

Abstract
Adhesin P1 (aka AgI/II) plays a pivotal role in mediating Streptococcus mutans attachment in the oral cavity, as well as in regulating biofilm development and maturation. P1’s naturally occurring truncation product, Antigen II (AgII), adopts both soluble, monomeric and insoluble, amyloidogenic forms within the bacterial life cycle. Monomers are involved in important quaternary interactions that promote cell adhesion and the functional amyloid form promotes detachment of mature biofilms. The heterologous, 51-kD C123 construct comprises most of AgII and was previously characterized by X-ray crystallography. C123 contains three structurally homologous domains, C1, C2, and C3. NMR samples made using the original C123 construct, or its C3 domain, yielded moderately resolved NMR spectra. Using Alphafold, we re-analyzed the P1 sequence to better identify domain boundaries for C123, and in particular the C3 domain. We then generated a more tractable construct for NMR studies of the monomeric form, including quaternary interactions with other proteins. The addition of seven amino acids at the C-terminus greatly improved the spectral dispersion for C3 relative to the prior construct. Here we report the backbone NMR resonance assignments for the new construct and characterize some of its quaternary interactions. These data are in good agreement with the structure predicted by Alphafold, which contains additional β-sheet secondary structure compared to the C3 domain in the C123 crystal structure for a construct lacking the seven C-terminal amino acids. Its quaternary interactions with known protein partners are in good agreement with prior competitive binding assays. This construct can be used for further NMR studies, including protein-protein interaction studies and assessing the impact of environmental conditions on C3 structure and dynamics within C123 as it transitions from monomer to amyloid form.
Keywords: Streptococcus mutans adhesin P1, C3 domain, NMR, quaternary interaction
Biological Context
Streptococcus mutans (S. mutans) is a gram-positive bacterium commonly associated with dental caries, one of the most prevalent human diseases in the world (Lemos et al. 2019). The cell surface-localized protein adhesin called P1 (aka Antigen I/II, AgI/II) mediates sucrose-independent S. mutans adhesion, which facilitates bacterial colonization and biofilm formation on the tooth surface (Russell et al. 1980; Brady et al. 2010; Abranches et al. 2018). P1’s C- terminus includes three tandem globular domains (C123) upstream of the LPxTG consensus motif recognized by the transpeptidase SortaseA for attachment of P1 to the peptidoglycan matrix. Ag II is a naturally occurring C-terminal truncation product of P1. Cell wall attached, full-length P1 is inaccessible to several monoclonal antibodies (MAbs) whose epitopes lie within the AgII segment, but the extracellular AgII derivative is readily detectable within culture supernatants and is recognized by these antibodies (Heim et al. 2015). Extracellular AgII contributes to the adhesive properties of cell wall-attached P1 via its quaternary interactions with P1’s globular head-containing segment, called A3VP1, which projects outward from the cell wall (Larson et al. 2010; Heim et al. 2015). We utilize a recombinant C123 polypeptide as a surrogate for AgII in our in vitro experiments. Upon mechanical stirring, both C123 and full-length P1 form insoluble aggregates that exhibit classic amyloid properties (Oli et al. 2012; Besingi et al. 2017; Barran-Berdon et al. 2020). Recently we demonstrated that S. mutans amyloid aggregates are localized to the non-adherent fraction of aging biofilms, and that amyloid formation diminishes S. mutans adhesion (Yarmola et al. 2022). Thus, the C-terminal derivative of P1 plays a major regulatory role not only in initial adhesion and biofilm genesis through quaternary interactions of its monomeric form with other P1 domains, but also in biofilm maturation and detachment through the formation of functional amyloid. Identifying environmental cues that trigger the conversion of AgII from monomeric to amyloid form are the subject of ongoing research.
In our initial structural studies of recombinant C123, this heterologously expressed construct was crystallized and found to contain three structurally similar domains, C1, C2, and C3 (Larson et al. 2011). Each domain exhibited a DE-variant IgG-fold whose beta-sheet structure was stabilized by an intramolecular isopeptide bond. Individually cloned and expressed C1, C2, and C3 constructs were also characterized. In order to use NMR to evaluate C123 quaternary interactions with other segments of P1, and possible structural changes consequent to such interaction, we started with the assignment of the previous C3 construct, since the complete C123 construct was >50 kD and prone to aggregation (Rivière et al. 2019). In that earlier work, we were able to assign ~60% of the residues within C3 and confirmed via chemical shift analysis that its secondary structure was consistent with the available C123 crystal structure (BMRB Entry 27935). Importantly, we were also able to map C3’s interactions with P1’s A3VP1 and C12 segments as well as with a MAb 6–8C, which binds to C123. Despite its tractable 17 kD size, and high stability in solution, we were unsuccessful in finding NMR conditions that enabled full assignment of this older C3 construct.
The original molecular C123 construct was designed to enhance the likelihood of crystallization. The resulting structural model (1) identified likely boundaries of the individual C1, C2, and C3 domains, and (2) eliminated the C-terminal cell wall and membrane-spanning segments of intact P1 including the LPxTG sortase motif, which are predicted to be unstructured. In the current work we sought to improve characterization of C3 by NMR in order to better define structural changes likely to occur within the P1-AgII adhesin complex during the process of biofilm formation and detachment. Because structure prediction programs have improved considerably over the past fifteen years, we sought to develop a more robust construct using modern bioinformatics tools. We used AlphaFold 2.0 to re-examine the predicted structural domain boundaries within full-length P1. This analysis revealed that P1’s C3 domain is predicted to contain an additional seven amino acids compared to the original construct. These additional residues extend the beta-sheet structure at C3’s C-terminus and increase its structural homology with C1 and C2.
We report herein the backbone (13Cα, 13Cβ, 13CO, 15NH, 1HN, 1Hα, 1Hβ) assignments for this new C3 construct. The additional seven amino acids greatly increased the spectral dispersion of the C3 resonances and enabled us to assign 92% of the protein backbone amide resonances. Because of the substantially improved resolution and extent of assignments, we re-examined the interaction of C3 with both A3VP1 and MAb 6–8C using this new construct. We note that C3’s binding interactions with these proteins are consistent with our prior work, but we are now able to more completely assign the residues likely to directly mediate those interactions. Our new results show that the binding perturbations induced by A3VP1 very closely mimic those induced by MAb 6–8C. Because C3 is contained within AgII, and A3VP1 is contained within P1, this new information provides a more complete mechanistic explanation for the ability of 6–8C to interfere with AgII’s interaction with cell-associated P1 and thereby inhibit S. mutans adhesion to its physiologic substrate in saliva (Heim et al. 2014). Our new data will also provide a useful foundation to begin the assignment of a new complete C123 construct and will lend insight as we track structural changes within P1 and AgII in response to environmental triggers such as pH, bivalent cations, or quaternary interactions with other biomolecules.
Methods and experiments
Protein expression and purification
The DNA sequence coding for the C3 domain of adhesin P1 (residues 1328–1490; Uniprot accession number P23504) was synthesized, with codon optimization for E. coli, by Genescript and inserted into pET21a(+) plasmid to yield the pET21a-C3-His6 plasmid with a C-terminal His tag. The pET21a-C3-His6 plasmid was transformed into E. coli BL21(DE3) cells for overexpression. A single colony was grown in 5 mL of terrific broth supplemented with 100 μg/mL ampicillin at 37 °C overnight. This pre-culture was added to 1 liter of an optimized M9 minimal medium containing 2.7 g/L 15NH4Cl, 4 g/L 13C6-D-glucose, 7.1 g/L Na2HPO4, 6.8 g/L KH2PO4, and 0.7 g/L Na2SO4 with 100 μg/mL ampicillin. Cells were grown at 37 °C to an OD600 of ~ 0.8. The incubation temperature was then reduced to 30 °C and protein expression was induced by addition of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG). After 16 h at 30 °C, the cells were pelleted by centrifugation at 6,000 × g at 4 °C for 30 min. The cells were resuspended in 40 mL of lysis/binding buffer (30 mM Tris pH 7.4 containing 100 mM NaCl, 20 mM Imidazole and 1 mM PMSF). Resuspended cells were sonicated, and cellular debris was removed via centrifugation at 39,000 × g for 90 min at 4 °C. The supernatant containing recombinant C3 was filtered using a 0.45 μm syringe filter and loaded onto a 5 mL HisTrap-HP column (GE Healthcare). The column was washed with 10 volumes of lysis/binding buffer. C3 was eluted from the column with 10 volumes of elution buffer (30 mM Tris pH 7.4 containing 100 mM NaCl, 300 mM Imidazole and 1 mM PMS). The elution fractions were analyzed by SDS-PAGE with Coomassie staining and protein purity was verified by observation of a single intense band at the expected molecular weight. Fractions containing protein were combined and dialyzed against 50 mM phosphate buffer pH 8 containing 100 mM NaCl at 4 °C overnight. The dialyzed supernatant was concentrated using a 10 kDa MWCO centrifugal concentrator (Amicon Ultra-15 10 K). Prior to NMR experiments, samples were exchanged into NMR buffer (50 mM sodium phosphate, 50 mM NaCl, 10% (v/v) D2O, 1 mM sodium trimethylsilyl proprionate (TSP) at pH 6).
NMR spectroscopy
NMR experiments for backbone assignments were recorded at 298 K, in a 800 MHz Bruker spectrometer (Avance II) equipped with a TCI cryoprobe using a 440 μM sample of 15N,13C-enriched C3. Backbone assignments were obtained from 3D experiments using standard Bruker pulse sequences for HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH, and HBHA(CO)NH correlations. The data were acquired and processed using Topspin 3.6.5 (Bruker Biospin). TSP was used as the 1H chemical shift reference, and the 15N and 13C chemical shifts were indirectly referenced based on 1H chemical shifts. The NMR data were analyzed using CcpNmr Analysis V3 (Vranken et al. 2005).
Chemical shift perturbation measurements
Protein-protein interaction surfaces on uniformly 15N-enriched C3 were characterized by CSPs measured using 1H-15N-TROSY experiments recorded with a fixed molar ratio of ligand candidates: A3VP1 (protein ratio 1:1 C3:A3VP1) or MAb 6–8C (protein ratio 6:1 C3:MAb 6–8C). Samples contained 130 μM 15N-enriched C3 in NMR buffer. NMR data were processed and analyzed using Topspin software. The spectral perturbations were quantified as the combined amide CSPs, specifically for each amide resonance the CSP was calculated as:
Where and are the change in chemical shift for an amide resonance in the 1H and 15N dimensions, respectively. The change in 15N chemical shift is scaled in calculating the combined CSP to reflect the different chemical shift ranges observed for the 15N resonances (~25 ppm) compared to the 1H resonances (~5 ppm).
Extent of assignment and data deposition
The 2D 1H, 15N TROSY spectrum of the C3 domain (residues 1328–1490 of the full-length adhesin P1 protein, Uniprot accession number P23504) displays well-dispersed resonances with minimal overlap (Figure 1). Sequence-specific resonance assignments for 144 of 156 possible C3 1HN-15N correlations (92%) were unambiguously determined using heteronuclear multidimensional NMR methods (Figure 1a). Using these 144 correlations, 150 13Cα resonances (96%), 141 13Cβ resonances (95%), 150 13CO resonances (96%), and 149 1Hα resonances (91%) were assigned. The unassigned residues in C3 primarily correspond to eight amino acids forming a third β-strand in the AlphaFold predicted structure and unstructured N- and C-terminal segments for the pET21 construct (Figure 1b). In order to verify the secondary structural elements of C3, the chemical shift assignments of backbone atoms (HN, Hα, Cα, Cβ, CO, and N) were analyzed with the CSI 3.0 (Hafsa et al. 2015). The returned secondary structure predictions (Figure 2) agree well with the AlphaFold predicted structure. Notably, the additional seven C-terminal amino acids in the new C3 construct resulted in more extensive β-sheet structure at the C-terminus of C3 compared to the X-ray structure for C123 (PDB 3QE5), as predicted by Alphafold. This substantially improved the extent of assignments possible compared to the prior C3 construct (Figures 1c and 1d) (Rivière et al. 2019). The backbone resonance assignments, along with characterization of secondary structural features of C3, will facilitate solution-phase structural and biochemical studies of this polypeptide, and of the larger C123 construct since C3 is an integral domain within the larger protein. While we did not remove the C-terminal His6 tag from the construct prior to NMR measurements, it was too dynamic to assign and is unlikely to contribute to the improved resolution and observed structure.
Figure 1.

(a) Annotated 2D 1H,15N TROSY spectrum of Adhesin P1 C3 domain (BMRB 52097) collected in an 800 MHz spectrometer at 25 °C in phosphate buffer pH 6. Resonance assignments are shown with black labels; (b) Alphafold structural model for the new, longer C3 construct with the amino acid residues that are currently assigned shown in blue; (c) C3 domain structure (PDB 3QE5) with the residues assigned using the previous, shorter C3 construct (BMRB 27935) shown in red; (d) comparison of 2D 1H,15N TROSY spectra for the previous, shorter C3 construct (red) and the new, longer construct for the AlphaFold-predicted C3 domain (blue). Disordered, poorly resolved resonances observed for the prior C3 construct are now well resolved for the new C3 construct by addition of the seven C-terminal amino acids.
Figure 2:

Comparison of observed and predicted secondary structures for the C3 domain. Shown are alignments of the observed secondary structure for the C3 domain within the X-ray structure of the C123 construct (PDB 3QE5) which lacked the seven C-terminal amino acids added in the present study, the predicted secondary structure (using AlphaFold2.0) for the C3 domain within the intact P1 protein, and the predicted secondary structure based on C3 chemical shifts (using CSI 3.0) from the NMR assignments for the new C3 construct in the present study (BMRB 52097).
Interactions of C3 with A3VP1 and MAb 6–8C
The interaction of C3 with either A3VP1 or MAb 6–8C resulted in significant chemical shift changes in specific regions of C3, providing confirmation of the specific and direct binding of A3VP1 or MAb 6–8C to C3 (Figure 3a). In comparing CSPs for the new C3 construct interacting with these two proteins to those for the old C3 construct, we note the overlap in perturbations caused by the two binding partners is significantly improved with the new construct and that the C3 CSPs induced by A3VP1 are substantially larger, making them now of the same magnitude as the CSPs induced by MAb 6–8C (albeit A3VP1 is at a six-fold higher concentration to account for differences in binding affinity) (Rivière et al. 2019). This observation suggests stabilization of the β-sheet structure at the C-terminus of C3 substantially improves its binding interaction with A3VP1. The residues in the C3 domain most shifted by binding either A3VP1 or MAb 6–8C were H48, S49, E50, Y68, M135, and K136. Binding to MAb 6–8C also significantly shifted residues Y61 and V102. Residues 61, 68, 102, 135, and 136 correspond to β-sheet segments that fall on one face of the protein distal to C3’s interface with C2 (Figure 3b and 3c) and are thus the likely binding interface for intact AgII with P1 on the cell surface of S. mutans (Heim et al. 2015). Interestingly, residues 48–50 lie at the interface of C3 with C2 within intact AgII. With the prior C3 construct, we observed this HSE triad is still affected by A3VP1 binding even when C3 is already in complex with C2, but that binding of MAb 6–8c did not substantially perturb these residues, even in the absence of C2 (Rivière et al. 2019). With the new C3 construct, the CSPs induced by MAb 6–8C for the C3 HSE triad match those for A3VP1. This again suggests that stabilization of C3 domain structure by addition of the missing C-terminal residues improves its utility as a tractable surrogate for intact AgII when examining protein-protein interactions as we have observed that MAb 6–8C effectively competes with C123 for P1 interactions on the surface of S. mutans in situ (Heim et al. 2015). The conservation of a distinct β-sheet binding surface on C3 for A3VP1 and MAb interactions that is distal from the C3/C2 interface coupled with a shift for the HSE triad at this C3/C2 interface on binding suggests that protein-protein interactions lead to allosteric modulation of the internal C123 structure. Given the role of monomeric C123 binding to intact P1 on the S. mutans cell wall surface in regulating cellular adhesion and the observation that its transition from a monomeric to amyloid form correlates with biofilm detachment (Heim et al. 2015; Yarmola et al. 2022), we our currently pursuing NMR experiments to structurally characterize both forms of the protein and environmental triggers of dynamics or structural transitions. These CSPs provide guidance for specific residues that can be isotopically enriched for querying changes in C123 structure via lower resolution solid-state NMR measurements within the context of intact cell walls and/or functional amyloid formation (Tang et al. 2016).
Figure 3:
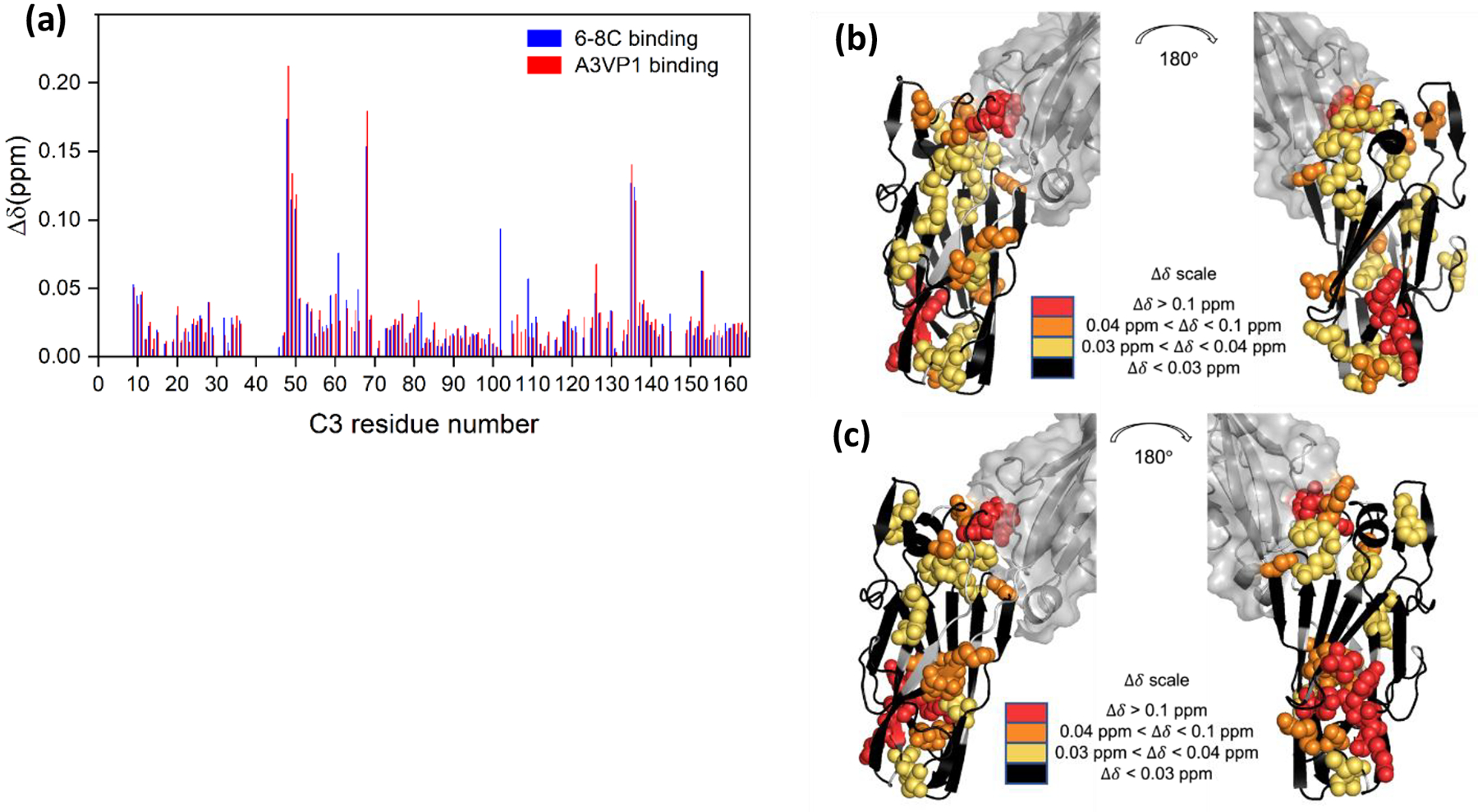
Mapping of A3VP1 and MAb 6–8C interactions with the new C3 domain construct. (a) Δδ for specific C3 amide resonances upon addition of either A3VP1 (red; protein ratio 1:1) or MAb 6–8C (blue; protein ratio ~6:1 C3:6–8C) at pH 6.5. (b) Mapping of C3 residues perturbed by A3VP1 binding onto the C123 structure predicted by Alphafold. (c) Mapping of C3 residues perturbed by 6–8C binding onto the C123 structure predicted by Alphafold.
Table 1.
List of NMR experiments acquired at 800 MHz and corresponding parameters used for C3 assignment
| Time domain size (pts) | Spectral width/Carrier Frequency (ppm) | Delay | |||||
|---|---|---|---|---|---|---|---|
| Experiment | T1 | T2 | T3 | F1 | F2 | F3 | |
| 15N TROSY | 2048 | 128 | 12/4.7 | 30/120 | 1 sec | ||
| CBCA(CO)NH | 2048 | 64 | 88 | 12/4.7 | 33/120 | 64/42.6 | 1 |
| HNCACB | 2048 | 64 | 88 | 12/4.7 | 33/120 | 64/42.6 | 1 |
| HNCO | 2048 | 40 | 64 | 12/4.7 | 33/120 | 12/173 | 1 |
| HN(CA)CO | 2048 | 40 | 64 | 12/4.7 | 33/120 | 12/173 | 1 |
| HN(CO)CA | 2048 | 40 | 88 | 12/4.7 | 33/120 | 30/45.85 | 1 |
| HNCA | 2048 | 40 | 64 | 12/4.7 | 33/120 | 30/45.85 | 1 |
| HBHA(CO)NH | 2048 | 64 | 128 | 12/4.7 | 36/120 | 10/4.7 | 1 |
Acknowledgments
We thank James Collins and James Rocca for technical assistance with NMR experiments.
Funding
This work was supported by NIH/NIDCR R01 DE021789 to L.J.B. and J.R.L. A portion of this work was performed in the McKnight Brain Institute at the National High Magnetic Field Laboratory’s AMRIS Facility, which is supported by National Science Foundation Cooperative Agreements DMR-1644779 and DMR-2128556, the State of Florida, and an NIH award, S10 OD028753, for magnetic resonance instrumentation.
Abbreviations:
- CSI
Chemical Shift Index
- Δδ
Chemical shift perturbation
Footnotes
Consent for publication
All the authors have seen and approved the submission of this manuscript.
Competing InterestsThe authors declare that they have no conflict of interest.
Availability of data and material
The chemical shift values for P1 Adhesin C3 domain were deposited in the Biological Magnetic Resonance Data Bank (BMRB) under accession code 52097.
References
- Abranches J, Zeng L, Kajfasz JK, et al. (2018) Biology of oral streptococci. Microbiol Spectr 6:5–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barran-Berdon AL, Ocampo S, Haider M, et al. (2020) Enhanced purification coupled with biophysical analyses shows cross-β structure as a core building block for Streptococcus mutans functional amyloids. Sci Rep 10:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besingi RN, Wenderska IB, Senadheera DB, et al. (2017) Functional amyloids in streptococcus mutans, their use as targets of biofilm inhibition and initial characterization of SMU_63c. Microbiology (United Kingdom) 163:488–501. 10.1099/mic.0.000443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady LJ, Maddocks SE, Larson MR, et al. (2010) The changing faces of Streptococcus antigen I/II polypeptide family adhesins. Mol Microbiol 77:276–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafsa NE, Arndt D, Wishart DS (2015) CSI 3.0: a web server for identifying secondary and super-secondary structure in proteins using NMR chemical shifts. Nucleic Acids Res 43:W370–W377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim KP, Crowley PJ, Long JR, et al. (2014) An intramolecular lock facilitates folding and stabilizes the tertiary structure of streptococcus mutans adhesin p1. Proc Natl Acad Sci U S A 111:15711–15716. 10.1073/pnas.1413018111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim KP, Sullan RMA, Crowley PJ, et al. (2015) Identification of a supramolecular functional architecture of Streptococcus mutans adhesin P1 on the bacterial cell. Journal of Biological Chemistry 290:9002–9019. 10.1074/jbc.M114.626663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson MR, Rajashankar KR, Crowley PJ, et al. (2011) Crystal structure of the C-terminal region of Streptococcus mutans antigen I/II and characterization of salivary agglutinin adherence domains. Journal of Biological Chemistry 286:21657–21666. 10.1074/jbc.M111.231100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson MR, Rajashankar KR, Patel MH, et al. (2010) Elongated fibrillar structure of a streptococcal adhesin assembled by the high-affinity association of α-and PPII-helices. Proceedings of the National Academy of Sciences 107:5983–5988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JA, Palmer SR, Zeng L, et al. (2019) The biology of Streptococcus mutans. Microbiol Spectr 7 (1): 10.1128/microbiolspec.GPP3-0051-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oli MW, Otoo HN, Crowley PJ, et al. (2012) Functional amyloid formation by Streptococcus mutans. Microbiology (N Y) 158:2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière G, Peng EQ, Brotgandel A, et al. (2019) Characterization of an intermolecular quaternary interaction between discrete segments of the Streptococcus mutans adhesin P1 by NMR spectroscopy. FEBS Journal. 10.1111/febs.15158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MW, Bergmeier LA, Zanders ED, Lehner T (1980) Protein antigens of Streptococcus mutans: purification and properties of a double antigen and its protease-resistant component. Infect Immun 28:486–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Bhatt A, Smith AN, et al. (2016) Specific binding of a naturally occurring amyloidogenic fragment of Streptococcus mutans adhesin P1 to intact P1 on the cell surface characterized by solid state NMR spectroscopy. J Biomol NMR 64:153–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, et al. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins: structure, function, and bioinformatics 59:687–696 [DOI] [PubMed] [Google Scholar]
- Yarmola E, Ishkov IP, di Cologna NM, et al. (2022) Amyloid Aggregates Are Localized to the Nonadherent Detached Fraction of Aging Streptococcus mutans Biofilms. Microbiol Spectr 10:e01661–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The chemical shift values for P1 Adhesin C3 domain were deposited in the Biological Magnetic Resonance Data Bank (BMRB) under accession code 52097.