

Abstract

Drug failure during experimental procedures due to low bioactivity presents a significant challenge. To mitigate this risk and enhance compound bioactivities, predicting bioactivity classes during lead optimization is essential. The existing studies on structure–activity relationships have highlighted the connection between the chemical structures of compounds and their bioactivity. However, these studies often overlook the intricate relationship between drugs and bioactivity, which encompasses multiple factors beyond the chemical structure alone. To address this issue, we propose the BioAct-Het model, employing a heterogeneous siamese neural network to model the complex relationship between drugs and bioactivity classes, bringing them into a unified latent space. In particular, we introduce a novel representation for the bioactivity classes, called Bio-Prof, and enhance the original bioactivity data sets to tackle data scarcity. These innovative approaches resulted in our model outperforming the previous ones. The evaluation of BioAct-Het is conducted through three distinct strategies: association-based, bioactivity class-based, and compound-based. The association-based strategy utilizes supervised learning classification, while the bioactivity class-based strategy adopts a retrospective study evaluation approach. On the other hand, the compound-based strategy demonstrates similarities to the concept of meta-learning. Furthermore, the model’s effectiveness in addressing real-world problems is analyzed through a case study on the application of vancomycin and oseltamivir for COVID-19 treatment as well as molnupiravir’s potential efficacy in treating COVID-19 patients. The data and code underlying this article are available on https://github.com/CBRC-lab/BioAct-Het. However, data sets were derived from sources in the public domain.
1. Introduction
Identifying and optimizing the druglikeness of a compound is an arduous and time-consuming process. In fact, bringing a new chemical compound to the market as a drug can typically take more than a decade and cost billions of dollars.1,2 Therefore, predicting the bioactivities of a compound is a critical component of the drug discovery process. Bioactivity refers to the effects of chemical compounds, or leads, on living organisms, including both favorable and unfavorable outcomes such as drug side effects, toxicity, solubility, and permeability.3 During the process of lead optimization, the goal is to enhance the structure of leads while maintaining their therapeutic properties, in order to improve their bioactivity.4 However, experimentally determining the bioactivity of a new compound can be challenging due to the vast number of possible analogues and the high cost of screening procedures. It has been estimated that there are approximately 1060 ensembles with the same atoms but different bioactivities.5 As a result, computational methods have gained much attention as a promising alternative to high-throughput screening methods.6
Computational studies have been extensively used to investigate the structure–activity relationship (SAR) and have demonstrated the potential of the compound structural properties to predict their bioactivity.7 These studies suggest that compounds with similar chemical structures often exhibit similar bioactivities.8,9 This hypothesis was first proposed by Brown and Fraser in 1868 when they examined the relationship between the molecular structure of a compound and its biological activity. They found that compounds with certain substructures or radicals possess a common biological action.10 This idea forms the foundation of modern SAR studies, which leverage computational methods to predict the bioactivity of new compounds based on their structural properties.
Predicting the bioactivity of new compounds via computational methods faces two main challenges: compound representation and data scarcity. The former, i.e., compound representation, refers to the selection of an appropriate chemical structure representation for the compounds. Traditionally, existing algorithms employ SMILES,11 fingerprints,12 and graphs to represent a compound in the bioactivity class prediction problem. However, selecting the optimal representation of a material can be challenging. The latter, i.e., the data scarcity challenge, arises due to the lack of sufficient data for training an accurate model. Therefore, preparing a proper data set to improve the performance of these algorithms is of crucial importance. The optimal data set should contain a diverse set of compounds with varying bioactivities to ensure that the model can accurately predict the bioactivity of new compounds. It is worth noting that the ratio of compounds exhibiting a specific bioactivity class (positive data) to those that do not (negative data) is often imbalanced, with severity affecting the performance of prediction. This can cause predictive models to focus on the majority class and make inaccurate predictions for the minority class, as highlighted by previous studies.13
Retrospective studies in drug discovery and bioinformatics have proposed useful approaches for bioactivity discovery, which are reviewed in the following. Altae-Tran et al.14 introduced a one-shot learning approach called IterRefLSTM to address the challenge of limited data in bioactivity prediction. The one-shot learning approach is designed to learn the similarity between pairs of samples and has demonstrated high accuracy in various applications. In drug discovery, they adapted the one-shot learning approach to estimate the behavior of a molecule in a new experimental assay, which is critical for predicting the bioactivity of a compound. This innovative approach provides a promising solution for overcoming the data scarcity challenge in drug discovery.14 To generate an accurate chemical structure representation of a compound, IterRefLSTM utilizes an iterative approach that refines the compound’s embedding by a long short-term memory (LSTM) model. The refinement process involves using a matching network and a residual graph convolutional network (GCN) architecture to learn meaningful distance metrics for a few small molecules by feeding their graph structures as input. This model is applied on SIDER,15 Tox21,16 and MUV17 databases to consider side effects, toxicity, and maximum unbiased validation classes as bioactivities.
Torres et al.18 proposed another one-shot classification approach to tackle the limited data challenge based on a siamese neural network (SNN) architecture using a convolutional neural network (CNN) in the middle layers. The advantage of this strategy in drug discovery is to identify novel compound features whose classes are less-represented.18 The model selects drug toxicity as a bioactivity property utilizing Tox2116 as the database. To represent a compound, a matrix is constructed in which each row contains the one-hot encoding of the SMILES letters. Later, the model groups the chemical compound based on their chemical structures to pair the compounds based on their corresponding groups for feeding to model. Therefore, it considers half of the pairs in the same class as positive data and half of the pairs from different classes as negative data for training the model.
Fernández-Llaneza et al.19 proposed an N-shot classification approach based on a deep SNN named SiameseCHEM using a bidirectional LSTM (BiLSTM) with a self-attention mechanism to tackle the biological data scarcity problem. The SiameseCHEM presumes the pXC50 as a bioactivity property, and it is applied to five data sets collected from ChEMBL20 and ExCAPE-DB.21 First, the model classifies the drugs as active or inactive by a threshold in pXC50. Next, SiameseCHEM presents the drugs by learning a task-specific fingerprint representation. Moreover, it performs a data augmentation process similar to oversampling to tackle the imbalanced data problem.
Lately, Vella and Ebejer22 extended the IterRefLSTM model with two new metric-based techniques, namely, prototypical and relation networks. To do so, the model assesses two different embeddings for the compounds: extended-connectivity fingerprints (ECFP) and GCNs. It then examines the effectiveness of different few-shot learning models such as SNNs, matching networks, prototypical networks, and relation networks. The study is evaluated based on three public databases: Tox21,16 MUV,17 and a subset of DUD-E, namely, GPCR.23 The evaluation scores of the model attest to the effectiveness of the learned embedding using GCNs compared to ECFP. Additionally, the introduced prototypical network, which is similar to matching network and considers the mean vector of embeddings for each class instead of using individual support set embeddings, outperforms IterRefLSTM results and has better capability for generalizing. The summary of the reviewed methods is shown in Table 1.
Table 1. Summary of Reviewed Methods.
| model name | representation | database | main idea |
|---|---|---|---|
| IterRefLSTM14 | graph | SIDER, Tox21, MUV | refined LSTM iteratively/one-shot learning |
| Torres’s model18 | SMILES with one-hot | Tox21 | SNN/one-shot learning |
| SiameseCHEM19 | task-specific fingerprint | ChEMBL, ExCAPE-DB | SNN/N-shot learning |
| Vella’s model22 | graph, ECFP | Tox21, MUV, GPCR | prototypical network/few-shot learning |
While several research efforts have been devoted to the bioactivity class prediction (BCP) problem, existing methods are based on the main hypothesis of SAR studies. These methods attempt to distinguish patterns in the chemical structures of drugs that are related to a particular bioactivity class. They are trained using pairs of compounds with the same bioactivity class to learn their similarities and pairs of compounds with different bioactivities to learn their differences and then used to predict whether new compounds belong to the same bioactivity class.
However, these studies refer to few-shot learning for predicting the behavior of a molecule in a new experimental assay that is different from the model on which it is trained on. In other words, the model has seen the chemical structure of a compound during training but not for the test experiment, which is excluded for model evaluation. Therefore, these models may not fully meet the criterion of normal few-shot learning, which requires generalizing for recognition of new classes using unseen data.22
Furthermore, detecting the bioactivity classes of a chemical compound is a nontrivial problem, as there can be conflicts in defining positive and negative data. For instance, if two compounds belong to multiple bioactivity classes, sharing some but differing in others, then it can be challenging to determine whether they should be considered positive or negative samples. This complexity in defining positive and negative data can make the construction of data sets challenging.
The evaluation of previous studies demonstrates that the relationship between chemical compounds and bioactivities is complex and intricate, posing a significant relationship challenge in the BCP problem.
To address these challenges, this paper proposes a new method called BioAct-Het, which aims to determine the likelihood of association between a compound and a bioactivity class, rather than learning the similarity between two compounds, which can be complicated due to a relationship challenge. BioAct-Het exploits a heterogeneous SNN to map chemical compounds and bioactivity classes into a unified latent space that is capable of representing bioactivity classes based on related compounds. The performance of BioAct-Het is evaluated using both supervised learning and meta-learning approaches on three databases: SIDER,15 Tox21,16 and MUV.17
The main contributions of BioAct-Het are listed as follows:
To construct the data set, the proposed model considers a compound–bioactivity class pair (⟨d, b⟩) as a positive pair if d activates b and a negative pair otherwise (see the Section 2.2).
To define the problem, we introduce a novel bioactivity representation model, which takes into account the role of bioactivities (see the Section 2.3).
To model the complex relationship between compounds and bioactivity classes, we aim at learning a unified latent space via a heterogeneous SNN (see the Section 2.4).
To infer the association between a chemical compound and a bioactivity class, the paper computes the likelihood of association between the compound and the given bioactivity in the unified latent space instead of relying solely on the similarity between two chemical compounds, as is done in previous studies (see the Section 2.4).
2. Materials and Methods
The task at hand is to predict whether a newly introduced compound causes some bioactivity classes before entering any development or marketing activity. Intuitively, we aim to build a computational model that permits forecasting the potential bioactivity of small molecules during virtual screening to reduce drug development time.
2.1. Definition of the BCP Problem
Let  denote a set of p different
compounds and
denote a set of p different
compounds and  show the set of t different
bioactivity classes. The BCP problem is the task of predicting whether
the compound
show the set of t different
bioactivity classes. The BCP problem is the task of predicting whether
the compound  may cause the bioactivity class
may cause the bioactivity class  . More specifically, we model the problem
as a binary classification where a pair of compound and bioactivity
class such as ⟨d, b⟩
is given to the model as input, and the model predicts 1 if compound d exhibits bioactivity class b, otherwise
0.
. More specifically, we model the problem
as a binary classification where a pair of compound and bioactivity
class such as ⟨d, b⟩
is given to the model as input, and the model predicts 1 if compound d exhibits bioactivity class b, otherwise
0.
2.2. Data Preparation
As aforementioned,
there is limited data about the activated or inactivated chemical
compounds relating to the bioactivity classes. Learning from the limited
available data is a challenging issue that may impact the efficiency
of the machine learning approaches. To overcome this issue, the retrospective
works14,18,19 apply few-shot
learning and metric-based approaches to compute the distance between
compounds d and d′ and predict
bioactivity classes, where  . These models often make a similarity function
that satisfies the following condition
. These models often make a similarity function
that satisfies the following condition
| 1 |
where 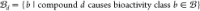 . In other words, the model attempts to
bring the structural information of the compounds in a latent space
close together as they share a common bioactivity class, while increasing
their distance as they cause different bioactivity classes. Sometimes
this function (see eq 1) can produce positive and negative pairs simultaneously when both
conditions are satisfied, as a compound may belong to different bioactivity
classes. Therefore, defining positive and negative pairs is a key
challenge of these methods. To address this issue, some studies14,22 define the problem as a task-specific one. This involves considering
each bioactivity class separately during the training, which can result
in a loss of generality.
. In other words, the model attempts to
bring the structural information of the compounds in a latent space
close together as they share a common bioactivity class, while increasing
their distance as they cause different bioactivity classes. Sometimes
this function (see eq 1) can produce positive and negative pairs simultaneously when both
conditions are satisfied, as a compound may belong to different bioactivity
classes. Therefore, defining positive and negative pairs is a key
challenge of these methods. To address this issue, some studies14,22 define the problem as a task-specific one. This involves considering
each bioactivity class separately during the training, which can result
in a loss of generality.
To address the aforementioned challenge, our study proposes a different approach to data set preparation. Unlike retrospective studies that only feed the model with ⟨d, d′⟩, we include the bioactivity as an input by feeding ⟨d, b⟩ to the model. The advantage of this approach over prior methods is that it trains the model on all experiments, rather than being limited to task-specific bioactivity classes. So, the data set Δis prepared as follows
| 2 |
where
| 3 |
and
| 4 |
Here, X is the set of compound–bioactivity class pairs and Yx is defined for every x ∈ X as a label to show the association of compound and bioactivity class.
2.3. Data Representation
This section explains the representation of the data set Δ to feed the model. As Δ consists of pairs of ⟨d, b⟩ ∈ X, it is necessary to have a representation for both chemical compound d and bioactivity class b. These representations are explained in detail below
To represent the chemical structure of compounds, we use the pretrained GCN models. To do so, two different models are extracted from the DGL-Life24 library named GCN-Canonical and GCN-AttentiveFP, which have been pretrained on the intended bioactivity using either Canonical featurization25 or AttentiveFP26 featurization of atoms, respectively. These pretrained models are used as feature extractors to present the chemical compounds. The output of GCN-Canonical and GCN-AttentiveFP for compound
 is a continuous vector shown by Fmd of length lm, where m ∈
{GCN–Canonical, GCN–AttentiveFP}. Figure 1 demonstrates the chemical compound representation
steps.
is a continuous vector shown by Fmd of length lm, where m ∈
{GCN–Canonical, GCN–AttentiveFP}. Figure 1 demonstrates the chemical compound representation
steps.
Figure 1.

Illustration of chemical compounds. The SMILES representation of chemical compounds is collected, their 2D structure then is retrieved, and a numerical representation using GCN-Canonical or GCN-AttentiveFP model is generated.
-
To show the bioactivity class representation, we introduce a novel method named Bio-Prof based on the profile of the chemical structure of related compounds (see Figure 2). Assume that:
is a set of compounds that are known for causing the bioactivity class
5  and
and
is a set of compounds without any association with bioactivity class
6  . Since there is a relationship between
having the specific chemical substructures and causing the bioactivities,7 first, the fingerprints of compounds in
. Since there is a relationship between
having the specific chemical substructures and causing the bioactivities,7 first, the fingerprints of compounds in 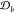 and
and 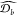 are collected. For this aim, the Morgan
fingerprint,27 which is a binary string
representation encoding functional groups and substructure of the
compounds, is extracted and shown by rd = [r1d, ···, r512d], where rid ∈
{0,1},
are collected. For this aim, the Morgan
fingerprint,27 which is a binary string
representation encoding functional groups and substructure of the
compounds, is extracted and shown by rd = [r1d, ···, r512d], where rid ∈
{0,1},  , and the length is considered as 512 with
a radius of 2. Therefore, the bioactivity class b is represented by Gb = [G1b,···,G512b] and defined as follows
, and the length is considered as 512 with
a radius of 2. Therefore, the bioactivity class b is represented by Gb = [G1b,···,G512b] and defined as follows
where
7 
8 Figure 2 illustrates the proposed approach for representing the bioactivity classes, Bio-Prof, based on the related drugs.
Figure 2.

Illustration of bioactivity representation (Bio-Prof). Two sets of drug fingerprints, one for active and one for nonactive class b drugs, are extracted as Pb and Nb, respectively. For each fingerprint, two probabilities of occurrence of substructure k in the active and nonactive class b drugs are computed as Pkb and Nkb, respectively. The importance of substructure k of drugs in class b is then calculated as the logarithm of Pkb to Nkb.
2.4. BioAct-Het Model
Recall that we aim to propose a model that brings the similar concept of bioactivity classes and chemical compounds close together in the latent space and then infers the likelihood of association through their distances. To achieve this end, we introduce BioAct-Het, a heterogeneous SNN that consists of two branches for embedding compounds and bioactivity classes, respectively. The reason for applying a heterogeneous SNN is the complexity and diversity of chemical compounds and bioactivity classes, which requires them to be transformed into a unified latent space for accurate predictions. Then, BioAct-Het subtracts the corresponding vectors and passes them through layers to obtain the final prediction (Figure 3). The details of the BioAct-Het model are explained in the following sections.
Figure 3.

Architecture of the BioAct-Het model. The upper and lower branches of the network are responsible for transforming the representation of chemical compounds and bioactivity classes into a unified latent space. The representation of drug d is shown as Fmd by using the GCN model m ∈ {GCN–Canonical, GCN–AttentiveFP}. The bioactivity class b is described by the Bio-Prof approach as Gb. Two functions f and g are two neural networks which transform Fmd and Gb in the unified latent space named Emd and Eb, respectively. The model then subtracts the corresponding vectors and passes them through several dense layers to predict the activation of bioactivity by the given chemical compound.
Figure 3 provides an illustration of the BioAct-Het model and its components, which include the embedding layers for compounds and bioactivity classes, the subtraction layer, and the fully connected layers.
The main
idea applied by the previous studies is based on homogeneous
SNNs28 where they consider the model input
as tuple ⟨d, d′⟩
and  . Homogeneous SNNs are dual-branch networks
with tied weights. They consist of the same network replicated in
two various branches and a similarity learning component. Since BioAct-Het
regards inputs as ⟨d, b⟩,
where
. Homogeneous SNNs are dual-branch networks
with tied weights. They consist of the same network replicated in
two various branches and a similarity learning component. Since BioAct-Het
regards inputs as ⟨d, b⟩,
where 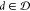 and
and  are different components, it employs heterogeneous
SNNs. The heterogeneous SNN applies two different network branches
to get two types of inputs (i.e., the compound chemical structure
and the bioactivity class). Each branch transforms the initial representation
of the chemical compounds and bioactivity classes into a unified latent
space, which enables them to share properties and become comparable.
In the training process, each branch simultaneously learns the embedding
of its input, and then, a similarity function is exploited to impose
the embeddings of similar concepts close together.
are different components, it employs heterogeneous
SNNs. The heterogeneous SNN applies two different network branches
to get two types of inputs (i.e., the compound chemical structure
and the bioactivity class). Each branch transforms the initial representation
of the chemical compounds and bioactivity classes into a unified latent
space, which enables them to share properties and become comparable.
In the training process, each branch simultaneously learns the embedding
of its input, and then, a similarity function is exploited to impose
the embeddings of similar concepts close together.
We hypothesize
that a chemical structure of a compound can implicitly
indicate the bioactivities of the corresponding compound. Moreover,
retrospective studies demonstrate that neural networks are effective
for extracting discriminative features. Inspired by these studies,
we define the BioAct–Het(Fmd, Gb) model to predict
the association between compound  represented by Fmd and bioactivity
class
represented by Fmd and bioactivity
class  shown by Gb, where m shows applying GCN–Canonical
or GCN–AttentiveFP for representing chemical compounds. The
BioAct-Het model consists of two distinct branches: chemical compound
embedding and bioactivity class embedding. These branches learn two
nonlinear functions: f(Fmd): Rlm → Rn for the chemical compound embedding and g(Gb): R512 → Rn for the bioactivity class embedding. Here, lm denotes the length of the
representation generated by GCN m, while 512 represents
the length of the drug fingerprint. These functions, f(Fmd) and g(Gb), produce Emd and Eb as the representation vectors of chemical compounds and bioactivity
classes in the unified latent space, where the chemical compounds
with a shared bioactivity class have similar distribution (see Figure 3). Moreover, if compound d causes bioactivity class b, their embedded
vectors would be near each other in the latent space and vice versa.
shown by Gb, where m shows applying GCN–Canonical
or GCN–AttentiveFP for representing chemical compounds. The
BioAct-Het model consists of two distinct branches: chemical compound
embedding and bioactivity class embedding. These branches learn two
nonlinear functions: f(Fmd): Rlm → Rn for the chemical compound embedding and g(Gb): R512 → Rn for the bioactivity class embedding. Here, lm denotes the length of the
representation generated by GCN m, while 512 represents
the length of the drug fingerprint. These functions, f(Fmd) and g(Gb), produce Emd and Eb as the representation vectors of chemical compounds and bioactivity
classes in the unified latent space, where the chemical compounds
with a shared bioactivity class have similar distribution (see Figure 3). Moreover, if compound d causes bioactivity class b, their embedded
vectors would be near each other in the latent space and vice versa.
In the second step of the model, BioAct-Het predicts the association between compound d and bioactivity class b based on their distance in the latent space. For this aim, the vector E is built using Emd and Eb as follows
| 9 |
Then, a nonlinear function h(E): Rn → [0,1] predicts the likelihood of association between a pair of ⟨d, b⟩, i.e., models BCP as a classification problem. As the output of the h(E) is the association probability of ⟨d, b⟩ in the range of [0,1], we consider probability greater than 0.5 to be 1 and otherwise 0. Thus, if the output is greater than 0.5, bioactivity class b is more likely to be caused by chemical compound d and vice versa. The details of the model architecture are provided in the Section 3.2.
3. Results
In this section, the performance of the BioAct-Het model is evaluated and compared to the state-of-the-art algorithms. Moreover, we assess the power of the model in facing real-world problems and conducting a case study on vancomycin, oseltamivir, and molnupiravir.
3.1. Databases
We assess the BioAct-Het performance in three publicly available databases: SIDER4.1,15 Tox21,16 and MUV,17 which are commonly utilized in computational drug discovery to assess the performance of models.14,22 SIDER database evaluates the ability of models to predict known side effects of drugs, whereas the Tox21 database is used to assess the capacity of approaches to predict the toxicological properties of chemicals. On the other hand, the MUV database evaluates the predictive capabilities of models in determining the biological activity of small molecules across multiple targets. The details of each database are explained in the following.
SIDER: The SIDER4.1 database is a comprehensive collection of observed side effects associated with marketed drugs.15 Side effects of SIDER are grouped into 27 primary classes using the MedDRA classification system based on system organ classes.
Tox21: The Tox21 database is designed to assess the toxicity of chemical compounds. It contains the results of 12 toxicological assays based on nuclear receptors, which are used to evaluate the potential risks associated with exposure to these compounds.16
MUV: The MUV database is a widely used benchmark for evaluating the performance of virtual screening methods, comprising 17 challenging tasks. One of the key strengths of this data set is its ability to mitigate analogue bias, as the positive samples in MUV are structurally diverse, thus minimizing the risk of overfitting to similar compounds.17
Table 2 demonstrates the statistics of the applied databases in this paper, such as the number of chemical compounds, bioactivity classes, and positive and negative samples. The term “number of positive and negative samples” refers to the number of positive and negative chemical compound–bioactivity class pairs, as defined in eq 4. Table 2 indicates a significant imbalance in the negative-to-positive sample ratio for Tox21 and MUV data sets. To address this issue, a random down sampling of negative data is performed, followed by upsampling of positive samples in order to achieve a more balanced training set.29
Table 2. Statistics of Applied Data Sets.
| database | # compounds
(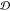 ) ) |
# bioactivity
classes (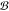 ) ) |
# positive samples | # negative samples |
|---|---|---|---|---|
| SIDER | 1427 | 27 | 21,868 | 16,661 |
| Tox21 | 7831 | 12 | 5862 | 72,084 |
| MUV | 93,087 | 17 | 489 | 1,332,593 |
3.2. BioAct-Het Model Architecture
Since we apply the BioAct-Het model to three different databases, each with distinct characteristics and exhibiting different types of biological activity, the model parameters vary depending on the specific database and biological activity under consideration. In this section, we specify the model parameters based on the intended database.
The Section 2.4 introduces
the model BioAct–Het(Fmd, Gb), where compound 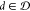 is represented by Fmd and
bioactivity class
is represented by Fmd and
bioactivity class  is shown by Gb. This model includes three main components: f(Fmd), g(Gb), and h(E). The architecture of f(Fmd) consists of ef dense hidden layers activated by using
the ReLU function. In addition, a dropout probability of of is used after every dense layer to
prevent overfitting. The architecture of g(Gb) also consists of eg dense hidden layers with a ReLU activation function
and a dropout probability of og.
is shown by Gb. This model includes three main components: f(Fmd), g(Gb), and h(E). The architecture of f(Fmd) consists of ef dense hidden layers activated by using
the ReLU function. In addition, a dropout probability of of is used after every dense layer to
prevent overfitting. The architecture of g(Gb) also consists of eg dense hidden layers with a ReLU activation function
and a dropout probability of og.
Since the main aim of f(Fmd) and g(Gb) is to make a unified latent space for chemical compounds and bioactivities, we apply the kl-divergence function as the loss function for producing a distribution.
Additionally, the architecture of h(E), E = |Emd – Eb|, includes eh dense layers with a ReLU activation function and a dropout probability of oh. The last layer of h(E) makes the final prediction with one unit and a sigmoid activation function. While the BCP is a classification task, the loss function of h(E) is the binary cross-entropy function. Table 3 includes the information on hidden layer numbers and dropout probabilities.
Table 3. Hyperparameters of BioAct-Het Based on Each Databasea.
| data set | components | input layer dimension | number of hidden layers {dimensions} | the dropout probability | |
|---|---|---|---|---|---|
| SIDER | f(Fmd) | m = GCN-Canonical | 512 | ef = 2 {256, 128} | of = 0.2, 0.2 |
| m = GCN-AttentiveFP | 1024 | ef = 3 {512, 256, 128} | of = 0.2, 0.2, 0.2 | ||
| g(Gb) | 512 | eg = 2 {256, 128} | og = 0.2, 0.2 | ||
| h(E) | 128 | eh = 4 {64, 32, 16, 8, 1} | oh = 0.2, 0.2, 0.2, 0.2 | ||
| Tox21 | f(Fmd) | m = GCN-Canonical | 128 | ef = 1 {64} | of = 0.3 |
| m = GCN-AttentiveFP | 512 | ef = 3 {256, 128, 64} | of = 0.3, 0.3 | ||
| g(Gb) | 512 | eg = 3 {256, 128, 64} | og = 0.3, 0.3 | ||
| h(E) | 64 | eh = 3 {32, 16, 8, 1} | oh = 0.3, 0.3, 0.3 | ||
| MUV | f(Fmd) | m = GCN-Canonical | 128 | ef = 1 {64} | of = 0.3 |
| m = GCN-AttentiveFP | 128 | ef = 1 {64} | of = 0.3 | ||
| g(Gb) | 512 | eg = 3 {256, 128, 64} | og = 0.3, 0.3 | ||
| h(E) | 64 | eh = 3 {32, 16, 8, 1} | oh = 0.3, 0.3, 0.3 | ||
For three components of the model, f( Fmd), g(Gb), and h(E), the input layer dimension, number of hidden layers and the number of their neurons in brackets, and dropout probability are defined based on each applied data set. For each drug d, Fmd shows a vector for representing the drug using the pretrained GCN model m. For each bioactivity class b, a vector named Gb is constructed using Bio-Prof with a length of 512. The fourth column labeled “{dimension}” indicates the dimension of each layer in a sequential order.
3.3. Evaluation Metrics
We select the area under the receiver operating characteristic curve (AUC-ROC) to assess how accurately BioAct-Het performs on the detection of bioactivities for a compound. The AUC-ROC criterion measures the true-positive rate (TPR) against the false-positive rate (FPR) based on different ranking cutoffs,30 where
| 10 |
The definition of true positive (TP), true negative (TN), false positive (FP), and false negative (FN) is given in Table 4. It is worth noting that the AUC-ROC is a better evaluation measure when the data set is imbalanced.
Table 4. Definition of TP, TN, FP, and FN.
| prediction | definition |
|---|---|
| TP | the number of activated bioactivity classes for a drug predicted correctly by the BioAct-Het |
| TN | the number of nonactivated bioactivity classes for a drug predicted correctly by the BioAct-Het |
| FP | the number of activated bioactivity classes for a drug predicted wrongly by the BioAct-Het |
| FN | the number of nonactivated bioactivity classes for a drug predicted wrongly by the BioAct-Het |
3.4. Model Training, Evaluation, and Comparison
This section introduces our training strategies and explains the evaluation approach. We perform three strategies to split training and test sets and evaluate the model as follows
The association-based strategy examines the supervised classification form of the model by splitting the compound–bioactivity class pairs to the training and test set. It is applicable when the aim is to find the association between a bioactivity class and a chemical compound in the data set.
The bioactivity class-based strategy is a similar approach to other previously mentioned methods that keeps out some bioactivity classes during the training and evaluates the performance of BioAct-Het based on unseen bioactivity classes. The approach aims to uncover chemical compounds that demonstrate a new and uncommon bioactivity.
The compound-based strategy utilizes the meta-learning approach by excluding some chemical compounds completely during the training and even making the bioactivity class presentations. Accurately predicting the bioactivity classes of a newly found chemical compound is a crucial step before its market release. The strategy is employed to evaluate the BioAct-Het model’s ability to forecast the bioactivity classes of a new compound.
Furthermore, to assess the effectiveness of the chemical compound representation using two pretrained GCN models named GCN-Canonical and GCN-AttentiveFp, BioAct-Het is examined based on each separately. The details of each strategy and the comparison of the model with the state-of-the-art algorithms are available in the following subsections.
3.4.1. Association-Based Strategy
The association-based strategy is helpful when the aim is to predict the unknown association between a chemical compound and a bioactivity class. This strategy splits the primary data set Δ into the training and test sets as follows
To evaluate the BioAct-Het model based on the association strategy, we perform 10-fold cross-validation. The corresponding results for average of folds using GCN-Canonical and GCN-AttentiveFP chemical structure representation on SIDER, Tox21, and MUV databases are available in Table 5.
Table 5. Average AUC-ROC Scores on 10-Fold Cross-Validation for the Association-Based Strategy Generated by BioAct-Het on Three Databases and Two Different GCN Models for Compound Representation.
| database | GCN-Canonical model AUC-ROC | GCN-AttentiveFP model AUC-ROC |
|---|---|---|
| SIDER | 90.34% ± 0.0053 | 91.11% ± 0.0056 |
| Tox21 | 86.20% ± 0.0187 | 89.80% ± 0.0043 |
| MUV | 68.34% ± 0.0331 | 69.47% ± 0.0447 |
The BioAct-Het method uses the association-based strategy to analyze the supervised classification form of the model. During the training process, certain compound–bioactivity class pairs such as ⟨d, b⟩ are excluded, and the performance of BioAct-Het is evaluated based on these exclusions. In other words, this strategy involves evaluating BioAct-Het using ⟨d, b⟩ pairs, while the chemical compound d may be associated with classes other than b, or the class b may be activated by chemical compounds other than d during model training.
According to Table 5, the performance of the model using GCN-AttentiveFP is better than that of the GCN-Canonical model for representing chemical compounds. Additionally, the model effectively performs on SIDER and Tox21. Since the MUV is an unbiased database with diverse chemical compounds, the AUC-ROC score of the model is lower than two other data sources. However, the model should be compared with other studies to assess how it performs accurately.
The results of BioAct-Het using GCN-AttentiveFP model is compared with four descriptor-based and four graph-based algorithms of the Jiang et al. study31 (see Table 6). The descriptor-based algorithms include random forest (RF), extreme gradient boosting (XGBoost), deep neural network (DNN), and support vector machine (SVM), and graph-based algorithms consider the message passing neural network (MPNN), GCN, graph attention neural network (GAT), and AttentiveFP to classify bioactivity classes.
Table 6. Comparison of BioAct-Het While Compounds Are Represented by the GCN-AttentiveFP Model and Jiang et al. Model Based on the Association Strategy.
| data set | model name | algorithm | AUC-ROC (%) |
|---|---|---|---|
| SIDER | Jiang et al. model | RF | 64.6 |
| XGBoost | 64.2 | ||
| DNN | 63.1 | ||
| SVM | 63 | ||
| MPNN | 59.8 | ||
| GCN | 63.4 | ||
| GAT | 62.7 | ||
| AttentiveFP | 62.3 | ||
| BioAct-Het | association-based | 91.11 | |
| Tox21 | Jiang et al. model | RF | 83.8 |
| XGBoost | 83.6 | ||
| DNN | 84 | ||
| SVM | 81.7 | ||
| MPNN | 80.9 | ||
| GCN | 83.6 | ||
| GAT | 83.5 | ||
| AttentiveFP | 85.2 | ||
| BioAct-Het | association-based | 89.80 | |
| MUV | Jiang et al. model | RF | 6.1 |
| XGBoost | 6.8 | ||
| DNN | 2.1 | ||
| SVM | 11.2 | ||
| MPNN | 1.6 | ||
| GCN | 6.1 | ||
| GAT | 5.7 | ||
| AttentiveFP | 3.8 | ||
| BioAct-Het | association-based | 69.47 |
According to Table 6, the results of BioAct-Het significantly outperform the results in the study of Jiang32 based on each data set. While it may be due to the definition of data sets, it underscores predictive power of the proposed heterogeneous SNN model, BioAct-Het. Moreover, since the MUV consists of unbiased chemical compounds, BioAct-Het gets better level of confidence versus Jiang’s study.32
3.4.2. Bioactivity Class-Based Strategy
The bioactivity class-based strategy is designed to identify chemical compounds that exhibit a novel type of bioactivity. This approach is similar to previous studies that aim to predict how a compound will perform in an experimental assay that differs from the one it was originally trained on and considered as a type of meta-learning.14,22 In this strategy, certain bioactivity classes are excluded from the training set and used as a test set to evaluate the performance of BioAct-Het in predicting the behavior of compounds in those bioactivity classes. For making the training and test sets of the main data set Δ, we perform the following method

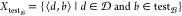

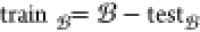
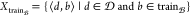

After training the model using  , we examine
the accuracy of BioAct-Het
based on each bioactivity class
, we examine
the accuracy of BioAct-Het
based on each bioactivity class 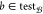 . Table 7 provides the performance of the model based
on each
excluded bioactivity class of the SIDER, Tox21, and MUV using GCN-Canonical
and GCN-AttentiveFP chemical structure representations. To compare
our model with state-of-the-art methods, we select IterRefLSTM14 and Vella’s model,22 both of which also exclude certain bioactivity classes
for evaluating their model. Since the IterRefLSTM utilizes our applied
databases and Vella’s also uses Tox21 and MUV, BioAct-Het is
compared with these models based on their best average performance
on the applied data sets (5+/10−) strategy. Moreover, since
Vella’s model using the prototypical network gets better results,
we use the results of this approach to compare the models. According
to Table 7, BioAct-Het
outperforms IterRefLSTM and Vella’s models using GCN-AttentiveFP
representation for each data set.
. Table 7 provides the performance of the model based
on each
excluded bioactivity class of the SIDER, Tox21, and MUV using GCN-Canonical
and GCN-AttentiveFP chemical structure representations. To compare
our model with state-of-the-art methods, we select IterRefLSTM14 and Vella’s model,22 both of which also exclude certain bioactivity classes
for evaluating their model. Since the IterRefLSTM utilizes our applied
databases and Vella’s also uses Tox21 and MUV, BioAct-Het is
compared with these models based on their best average performance
on the applied data sets (5+/10−) strategy. Moreover, since
Vella’s model using the prototypical network gets better results,
we use the results of this approach to compare the models. According
to Table 7, BioAct-Het
outperforms IterRefLSTM and Vella’s models using GCN-AttentiveFP
representation for each data set.
Table 7. AUC-ROC Scores for the Bioactivity Class Strategy on BioAct-Het While Compounds Are Represented by the GCN-AttentiveFP Model and Comparison with State-of-the-Art Algorithms.
| data set | bioactivity class | BioAct-Het GCN-Canonical model | BioAct-Het GCN-attentiveFP model | IterRefLSTM (%) | Vella’s model (%) |
|---|---|---|---|---|---|
| SIDER | renal and urinary disorders | 83.86% | 84.06% | 71.0 | |
| pregnancy, puerperium and perinatal conditions | 76.94% | 80.12% | 71.4 | ||
| ear and labyrinth disorders | 83.05% | 82.66% | 68.0 | ||
| cardiac disorders | 87.88% | 87.89% | 70.4 | ||
| nervous system disorders | 89.73% | 90.79% | 80.3 | ||
| injury, poisoning and procedural complications | 81.71% | 81.60% | 68.8 | ||
| mean | 83.86% | 84.52% | 71.65 | ||
| variance | 0.0455 | 0.04055 | |||
| Tox21 | SR-HSE | 80.20% | 80.65% | 77.1 | 77.2 |
| SR-MMP | 84.66% | 86.42% | 84.7 | 84.6 | |
| SR-p53 | 84.12% | 84.98% | 83.0 | 85.2 | |
| mean | 82.99% | 84.02% | 81.6 | 82.3 | |
| variance | 0.01987 | 0.02452 | |||
| MUV | MUV-832 | 62.20% | 70.56% | 72.6 | 65.6 |
| MUV-846 | 71.06% | 77.09% | 66.3 | 54.9 | |
| MUV-852 | 61.96% | 73.91% | 75.5 | 45.3 | |
| MUV-858 | 64.50% | 60.79% | 62.9 | 46.9 | |
| MUV-859 | 49.93% | 45.27% | 38.6 | 48.1 | |
| mean | 61.93% | 65.55% | 63.18 | 52.16 | |
| variance | 0.068415 | 0.11507 |
The strategy is particularly useful when a new bioactivity class is identified while training the data. For instance, SIDER is classified into 27 classes using MedDRA, but MedDRA Version 16.0 only contained 26 classes32 of system organs, while Version 19.1 included 27 classes.33 Therefore, it is possible to encounter a new bioactivity class that is not presented in the training data, and this strategy can help address this issue.
3.4.3. Compound-Based Strategy
It is essential to predict the bioactivity classes of a newly found chemical compound before it is released to markets. The compound-based strategy aims to assess how accurately BioAct-Het can forecast the bioactivity classes of a new compound. Thus, we excluded some chemical compounds as a test set. For making the training and test sets of the main data set Δ, we perform the following method

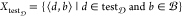
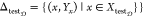



It is important to note that BioAct-Het relies on the Bio-Prof approach, which considers the related chemical compounds to create the bioactivity class representation. To ensure a fair evaluation of the BioAct-Het model, it is necessary to exclude any compounds that are kept out for the test set when applying Bio-Prof. This ensures that the evaluation is conducted solely on the training set.
We believe that this approach of evaluation, which involves testing
the model’s performance without any knowledge of certain compounds,
is more similar to the concept of meta-learning. By excluding the
test set compounds from the bioactivity representation vector, we
can evaluate the generalization ability of the model for new compounds
on which it has not been trained on. This approach enhances the reliability
and robustness of the evaluation process for BioAct-Het. Moreover,
we perform 10-fold cross-validation based on the drug set  . Table 8 shows the average
of the corresponding results of
applying the compound-based strategy using GCN-Canonical and GCN-AttentiveFP
models for bioactivity prediction. Since we cannot find any methods
evaluated similar to our approach, BioAct-Het is not compared to other
models based on this strategy.
. Table 8 shows the average
of the corresponding results of
applying the compound-based strategy using GCN-Canonical and GCN-AttentiveFP
models for bioactivity prediction. Since we cannot find any methods
evaluated similar to our approach, BioAct-Het is not compared to other
models based on this strategy.
Table 8. Average AUC-ROC Scores for the Compound-Based Strategy Using 10-Fold Cross-Validation Generated by BioAct-Het on Three Databases and Two Different GCN Models for Compound Representation.
| Data set | GCN-Canonical model AUC-ROC (%) | GCN-AttentiveFP model AUC-ROC (%) |
|---|---|---|
| SIDER | 81.91 | 84.83 |
| Tox21 | 78.87 | 79.10 |
| MUV | 64.12 | 66.30 |
3.5. Assessment of Chemical Representation on the BioAct-Het Performance
In this section, we aim to investigate the impact of chemical representation on the performance of the BioAct-Het model. To begin, we assess which pretrained GCN-Canonical or GCN-AttentiveFP representations are more effective in addressing the BCP problem. We then analyze the distribution of the extracted vectors from the pretrained GCN model in the latent space, both before and after training the BioAct-Het model. This will indicate whether the BioAct-Het model is able to improve the representation of chemical compounds and enable their separation based on their associated bioactivity classes. To accomplish this, we use the T-distributed stochastic neighbor embedding35 (t-SNE) technique for visualizing the representations of chemical compounds. Finally, we conduct an experiment to determine whether the good separation of data is solely due to the pretrained GCN models or the BioAct-Het model itself.
3.5.1. Comparison of Chemical Compound Representation
BioAct-Het utilizes pretrained models based on the SIDER, Tox21, and MUV databases to represent chemical compounds by employing both GCN-AttentiveFP and GCN-Canonical models. Moreover, to evaluate the performance of the model, three different strategies were conducted as mentioned above. To select which representation is more appropriate in facing the BCP problem, we compare the evaluation score of applying GCN-Canonical and GCN-AttentiveFP representations on each strategy for every database. Figure 4 shows that the evaluation strategies achieve higher scores using GCN-AttentiveFP than using GCN-Canonical, indicating that the former more accurately extracts functional and structural information from chemical compounds in addressing the BCP problem. These findings suggest that GCN-AttentiveFP generates more effective representation for chemical compounds in the context of BCP and improve the accuracy of predictive models for drug discovery and development.
Figure 4.

Assessment of chemical compound representation based on each evaluation strategy.
3.5.2. Distribution Analysis of Chemical Compound Representation
To assess the effectiveness of the proposed model in bringing compounds with similar bioactivity classes closer together in the latent space, we conduct an experiment to visualize the distribution of chemical compounds before and after applying BioAct-Het, using the t-SNE technique. t-SNE is a dimensionality reduction and data visualization technique that uses an association-based strategy.
Recall that BioAct-Het comprises two networks for embedding of chemical compounds and bioactivity classes, respectively, that make the representation of compounds and bioactivities comparable in the unified latent space.
Figure 5 preserves the effectiveness of our model in capturing the similarities of chemical compounds with shared bioactivity classes in the latent space using t-SNE. Specifically, Figure 5A illustrates the distribution of chemical compound representations extracted from pretrained GCN-AttentiveFP on the SIDER before training the BioAct-Het model, while Figure 5B shows their distribution after training with the SIDER database.
Figure 5.

Illustration of bringing similar concepts close together using a heterogeneous SNN. (A) Distribution of chemical compound representations extracted from pretrained GCN-AttentiveFP on the SIDER data set prior to training the BioAct-Het model. (B) Distribution after training the model with the SIDER database.
Furthermore, after fitting the BioAct-Het model on the training data, we assess its performance on the distribution of test set compounds. We find that the compounds in the test set lack in exhibiting any meaningful relationship in the space before training (see Figure 6A). However, after fitting the model, compounds with similar bioactivity classes are brought closer together (Figure 6B). Figure 6 illustrates this improvement.
Figure 6.

Test set distribution before (A) and after embedding (B) with BioAct-Het.
3.5.3. Dependency Assessment of the Model to the Pretrained GCN Models
The previous subsection showcased the effectiveness of our model in improving the representation of the chemical compounds. In this section, we aim to demonstrate the extent to which the model’s performance is due to the extracted features from the pretrained model on the intended bioactivities. In other words, while the previous section highlighted the ability of BioAct-Het to create an accurate representation of chemical compounds, we want to conduct further analysis to ensure that the good separation of information achieved after training BioAct-Het is not solely due to the use of pretrained GCN models for compound representation under the bioactivity of consideration.
To do so, we employ a transfer learning approach that involves using a model trained on a specific task and transferring its benefits to a different but related task.
For this aim, we use the GCN-AttentiveFP model pretrained on the SIDER database to represent the chemical compounds in the Tox21 database. We also remove the common chemical structures between SIDER and Tox21. BioAct-Het is then trained on Tox21 using the chemical representations obtained from GCN-AttentiveFP pretrained on SIDER.
Based on the results presented in Figure 7, we conclude that using GCN-AttentiveFP pretrained on SIDER to represent compounds of Tox21, as opposed to using GCN-AttentiveFP pretrained on Tox21, did not result in a significant difference in performance across all evaluation strategies. However, it is still recommended to use a pretrained model that is tailored to the specific features of interest to achieve optimal performance.
Figure 7.

Assessment of the pretrained GCN model role on the performance of the model for Tox21. The blue bars are results obtained by using the pretrained model on SIDER for representing compounds of Tox21, and the green bars are the results of using the pretrained model on the Tox21.
4. Discussion
In this section, we aim to evaluate the effectiveness of the proposed model in determining its potential usefulness in real-world applications, particularly in discovering the bioactivities of newly developed chemical compounds. To the best of our knowledge, this case study is the first of its kind. Obtaining high-quality bioactivity data, such as toxicological experiment data or MUV experiments, can be challenging, but we use side effects as a reasonable approach due to the availability of these data in various resources.
Furthermore, we focus on COVID-19, a newly emerging disease that has caused a significant global health crisis, prompting researchers to investigate effective treatments and vaccines. As physicians prescribe different medications or newly introduced drugs, it is crucial to evaluate their potential bioactivities, including side effects, accurately before administering them to patients to ensure patient safety and improve treatment outcomes. The proposed model can assist in identifying the bioactivities of such drugs and aid in the decision-making process for their use in clinical practice.
In the early stages of the pandemic, many drugs were repurposed for treating COVID-19, including hydroxychloroquine, an antimalarial and immunosuppressive drug.34 However, further studies have shown that hydroxychloroquine can cause severe side effects, including cardiac toxicity, such as arrhythmias and sudden cardiac arrest.35,36 As a result, the use of hydroxychloroquine in COVID-19 patients has been discontinued in most countries. Except this, some researchers suggested that using antibiotic or other antiviral37,38 drugs such as vancomycin (DB00512) and oseltamivir (DB00198), which were originally prescribed for treating MRSA39,40 and influenza type A and B,41,42 may treat COVID-19 patients. More recently, molnupiravir (DB15661) has emerged as a promising treatment for COVID-19.43,44 It is an FDA-approved drug that has been shown to be effective in preventing severe outcomes and hospitalization in COVID-19 patients.44
In this study, we focus on these three drugs, namely, vancomycin (DB00512), oseltamivir (DB00198), and molnupiravir (DB15661), which are not included in the SIDER database. This approach, which is compatible with our compound-based strategy, will help us determine whether the model can accurately predict bioactivities in real-world scenarios and assist in the discovery of new drug candidate’s bioactivities, specifically side effects.
To do so, we employ the pretrained GCN-AttentiveFP model on SIDER to get the chemical compounds representations. These representations are then fed to the BioAct-Het model, paired each side effect class representation which was obtained based on eq 7. The model predicts the probability of each drug exhibiting side effect classes in the range of 0 to 1. The side effect classes are obtained from the medical dictionary for regulatory activities (MedDRA), a standardized medical terminology that classifies and codes medical history, clinical trial data, and adverse events in drug development and postmarket surveillance. MedDRA comprises more than 20,000 preferred terms (PT), which are the low-level basic classes, organized into 27 system organ classifications.45
In the next step, we investigate an analysis of 27,699 side effect case reports associated with vancomycin, 13,374 reports associated with oseltamivir, and 2933 reports for molnupiravir recorded in the FDA adverse event reporting system (FAERS)46 database since 1978, 1999, and 2022, respectively.46 It indicates that while the first two drugs have long-standing uses and their most side effects are known, as molnupiravir is recently released to markets, its side effects need to be completed during the time and using postmarketing analysis. In this analysis, we consider only side effects that are reported in >1% of cases. Tables S1–S3 in the appendix file shows these side effects and their belonging to each side effect classes based on MedDRA organ system classification.
We then compare the model’s predicted probabilities with the reported side effect with a frequency of more than 1% for these drugs. To evaluate the accuracy of the model for these drugs, we define TP, TN, FP, and FN according to Table 4. It should be noted about FP that it may report some PTs for any drug which is less than 1% frequent.
Table S4 in the appendix file demonstrates the performance of the model for each drug separately and indicates the accuracy of 85.18, 70.37, and 70.37% for vancomycin, oseltamivir, and molnupiravir, respectively.
Since the selected drugs are not included in the SIDER database, we can be certain that our model does not see them during training such as the compound-based strategy, where the goal is to predict bioactivity classes for a new chemical compound. Notably, the BioAct-Het model successfully predicts the bioactivity classes of all reported side effects, confirming the efficacy of the compound-based evaluation strategy. Moreover, the study suggests possible associations between vancomycin and side effects on metabolism and nutrition disorders, musculoskeletal and connective tissue disorders, psychiatric disorders, and nervous system disorders, although their prevalence in FAERS is below 1%. It is worth considering that other studies have reported side effects on these organ systems,47−50 indicating potential variations in reporting across diverse data sources. In addition, the model estimates possible exhibition of side effect classes metabolism and nutrition disorders, eye disorder, musculoskeletal and connective tissue disorders, immune system disorders, reproductive system and breast disorders, vascular disorders, and blood and lymphatic system disorders for oseltamivir, which are also reported in other research studies.51−59 Among the selected drugs, molnupiravir has recently been developed and released to the markets. So, it is highly possible to report any side effect classes later. Moreover, BioAct-Het suggests to investigate the potential association between molnupiravir and investigations, musculoskeletal and connective tissue disorders, immune system disorders, vascular disorders, blood and lymphatic system disorders, and cardiac disorders, which are all reported in a new survey that was recently published.60 However, due to limited studies on this drug, further research is needed to fully understand its potential side effects and evaluate its safety and efficacy for treating COVID-19 and other diseases.
Figure 8 presents a bubble chart that elucidates the performance of our model in predicting the side effect classes for the selected drugs. The X-axis represents the observed side effect classes associated with the drug, while the Y-axis corresponds to the model’s predictive values. The bubble size in this chart is indicative of the model’s confidence in its predictions, with larger bubbles corresponding to higher probabilities. Thus, larger bubbles denote the side effect classes that the model most frequently predicts, while smaller bubbles signify those with lower probabilities. Interpreting the chart, we anticipate seeing the observed side effects predominantly in the upper right quadrant, representing higher probabilities and positive class prediction. In contrast, the lower left quadrant is likely to feature side effect classes that are not commonly observed, highlighting the model’s ability to discern between frequently and infrequently occurring side effects.
Figure 8.

Bubble charts show the performance of the proposed model in predicting side effect classes of vancomycin, oseltamivir, and molnupiravir, which are not included in the SIDER database. The X-axis represents the real state of drug and side effect class association based on reported side effects in FAERS with more than 1% frequency, while the Y-axis represents the predicted probability of these associations. The ideal outcome is for most of the bubbles to be located in the right-up and left-down corners, indicating true positives (TPs) and true negatives (TNs), respectively. However, due to limited case reports for molnupiravir, a new drug, the number of false positives (FPs) is higher than those of the other two drugs. Nonetheless, a recent study has shown that molnupiravir causes all of these side effects, which our model correctly predicts as positive.60
5. Conclusions
This paper introduces the BioAct-Het model for addressing the BCP problem. The main contributions of this model can be attributed to two key factors: first, our approach of introducing Bio-Prof to represent bioactivity classes as input and second, our use of a heterogeneous SNN named BioAct-Het instead of a homogeneous one, as retrospective studies employed that. The use of a heterogeneous SNN in our approach is motivated by the complex and diverse nature of the relationships between chemical compounds and bioactivity classes. Since their vectors cannot be directly compared, we develop a representation that shares common properties between the two through the use of a heterogeneous SNN known as BioAct-Het. The model consists of two branches for embedding compounds and bioactivity classes with the aim of capturing similar concepts in a unified latent space.
While BioAct-Het relied on Bio-Prof for representing bioactivity classes, it benefited from the pretrained GCN model on the intended bioactivity to represent the chemical structure of compounds. According to the experimental results, the GCN-AttentiveFP model represented the chemical structure of the chemical compounds more accurately than GCN-Canonical. Moreover, based on conducted experiments such as visualizing the distribution of chemical compounds before and after fitting the model using the t-SNE technique, while using the pretrained GCN model as the compound representation is suggested, the good performance of the model is not solely based on them, which highlight the role of preparing the data set and the applied heterogeneous SNN model.
Furthermore, we evaluated the performance of the BioAct-Het model in three strategies: association-based as a supervised classification, bioactivity class-based as a similar approach to previous studies, and finally, the compound-based as a meta-learning approach. The association-based strategy kept some compound–bioactivity class associations out during training and estimated their association while evaluation. The bioactivity class-based strategy excluded some bioactivity classes during the training completely. It showed the ability of the model when there is limited information about a bioactivity class. The compound-based strategy kept out some compounds during training and made the bioactivity class representation and demonstrated the power of the method for predicting the potential bioactivity of a new chemical compound. To benchmark the model, it utilized the SIDER, Tox21, and MUV databases.
In addition, BioAct-Het was compared with IterRefLSTM and Vella’s and Jiang’s study. The results showed that the BioAct-Het was significantly more accurate than other methods that addressed the BCP problem. Finally, we assessed the ability of our model to address a real-world problem by predicting side effect classes for vancomycin, oseltamivir, and molnupiravir. This analysis demonstrated its potential practical applications.
A significant limitation of this study is its focus on small molecules, which may not encompass larger and more complex molecules, in particular, macrocycles and their stereoisomers, such as calix[4]arene-pyrazole.61 Stereoisomers, molecules sharing identical molecular formulas but differing in spatial arrangements, can present significant challenges, especially when they display varying bioactivity. This factor should be carefully considered in future research endeavors.
Glossary
Table of notations
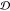
set of chemical compounds
- d
compound
- p
size of

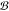
set of bioactivity classes
- b
bioactivity class
- m
canonical or AttentiveFP model (from
 )
)- lm
length of compound’s representation vector regarding model m
- Fmd
representation vector for drug d by regarding m approach
- rd = [rd1,···, rdk]
Morgan fingerprint representation
- k
length of fingerprint
- Gb = [Gb[1],···,Gb[k]]
representation of bioactivity class b
- f(Fmd):Rlm → Rn)
chemical compound embedding function
- g(Gb): Rk → Rn)
bioactivity class embedding function
- n
dimension of the unified latent space
- Emd
embedded vector of a chemical compound
- Eb
embedded vector of chemical bioactivity
- E
subtraction of Emd and Eb
- h
subtraction function
- ef
number of dense hidden layers for branch f(Fmd)
- eg
number of dense hidden layers for branch g(Gb)
- eh
number of dense hidden layers for branch h(E)
- of
dropout probability of f(Fmd)layers
- og
dropout probability of g(Gb)layers
- oh
dropout probability of h(E) layers
Glossary
Abbreviations
- SAR
structure–activity relationship
- GCN
graph convolutional neural network
- CNN
convolutional neural network
- SNN
siamese neural network
- ECFP
extended-connectivity fingerprint
- BCP
bioactivity class prediction
- TP
true positive
- TN
true negative
- FP
false positive
- FN
false negative
- DNN
deep neural network
- MPNN
message passing neural network
- GAT
graph attention neural network
- PT
preferred term
- t-SNE
T-distributed stochastic neighbor embedding
Data Availability Statement
The data and code underlying this article are available publicly at https://github.com/CBRC-lab/BioAct-Het. However, data sets were derived from sources in the public domain.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c05778.
Most frequent reported side effects for vancomycin using FAERS portal from 1978 to 31 March 2023 (Table S1); most frequent reported side effects for Oseltamivir using FAERS portal from 1999 to 31 March 2023 (Table S2); most frequent reported side effects for Molnupiravir using FAERS portal from 2022 to 31 March 2023 (Table S3); and performance of model for selected drugs (Table S4) (PDF)
Author Contributions
M.P.H. developed and implemented the method and conducted the experiments. Z.G. interpreted the results and wrote the manuscript. F.Z. conceptualized the study, interpreted the results, supervised the work, administered the project, and edited the manuscript. M.A. conceptualized using the siamese neural network and edited the manuscript. G.P. confirmed the medical information related to COVID-19.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The authors declare no competing financial interest.
Supplementary Material
References
- Wouters O. J.; McKee M.; Luyten J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853. 10.1001/jama.2020.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waring M. J.; Arrowsmith J.; Leach A. R.; Leeson P. D.; Mandrell S.; Owen R. M.; Pairaudeau G.; Pennie W. D.; Pickett S. D.; Wang J.; Wallace O.; Weir A. An Analysis of the Attrition of Drug Candidates from Four Major Pharmaceutical Companies. Nat. Rev. Drug Discovery 2015, 14 (7), 475–486. 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
- Miller B. F.; O’Toole M. T.. Encyclopedia & Dictionary of Medicine, Nursing, and Allied Health; Saunders, 2003. [Google Scholar]
- Hughes J. P.; Rees S. S.; Kalindjian S. B.; Philpott K. L. Principles of Early Drug Discovery. Br. J. Pharmacol. 2011, 162 (6), 1239–1249. 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond J. L.; Ruddigkeit L.; Blum L.; van Deursen R. The Enumeration of Chemical Space. WIREs Comput. Mol. Sci. 2012, 2 (5), 717–733. 10.1002/wcms.1104. [DOI] [Google Scholar]
- Ghorbanali Z.; Zare-Mirakabad F.; Akbari M.; Salehi N.; Masoudi-Nejad A. DrugRep-KG: Toward Learning a Unified Latent Space for Drug Repurposing Using Knowledge Graphs. J. Chem. Inf. Model. 2023, 63, 2532–2545. 10.1021/acs.jcim.2c01291. [DOI] [PubMed] [Google Scholar]
- Nantasenamat C.; Isarankura-Na-Ayudhya C.; Prachayasittikul V. Advances in Computational Methods to Predict the Biological Activity of Compounds. Expert Opin. Drug Discovery 2010, 5 (7), 633–654. 10.1517/17460441.2010.492827. [DOI] [PubMed] [Google Scholar]
- Bertoni M.; Duran-Frigola M.; Badia-i-Mompel P.; Pauls E.; Orozco-Ruiz M.; Guitart-Pla O.; Alcalde V.; Diaz V. M.; Berenguer-Llergo A.; Brun-Heath I.; Villegas N.; de Herreros A. G.; Aloy P. Bioactivity Descriptors for Uncharacterized Chemical Compounds. Nat. Commun. 2021, 12 (1), 3932 10.1038/s41467-021-24150-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Greenside P.; Paik H.; Sirota M.; Hadley D.; Butte A. J. Relating Chemical Structure to Cellular Response: An Integrative Analysis of Gene Expression, Bioactivity, and Structural Data Across 11,000 Compounds. CPT: Pharmacometrics Syst. Pharmacol. 2015, 4 (10), 576–584. 10.1002/psp4.12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. C.; Fraser T. R. On the Connection between Chemical Constitution and Physiological Action; with Special Reference to the Physiological Action of the Salts of the Ammonium Bases Derived from Strychnia, Brucia, Thebaia, Codeia, Morphia, and Nicotia. J. Anat. Physiol. 1868, 2 (2), 224–242. 10.1017/S0080456800028155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weininger D. SMILES, a Chemical Language and Information System: 1: Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28 (1), 31–36. 10.1021/ci00057a005. [DOI] [Google Scholar]
- Pattanaik L.; Coley C. W. Molecular Representation: Going Long on Fingerprints. Chem 2020, 6 (6), 1204–1207. 10.1016/j.chempr.2020.05.002. [DOI] [Google Scholar]
- Krawczyk B. Learning from Imbalanced Data: Open Challenges and Future Directions. Prog. Artif. Intell. 2016, 5 (4), 221–232. 10.1007/s13748-016-0094-0. [DOI] [Google Scholar]
- Altae-Tran H.; Ramsundar B.; Pappu A. S.; Pande V. Low Data Drug Discovery with One-Shot Learning. ACS Cent. Sci. 2017, 3 (4), 283–293. 10.1021/acscentsci.6b00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M.; Letunic I.; Jensen L. J.; Bork P. The SIDER Database of Drugs and Side Effects. Nucleic Acids Res. 2016, 44, D1075–D1079. 10.1093/NAR/GKV1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard A. M.; Huang R.; Waidyanatha S.; Shinn P.; Collins B. J.; Thillainadarajah I.; Grulke C. M.; Williams A. J.; Lougee R. R.; Judson R. S.; Houck K. A.; Shobair M.; Yang C.; Rathman J. F.; Yasgar A.; Fitzpatrick S. C.; Simeonov A.; Thomas R. S.; Crofton K. M.; Paules R. S.; Bucher J. R.; Austin C. P.; Kavlock R. J.; Tice R. R. The Tox21 10K Compound Library: Collaborative Chemistry Advancing Toxicology. Chem. Res. Toxicol. 2021, 34 (2), 189–216. 10.1021/acs.chemrestox.0c00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer S. G.; Baumann K. Maximum Unbiased Validation (MUV) Data Sets for Virtual Screening Based on PubChem Bioactivity Data. J. Chem. Inf Model 2009, 49 (2), 169–184. 10.1021/ci8002649. [DOI] [PubMed] [Google Scholar]
- Torres L.; Monteiro N.; Oliveira J.; Arrais J.; Ribeiro B. In Exploring a Siamese Neural Network Architecture for One-Shot Drug Discovery, Proceedings - IEEE 20th International Conference on Bioinformatics and Bioengineering, BIBE 2020; IEEE, 2020; pp 168–175.
- Fernández-Llaneza D.; Ulander S.; Gogishvili D.; Nittinger E.; Zhao H.; Tyrchan C. Siamese Recurrent Neural Network with a Self-Attention Mechanism for Bioactivity Prediction. ACS Omega 2021, 6 (16), 11086–11094. 10.1021/acsomega.1C01266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez D.; Gaulton A.; Bento A. P.; Chambers J.; De Veij M.; Félix E.; Magariños M. P.; Mosquera J. F.; Mutowo P.; Nowotka M.; Gordillo-Marañón M.; Hunter F.; Junco L.; Mugumbate G.; Rodriguez-Lopez M.; Atkinson F.; Bosc N.; Radoux C. J.; Segura-Cabrera A.; Hersey A.; Leach A. R. ChEMBL: Towards Direct Deposition of Bioassay Data. Nucleic Acids Res. 2019, 47 (D1), D930–D940. 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ExCAPE-DB: ExCAPE Chemogenomics Database, https://solr.ideaconsult.net/search/excape/. (accessed July 15, 2023).
- Vella D.; Ebejer J. P. Few-Shot Learning for Low-Data Drug Discovery. J. Chem. Inf. Model. 2023, 63 (1), 27–42. 10.1021/acs.jcim.2c00779. [DOI] [PubMed] [Google Scholar]
- Mysinger M. M.; Carchia M.; Irwin J. J.; Shoichet B. K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55 (14), 6582–6594. 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Zhou J.; Hu J.; Fan W.; Zhang Y.; Gu Y.; Karypis G. DGL-LifeSci: An Open-Source Toolkit for Deep Learning on Graphs in Life Science. ACS Omega 2021, 6 (41), 27233–27238. 10.1021/acsomega.1c04017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipf T. N.; Welling M. In Semi-Supervised Classification with Graph Convolutional Networks; 5th International Conference on Learning Representations, ICLR 2017 - Conference Track Proceedings, 2016.
- Xiong Z.; Wang D.; Liu X.; Zhong F.; Wan X.; Li X.; Li Z.; Luo X.; Chen K.; Jiang H.; Zheng M. Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism. J. Med. Chem. 2020, 63 (16), 8749–8760. 10.1021/acs.jmedchem.9b00959. [DOI] [PubMed] [Google Scholar]
- Morgan H. L. The Generation of a Unique Machine Description for Chemical Structures—A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5 (2), 107–113. 10.1021/C160017A018. [DOI] [Google Scholar]
- Chicco D. Siamese Neural Networks: An Overview. Methods Mol. Biol. 2021, 2190, 73–94. 10.1007/978-1-0716-0826-5_3/COVER. [DOI] [PubMed] [Google Scholar]
- Chawla N. V.; Bowyer K. W.; Hall L. O.; Kegelmeyer W. P. SMOTE: Synthetic Minority Over-Sampling Technique. J. Artifi. Intell. Res. 2002, 16, 321–357. 10.1613/jair.953. [DOI] [Google Scholar]
- Powers D. M. W.Evaluation: From Precision, Recall and F-Measure to ROC, Informedness, Markedness and Correlation. 2020, arXiv:2010.16061. arXiv.org e-Printarchive. https://arxiv.org/abs/2010.16061.
- Jiang D.; Wu Z.; Hsieh C. Y.; Chen G.; Liao B.; Wang Z.; Shen C.; Cao D.; Wu J.; Hou T. Could Graph Neural Networks Learn Better Molecular Representation for Drug Discovery? A Comparison Study of Descriptor-Based and Graph-Based Models. J. Cheminf. 2021, 13 (1), 12 10.1186/s13321-020-00479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’hare B. J.; Links Q.. Published by MedDRA MSSO for Our Users What’s New for MedDRA Version 16.0 Behind the Scenes: The Points to Consider Documents New Developments in MedDRA Version 16.0 MedDRA Training Did You Know··· MedDRA Training (Courses and Schedule). 2013.
- Introductory Guide MedDRA Version 19.1. 2016.
- Shih R. D.; Johnson H. M.; Maki D. G.; Hennekens C. H. Hydroxychloroquine for Coronavirus: The Urgent Need for a Moratorium on Prescriptions. Am. J. Med. 2020, 133 (9), 1007–1008. 10.1016/j.amjmed.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fram G.; Wang D. D.; Malette K.; Villablanca P.; Kang G.; So K.; Basir M. B.; Khan A.; McKinnon J. E.; Zervos M.; O’Neill W. W. Cardiac Complications Attributed to Hydroxychloroquine: A Systematic Review of the Literature Pre-COVID-19. Curr. Cardiol. Rev. 2021, 17 (3), 319–327. 10.2174/1573403X16666201014144022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pers Y. M.; Padern G. Revisiting the Cardiovascular Risk of Hydroxychloroquine in RA. Nat. Rev. Rheumatol. 2020, 16 (12), 671–672. 10.1038/s41584-020-00521-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford-Faucher A. Antibiotics for the Treatment of COVID-19. Am. Fam. Phys. 2022, 105 (3), 237–238. [PubMed] [Google Scholar]
- Beovic B.; Dousak M.; Ferreira-Coimbra J.; Nadrah K.; Rubulotta F.; Belliato M.; Berger-Estilita J.; Ayoade F.; Rello J.; Erdem H. Antibiotic Use in Patients with COVID-19: A ‘Snapshot’ Infectious Diseases International Research Initiative (ID-IRI) Survey. J. Antimicrob. Chemother. 2020, 75 (11), 3386–3390. 10.1093/jac/dkaa326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei E.; Tao H.; Jiao S.; Yang A.; Zhou Y.; Wang M.; Wen K.; Wang Y.; Chen Z.; Chen X.; Song J.; Zhou C.; Huang W.; Xu L.; Guan D.; Tan C.; Liu H.; Cai Q.; Zhou K.; Modica J.; Huang S. Y.; Huang W.; Feng X. Potentiation of Vancomycin: Creating Cooperative Membrane Lysis through a “Derivatization-for-Sensitization” Approach. J. Am. Chem. Soc. 2022, 144 (23), 10622–10639. 10.1021/JACS.2C03784. [DOI] [PubMed] [Google Scholar]
- General Information | MRSA | CDC. https://www.cdc.gov/mrsa/community/index.html. (accessed July 13, 2023).
- McClellan K.; Perry C. M. Oseltamivir: A Review of Its Use in Influenza. Drugs 2001, 61 (2), 263–283. 10.2165/00003495-200161020-00011. [DOI] [PubMed] [Google Scholar]
- Jefferson T.; Jones M.; Doshi P.; Spencer E. A.; Onakpoya I.; Heneghan C. J. Oseltamivir for Influenza in Adults and Children: Systematic Review of Clinical Study Reports and Summary of Regulatory Comments. BMJ 2014, 348, g2545 10.1136/BMJ.G2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W.; Eron J. J.; Holman W.; Cohen M. S.; Fang L.; Szewczyk L. J.; Sheahan T. P.; Baric R.; Mollan K. R.; Wolfe C. R.; Duke E. R.; Azizad M. M.; Borroto-Esoda K.; Wohl D. A.; Loftis A. J.; Alabanza P.; Lipansky F.; Painter W. P.. Molnupiravir, an Oral Antiviral Treatment for COVID-19 medRxiv 2021 10.1101/2021.06.17.21258639. [DOI]
- Jayk Bernal A.; Gomes da Silva M. M.; Musungaie D. B.; Kovalchuk E.; Gonzalez A.; Delos Reyes V.; Martín-Quirós A.; Caraco Y.; Williams-Diaz A.; Brown M. L.; Du J.; Pedley A.; Assaid C.; Strizki J.; Grobler J. A.; Shamsuddin H. H.; Tipping R.; Wan H.; Paschke A.; Butterton J. R.; Johnson M. G.; De Anda C. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386 (6), 509–520. 10.1056/NEJMoa2116044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Medical Dictionary for Regulatory Activities Understanding MedDRA.
- FDA Adverse Events Reporting System (FAERS) Public Dashboard - FDA Adverse Events Reporting System (FAERS) Public Dashboard | Sheet - Qlik Sense. https://fis.fda.gov/sense/app/95239e26-e0be-42d9-a960-9a5f7f1c25ee/sheet/8eef7d83-7945-4091-b349-e5c41ed49f99/state/analysis. (accessed July 13, 2023).
- Vickers R. J.; Tillotson G. S.; Nathan R.; Hazan S.; Pullman J.; Lucasti C.; Deck K.; Yacyshyn B.; Maliakkal B.; Pesant Y.; Tejura B.; Roblin D.; Gerding D. N.; Wilcox M. H.; Bhan A.; Campbell W.; Chopra T.; Deck K.; Golan Y.; Gordon I.; Kamepalli R.; Khanna S.; Lee C.; Lucasti C.; Maliakkal B.; Minang I.; Mullane K.; Nathan R.; Oughton M.; Pesant Y.; Phillips J.; Pullman J.; Riska P.; Schrock C.; Siegel J.; Steinberg A.; Talan D.; Tamang S.; Tan M.; Weiss K.; Wang C.; Yacyshyn B.; Young J. A.; Zenilman J. Efficacy and Safety of Ridinilazole Compared with Vancomycin for the Treatment of Clostridium Difficile Infection: A Phase 2, Randomised, Double-Blind, Active-Controlled, Non-Inferiority Study. Lancet Infect. Dis. 2017, 17 (7), 735–744. 10.1016/S1473-3099(17)30235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essali N.; Miller B. J. Psychosis as an Adverse Effect of Antibiotics. Brain, Behav., Immun.: Health 2020, 9, 100148 10.1016/j.bbih.2020.100148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagami J.; Nakamura Y.; Nagao K.; Funakoshi T.; Takahashi H.; Tanikawa A.; Hachiya T.; Yamamoto T.; Ishida-Yamamoto A.; Tanaka T.; Fujimoto N.; Nishigori C.; Yoshida T.; Ishii N.; Hashimoto T.; Amagai M. Vancomycin Mediates IgA Autoreactivity in Drug-Induced Linear IgA Bullous Dermatosis. J. Invest. Dermatol. 2018, 138 (7), 1473–1480. 10.1016/j.jid.2017.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basolo A.; Hohenadel M.; Ang Q. Y.; Piaggi P.; Heinitz S.; Walter M.; Walter P.; Parrington S.; Trinidad D. D.; von Schwartzenberg R. J.; Turnbaugh P. J.; Krakoff J. Effects of Underfeeding and Oral Vancomycin on Gut Microbiome and Nutrient Absorption in Humans. Nat. Med. 2020, 26 (4), 589–598. 10.1038/S41591-020-0801-Z. [DOI] [PubMed] [Google Scholar]
- Han N.; Oh J. M.; Kim I. W. Assessment of Adverse Events Related to Anti-Influenza Neuraminidase Inhibitors Using the FDA Adverse Event Reporting System and Online Patient Reviews. Sci. Rep. 2020, 10 (1), 3116 10.1038/s41598-020-60068-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Oliveira J. T.; Santos A. L.; Gomes C.; Barros R.; Ribeiro C.; Mendes N.; De Matos A. J.; Vasconcelos M. H.; Oliveira M. J.; Reis C. A.; Gärtner F. Anti-Influenza Neuraminidase Inhibitor Oseltamivir Phosphate Induces Canine Mammary Cancer Cell Aggressiveness. PLoS One 2015, 10 (4), e0121590 10.1371/JOURNAL.PONE.0121590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger R. A.; Billingsley J. L.; Huffman J. H.; Bailey K. W.; Kim C. U.; Sidwell R. W. Immunological Effects of the Orally Administered Neuraminidase Inhibitor Oseltamivir in Influenza Virus-Infected and Uninfected Mice. Immunopharmacology 2000, 47 (1), 45–52. 10.1016/S0162-3109(99)00184-8. [DOI] [PubMed] [Google Scholar]
- Bird N. L.; Olson M. R.; Hurt A. C.; Oshansky C. M.; Oh D. Y.; Reading P. C.; Chua B. Y.; Sun Y.; Tang L.; Handel A.; Jackson D. C.; Turner S. J.; Thomas P. G.; Kedzierska K. Oseltamivir Prophylaxis Reduces Inflammation and Facilitates Establishment of Cross-Strain Protective T Cell Memory to Influenza Viruses. PLoS One 2015, 10 (6), e0129768 10.1371/JOURNAL.PONE.0129768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han N.; Oh J. M.; Kim I. W. Assessment of Adverse Events Related to Anti-Influenza Neuraminidase Inhibitors Using the FDA Adverse Event Reporting System and Online Patient Reviews. Sci. Rep. 2020, 10 (1), 3116 10.1038/s41598-020-60068-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. W.; Lee J. E.; Choi H. Y.; Lee J. S. Oseltamivir (Tamiflu)-Induced Bilateral Acute Angle Closure Glaucoma and Transient Myopia. Indian J. Ophthalmol. 2014, 62 (12), 1165–1167. 10.4103/0301-4738.109531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson L.; Price J. E. Temporary Central Vision Blindness After Oseltamivir Administration in A-Year-Old Pediatric Male Positive for Influenza A. Hosp. Pharm. 2021, 56 (6), 678–680. 10.1177/0018578720942225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara K.; Yamamoto Y.; Saita T.; Matsufuji S. Metabolism and Disposition of Oseltamivir (OS) in Rats, Determined by Immunohistochemistry with Monospecific Antibody for OS or Its Active Metabolite Oseltamivir Carboxylate (OC): A Possibility of Transporters Dividing the Drugs’ Excretion into the Bile and Kidney. Pharmacol Res. Perspect. 2020, 8 (3), e00597 10.1002/prp2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocquet O.; Wahart A.; Sarazin T.; Vincent E.; Schneider C.; Fougerat A.; Gayral S.; Henry A.; Blaise S.; Romier-Crouzet B.; Boulagnon C.; Jaisson S.; Gillery P.; Bennasroune A.; Sartelet H.; Laffargue M.; Martiny L.; Duca L.; Maurice P. Adverse Effects of Oseltamivir Phosphate Therapy on the Liver of LDLR–/– Mice Without Any Benefit on Atherosclerosis and Thrombosis. J. Cardiovasc. Pharmacol. 2021, 77 (5), 660–672. 10.1097/FJC.0000000000001002. [DOI] [PubMed] [Google Scholar]
- Khoo S. H.; FitzGerald R.; Saunders G.; Middleton C.; Ahmad S.; Edwards C. J.; Hadjiyiannakis D.; Walker L.; Lyon R.; Shaw V.; Mozgunov P.; Periselneris J.; Woods C.; Bullock K.; Hale C.; Reynolds H.; Downs N.; Ewings S.; Buadi A.; Cameron D.; Edwards T.; Knox E.; Donovan-Banfield I.; Greenhalf W.; Chiong J.; Lavelle-Langham L.; Jacobs M.; Northey J.; Painter W.; Holman W.; Lalloo D. G.; Tetlow M.; Hiscox J. A.; Jaki T.; Fletcher T.; Griffiths G.; Paton N.; Hayden F.; Darbyshire J.; Lucas A.; Lorch U.; Freedman A.; Knight R.; Julious S.; Byrne R.; Cubas Atienzar A.; Jones J.; Williams C.; Song A.; Dixon J.; Alexandersson A.; Hatchard P.; Tilt E.; Titman A.; Doce Carracedo A.; Chandran Gorner V.; Davies A.; Woodhouse L.; Carlucci N.; Okenyi E.; Bula M.; Dodd K.; Gibney J.; Dry L.; Rashid Gardner Z.; Sammour A.; Cole C.; Rowland T.; Tsakiroglu M.; Yip V.; Osanlou R.; Stewart A.; Parker B.; Turgut T.; Ahmed A.; Starkey K.; Subin S.; Stockdale J.; Herring L.; Baker J.; Oliver A.; Pacurar M.; Owens D.; Munro A.; Babbage G.; Faust S.; Harvey M.; Pratt D.; Nagra D.; Vyas A. Molnupiravir versus Placebo in Unvaccinated and Vaccinated Patients with Early SARS-CoV-2 Infection in the UK (AGILE CST-2): A Randomised, Placebo-Controlled, Double-Blind, Phase 2 Trial. Lancet Infect. Dis. 2023, 23 (2), 183–195. 10.1016/S1473-3099(22)00644-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muravev A. A.; Voloshina A. D.; Sapunova A. S.; Gabdrakhmanova F. B.; Lenina O. A.; Petrov K. A.; Shityakov S. V.; Skorb E. V.; Solovieva S. E.; Antipin I. S. Calix[4]Arene-Pyrazole Conjugates as Potential Cancer Therapeutics. Bioorg. Chem. 2023, 139, 106742 10.1016/j.bioorg.2023.106742. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and code underlying this article are available publicly at https://github.com/CBRC-lab/BioAct-Het. However, data sets were derived from sources in the public domain.