-BM4045, we performed non-enzymatic incubation with Cellstripper (Corning) for 1 min to bring cells into a single cell suspension prior to the PBS wash step.
-
ii.Bring cells to a concentration of 1.5 × 106 cells per mL and aliquot 200 μL (equals 0.3 × 106) into each well of a round-bottom 96-well plate using a multi-channel pipette.
-
iii.Centrifuge the plate at 335 × g for 5 min and discard supernatant (referred to hereafter as “centrifugation step”), and wash cells twice with 200 μL of FACS buffer using a multi-channel pipette.
-
i.
-
c.Stain cells and acquire data.
-
i.Add 100 μL of antibody mix to cells as directed on the plate map, mix well by pipetting up and down, and stain for 30 min on ice in the dark.
-
ii.After the incubation, perform centrifugation step and wash cells twice with 200 μL of FACS buffer.
-
iii.Resuspend cells in 100 μL of FACS buffer and run samples on flow cytometer in plate mode. Ensure that the positive population is within the range of detection of the instrument.
-
iv.For antibodies used in antigen density quantitation, analyze results to determine saturating concentrations of each antibody in positive control specimens in the absence of significant background staining in negative control cell lines for the use in the experiment. For other antibodies, optimize stain index. See Table 2 for the final panel used.
-
i.
-
a.
CRITICAL: Antibody conjugates are light and temperature sensitive and must be stored in the dark to preserve their stability.
Table 2.
Antibody panel used for quantification of neuroblastoma cell surface antigens in patient bone marrow samples
| Laser | Filter | Fluorophore | Antibody | Clone | Isotype | Purpose | LOT | mAB conc. | Use per testd | Staining conc. | F:P assessment | F:P ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Red 640 nM |
780/60 | eFluor 780 | fixable viability dye |
Gate out dead cells | 0.1 μL | N/A | N/A | N/A | ||||
| Red 640 nM | 670/30 | APC | ALKb | ALK-48 | Mouse IgG2a | Tumor marker | custom | 1 mg/mL | 0.2 μL | 0.2 mg/mL | determined with PE-clonea | 3.34 |
| Violet 405 nM |
525/50 | BV510 | GD2 | 14g2a | Mouse IgG2a, κ | Tumor marker | B280958 | 0.1 mg/mL | 5 μL | 0.5 mg/mL | provided by BioLegend | 3.1 |
| Violet 405 nM |
610/20 | BV605 | NCAM (CD56) | 5.1H11 | Mouse IgG1, κ | Tumor marker | B280386 | 0.1 mg/mL | 1 μL | 0.1 mg/mL | provided by BioLegend | 3.4 |
| Violet 405 nM |
450/50 | BV421 | L1CAM (CD171) | 5G3 | Mouse IgG2a | Tumor marker | 9241038 | 0.4 mg/mL | 0.5 μL | 0.2 mg/mL | determined with PE-clonec | 2.21 |
| Blue 488 nM |
780/60 | PE-Cy7 | B7H3 | MIH42 | Mouse IgG1, κ | Tumor marker | B293754 | 0.4 mg/mL | 2.5 μL | 1 mg/mL | provided by BioLegend | 0.92 |
| Blue 488 nM |
575/26 | PE | GPC2a | D3 | Human IgG1 | Tumor marker | custom | 1 mg/mL | 0.5 μL | 0.5 mg/mL | 1:1 | |
| Blue 488 nM |
710/50 | PerCP-Cy5.5 | CD45 | HI30 | Mouse IgG2b,κ | Gate out hematopoietic cells | 0.025 mg/mL | 4 μL | 0.1 mg/mL | N/A | N/A | |
| UV 355 nM | 740/35 | BUV737 | CD13 | L138 | Mouse IgG1, κ | Gate out bone marrow stroma cells | 0.2 mg/mL | 2.5 μL | 0.5 mg/mL | N/A | N/A |
PE-conjugated using Abcam PE Conjugation kit Lightning-Link.
APC-conjugated using Abcam APC Conjugation kit Lightning-Link.
PE-conjugated clone from BD Biosciences (Cat# 564193, LOT: 0133728).
saturating antibody per titration results, volume to add for 100 μL total staining volume.
Test antibody panel on spike-in control bone marrow samples
Prior to running patient samples, the sample preparation, staining panel, instrument setup, compensation scheme, and gating strategy to distinctively detect the tumor cell population must be tested on samples less precious than clinical tumor specimens.
Note: Given the expected rare tumor cell infiltration in some bone marrow patient samples, control samples must be supplied using alternative specimens. To mimic such samples, we use tumor-uninvolved bone marrow samples derived from patients with B-cell acute lymphoblastic leukemia at remission time points with and without spike-in of ST16-BM4045 neuroblastoma to test the entire panel and for florescence minus one control samples (FMO: tube containing the entire panel, except one target antibody, to understand fluorescent spreading and spillover). ST16-4045 was also used as single-stain controls for compensation of biologically relevant signal for high-expressing antigens alongside compensation beads for low/moderate expressing antigens. In addition, we tested the panel on neuroblastoma cell lines with known antigen density of GPC2 ranging from low to high (SMS-SAN, NB-SD, NGP-GPC2) as positive and CHO-K1 as a negative control specimen. An example of the controls, compensation, and sample preparation is provided in Figure 2, and the completed panel run on the ST16-BM4045 spiked into healthy bone marrow is shown in Figure 3.
Figure 2.

Experimental setup with all control and sample tubes used
(A) Patient-derived neuroblastoma cell line ST16-4045 was used for single-stain compensation controls, as were UltraComp eBeads PLUS. A control bone marrow sample from a B-ALL patient during remission (with or without spike-in of ST16-4045) was stained with the entire 9-color antibody panel, as were CHO cells (a negative control) and neuroblastoma cell lines (used as benchmark controls for antigen density). Experimental samples were stained with the full antibody panel.
(B) The staining procedure involves pre-incubating samples with Fc Block. It is important to use Brilliant Stain Buffer and to add the additional BV antibodies separately, to avoid the antibodies binding to each other instead of the cells.
Figure 3.

Gating strategy using tumor-negative control bone marrow samples with and without neuroblastoma tumor cell spike-in
(A) control bone marrow sample from a B-ALL patient during remission was used to test the entire 9-color antibody panel either without or with 2% or 25% spike-in of patient-derived neuroblastoma cells ST16-4045. The gating strategy used (A, from left to right) was established to capture the cell population without debris via FSC-A/SSC-A, singlets via FSC-A/FSC-H, and viable cells using fixable viability dye. Subsequently, in (B) no-spike-in controls as well as (C) 2% spike-in and (D) 25% spike-in, hematopoietic cells and bone-marrow stromal cells were excluded using the markers CD45 and CD13. The tumor cell population is double-positive for the neuroblastoma tumor marker NCAM and GD2.
-
5.Carefully test the full antibody panel using the concentrations determined in step 4 on control samples.
-
a.Harvest and prepare cultured positive and negative control cell lines as outlined in Step 4b above and aliquot into FACS tubes.
-
b.Prepare spike-in control bone marrow samples.
-
i.Thaw three replicates of tumor uninvolved bone marrow, assess cell count, and add into three FACS tubes.
-
ii.Spike-in 2% and 25% patient-derived neuroblastoma tumor cells and leave the third tube without spike-in.
-
i.
-
c.Prepare initial antibody staining mix.
-
i.Collect all antibodies needed for the panel (Table 2), vortex and centrifuge each vial and place on ice.
-
ii.Prepare staining mix for all samples using viability dye and all antibodies listed in Table 2 but only one of the three BV antibodies.Note: Because this staining panel uses multiple BD Horizon Brilliant Violet (BV) conjugated antibodies, a BD Horizon Brilliant Stain Buffer must be used to avoid staining artifacts by fluorescent dye interactions. All BV antibodies are added separately, only one BV antibody is added to the initial staining mix, the other two are added separately thereafter.
-
i.
-
d.Stain all single-cell controls, negative and FMO controls, and sample tubes (neuroblastoma cell lines and spike-in bone marrow samples) with the respective panel (see Figure 2A).
-
i.Perform centrifugation step and wash cells twice with 2 mL of FACS buffer.
-
ii.After the second wash, resuspend the pellet in a total volume of 30 μL FACS buffer containing 5 μL Fc Block per 1 × 106 cells and incubate samples for 10 min at a temperature of 18°C–22°C. The Fc Block is necessary to prevent nonspecific staining by Fc receptor expressing cells.CRITICAL: Do NOT wash after this step!
-
iii.Once the 10-min Fc Block incubation period is complete, add 50 μL of BD Horizon Brilliant Stain Buffer (BD Biosciences, San Jose, CA, USA).
-
iv.Add the antibody mix that contains all the antibodies (without the two remaining BV antibodies) and mix well by pipetting up and down.
-
v.Separately add each of the two remaining BV antibodies to the tubes and mix well by pipetting up and down.
-
vi.Incubate the samples for 30 min on ice in the dark.
-
vii.After the incubation, perform centrifugation step and wash cells twice with 2 mL of FACS buffer.
-
viii.Resuspend cells in 300 μL FACS buffer and pass through FACS tube strainer cap.
-
ix.Place cells on ice in the dark until ready to run samples.
-
i.
-
a.
Prepare quantitation beads
-
6.Prepare BD quantification beads for the assay.
-
a.Remove one tube of BD Quantibrite PE beads and one tube of BD Custom Quantitation Beads per color (PE, PE-Cy7, APC, BV605, BV510, and BV421) from the foil pouch packaging just prior to use. Each tube contains a dried pellet of beads comprising 4–6 levels of intensities.
-
b.Resuspend beads in 300 μL of FACS buffer, vortex for a few seconds and place on ice in the dark until ready to run.
-
a.
Note: Permissible buffer systems for reconstitution of the beads should have a pH around neutrality (pH 7.0–7.5), and FACS buffer as prepared in this protocol is commonly used. The use of any ionic, non-ionic detergents or surfactants of any kind is prohibited for reconstitution of the dried quantitation beads and may result in failure of beads performance.
Set up the flow cytometer including compensation and run control samples and quantitation beads
The purpose of this step is to set up ensure optimal instrument performance, select the acquisition settings on the flow cytometer, run compensation, set up the gating strategy for the experiment and run all control samples and quantitation beads.
-
7.Set up the Flow Cytometer to be used and test for optimal performance.
-
a.Turn on the instrument and allow the lasers to warm up.
-
b.Follow standard lab operating procedures for startup, cleaning, and instrument quality control with bead standards such as CS&T or 8-peak rainbow beads to ensure optimal performance.
-
a.
-
8.Set up the PMT voltages and acquisition settings on the flow cytometer.
-
a.Open a new experiment and select the channels for visualizing the antibody panel used in the experiment and select FSC and SSC height and area as parameters.
-
b.Choose optimal values for FSC and SSC to visualize the cell populations.
-
c.Optimize voltages for optimal signal-to-noise ratio within the cell assay for which the quantitation of the surface markers is the main goal. Test-run single-color compensation controls and Quantitation Beads to make sure negative populations are visible above noise and positive populations are on-scale within linear range.
-
d.Optimized results can be saved as “application settings” for the flow cytometry assay.
-
a.
-
9.Set up compensation and gating strategy.
-
a.At set voltages, run compensation tubes, and calculate compensation.
-
b.Set up the gating strategy (Figure 3) and run the fully stained control samples on the instrument.
-
a.
Note: In this protocol, we use single stains of ST16-4045 for the high-expressing antigens NCAM and GD2 to reflect their biologically relevant signal and compensation beads for all other antibodies in the panel to define compensation. We used UltraComp eBeads PLUS to enable binding of the GPC2 antibody, which contains a human Fc domain. Once the setup and compensation are adjusted, the quantitation beads need to be run (see below) under the same instrument setup and compensation optimized for the assay.
-
10.Run Quantitation Beads.
-
a.Quantitation beads must be acquired at the same instrument and compensation settings established for the experiment.
-
b.Vortex the resuspended quantitation bead tube for a few seconds prior to acquisition. Identify the bead population by FSC-A vs. SSC-A and set a gate around the singlet bead population.
-
c.Run Quantitation Beads and collect 10,000 events in the bead population gate. The singlet bead population is analyzed using a histogram plot of fluorophore intensity.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human CD13-BUV737 (clone L138, 1:40) | BD Biosciences | Cat# 749265 |
| Human CD45-PerCp-Cy5.5 (clone HI30, 1:25) | Thermo Fisher Scientific | Cat# 45-0459-42 |
| Human GPC2 (clone D3, PE labeled, 1:200) | Dr. Dimiter S. Dimitrov (University of Pittsburgh) | N/A |
| Human GD2-BV510 (clone 14g2a, 1:20) | BioLegend | Cat# 357316 |
| Human NCAM-BV605 (clone 5.1H11, 1:100) | BioLegend | Cat# 362538 |
| Human L1CAM-BV421 (clone 5G3, 1:200) | BD Biosciences | Cat# 565732 |
| Human L1CAM-PE (clone 5G3, 1:200) | BD Biosciences | Cat# 564193 |
| Human ALK (clone 48, APC labeled, 1:500) | Mark Vigny | N/A |
| Human B7-H3-PE-Cy7 (clone MIH42, 1:40) | BioLegend | Cat# 351008 |
| Biological samples | ||
| Human neuroblastoma-infiltrated bone marrow samples | Stanford Pediatrics Bass Center Tissue Bank (IRB: #45458 and #56619) | N/A |
| Human bone marrow samples from B-ALL patients (uninvolved tumor-free at remission) | Stanford Pediatrics Bass Center Tissue Bank (IRB: #45458 and #56619) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Abcam PE Conjugation Kit Lightning-Link | Abcam | Cat# ab102918 |
| Abcam APC Conjugation Kit Lightning-Link | Abcam | Cat# ab201807 |
| BD Quantibrite PE beads | BD Biosciences | Cat# 340495 |
| BD custom quantitation beads in APC | BD Biosciences | Custom 626425 |
| BD custom quantitation beads in PE-Cy7 | BD Biosciences | Custom 625639 |
| BD custom quantitation beads in BV421 | BD Biosciences | Custom 625508 |
| BD custom quantitation beads in BV510 | BD Biosciences | Custom 625594 |
| BD custom quantitation beads in BV605 | BD Biosciences | Custom 625638 |
| UltraComp eBeads Plus compensation beads | Thermo Fisher Scientific | Cat# 01-3333-41 |
| Experimental models: Human cell lines and PDX | ||
| CHO-K1 (female) | ATCC | RRID:CVCL_0213 |
| NGP (male) | Dr. John Maris (CHOP) | RRID:CVCL_2141 |
| NGP-NPC2 (male) | Dr. John Maris (CHOP) | N/A |
| SMS-SAN (female) | Dr. John Maris (CHOP) | RRID:CVCL_7136 |
| NBSD (female) | Dr. John Maris (CHOP) | RRID:CVCL_LF68 |
| ST16-BM4045 (male, diagnostic) | This manuscript | N/A |
| Software and algorithms | ||
| FlowJo v10.7.1 | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads |
| GraphPad Prism v8.4 | GraphPad Software, Inc. | https://www.graphpad.com/updates |
| BioRender | BioRender | https://www.biorender.com |
| Other | ||
| LSRFortessa X-20 4 lasers (405 nM violet, 488 nM blue, 640 nM red, and 355 nM UV) | BD Biosciences | N/A |
| Fixable viability dye eFluor 780 | Thermo Fisher Scientific (eBioscience) | Cat# 65-0865-14 |
| Brilliant stain buffer | BD Biosciences | Cat# 566349 |
| Human BD Fc Block | BD Biosciences | Cat# 564219 |
| Corning Cellstripper dissociation reagent | Corning | Cat# MT25056CI |
| Ficoll-Paque PLUS density gradient media | Cytiva | Cat# 17144002 |
| Gibco DPBS, no calcium, no magnesium | Thermo Fisher Scientific | Cat# 14190144 |
| Invitrogen EDTA (0.5 M), pH 8.0, RNase-free | Thermo Fisher Scientific | Cat# AM9260G |
| Fetal bovine serum, qualified, heat inactivated | Thermo Fisher Scientific | Cat# 16140071 |
| Corning Falcon round-bottom polystyrene test tubes with cell strainer snap cap | Fisher Scientific | Cat# 08-771-23 |
| Nalgene Mr. Frosty freezing container | Thermo Fisher Scientific | Cat# 5100-0001 |
| Corning Costar 96-well, cell culture treated, U-shaped-bottom microplate | Fisher Scientific | Cat# 07-200-95 |
| Corning Falcon 15 mL conical centrifuge tubes | Fisher Scientific | Cat# 14-959-49D |
| RPMI 1640 medium, no glutamine | Thermo Fisher Scientific | Cat# 21870092 |
Materials and equipment
Flow cytometer
The flow cytometer used in this protocol was the BD LSRFortessa X-20 equipped with 4 lasers (405 nm Violet, 488 nm Blue, 640 nm Red and 355 nm UV) and 19 fluorescent detectors. The instrument configuration used in this protocol as well as detailed information on antibodies used for each channel are shown in Table 2.
FACS buffer
Prepare FACS buffer according to the (Stanford) recipe below and store up to one month in the fridge at 4°C.
FACS buffer
| Component | Final concentration | Amount |
|---|---|---|
| 1X PBS | 488 mL | |
| FBS | 2% | 10 mL |
| 0.5 M EDTA | 0.4% | 2 mL |
| Total | N/A | 500 mL |
Step-by-step method details
This section defines the activities on the day of actual experiments with patient specimens, assuming the assay is fully developed as outlined in the sections above.
Prepare and run control samples, compensation, and quantitation bead tubes for proper instrument setup
In order to keep the processing time for clinical samples to a minimum (from thawing to data acquisition) in this step, the instrument setup including compensation and acquisition for all control samples is performed prior to thawing any patient samples.
-
1.Prepare all sample tubes required for this step.
-
a.Stain all positive and negative control cell lines and spike-in controls samples according to steps outlined in section ‘test antibody panel on spike-in control bone marrow samples’ above.
-
b.Prepare compensation tubes containing compensation beads or ST16-BM4045 cells as outlined in the same section above.
-
c.Prepare BD quantitation beads for the assay as outlined in the ‘prepare quantitation beads’
-
a.
-
2.
Set up flow cytometer and run compensation control samples as well as quantitation beads as outlined in section ‘set up the flow cytometer including compensation and run control samples and quantitation beads’
Thaw patient samples and stain with antibody panel
This step serves to thaw, process, stain, and wash the patient samples with the entire antibody panel.
-
3.Thaw the bone marrow samples and bring into single-cell suspensions.
-
a.Get the samples from the liquid nitrogen and place on dry ice.
-
b.Place samples in the 37°C water bath and thaw quickly.
-
c.Transfer each sample to a 15 mL conical tube and resuspend in 10 mL of 4°C cold RPMI complete media.
-
d.Perform centrifugation step.
-
e.Resuspend cell pellets in 1 mL of Cellstripper a non-enzymatic dissociation reagent to break up tumor cell spheres and incubate samples for 2 min.
-
f.Add 2 mL of FACS buffer, perform centrifugation step, and wash cells twice.
-
a.
-
4.Stain patient samples with the entire antibody panel.
-
a.Follow the staining procedure specified above under ‘Stain control cell lines and compensation tubes’.
-
b.Pass cells through FACS tube strainer caps before running.
-
a.
Note: The bone marrow patient samples used for this assay contained a total of 1‒5 × 106 frozen cells and viability can be quite low (∼20%) due to Ficoll-processing before freezing and decreases post thaw. The tumor infiltration based on the pathology report in our samples ranged between 1% and 80%. We generally stain the entire sample with our antibody panel, yet if cells are viable and abundant and the expected tumor burden is high, we recommend aliquoting 1 × 106 cells for staining.
Run patient samples on the flow cytometer and analyze data
In this step data acquisition and analysis of antigen density quantification of the patient samples are described.
-
5.Run patient samples and acquire data.
-
a.Confirm that gates capture the cell population without debris (FSC-A vs. SSC-A) and include single cells rather than doublets (FSC-A vs. FSC-H) and capture viable cells (SSC-A vs. eF780) only.
-
i.Exclude CD45+ hematopoietic cells and CD13+ neuroblastoma-mimicking bone marrow stromal cells by gating on the CD45/CD13 double-negative population.
-
ii.Identify the tumor cell population by further sub-gating on NCAM+ and GD2+ (Figure 3).
-
i.
-
b.Run the entire tube of each sample as opposed to collecting a certain number of events to ensure all tumors cells present in the sample are captured.
-
a.
-
6.Export data and perform data analysis including quantitation.
-
a.Export the data and open FCS files in FlowJo software (BD Biosciences). Gate on the tumor cell population as outlined previously and shown in Figure 3.
-
b.In each Quantitation Bead Tube, gate on the singlet population of the beads by FSC-A vs. SSC-A and create a fluorophore histogram plot. In the histogram, create a separate gate for each of the bead populations present in the sample.
-
c.Using the ‘Table Editor’ function in FlowJo software, export the geometric mean values of the fluorescent intensity of each antigen and the respective quantitation bead populations.
-
d.For manual data analysis, use an Excel spreadsheet.
-
i.enter the exported MFI values of each quantitation bead population in an X-axis column and the lot-specific number of fluorophore molecules provided in the Technical Data Sheet (TDS) in a Y-axis column.
-
ii.Plot a linear regression of MFI (X-axis) vs. Fluorophore Molecules (Y-axis) using the linear equation ‘Y = aX + c’ where Y equals the number of fluorophore molecules and X equals MFI value (Figure 4).
-
iii.To determine the number of fluorophores for an unknown cell population, substitute the cell population MFI value in the equation and solve for the number of fluorophore molecules and divide the resulting value by the F:P ratio (Table 3). Given that antibody concentrations are saturating, the assumption is made that antibodies bound per cell provide a reasonable estimate of antigens per cell.
-
i.
-
a.
Note: For example, in the case of ALK-APC, if the MFI value of the patient’s tumor cell population is 4057, and using the equation generated for APC fluorescence quantitation, the formula is ‘Y = 6.2572 × (4057) – 3063.1’, resulting in 22322 molecules of APC bound per cell for the cell population of the interest. To arrive at antibodies bound per cell, this value must be divided by the F:P ratio (which is 3.34 for ALK-APC), resulting in 22322 molecules of APC divided by 3.34 APC per antibody equals 6683 antibodies bound per cell (Table 3; Figure 4).
Figure 4.

Conversion of mean fluorescent intensity of quantitation bead populations into molecules per cell
Representative Histograms of Quantitation Beads of each fluorophore (first panel from the left), mean fluorescent intensity of each bead population and pre-defined LOT-specific fluorescent molecules/bead for each population (second panel) to set up the equation (third panel) for converting mean fluorescent intensity of each antibody signal into molecules of fluorophore/cell. Representative histograms of each antigen of a patient neuroblastoma-infiltrated bone marrow sample gated according to Figure 3 (fourth panel) used for example calculation adjusted for F:P ratios in Table 3.
Table 3.
Example determination of antigen molecules per cell from MFI and F:P ratio
| Fluorophore | Antigen | MFI sample | F:P ratio | Mol./Cell |
|---|---|---|---|---|
| PE | GPC2 | 4776 | 1 | 6041 |
| PE-Cy7 | B7H3 | 2290 | 0.92 | 4831 |
| BV510 | GD2 | 28258 | 3.1 | 683694 |
| APC | ALK | 4057 | 3.34 | 6683 |
| BV605 | NCAM | 7526 | 3.4 | 19684 |
| BV421 | L1CAM | 9112 | 2.21 | 22924 |
Representative data of one patient sample according to the equations shown in Figure 4.
Expected outcomes
This protocol developed at Stanford University and described here allows the quantification of antigen densities of immunotherapy-relevant cell-surface antigens on neuroblastoma cells metastatic to the bone marrow. The protocol was used to analyze patient samples at Stanford and also at a collaborating institution (Children’s Cancer Research Institute Vienna, Austria), using the same positive and negative reference cell lines to confirm reproducible performance. Figure 4 shows representative flow cytometry histograms of a patient sample analyzed according to this protocol. An example conversion of the fluorescent signal into antigen density quantification for the 6 putative targetable markers of interest is shown in Table 3.
Quantification and statistical analysis
The resulting antigen quantification data (molecules/cell) for each cell-surface target on bone-marrow infiltrating neuroblastoma cells can uncover biological differences between groups. If those are of unequal sample size as in the main study using this protocol,1 where clinical specimens were compared to cell lines and patient-derived xenograft samples, those should be compared using a Welch’s t test for statistical analysis with considering a P < 0.05 as statistically significant.
Limitations
Antigen density assessment as outlined here represents a best estimate, based on measuring the number of antibodies bound per cell (ABC) at saturating concentrations. Some key factors for accurate assessment are the following: using antibodies at saturating concentrations and the assumption that saturating antibodies reflect the underlying number of antigens; the measurement of F:P ratio by the vendor or empirically in the lab; the condition of the sample at the time of analysis and whether it is representative of the original cells prior to specimen acquisition due to sample processing and storage methods; an appropriate flow cytometry panel design as well as the quality of the instrument and compensation setup.
Each fluorescence channel used for antigen density measurement has a limit of detection that translates to the lowest number of antigens that can be detected. Measurement must be made in the linear range and the linear regression (Figure 4) intercepts either on X- or Y-axis, indicating the limit of flow cytometer detection either on the X-axis (MFI value) or on Y-axis (number of fluorophore molecules). Depending on the complexity of the flow cytometry panel, additional minor losses in limit of detection may occur due to spillover and compensation issues. An upper limit might occur if antigen density is locally greater than the maximum density of antibodies able to bind (e.g., given steric, size, and other factors). If the ratio of antibody to antigen is expected to be other than 1:1 for a particular case (e.g., for antigens that possess more than one binding site per molecule), a correction may be considered. Regardless, this semiquantitative relationship is expected to be proportional, representative of the functional epitopes accessible, and as such informative. Access to clinical specimens can be especially challenging and care should be taken to use the highest quality clinical specimens available and, if possible, to characterize the effect of specimen storage time or processing on antigen expression. Despite these limitations and considerations, this protocol provides significant value over simply reporting MFI because it allows a consistent approach to quantitation irrespective of instrument, setup or laboratory-specific factors. This protocol is for research purposes only, not for use in diagnostic procedures.
Troubleshooting
Problem 1
Patient-derived samples may have unpredictably high or low cell-surface expression levels of the antigens in question, resulting in off-scale signal and/or non-optimum performance when using pre-designed reagent panels, antibody titrations, and compensation schemes. For instance, in our experiment many of the patient samples expressed GD2 and NCAM at much higher levels than signal emanating from compensation with bead-bound GD2 and NCAM antibodies. This might lead to complications when samples are run on the flow cytometer (Steps 1–4 (step-by-step method details): prepare and run control samples, compensation, and quantitation bead tubes for proper instrument setup).
Potential solution
To avoid complications and any time delays once precious patient samples are processed and stained, it is critical to establish instrument setup and compensation in advance using the best available controls. We recommend comparing compensation beads to a biologically relevant positive control sample stained simultaneously and carefully choosing the superior sample for compensation setup, which reflects the anticipated biological signal. We stained both NB ST16-BM4045 and compensation beads in our protocol and found that the MFI of certain antibodies, such as GD2 and NCAM on NB ST16-BM4045, was much higher than the MFI of the compensation beads (Figure 5). Using NB ST16-BM4045 cells for single-color stains to set up the instrument allowed us proper voltage adjustment and compensation prior to the experiment using patient sample.
Figure 5.

Comparison of single-color stains for compensation setup on NB PDX cells compared to compensation beads
Both UltraComp eBeads PLUS as well as the neuroblastoma PDX cells ST16-4045, which closely resemble the biological signal on patient tumor specimens, were stained simultaneously with the same concentration of (A) GPC2-PE (B) B7H3-PE-Cy7 (C) GD2-BV510 (D) ALK-APC (E) NCAM-BV605 or (F) L1CAM-BV421 mABs used in the quantification panel.
Problem 2
Non-tumor cell populations may be present in patient samples in large numbers, and these populations will be specific to the tissue location from which a given sample was taken. The experimental antibody staining panel must be optimized and adjusted accordingly to gate out non-tumor populations expected to be present. Ideally, markers to positively select the tumor cell population should be included if antigens that identify tumor cells in a distinct and homogenous manner can be identified. (Step 2 (before you begin): flow cytometry panel development for antigen density quantitation).
Potential solution
For bone marrow-derived neuroblastoma patient samples, we gate out stromal cells and hematopoietic stem cells using CD13 and CD45, and positively gate on the tumor cell population using NCAM and GD2. Sufficient sample must be obtained and analyzed to account for potential scarcity of tumor cells.
Problem 3
This assay depends on accurate known fluorophore-to-protein ratios of each monoclonal antibody in the panel, to accurately convert the number of fluorophore molecules bound per cell into antibodies bound per cell. Commercially available antibodies that work well in the panel might not have information on F:P ratios available from the manufacturer and unconjugated custom antibodies might be utilized for novel antigens of interest. In our protocol, determining the F:P ratio of the commercially available L1CAM-BV421 mAB and a custom-made unconjugated ALK mAB, which we conjugated to APC, were necessary (Step 2 (before you begin): flow cytometry panel development for antigen density quantitation).
Potential solution
To estimate the F:P for an unknown conjugate, we compared the staining to the PE version of the same clone at the same titrated concentration. The PE-conjugated mABs possess 1:1 F:P ratios and by running quantitation beads in both colors alongside a biological sample individually stained in both colors (Figure 6) allows determination of the number of antibodies bound per cell (ABC). See Table 1 as an example for calculations of the F:P ratios for the ALK-APC and L1CAM-BV421 antibody conjugates used in our study.
Figure 6.

Determination of fluorophore-to-protein ratios using a PE-conjugated version of the respective antibody clone
Neuroblastoma cells (SMS-SAN) stained with (A) the ALK-48 mAB conjugated either to APC or PE and (B) the L1CAM mAB clone 5G3 conjugated to either BV421 or PE. The PE-versions of the antibodies possess a 1:1 Fluorophore-to-Protein ratio and allow determination of the respective ratio of the other clone by running Quantitation Beads in both colors according to Table 1.
Resource availability
Lead contact
Requests for further information and resources should be directed to the lead contacts, Sabine Heitzeneder (sabineh@stanford.edu) and Scott J. Bornheimer (scott_bornheimer@bd.com).
Materials availability
This study did not generate new unique reagents. Most materials and reagents used in this study are commercially available. The human GPC2 and ALK antibody are custom reagents developed by collaborators (see key resources table). Inquiries for BD Custom Quantitation Beads should be directed to BD Biosciences Custom Orders (bdb_custom_orders@bd.com).
Data and code availability
This assay did not generate new code and the data are published in Heitzeneder et al.1
Acknowledgments
This work was supported by the NIH U54 CA232568-01 (C.L.M.), P01CA217959-01 (C.L.M. and S.H.), and a St. Baldrick’s/Stand Up To Cancer (SU2C) Pediatric Dream Team Translational Cancer Research Grant (SU2CAACR-DT1113; C.L.M. and S.H.). SU2C is a program of the Entertainment Industry Foundation administered by the AACR. C.L.M. is a member of the Parker Institute for Cancer Immunotherapy (PICI), which supports the Stanford University Cancer Immunotherapy Program and is supported by the Virginia and D.K. Ludwig Fund for Cancer Research. The authors thank the staff of the pediatric hematology/oncology department at the Lucile Packard Children’s Hospital for collecting BM samples and the patients and their parents for providing their informed consent. We thank Garry L. Coles, Julien Sage, and Min Huang for providing the ST16-BM4045 patient-derived cell line. We thank Robbie G. Majzner and Elena Sotillo from the Center for Cell Therapy at the Stanford Cancer Center for input on assay development and Meena Malipatlolla for careful BD LSRFortessa X-20 flow cytometer maintenance. We thank BioLegend for providing fluorophore-to-protein ratios of the commercially available antibodies used for quantitation in the panel. All cartoons were created using BioRender.com.
Author contributions
S.H. designed and conceptualized the assay together with C.L.M., S.J.B., M.M., J.S.O., and B.S. S.H. and M.T.R. performed the experiments and collected and analyzed the data. M.T.R., S.J.B., M.M., and S.H. wrote the manuscript. S.H. and C.L.M. supervised the study. C.L.M. provided funding. All other authors commented on and revised the manuscript.
Declaration of interests
S.H. and C.L.M. are co-inventors on patents relevant to CAR T cells targeting GPC2. C.L.M. has multiple patents pertinent to CAR T cells and is a co-founder of Lyell Immunopharma and CARGO Therapeutics, formerly Syncopation Life Sciences, which develop CAR-based therapies, and consults for Lyell, NeoImmune Tech, Apricity, Nektar, and Immatics. S.J.B. and M.M. are employees of BD Biosciences.
References
- 1.Heitzeneder S., Bosse K.R., Zhu Z., Zhelev D., Majzner R.G., Radosevich M.T., Dhingra S., Sotillo E., Buongervino S., Pascual-Pasto G., et al. GPC2-CAR T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell. 2022;40:53–69.e9. doi: 10.1016/j.ccell.2021.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Majzner R.G., Rietberg S.P., Sotillo E., Dong R., Vachharajani V.T., Labanieh L., Myklebust J.H., Kadapakkam M., Weber E.W., Tousley A.M., et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020;10:702–723. doi: 10.1158/2159-8290.CD-19-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fry T.J., Shah N.N., Orentas R.J., Stetler-Stevenson M., Yuan C.M., Ramakrishna S., Wolters P., Martin S., Delbrook C., Yates B., et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018;24:20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spiegel J.Y., Patel S., Muffly L., Hossain N.M., Oak J., Baird J.H., Frank M.J., Shiraz P., Sahaf B., Craig J., et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat. Med. 2021;27:1419–1431. doi: 10.1038/s41591-021-01436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards R.M., Sotillo E., Majzner R.G. CAR T Cell Therapy for Neuroblastoma. Front. Immunol. 2018;9:2380. doi: 10.3389/fimmu.2018.02380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis K.A., Abrams B., Iyer S.B., Hoffman R.A., Bishop J.E. Determination of CD4 antigen density on cells: role of antibody valency, avidity, clones, and conjugation. Cytometry. 1998;33:197–205. doi: 10.1002/(sici)1097-0320(19981001)33:2<197::aid-cyto14>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 7.Iyer S.B., Hultin L.E., Zawadzki J.A., Davis K.A., Giorgi J.V. Quantitation of CD38 expression using QuantiBRITE beads. Cytometry. 1998;33:206–212. doi: 10.1002/(sici)1097-0320(19981001)33:2<206::aid-cyto15>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 8.Wang L., Abbasi F., Gaigalas A.K., Vogt R.F., Marti G.E. Comparison of fluorescein and phycoerythrin conjugates for quantifying CD20 expression on normal and leukemic B-cells. Cytometry B Clin. Cytom. 2006;70:410–415. doi: 10.1002/cyto.b.20140. [DOI] [PubMed] [Google Scholar]
- 9.Wang L., Abbasi F., Jasper G.A., Kreitman R.J., Liewehr D.J., Marti G.E., Stetler-Stevenson M. Variables in the quantification of CD4 in normals and hairy cell leukemia patients. Cytometry B Clin. Cytom. 2011;80:51–56. doi: 10.1002/cyto.b.20541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L., Abbasi F., Ornatsky O., Cole K.D., Misakian M., Gaigalas A.K., He H.J., Marti G.E., Tanner S., Stebbings R. Human CD4+ lymphocytes for antigen quantification: characterization using conventional flow cytometry and mass cytometry. Cytometry A. 2012;81:567–575. doi: 10.1002/cyto.a.22060. [DOI] [PubMed] [Google Scholar]
- 11.Brinkley M. A brief survey of methods for preparing protein conjugates with dyes, haptens, and cross-linking reagents. Bioconjug. Chem. 1992;3:2–13. doi: 10.1021/bc00013a001. [DOI] [PubMed] [Google Scholar]
- 12.Wang L., Abbasi F., Gaigalas A.K., Hoffman R.A., Flagler D., Marti G.E. Discrepancy in measuring CD4 expression on T-lymphocytes using fluorescein conjugates in comparison with unimolar CD4-phycoerythrin conjugates. Cytometry B Clin. Cytom. 2007;72:442–449. doi: 10.1002/cyto.b.20354. [DOI] [PubMed] [Google Scholar]
- 13.Wang L., Gaigalas A.K., Wood J. Quantitative Fluorescence Measurements with Multicolor Flow Cytometry. Methods Mol. Biol. 2018;1678:93–110. doi: 10.1007/978-1-4939-7346-0_6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This assay did not generate new code and the data are published in Heitzeneder et al.1