

Abstract
Cardiovascular disease remains the leading cause of mortality worldwide. Cardiomyocytes are irreversibly lost due to cardiac ischemia secondary to disease. This leads to increased cardiac fibrosis, poor contractility, cardiac hypertrophy, and subsequent life-threatening heart failure. Adult mammalian hearts exhibit notoriously low regenerative potential, further compounding the calamities described above. Neonatal mammalian hearts, on the other hand, display robust regenerative capacities. Lower vertebrates such as zebrafish and salamanders retain the ability to replenish lost cardiomyocytes throughout life. It is critical to understand the varying mechanisms that are responsible for these differences in cardiac regeneration across phylogeny and ontogeny. Adult mammalian cardiomyocyte cell cycle arrest and polyploidization have been proposed as major barriers to heart regeneration. Here we review current models about why adult mammalian cardiac regenerative potential is lost, discuss recent progress and highlight conflicting reports pertaining to extrinsic and intrinsic signaling pathways that control cardiomyocyte proliferation and polyploidization in growth and regeneration. Uncovering the physiological brakes of cardiac regeneration could illuminate novel molecular targets and offer promising therapeutic strategies to treat heart failure.
Keywords: Heart regeneration, cardiomyocyte, polyploidization, ontogeny, phylogeny
1. Introduction
Heart attack is one common cardiovascular disease which arises from coronary vessel blockade and cardiac ischemia [1]. Cardiomyocytes in the ischemic area are rapidly deprived of oxygen and subsequently perish. The mammalian heart will attempt to repair the ischemic injury by replacing damaged areas of the heart with a dense mesh of extracellular matrix left behind by fibroblasts [1–3]. This fibrotic tissue prevents cardiac rupture but will paradoxically compromise the integrity of the myocardium later; heart contractility declines substantially, leading to heart failure and even death [4]. Conventional treatments of acute myocardial infarction involve the use of a catheter to act as a balloon to expand the occluded coronary artery [5]. This method may restore circulation and perfusion to undamaged areas of the heart, but it does not stimulate the regeneration of lost myocardium. Regenerating injured hearts therefore remains one of the largest unmet clinical needs.
Even though adult mammalian hearts have limited regenerative capacity, heart regeneration of amphibians and certain fish can persist well into adulthood. Investigating the mechanisms of cardiac regenerative potential across phylogeny and ontogeny has therefore become of great interest. The underlying logic is to uncover the reasons for the disparity of heart regenerative potential and to translate these findings for therapeutic applications in the clinic. In this review, we cover pertinent information regarding what is currently known about the mechanisms that govern cardiac regeneration across phylogeny and ontogeny. We discuss the divergence in cardiomyocyte proliferation in the context of cardiomyocyte ploidy, current models about why mammals (namely mice and humans) lose cardiomyocyte regenerative potential after birth, endocrine factors and neural regulators of cardiomyocyte proliferation, as well as key signaling pathways that influence cardiomyocyte regeneration.
2. Divergence in cardiomyocyte proliferation, polyploidy and regeneration in phylogeny and ontogeny
2.1. Divergence in phylogeny
Earlier studies of heart regeneration in several adult frog and reptilian species (Rana temporaria, Agama caucasica and Lacerta armeniaca) showed that cardiomyocytes are capable of robust proliferation and significant regeneration following myocardial injury [6,7]. Similar findings were later observed in adult newts [8] and zebrafish [9]. There is an apparent correlation between cardiomyocyte ploidy and the propensity of the proliferative and regenerative capacity of cardiomyocytes, as illustrated in Figure 1. Zebrafish, frogs, and newts have >95% diploid, mononucleated cardiomyocytes [10–13]. In adult mammals, however, most cardiomyocytes become polypoid and permanently exit the cell cycle [10,11]. This is associated with limited regenerative capacity in adult mammalian hearts. To identify the transition of cardiomyocyte polyploidy in phylogeny, Hirose et al. recently analyzed cardiomyocyte ploidy among a number of under-studied vertebrate species, such as short-beaked echidnas, silky anteaters, bowhead whales, and brown-throated sloths [14]. Intriguingly, the change of cardiomyocyte ploidy in phylogeny seems to be in parallel with the ectotherm-to-endotherm transition [14]. Whether the mammalian species that possess abundant diploid cardiomyocytes in the adult heart retain significant cardiac regenerative potential deserves further investigation.
Figure 1. Correlation between heart regenerative potential, adult cardiomyocyte proliferative potential and proportion of polyploid cardiomyocyte across species.
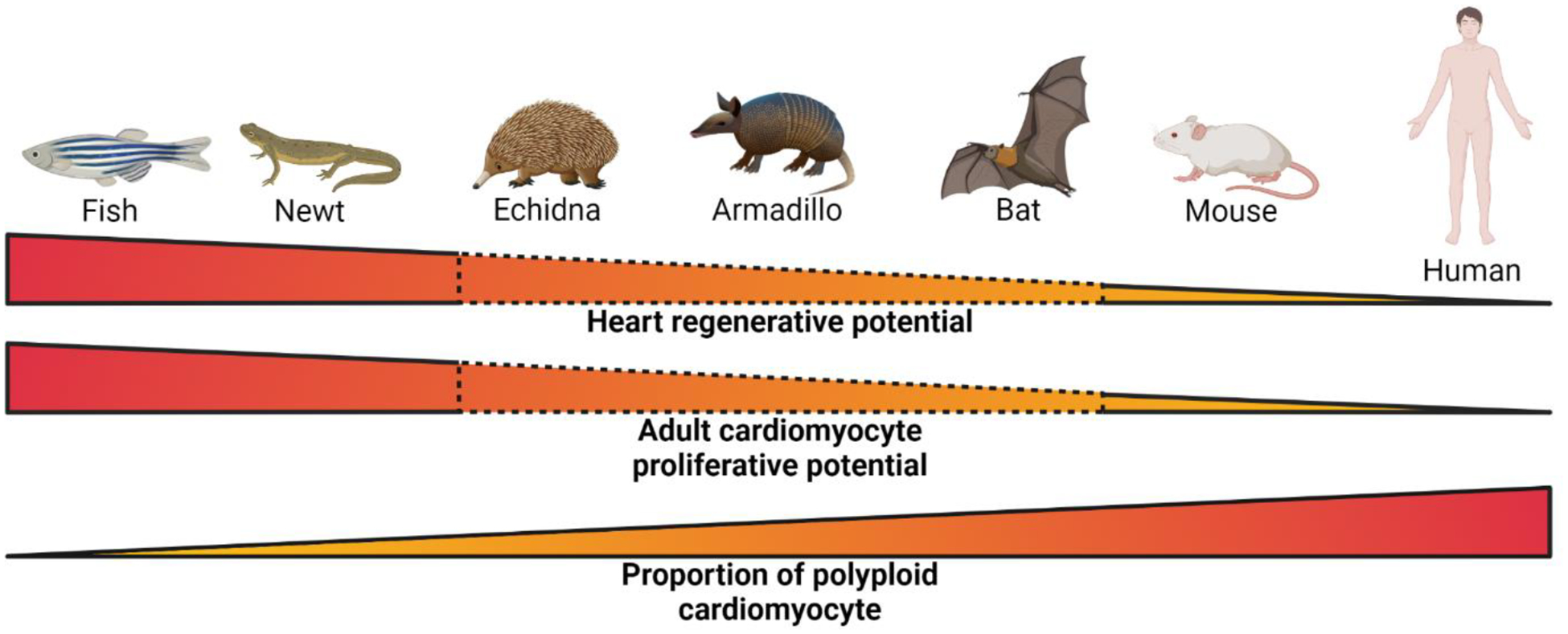
Species with low proportion of polyploid cardiomyocytes (fish and newt) display high heart regenerative potential and adult cardiomyocyte proliferative potential, whereas species with high proportion of polyploid cardiomyocytes (mouse and human) display low heart regenerative potential and adult cardiomyocyte proliferative potential. Species with intermediate proportion of polyploid cardiomyocytes (echidna, armadillo, and bat) are predicted to have intermediate or transient heart regenerative potential but further scientific validation is required. The image was created with BioRender.com.
It is important to stress that animals with abundant diploid and proliferationcompetent cardiomyocytes are not necessarily always capable of heart regeneration. This was best illustrated in comparative studies between two teleost fish species zebrafish and medaka. Although both can regrow lost tails efficiently, medaka fish, unlike zebrafish, cannot mount regenerative responses following cardiac injury [15]. Comparison of injury repair in these two species recently demonstrated that myocardial damage in medaka resulted in delayed and reduced recruitment of macrophages, and stimulation of Toll-like receptor signaling enhanced immune cell dynamics, cardiomyocyte proliferation, scar resolution, and heart regeneration in medaka (Lai et al., 2017). Therefore, although medaka cardiomyocytes are diploid and proliferation-competent, medaka don’t regenerate the heart naturally unless the innate immunity is stimulated. It is tempting to speculate whether a similar mechanism could explain the recent conflicting reports that adult frogs Xenopus laevis cannot, but Xenopus tropicalis can regenerate the heart [16,17].
2.2. Divergence in ontogeny
In most mammals, cardiomyocytes permanently exit the cell cycle and become polyploid due to failure in either cytokinesis or karyokinesis in the perinatal window [18–22]. This coincides with the closure of cardiac regenerative period [23–25]. Indeed, it has been shown that stimulation of cardiomyocyte proliferation enhances cardiac regeneration and reverse heart failure [26,27]. When heart regenerative potential is lost in humans has not been determined, but a previous case report showed cardiac function recovery in a newborn baby after a severe myocardial infarction [28]. In addition, the percentage of diploid cardiomyocytes in the adult human heart and the timing of human cardiomyocyte cell cycle exit are controversial [20–22,29]. This may be explained by the different analysis methods and the heterogeneous nature of human population. As a support, study of different mouse strains demonstrated an array of diploid cardiomyocyte abundance, and mouse strains with higher frequencies of diploid cardiomyocytes retain better cardiac regenerative capacity [12]. Altogether, these findings support that cardiomyocyte cell cycle arrest and polyploidization are major barriers to heart regeneration [27,30–32].
Although it is generally agreed that adult mammalian hearts have limited regenerative potential, it is intriguing that myocardial recovery has also been reported in a small number of patients who have received left ventricular assist devices (LVADs) [33]. LVADs are implanted in advanced heart failure patients to help improve cardiac output, systemic perfusion, and overall organ function. A study from 2015 sought to investigate the molecular basis of these observations, and found evidence of enhanced cardiomyocyte cycling and cytokinesis [34]. Despite its promising results, the findings of this study were not recapitulated in a recent study [35]. Luo et al. quantified cardiomyocyte cell cycle markers by imaging and flow cytometry using samples from patients undergoing LVAD implantation. But they found no evidence of increased cardiomyocyte proliferation or changes in cardiomyocyte nucleation changes although cardiomyocyte DNA content was reduced in unloaded hearts. Although it is possible that different methods used in these two studies contributed to the different outcomes, this recent controversy highlights the need of more independent investigations to examine whether LVAD implantation and unloading the heart could facilitate endogenous cardiomyocyte proliferation and regeneration.
3. Current models about why we lose cardiac regenerative potential
3.1. Oxygen-rich environment
An oxygen-rich postnatal environment has been proposed as a primary factor inducing mammalian cardiomyocyte cell cycle arrest and loss of cardiac regenerative potential [36]. The generation of reactive oxygen species and corresponding DNA damage in mouse cardiomyocytes throughout the first week of life parallels the reduction of neonatal mouse cardiac regeneration during the first week of life [36]. Scavenging reactive oxygen species from cardiomyocytes delayed postnatal cell cycle exit and slightly increased the percentage of mononuclear cardiomyocytes in neonatal mouse cardiomyocytes [36]. Increasing the levels of reactive oxygen species, on the other hand, accelerated cardiomyocyte cell cycle arrest in neonatal mice [36]. Chronic hypoxia was also shown to alleviate oxidative damage, induce mitosis in adult mouse cardiomyocytes, and promote cardiac regeneration after myocardial infarction in adult mice [37]. In sum, these observations support that the high oxygen level in the postnatal environment stimulates cardiomyocyte cell cycle withdrawal.
Most of the studies above were performed in altricial mammals such as mice. Cardiomyocytes from precocial mammals such as sheep, however, exhibit near-complete polyploidization and cell cycle arrest before birth [38]. This suggests that other major physiological mechanisms other than oxygen status impacts cardiomyocyte proliferative potential.
3.2. Acquisition of endothermy
Vertebrate thermogenesis appears to be inversely correlated with cardiac regenerative potential. Tissue regeneration in general may be an ancestral vertebrate trait that was lost later in evolution [39]. Pro-regenerative species such as zebrafish and newts, however, are not as efficient in thermogenesis [40]. Neonatal mammals are less able to thermoregulate when compared to adults [41]. Adult humans and rodents can efficiently thermoregulate, and this is associated with poor cardiac regeneration. This corroborates with the inverse relationship between thermogenesis and organ regenerative capacity.
A recent phylogenetic analysis of 41 vertebrate species showed that diploid CM content within the myocardium correlates negatively with standard metabolic rate [14]. Lower vertebrates with higher cardiac regenerative potential, such as zebrafish, newts, and reptiles, have standard metabolic rates that are an order of magnitude lower than those of endothermic mammals such as rodents and humans [14]. Mammalian body temperature is also negatively correlated with diploid cardiomyocyte percentage [14]. Furthermore, Payumo et al. followed the fractal nature of the cardiovascular system and uncovered that the increased thermogenesis during the acquisition of endothermy demands a proportional elevation of blood flow and cardiac output [42]. Inhibition of thyroid hormone and adrenergic receptor signaling, two major thermogenic pathways, was found to remarkably delay body temperature rise, reduce polyploid cardiomyocytes, and prolong the cardiac regenerative window in mice [42]. Thus, the physiological changes and cardiac adaptations during endothermy acquisition may induce cardiomyocyte polyploidization and cardiac regenerative potential loss [32]. This model may account for both altricial and precocial mammals which develop endothermy after and before birth, respectively. It would be interesting to test whether this model also explains the loss of regenerative capacity of other organs and tissues such as the brain and spinal cord, which occurs in the same time window when animals increase thermogenesis [43–45].
3.3. Altered immune system
The development of robust and complex adaptive immune systems across phylogeny and ontogeny also parallels the decline of tissue regenerative potential [46] [47]. It has also been proposed that efficient immune function is a trade-off for regenerative capacity [48]. Salamanders and frogs have high and robust cardiac regenerative potential when compared to that of mammals [16, 49, 50]. The immune response of these amphibians is more primitive than those of mammals. They are less specific, slower in onset, and are less adaptive than those of less-regenerative animals [47]. In mammals, CD4+ T-cells – critical players in the adaptive immune system are enriched in non-regenerative P8 mouse hearts when compared to pro-regenerative P3 hearts. Ablating CD4+ T-cells in P8 mice increases cardiomyocyte regenerative potential and ameliorates fibrosis after apical resection [51]. Recently, reconstitution of neonatal mice with adult T cells was shown to cause deficient cardiac regeneration [52]. Furthermore, interferon gamma signaling seems to be critical in regulating this process because newborn mice that received adult Ifng knockout T cells before myocardial infarction exhibited a comparable regenerative phenotype [52]. In both reports, macrophage recruitment and polarization are likely key cellular mediators of regenerative response [51,52].
The innate immune response also appears to be a critical determinant of cardiac regenerative potential [32]. Unresolved post-injury inflammatory responses in adult mice may contribute to their inability to regenerate hearts. TLR2 and TLR4 seem to be key players that mediate this phenomenon [53–55]. An early resolution of immune activity in the infarct site improves cardiac regeneration in adults [56,57]. Interestingly, acute inflammation in P1 mice is critical for their regenerative response [58]. Macrophage depletion negatively affects cardiac regenerative capacity in P1 mice [59]. Regenerative M2 macrophages in P1 mouse hearts upregulate the expression of soluble factors that support myogenic differentiation and growth. These factors include IGF-1, TGF-beta, activin-A, and Arg1. These factors are not present in the hearts of P14 mice [47]. The transition from M2 to M1 may also play a role in the developmental loss of regenerative potential in other organs. CCR2 inhibition in adult hearts improved angiogenesis and reduced inflammation after injury by inhibiting post-injury monocyte recruitment to the heart, thereby preserving embryonic macrophage activity [60]. Recently, transplantation of macrophages from neonatal mice to adult mice was shown to facilitate cardiomyocyte proliferation and cardiac regenerative repair [61]. This study would have been more convincing and impactful if dead neonatal macrophages had been transplanted in a control group and no beneficial effect had been observed. This is an important control especially given the previous controversies surrounding the cardiac stem cell therapy where injection of both live and dead mesenchymal stem cells in adult mouse hearts yielded comparable cardiac function improvement following injury, mainly through modulation of endogenous immune response [62].
3.4. Cancer risk trade-off
A prominent paradigm maintains that mammals have lost their regenerative potential to prevent the development of cancer [63]. The balance between cancer risk and organ regeneration is mediated by tumor suppressor genes that reduce the oncogenic risks of stem cells but also limit regenerative potential [32]. The p53 family of tumor suppressors arose in sea anemones to protect germline cells from DNA damage. p53 itself arose in early vertebrates like cartilaginous fish [64]. Vertebrate regenerative potential also declines with age as age-related DNA damage and cancer risks accumulate [65]. Permanent cell cycle exit in mammalian cardiomyocytes prevents their proliferation in the adult mammalian heart. This may explain the rarity of cardiac tumors [32].
In addition, an increase of cell ploidy has been proposed to reduce the risk of cancer formation because polyploid cells need to lose more copies of a tumor suppressor gene for a complete loss of function. This proposal has been supported by recent studies in hepatocytes [66]. Whether manipulations to enhance cardiomyocyte proliferative capacity and reduce cardiomyocyte ploidy also significantly increase the incidence of cardiac tumorigenesis awaits experimental evidence.
4. Molecular pathways that control cardiomyocyte proliferation, polyploidy and regeneration
4.1. Endocrine factors
4.1.1. Thyroid hormone signaling
Thyroid hormones are produced by the thyroid gland and secreted to the bloodstream. Thyroid hormone exists in two forms: L-thyroxine (T4) and 3,3,5-triiodo-Lthyroxine (T3) in mammals. T4 is the prohormone that is converted into its biologically active T3 form. In target cells, thyroid hormones bind to their receptor thyroid hormone receptor in the nucleus. Retinoid X receptor (RXR) is associated with thyroid hormone receptor, and this heterodimer is bound to DNA through thyroid hormone elements. Downstream target genes are subsequently expressed when ligand is bound to the receptor. These thyroid hormone-inducible genes have widespread functions in regulating metabolic balance and cell maturation [67–70].
Serum thyroid hormone levels are substantially lower in newts and zebrafish than in non-regenerative mammals, rising before birth in sheep and soon after birth in neonatal mice [38,71,72]. How thyroid hormones impact cardiomyocyte cell cycle activity and heart regeneration has been explored in certain vertebrate species [68,73,74]. Gerdes and colleagues investigated the consequence of T3 treatment in neonatal rats. They observed a 19% reduction of the total cardiomyocyte number and a 31% increase of the cardiomyocyte volume in T3-treated rats [75]. Similarly, Chattergoon et al. have shown that T3 administration to fetal sheep reduces cardiomyocyte proliferation and the total cardiomyocyte number, and increases cardiomyocyte binucleation and cell size [76]. In contrast, Naqvi et al. reported that the postnatal surge of thyroid hormone in mice stimulates preadolescent cardiomyocyte expansion [77]. However, this burst of cardiomyocytes was not found by other groups in two subsequent reports [78, 79]. Recently, Hirose et al have found that the percentage of diploid cardiomyocytes is inversely correlated with total plasma T4 levels in different vertebrate species [14]. Genetic expression of a dominant negative thyroid hormone receptor TRα in mouse cardiomyocytes increased heart size by 21% at postnatal day 14. This increase seems to be a result of an increase in cell number rather than cell size. Mutant mice also showed a higher frequency of diploid cardiomyocytes, increased cardiomyocyte proliferative capacity, and enhanced heart regenerative potential in the adult and diploid percentage [14]. All these findings together suggest that the high level of circulating thyroid hormones in adult mammals repress cardiomyocyte proliferation and heart regeneration.
4.1.2. Glucocorticoid receptor signaling
Glucocorticoids are steroid-derived hormones that, like thyroid hormone, are responsible for maintaining numerous homeostatic functions. The major glucocorticoid in humans is cortisol (also known as corticosterone in mice). The circadian rhythm and/or environmental/physiological stress can trigger the hypothalamus to secrete corticotropin-releasing hormone (CRH) which will stimulate the production and secretion of adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. ACTH will bind to its receptor on the adrenal gland to stimulate the production and release of glucocorticoids from the zona fasciculata [80]. Glucocorticoids will diffuse through their target cells directly to the nucleus to bind its receptor, GR, which is already bound to glucocorticoid response elements. Downstream targets activated from glucocorticoid signaling are increased mobilization of glucose, increasing blood pressure and heart contractility, and increasing lipid metabolism [80].
The role of GR in cardiomyocyte proliferation and heart regeneration is context dependent. Exogenous glucocorticoid hormone has an inhibitory effect on cardiomyocyte proliferation in vitro and in vivo by possibly regulating Cyclin D2 gene expression [32,81–83]. Both exogenous GR agonist exposure [84] and crowding-induced stress through GR activation impair regeneration in the zebrafish heart [85]. In a recent study by Pianca et al., genetic deletion and pharmacological inhibition of GR in mice promoted cardiomyocyte proliferation and heart regeneration after experimental myocardial infarction [86]. Moreover, Pianca et al. demonstrated that cardiomyocyte-specific GR ablation in conditional knockout mice showed delayed postnatal cardiomyocyte cell cycle exit. This result is in contrast with a previous report showing no change of cardiomyocyte cell cycle exit and polyploidization in mice with GR deletion in cardiomyocytes [32]. The circulating glucocorticoid hormone level plummets after birth and increases after the completion of cardiomyocyte cell cycle exit in mice and rats [87,88], therefore GR may not be the major physiological driver of postnatal cardiomyocyte cell cycle withdrawal and polyploidization.
4.1.3. Vitamin D receptor signaling
Vitamin D is a lipophilic sterol-derived hormone that binds to the nuclear receptor VDR (vitamin D receptor). VDR is bound to RXR as a heterodimer. When vitamin D binds to its receptor, downstream targets of the pathway play crucial roles in metabolism and homeostasis. These functions include but are not limited to maintaining calcium and bone homeostasis, boosting anti-bacterial immune responses, anti-tumorigenic activities in various tissues, and mitigating inflammation [89].
VDR signaling is also directly implicated in cardiomyocyte proliferation and heart regeneration. For instance, ablating VDR in cardiomyocytes induces hypertrophy in mice [32,90]. Vitamin D analogs were also shown to inhibit proliferation in primary mammalian cardiomyocytes [32,91–93]. However, in zebrafish, vitamin D agonists increase CM proliferation in embryos, and a VDR inhibitor decreases zebrafish CM proliferation [94]. Interestingly, while exogenous VDR stimulation suppresses the proliferation of neonatal mouse cardiomyocytes in vitro and in vivo, mice with cardiomyocyte-specific knockout of VDR didn’t show any difference in postnatal cardiomyocyte proliferation and ploidy [32], suggesting that VDR may have different physiological functions in zebrafish and mice in regulating cardiomyocyte proliferation.
4.2. Neural regulators
4.2.1. Sympathetic nerve-adrenergic receptor signaling
The sympathetic nervous system is one arm of the autonomic nervous system, known for evoking the “fight or flight” response. The overarching goal of the activated sympathetic nervous system is to maximize tissue oxygenation and to provide tissues with ample amounts of energy in effort to guide the organism out of stressful situations [95–97]. The neurons of the sympathetic nervous system are typically synapsed to effector tissues using epinephrine or norepinephrine as neurotransmitters [98]. These neurotransmitters act on alpha- or beta-adrenergic receptors, which are typically G-protein coupled receptors [95]. Serve to increase cardiac output and oxygen delivery, sympathetic activation will lead to an increased heart rate and greater force of contraction per beat [95].
Sympathetic nerves innervate the heart before birth [99] but they seem to only become active after birth in rodents [100], coinciding with cardiomyocyte cell cycle exit and polyploidization. Using chemical and genetic approaches to ablate sympathetic nerves, two reports independently showed that sympathetic nerves inhibit postnatal cardiomyocyte proliferation [101,102]. Two other groups investigated the role of beta-adrenergic receptor in postnatal cardiomyocyte cycling and division by studying mice lacking beta 1 and beta 2 adrenergic receptors as well as mice treated with beta-adrenergic receptor inhibitors [103,104]. Both groups demonstrated that beta-adrenergic receptor inhibition regulates Hippo/YAP pathway activation, delays postnatal cardiomyocyte cell cycle arrest, reduces cardiomyocyte ploidy, and improved cardiac functions after myocardial infarction [103,104]. These findings are consistent with previous reports that activation of the G-protein, Gα, by epinephrine inactivates YAP/TAZ signaling [105] and the Hippo/YAP pathway controls cardiomyocyte proliferation and heart regeneration (see below). A third group showed that inhibition of beta-adrenergic receptor alone had mild effects in the control of cardiomyocyte proliferation and binucleation, but combined treatment of chemical blockers of alpha-, beta-adrenergic receptor and thyroid hormone signaling yielded profound phenotypes in postnatal mice with 60% diploid cardiomyocytes, a 60-fold increase of cardiomyocyte proliferation after injury, robust cardiac functional recovery and minimal scar formation [42]. Although all these reports have some disagreements in the degree of phenotypes and the exact effector downstream of sympathetic nerves and adrenergic receptors, they all support that activation of sympathetic nerve and adrenergic receptor are among the physiological triggers that induce cardiomyocyte cell cycle withdrawal and loss of heart regenerative potential in postnatal mammals.
4.2.2. Parasympathetic nerve-cholinergic receptor signaling
The parasympathetic nervous system is another functionally distinct arm of the autonomic nervous system. It typically acts in opposition to the sympathetic nervous system to regulate the relaxed “rest and digest” state [96,106]. Neurons of the PNS use acetylcholine as its major neurotransmitter. Acetylcholine in the PNS will activate muscarinic cholinergic receptors for downstream signaling. Muscarinic cholinergic signaling is mainly involved in the following functions: smooth muscle relaxation, constriction of the eye pupils, secretion of digestion enzymes, vasodilation, and decreased heart rate. The vagus nerve in particular is used by the parasympathetic nervous system to relay acetylcholine directly to the heart [106].
It has been reported that nerves play a critical role in aiding the regenerative processes of tissues in numerous species [107]. Salamander limb regeneration studies from the 19th century have demonstrated the reliance of nerves in limb regeneration [107,108]. Additional studies have shown that cholinergic neurons are often responsible for regenerating severed limbs [107,109,110]. Mahmoud et al. has shown that cholinergic receptor signaling of the parasympathetic nervous system directly modulates cardiomyocyte cell cycle activity in zebrafish and neonatal mice. Disrupting cholinergic signaling reduced the proliferative capacity in each of the indicated organisms [107]. Mechanically ablating the vagus nerve in neonatal mice reduced expression of two trophic factors Nrg1 and Ngf, and inhibited heart regeneration in neonatal mice after apical resection and myocardial infarction. This disruption of cardiomyocyte regenerative potential after vagus nerve ablation is rescued after administering recombinant Nrg1 in neonatal mice [107]. Transcriptomic profiling of hearts from mice with mechanically resected vagus nerves was remarkable for a disruption in inflammatory responses that guide heart regeneration after apical resection [107]. This study establishes a direct role of parasympathetic nerve signaling in cardiomyocyte regeneration. However, how the vagus nerve activity induces cardiac Nrg1 expression and regulates inflammatory responses during neonatal heart regeneration remains intriguing yet unanswered questions.
4.3. Local pathways
4.3.1. Hippo signaling
The Hippo-YAP (Yes-associated protein) pathway is an evolutionarily conserved pathway that plays a crucial role in regulating organ development, organ size, cell proliferation, apoptosis, and differentiation [111,112]. Even though there is still much to be learned about the Hippo pathway, genetic and biochemical studies have provided many important insights into how signaling in this pathway works [111]. Core signaling components include serine/threonine kinases mammalian STE20-like protein kinases 1 and 2 (MST1/2), the scaffold proteins Salvador 1 (SAV1) and MOB1A/B (Mps one binder kinase activator 1A and 1B), large tumor suppressor homologue 1 and 2 (LATS1/2), and the transcriptional co-activating factors YAP (Yes-associated protein) and TAZ (transcriptional co-activator with PDZ-binding motif). In general, Hippo pathway activation results in the phosphorylation and cytosolic retention/degradation of YAP and TAZ. The kinase activity stems from a complex formed by MST1/2 and SAV1. This kinase complex phosphorylates LATS1/2, which in turn interacts with cofactors MOB1A/1B. This induces the phosphorylation of YAP/TAZ and its subsequent proteasomal degradation. When the Hippo pathway is inhibited, the kinase cascade involving MST1/2+SAV1 and LATS1/2+MOB1A/1B are absent. This allows for YAP/TAZ to accumulate in the nucleus, thereby perpetuating the expression of genes that regulate cell growth and survival. Interestingly, the nuclear localization of YAP/TAZ can be modulated by signaling mechanisms other than the Hippo pathway. These include GPCR signaling, the TGFβ/BMP pathways, the Wnt/β-catenin pathway, as well as IGF signaling [111,113–115].
There is an abundance of evidence that Hippo-YAP signaling plays a major cell-intrinsic role in heart development and regeneration [111,116–118]. For instance, the deletion of Salv leads to cardiomegaly in mice at birth which is a result of cardiac progenitor cell hyperproliferation in utero [111,116,119]. Conditional inactivation of other Hippo pathway genes such as Mst1, Mst2, and Lats2 also confer similar phenotypes [111,116]. Additionally, targeted cardiomyocyte deletion of YAP resulted in cardiac hypoplasia, heart failure, and early embryonic death. These data illustrate the importance of balanced Hippo-YAP signaling during heart development [117,118,120]. Furthermore, it has been shown that modulation of the Hippo/YAP pathway yields substantial benefits on cardiac regenerative capacity. Conditional deletion of Sav1 and expression of constitutively active Yap in cardiomyocytes have been shown to stimulate cardiomyocyte proliferation, reduce polyploid cardiomyocytes, promote heart regeneration and reverse heart failure [26,117,121–123]. Recently, viral depletion of Salv was demonstrated to facilitate regenerative repair and functional improvement in a pig model of myocardial infarction even without arrhythmogenesis which is often associated with uncontrolled cardiomyocyte proliferation [123,124]. Because pigs live longer than mice which have a maximal lifespan of four years, this gene therapy pig model could be valuable for assessing another major concern of chronic activation of cardiomyocyte proliferation, the long-term risk of tumorigenesis, before clinical trials in patients.
4.3.2. Wnt/β-catenin
The Wnt/β-catenin pathway is another evolutionarily conserved morphogenic pathway whose signaling effector proteins dictate a broad range of cellular functions. Like the Hippo pathway, Wnt/β-catenin signaling modulates cell proliferation, differentiation, survival, and apoptosis [125–128]. This pathway plays a significant role in embryonic development as well as maintaining adult tissues. In adult organs in particular, Wnt signaling is involved in stem cell renewal, differentiation and differentiation [125]. Active Wnt ligands interact with their Frizzled receptors, leading to β-catenin stabilization and nuclear translocation [129–131]. Once in the nucleus, β-catenin binds to the T-cell factor/lymphoid enhancer factor (Tcf/Lef) transcriptional complex and induces target gene expression [132–135].
Stimulation of the Wnt/β-catenin pathway is among the most potent triggers of cardiomyocyte proliferation in cell culture in vitro and in neonatal mice in vivo [136,137]. Surprisingly, activation of this pathway in adult mice failed to promote cardiomyocyte cycling and expansion [137]. Through comparative analyses, Quaife-Ryan et al. demonstrated that β-catenin engages a genetic circuity in adult cardiomyocytes that is distinct from the neonatal proliferative gene network [137]. Interestingly, although β-catenin activation does not promote cardiac regeneration, it confers cardiac protection against ischemic injury by inducing a neonatal glycolytic gene program [137]. This study underscores the difference between adult and neonatal cardiomyocytes, and suggests that simple activation of the pathways involved in fetal and neonatal cardiomyogenesis may not be sufficient to induce adult cardiomyocyte proliferation and heart regeneration.
4.3.3. Notch signaling
Notch ligands bind to their receptors (Notch1–4) [138–140]. When Notch ligands on adjacent cells bind to Notch receptors, cleavage sites on the cytosolic end of the transmembrane domain of Notch become exposed toto γ-secretase. The cleavage of Notch sets the Notch intracellular domain (NICD) free to translocate to the nucleus for further gene transcription [141].
Continual Notch signaling was reported in adult zebrafish hearts [142]. As such, zebrafish have become the model of choice for studying heart regeneration. This also suggests that Notch signaling plays a role in cardiac regeneration. It was later discovered that Notch signaling is most prominent in the endocardial cells of zebrafish hearts. Blockage of Notch signaling by a small molecule inhibitor and genetic ablation of Notch in the endocardial region was remarkable for impaired proliferation and transdifferentiation of cardiomyocytes [143]. In the follow-up study, Zhao et al. discovered that the rate of cardiomyocyte proliferation decreased dramatically after ventricular amputation when Notch signaling was blocked Tg (hsp70: dn-Maml) zebrafish [144]. In neonatal murine models, knocking out Notch1 worsened cardiac fibrosis and hypertrophy. This implies that Notch signaling may play a role in preventing maladaptive hypertrophy after acute heart injury [145]. Partial inhibition of Notch signaling by knocking out RBP-J also appears to be responsible for increased apoptosis of cardiomyocytes after cardiac injury in neonatal mice [146]. Overexpression of Notch can enhance neonatal mouse cardiomyocyte proliferation, but this does not appear to occur in the heart of adult mice. These findings maintain that Notch signaling is conserved in mammalian cardiac regeneration, but the mechanisms that regulate heart regeneration may be more complicated than those of zebrafish.
4.3.4. TGF-β signaling
The TGF-β superfamily consists of many soluble ligands including TGF-β’s, Bone Morphogenetic Proteins (BMPs), Growth Differentiation Factors (GDFs), Nodal, and Activins [147]. Ligands must be proteolytically cleaved after its production to initiate signaling. The ligand is then bound to Latency Associated Peptide (LAP) and Latent TGF-β-Binding Proteins 1–4 (LTBP1–4). This complex sequesters the ligand to the extracellular matrix of the cell that produced it [148,149]. The ligand then escapes the LAP and LTBP complex to bind to its receptor on receiving cells. TGF-β receptors are found on the plasma membranes of cells and are mainly classified into three types of receptors: Type 1, Type II, and Type III. TGF-β ligands become activated upon dimerization and bind to Type II receptors. This will recruit Type I receptors and allow for the cytoplasmic domain of the Type I receptor to be phosphorylated by the Type II receptor. The Type I receptor will then act as a kinase to phosphorylate SMAD effector proteins [150]. TGF-β and Activins typically signal through SMAD 2/3, while BMPs primarily signal through SMAD 1/5/8. Phosphorylated SMADs will bind to SMAD 4, which will translocate the entire complex into the nucleus to regulate gene expression [151].
Genes of cell cycle progression are among the major downstream targets of TGF-β signaling [152]. Interestingly, the core TGF-β ligands (TGF-β1, TGF-β2, and TGF-β3) do not appear to be essential for the activation of cardiomyocyte proliferation. Genetic knockouts of either of these ligands did not cause any developmental heart defects in mice [153–155]. This data suggests that TGF-βs 1–3 are not necessary for adequate cardiovascular development. Other ligands such as Inhbaa and Mstnb were shown to directly influence cardiomyocyte proliferation in zebrafish [156]. As the homolog of myostatin, Mstnb production is inhibited during cardiac injury while Inhbaa is upregulated to promote cardiomyocyte proliferation [156]. Myostatin also inhibits rat CM proliferation in vitro [157]. TGFBR1 is the most extensively studied TGF-β receptor that was shown to play a role in cardiomyocyte proliferation. Mice deficient in this receptor do not exhibit detectable cardiac defects [158]. Zebrafish that were treated with a drug that specifically inhibits TGFBR1 (SB-431542) exhibited poor cardiomyocyte proliferation in response to injury [159]. Ultimately, the extent of TGFBR1 activity in cardiomyocyte proliferation is yet to be clearly defined.
4.3.5. Neuregulin-ERBB signaling
Neuregulin 1 (Nrg1) plays a major role in the following processes regarding cardiovascular homeostasis and disease: atherosclerosis, myocardial infarction, heart failure, and ischemia reperfusion. Nrg1 can crosstalk with other related signaling mechanisms through the Nrg1/ErbB pathway to regulate inflammation, apoptosis, and oxidative stress [160]. Nrg1 signaling acts by Nrg1 binding to a receptor tyrosine kinase comprised of ErbB2, 3, and 4. Nrg1 binds to the extracellular domains of ErbB3 and ErbB4, which leads to a conformational change that allows for ErbB2 to bind either ErbB3 or ErbB4 [161,162]. Upon activation, ErbBs phosphorylate each other’s C-termini and initiate numerous downstream signaling cascades that mediate cell growth, adhesion, migration, and differentiation [163].
Nrg1 and ErbB signaling has also been shown to promote cardiomyocyte proliferation and heart regeneration after injury [164,165]. NRG1 promotes proliferation of mononucleated ventricular cardiomyocytes of adult rats in vitro and this effect was dependent on ERBB4 [164]. In addition, injecting NRG1 into adult mice promoted cardiomyocyte cell cycle activity and regeneration after myocardial infarction [164]. Furthermore, genetic inhibition and overexpression of ErbB4 inhibited and promoted postnatal cardiomyocyte proliferation, respectively [164]. Supporting the more prominent roles of NRG1 on promoting proliferation in mononucleated but not binucleated cardiomyocytes [164], recombinant NRG1 promotes proliferation and regeneration after injury more effectively when administered only during the short postnatal window, when more cardiomyocytes remain mononucleated [166]. Another group emphasized the role of ErbB2 in promoting cardiomyocyte proliferation and heart regeneration [167,168]. ErbB2 expression was substantially reduced during the first week of postnatal development [167], which correlates with the window that regenerative potential declines. Genetic inhibition of ErbB2 inhibited cardiomyocyte proliferation during embryonic and neonatal development, and expression of constitutively active ErbB2 induced cardiomyocyte dedifferentiation, proliferation, hypertrophy in neonatal, juvenile, and adult mice and results in cardiomegaly [167]. Interestingly, constitutively active ErbB2 also promoted cell division of binucleated cardiomyocytes, suggesting the possibility that Nrg1 might promote dedifferentiation and proliferation of binucleated cardiomyocytes in an ErbB2-dependent manner. Furthermore, overexpression of constitutively active Erb2 was shown to phosphorylate YAP at S352 and S274 in an ERK-dependent but not Hippo-dependent manner [168]. Recently, Nrg1-EbB4 was also strongly shown to regulate proliferation through the Hippo YAP pathway [160,169], highlighting the molecular crosstalk between various proliferative pathways. While long-term expression of constitutively active ErbBs in adult hearts may induce chronic cardiomyocyte dedifferentiation and cause detrimental outcomes [168], re-expression of wildtype ErbBs in the myocardium together with transient systemic delivery of Nrg1 could be a promising strategy for promoting cardiac regenerative repair.
4.3.6. Cell cycle regulators
A newly emerging perspective focuses on cytoskeletal structure of cardiomyocytes in relation to its regulation by the cell cycle. It was proposed that the sarcomere of adult cardiomyocytes inhibits proliferation because sarcomere structure cannot be rearranged at the adult stage [170]. In addition, this may be connected to cell cycle regulation, as cell cycle regulators can modulate cytoskeleton structure during mitosis [170]. Cell cycle regulators in particular are referred to as cyclins, cyclin-dependent kinases (CDKs), and CDK inhibitors (CDKis) [171]. Actin also undergoes structural changes during the cell cycle [172–174]. For instance, cyclin D-CDK4/CDK6 is involved with cell cycle progression while E-CDK2 initiates the G1/S checkpoint [175,176]. Actin assembly can activate the G1/S checkpoint by upregulating E-CDK2, which indicates that the cell cycle is progressing normally. Inhibiting actin proliferation, on the other hand, can delay mitosis and slow down the overall progression of the cell cycle. An actin-associated protein named cortactin is responsible for mediating the contact between actin and the centrosome during the centrosome separation stage of mitosis [177]. Cortactin was also shown to be a substrate for CDK1, which in turn phosphorylates the actin crosslinking protein filamin A. This is essential for the post-mitotic separation of daughter cells [178,179]. This serves as another body of evidence that establishes a link between cytoskeleton structure regulation and mitosis.
Studies in mice have shown that cdk and cycline overexpression promotes cardiomyocyte proliferation in adult hearts. Cyclin A2 is a cell cycle regulator and activates CDK1 and CDK2, and is normally silenced in postnatal cardiomyocytes [180,181]. These findings suggest the important roles of cyclin A2 in postnatal cell cycle withdrawal of cardiomyocytes. Supporting this idea, constitutive expression of cyclin A2 in cardiomyocytes promoted cardiomyocyte proliferation and resulted in heart hyperplasia [181]. Furthermore, constitutive expression of cyclin A2 promoted cardiac regeneration and functional recovery after myocardial infarction [182]. In addition, viral expression of cyclin A2 in infarcted hearts promoted regenerative and protective response in rats and pigs [183,184]. Another cell cycle regulator cyclin D1 and D2, activating partners of CDK4, have also been shown to promote cardiomyocyte proliferation [185,186]. Transgenic overexpression of cyclin D2 promoted infarct regression, suggesting the regenerative potential of cyclin D2 [186]. Recent studies have also shown that combining cell cycle regulators such as cyclin B1, CDK1, cyclin D1, and CDK4 can reactivate the cell cycle in adult cardiomyocytes [27].
Overexpressing this combination of genes caused a greater upregulation of phosphohistone 3 when compared to overexpressing factors individually [27]. In addition to the apparent loss of cell cycle activator expression, adult mammalian cardiomyocytes exhibit a relatively higher expression of cell cycle inhibitors (CKIs) such as p16, p21, p27, and p53 [187–189]. Defined roles of each of these CKIs have not been established yet but are of great therapeutic interest, nonetheless. While cell cycle regulator modulation yielded some success in enhancing mammalian cardiomyocyte regeneration, it is not entirely known how these pathways converge with respect to the regulation of cardiomyocyte cytoskeletal structure [170].
5. Conclusions and future perspectives
Heart failure is, unfortunately, the leading cause of death and disability throughout the entire world. Neonatal mammals and lower vertebrates have a robust and efficient means of regenerating their hearts. As such, there is a substantial thrust to understand the mechanisms involved with sustained cardiac regenerative potential. In this review, we summarized diverging regenerative capacities across the known phylogenetic species. We have also discussed some of the new and prevailing theories as to how mammals lose regenerative potential of their hearts. Also discussed are endocrine factors, neural regulators, and local signaling cascades that influence cardiomyocyte proliferation and cardiovascular health.
Current clinical developments maintain that LVAD placement can increase cardiomyocyte proliferation in human patients, thus contributing to improved clinical outcomes. However, these findings appear to be controversial given the evidence that suggests otherwise. This further contributes to the issue of what is driving polyploidization of mammalian cardiomyocytes. Current models purport that polyploidization results from cardiomyocytes exiting the cell cycle or the cardiomyocyte’s inability to exit the cell cycle, resulting in incomplete cytokinesis. In addition, increased metabolic activity is associated with diminished cardiac regeneration. This is evidenced by the negative correlation between increased core body temperature and abrogated cardiomyocyte proliferation. This appears to be due in part to increased levels of thyroid hormone and sympathetic nerve activity during the acquisition of endothermy.
Local signaling cascades may also give rise to potential pharmacological targets. Many canonical signaling pathways contribute to cardiomyocyte proliferation. This includes the YAP/TAZ pathway, the Wnt pathway, the Notch pathway, the TGFβ pathway, neuregulin signaling, and many factors pertaining to cell cycle control. Can the ligands of any of these individual pathways be sufficient to drive adequate regenerative programs, or will a combination of novel factors be required? It would also be interesting to examine the direct relationships between CDKs, CDK inhibitors, and polyploidization with respect to alterations in cardiomyocyte structure. It is also important to understand why endocrine signaling (i.e. thyroid hormone, glucocorticoid, and vitamin D signaling) are essential for life but paradoxically stifle cardiomyocyte regeneration. This knowledge will shed light on why such a seemingly beneficial trait as the ability to restore damaged tissues is ever lost in development and evolution. It could also help us design novel strategies to promote de novo heart regeneration to treat cardiovascular diseases.
Figure 2. Proposed models explaining differences in cardiomyocyte regeneration across species.
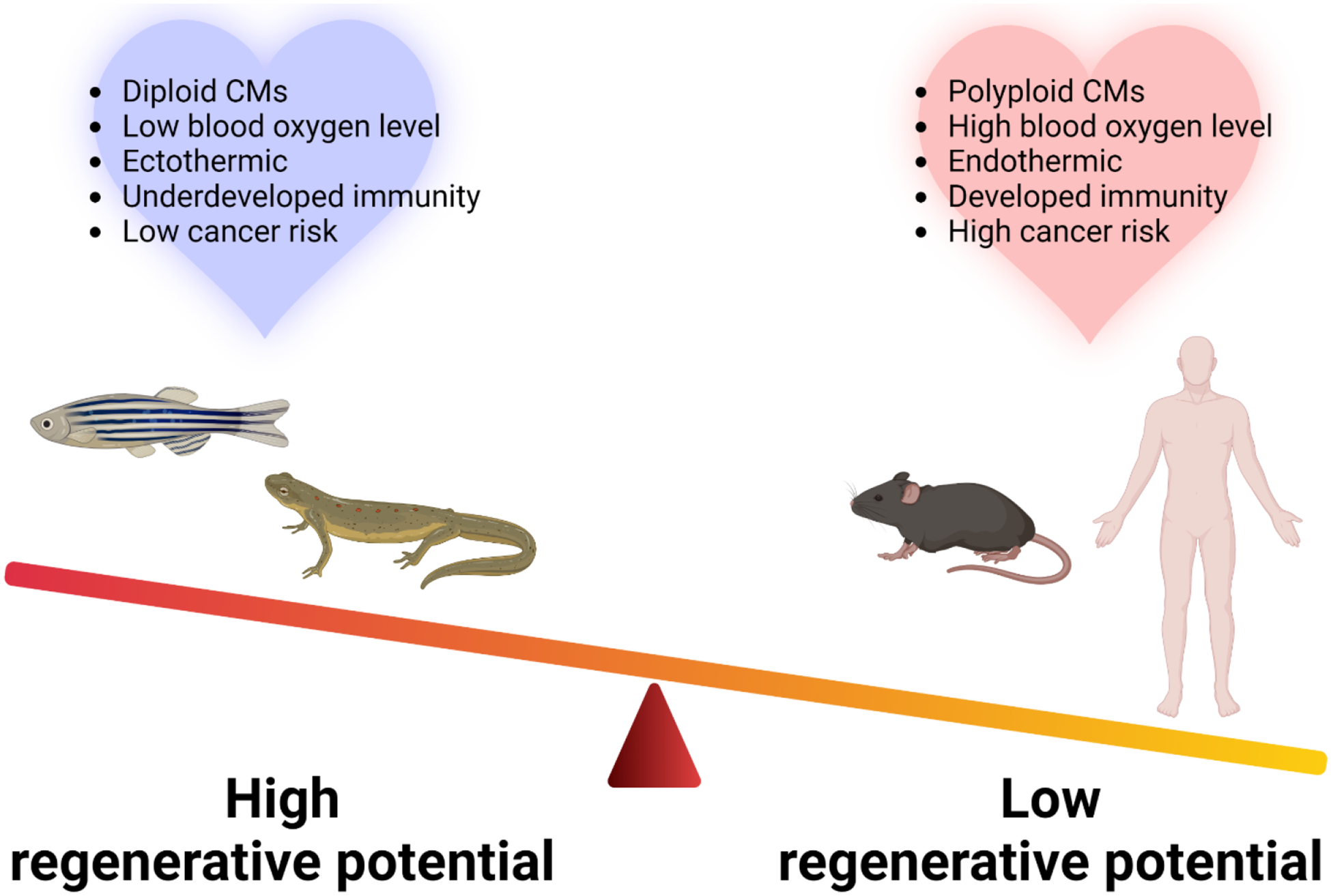
Hearts of species with robust regenerative potential typically have diploid cardiomyocytes (CMs), are exposed to low blood oxygen level. In addition, ectothermy, underdeveloped immunity, and low cancer risk is highly associated with the high regenerative potential. The opposite is apparent for species that exhibit poor cardiac regeneration. The image was created with BioRender.com.
Figure 3. Molecular pathways regulating cardiomyocyte proliferation.
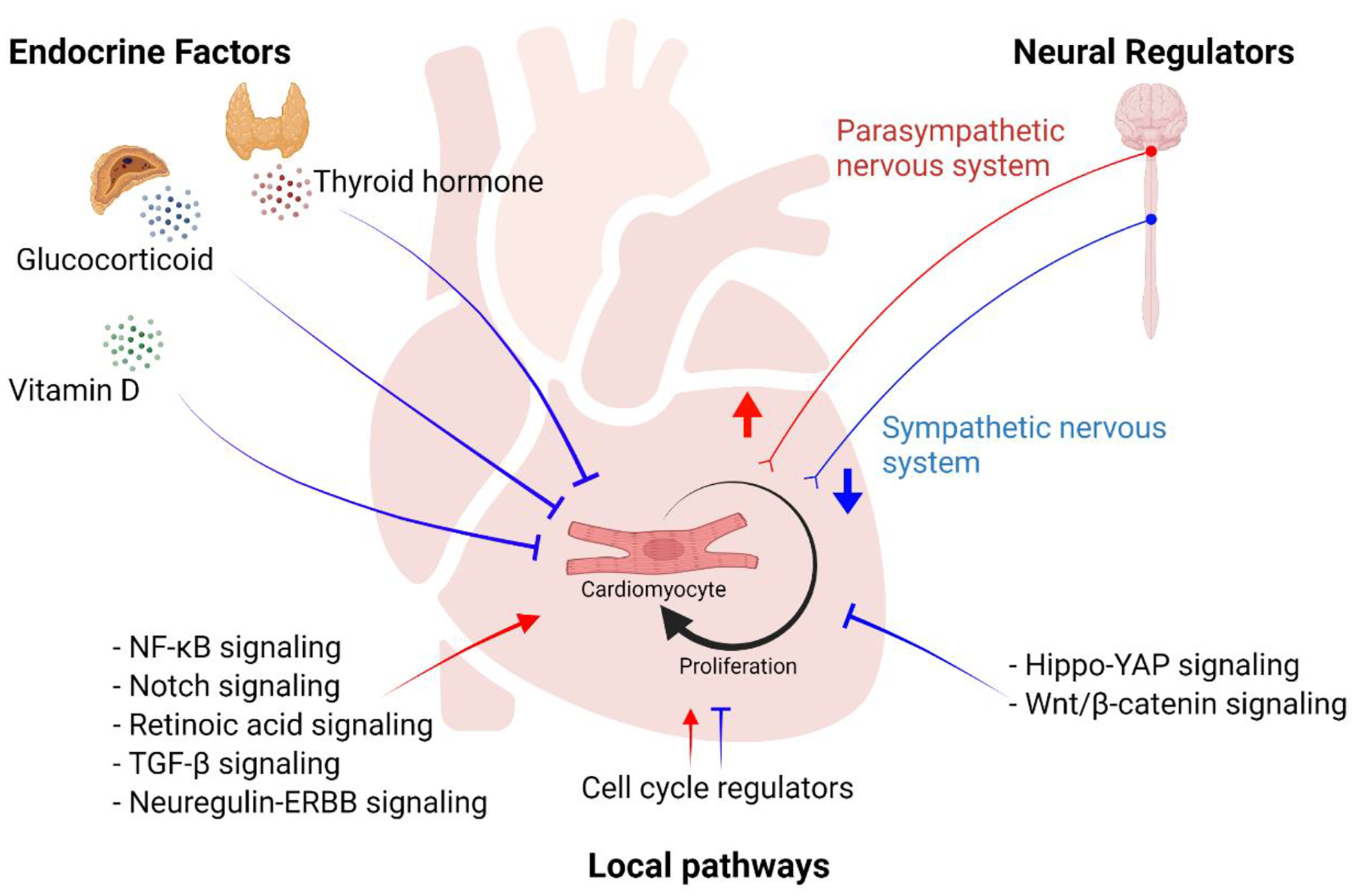
A schematic overview of molecular pathways regulating cardiomyocyte proliferation. Endocrine factors including thyroid hormone, glucocorticoid, and vitamin D inhibit cardiomyocyte proliferation. Neural signals from sympathetic nervous system inhibit cardiomyocyte proliferation whereas those from parasympathetic nervous system promote proliferation. Local pathways including paracrine and cell-intrinsic factors have distinct effects on regulating cardiomyocyte proliferation. The image was created with BioRender.com.
Highlights.
Heart regenerative capacity is lost as cardiomyocytes exit the cell cycle and become polyploid.
Loss of cardiac regenerative potential in adult mammals may be an ecological and physiological tradeoff.
Cardiomyocyte proliferation and polyploidization are regulated by neurohormonal and local signals.
Acknowledgements
We thank Huang laboratory members for helpful comments on the manuscript. This work was supported by the CVRI NIH T32 awarded to MM, NIH (R01HL138456 and R01HL157280), American Heart Association Transformation Award, Tobacco-Related Disease Research Program, and BARI New Investigator Award to GNH.
Footnotes
Declaration of interest
The authors declare no competing interests.
References
- [1].Vujic A, Natarajan N, Lee RT. Molecular mechanisms of heart regeneration. Semin Cell Dev Biol 2020;100:20–8. 10.1016/j.semcdb.2019.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].van den Borne SWM, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol 2010;7:30–7. 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- [3].Laflamme MA, Murry CE. Heart regeneration. Nature 2011;473:326–35. 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 2010;72:19–44. 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- [5].Verheugt FWA, Gersh BJ, Armstrong PW. Aborted myocardial infarction: a new target for reperfusion therapy. Eur Heart J 2006;27:901–4. 10.1093/eurheartj/ehi829. [DOI] [PubMed] [Google Scholar]
- [6].Rumyantsev PP. Evidence of regeneration of significant parts of myocardial fibers of frogs after trauma. Arkh Anat Gistol Embriol 1961;376:65–74. [PubMed] [Google Scholar]
- [7].Rumiantsev PP (Pavel P, Carlson BM. Growth and hyperplasia of cardiac muscle cells. London, U.K: Harwood Academic Publishers; 1991. [Google Scholar]
- [8].Oberpriller JO, Oberpriller JC. Response of the adult newt ventricle to injury. J Exp Zool 1974;187:249–53. 10.1002/jez.1401870208. [DOI] [PubMed] [Google Scholar]
- [9].Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science 2002;298:2188–90. 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- [10].Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol 2013;14:529–41. 10.1038/nrm3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Matrone G, Tucker CS, Denvir MA. Cardiomyocyte proliferation in zebrafish and mammals: lessons for human disease. Cell Mol Life Sci 2017;74:1367–78. 10.1007/s00018-016-2404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Patterson M, Barske L, Van Handel B, Rau CD, Gan P, Sharma A, et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet 2017;49:1346–53. 10.1038/ng.3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].González-Rosa JM, Sharpe M, Field D, Soonpaa MH, Field LJ, Burns CE, et al. Myocardial polyploidization creates a barrier to heart regeneration in zebrafish. Dev Cell 2018;44:433–446.e7. 10.1016/j.devcel.2018.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hirose K, Payumo AY, Cutie S, Hoang A, Zhang H, Guyot R, et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 2019;364:184–8. 10.1126/science.aar2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ito K, Morioka M, Kimura S, Tasaki M, Inohaya K, Kudo A. Differential reparative phenotypes between zebrafish and medaka after cardiac injury. Dev Dyn 2014;243:1106–15. 10.1002/dvdy.24154. [DOI] [PubMed] [Google Scholar]
- [16].Liao S, Dong W, Lv L, Guo H, Yang J, Zhao H, et al. Heart regeneration in adult Xenopus tropicalis after apical resection. Cell Biosci 2017;7:70. 10.1186/s13578-017-0199-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Marshall L, Vivien C, Girardot F, Péricard L, Demeneix BA, Coen L, et al. Persistent fibrosis, hypertrophy and sarcomere disorganisation after endoscopy-guided heart resection in adult Xenopus. PLoS ONE 2017;12:e0173418. 10.1371/journal.pone.0173418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol 1996;271:H2183–9. 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- [19].Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 1996;28:1737–46. 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- [20].Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park S-Y, et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci USA 2013;110:1446–51. 10.1073/pnas.1214608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, et al. Dynamics of cell generation and turnover in the human heart. Cell 2015;161:1566–75. 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]
- [22].Derks W, Bergmann O. Polyploidy in cardiomyocytes: roadblock to heart regeneration? Circ Res 2020;126:552–65. 10.1161/CIRCRESAHA.119.315408. [DOI] [PubMed] [Google Scholar]
- [23].Ye L, D’Agostino G, Loo SJ, Wang CX, Su LP, Tan SH, et al. Early regenerative capacity in the porcine heart. Circulation 2018;138:2798–808. 10.1161/CIRCULATIONAHA.117.031542. [DOI] [PubMed] [Google Scholar]
- [24].Zhu W, Zhang E, Zhao M, Chong Z, Fan C, Tang Y, et al. Regenerative potential of neonatal porcine hearts. Circulation 2018;138:2809–16. 10.1161/CIRCULATIONAHA.118.034886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science 2011;331:1078–80. 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, et al. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017;550:260–4. 10.1038/nature24045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mohamed TMA, Ang Y-S, Radzinsky E, Zhou P, Huang Y, Elfenbein A, et al. Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell 2018;173:104–116.e12. 10.1016/j.cell.2018.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Haubner BJ, Schneider J, Schweigmann U, Schuetz T, Dichtl W, Velik-Salchner C, et al. Functional recovery of a human neonatal heart after severe myocardial infarction. Circ Res 2016;118:216–21. 10.1161/CIRCRESAHA.115.307017. [DOI] [PubMed] [Google Scholar]
- [29].Anatskaya OV, Vinogradov AE. Paradoxical relationship between protein content and nucleolar activity in mammalian cardiomyocytes. Genome 2004;47:565–78. 10.1139/g04-015. [DOI] [PubMed] [Google Scholar]
- [30].Heallen TR, Kadow ZA, Kim JH, Wang J, Martin JF. Stimulating cardiogenesis as a treatment for heart failure. Circ Res 2019;124:1647–57. 10.1161/CIRCRESAHA.118.313573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gan P, Patterson M, Sucov HM. Cardiomyocyte polyploidy and implications for heart regeneration. Annu Rev Physiol 2020;82:45–61. 10.1146/annurev-physiol-021119-034618. [DOI] [PubMed] [Google Scholar]
- [32].Cutie S, Huang GN. Vertebrate cardiac regeneration: evolutionary and developmental perspectives. Cell Regen (Lond) 2021;10:6. 10.1186/s13619-020-00068-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Topkara VK, Garan AR, Fine B, Godier-Furnémont AF, Breskin A, Cagliostro B, et al. Myocardial recovery in patients receiving contemporary left ventricular assist devices: results from the interagency registry for mechanically assisted circulatory support (INTERMACS). Circ Heart Fail 2016;9. 10.1161/CIRCHEARTFAILURE.116.003157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Canseco DC, Kimura W, Garg S, Mukherjee S, Bhattacharya S, Abdisalaam S, et al. Human ventricular unloading induces cardiomyocyte proliferation. J Am Coll Cardiol 2015;65:892–900. 10.1016/j.jacc.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Luo J, Farris SD, Helterline D, Stempien-Otero A. Mechanical Unloading is Associated with Decreased DNA Content in Cardiomyocytes Independent of Nucleation State. MedRxiv 2020. [Google Scholar]
- [36].Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014;157:565–79. 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, et al. Hypoxia induces heart regeneration in adult mice. Nature 2017;541:222–7. 10.1038/nature20173. [DOI] [PubMed] [Google Scholar]
- [38].Jonker SS, Louey S, Giraud GD, Thornburg KL, Faber JJ. Timing of cardiomyocyte growth, maturation, and attrition in perinatal sheep. FASEB J 2015;29:4346–57. 10.1096/fj.15-272013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Goss RJ. The evolution of regeneration: adaptive or inherent? J Theor Biol 1992;159:241–60. 10.1016/s0022-5193(05)80704-0. [DOI] [PubMed] [Google Scholar]
- [40].Ivanov KP. The development of the concepts of homeothermy and thermoregulation. J Therm Biol 2006;31:24–9. 10.1016/j.jtherbio.2005.12.005. [DOI] [Google Scholar]
- [41].Tourneux P, Libert JP, Ghyselen L, Léké A, Delanaud S, Dégrugilliers L, et al. [Heat exchanges and thermoregulation in the neonate]. Arch Pediatr 2009;16:1057–62. 10.1016/j.arcped.2009.03.014. [DOI] [PubMed] [Google Scholar]
- [42].Payumo AY, Chen X, Hirose K, Chen X, Hoang A, Khyeam S, et al. Adrenergic-Thyroid Hormone Interactions Drive Postnatal Thermogenesis and Loss of Mammalian Heart Regenerative Capacity. Circulation 2021;144:1000–3. 10.1161/CIRCULATIONAHA.121.054846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Khyeam S, Lee S, Huang GN. Genetic, Epigenetic, and Post-Transcriptional Basis of Divergent Tissue Regenerative Capacities Among Vertebrates. Adv Genet (Hoboken) 2021;2. 10.1002/ggn2.10042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jinnou H, Sawada M, Kawase K, Kaneko N, Herranz-Pérez V, Miyamoto T, et al. Radial Glial Fibers Promote Neuronal Migration and Functional Recovery after Neonatal Brain Injury. Cell Stem Cell 2018;22:128–137.e9. 10.1016/j.stem.2017.11.005. [DOI] [PubMed] [Google Scholar]
- [45].Li Y, He X, Kawaguchi R, Zhang Y, Wang Q, Monavarfeshani A, et al. Microglia-organized scar-free spinal cord repair in neonatal mice. Nature 2020;587:613–8. 10.1038/s41586-020-2795-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mescher AL, Neff AW. Regenerative capacity and the developing immune system. Adv Biochem Eng Biotechnol 2005;93:39–66. 10.1007/b99966. [DOI] [PubMed] [Google Scholar]
- [47].Aurora AB, Olson EN. Immune modulation of stem cells and regeneration. Cell Stem Cell 2014;15:14–25. 10.1016/j.stem.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhao A, Qin H, Fu X. What determines the regenerative capacity in animals? Bioscience 2016;66:735–46. 10.1093/biosci/biw079. [DOI] [Google Scholar]
- [49].Witman N, Murtuza B, Davis B, Arner A, Morrison JI. Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev Biol 2011;354:67–76. 10.1016/j.ydbio.2011.03.021. [DOI] [PubMed] [Google Scholar]
- [50].Cano-Martínez A, Vargas-González A, Guarner-Lans V, Prado-Zayago E, LeónOleda M, Nieto-Lima B. Functional and structural regeneration in the axolotl heart (Ambystoma mexicanum) after partial ventricular amputation. Arch Cardiol Mex 2010;80:79–86. [PubMed] [Google Scholar]
- [51].Li J, Liang C, Yang KY, Huang X, Han MY, Li X, et al. Specific ablation of CD4+ T-cells promotes heart regeneration in juvenile mice. Theranostics 2020;10:8018–35. 10.7150/thno.42943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dolejsi T, Schuetz T, Delgobo M, Tortola L, Bauer A, Ruschitzka F, et al. Adult T-cells impair neonatal cardiac regeneration. Eur Heart J 2020;41. 10.1093/ehjci/ehaa946.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Arslan F, Keogh B, McGuirk P, Parker AE. TLR2 and TLR4 in ischemia reperfusion injury. Mediators Inflamm 2010;2010:704202. 10.1155/2010/704202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Oyama J, Blais C, Liu X, Pu M, Kobzik L, Kelly RA, et al. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation 2004;109:784–9. 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- [55].Timmers L, Sluijter JPG, van Keulen JK, Hoefer IE, Nederhoff MGJ, Goumans M-J, et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res 2008;102:257–64. 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- [56].Vandervelde S, van Amerongen MJ, Tio RA, Petersen AH, van Luyn MJA, Harmsen MC. Increased inflammatory response and neovascularization in reperfused vs. non-reperfused murine myocardial infarction. Cardiovasc Pathol 2006;15:83–90. 10.1016/j.carpath.2005.10.006. [DOI] [PubMed] [Google Scholar]
- [57].Kain V, Liu F, Kozlovskaya V, Ingle KA, Khedkar S, Prabhu SD, et al. Resolution Agonist 15-epi-Lipoxin A4 Directs FPR2 to Expedite Healing Phase Post-Myocardial Infarction. The FASEB Journal 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Han C, Nie Y, Lian H, Liu R, He F, Huang H, et al. Acute inflammation stimulates a regenerative response in the neonatal mouse heart. Cell Res 2015;25:1137–51. 10.1038/cr.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, et al. Macrophages are required for neonatal heart regeneration. J Clin Invest 2014;124:1382–92. 10.1172/JCI72181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci USA 2014;111:16029–34. 10.1073/pnas.1406508111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li Y, Li H, Pei J, Hu S, Nie Y. Transplantation of murine neonatal cardiac macrophage improves adult cardiac repair. Cell Mol Immunol 2021;18:492–4. 10.1038/s41423-020-0371-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Vagnozzi RJ, Maillet M, Sargent MA, Khalil H, Johansen AKZ, Schwanekamp JA, et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020;577:405–9. 10.1038/s41586-019-1802-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Pomerantz JH, Blau HM. Tumor suppressors: enhancers or suppressors of regeneration? Development 2013;140:2502–12. 10.1242/dev.084210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio-Kuter A, et al. The origins and evolution of the p53 family of genes. Cold Spring Harb Perspect Biol 2010;2:a001198. 10.1101/cshperspect.a001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Seifert AW, Voss SR. Revisiting the relationship between regenerative ability and aging. BMC Biol 2013;11:2. 10.1186/1741-7007-11-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zhang S, Zhou K, Luo X, Li L, Tu H-C, Sehgal A, et al. The Polyploid State Plays a Tumor-Suppressive Role in the Liver. Dev Cell 2018;44:447–459.e5. 10.1016/j.devcel.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Mullur R, Liu Y-Y, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev 2014;94:355–82. 10.1152/physrev.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ross I, Omengan DB, Huang GN, Payumo AY. Thyroid hormone-dependent regulation of metabolism and heart regeneration. J Endocrinol 2022;252:R71–82. 10.1530/JOE-21-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Mancino G, Miro C, Di Cicco E, Dentice M. Thyroid hormone action in epidermal development and homeostasis and its implications in the pathophysiology of the skin. J Endocrinol Invest 2021. 10.1007/s40618-020-01492-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bernal J Thyroid hormone receptors in brain development and function. Nat Clin Pract Endocrinol Metab 2007;3:249–59. 10.1038/ncpendmet0424. [DOI] [PubMed] [Google Scholar]
- [71].Chang J, Wang M, Gui W, Zhao Y, Yu L, Zhu G. Changes in thyroid hormone levels during zebrafish development. Zool Sci 2012;29:181–4. 10.2108/zsj.29.181. [DOI] [PubMed] [Google Scholar]
- [72].Liversage RA, Korneluk RG. Serum levels of thyroid hormone during forelimb regeneration in the adult newt,Notophthalmus viridescens. J Exp Zool 1978;206:223–7. 10.1002/jez.1402060212. [DOI] [Google Scholar]
- [73].Amram AV, Cutie S, Huang GN. Hormonal control of cardiac regenerative potential. Endocr Connect 2021;10:R25–35. 10.1530/EC-20-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Graham N, Huang GN. Endocrine influence on cardiac metabolism in development and regeneration. Endocrinology 2021;162. 10.1210/endocr/bqab081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Gerdes AM, Kriseman J, Bishop SP. Changes in myocardial cell size and number during the development and reversal of hyperthyroidism in neonatal rats. Lab Invest 1983;48:598–602. [PubMed] [Google Scholar]
- [76].Chattergoon NN, Giraud GD, Louey S, Stork P, Fowden AL, Thornburg KL. Thyroid hormone drives fetal cardiomyocyte maturation. FASEB J 2012;26:397–408. 10.1096/fj.10-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J, et al. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell 2014;157:795–807. 10.1016/j.cell.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Soonpaa MH, Zebrowski DC, Platt C, Rosenzweig A, Engel FB, Field LJ. Cardiomyocyte Cell-Cycle Activity during Preadolescence. Cell 2015;163:781–2. 10.1016/j.cell.2015.10.037. [DOI] [PubMed] [Google Scholar]
- [79].Alkass K, Panula J, Westman M, Wu T-D, Guerquin-Kern J-L, Bergmann O. No evidence for cardiomyocyte number expansion in preadolescent mice. Cell 2015;163:1026–36. 10.1016/j.cell.2015.10.035. [DOI] [PubMed] [Google Scholar]
- [80].Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci 2013;34:518–30. 10.1016/j.tips.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gay MS, Li Y, Xiong F, Lin T, Zhang L. Dexamethasone Treatment of Newborn Rats Decreases Cardiomyocyte Endowment in the Developing Heart through Epigenetic Modifications. PLoS ONE 2015;10:e0125033. 10.1371/journal.pone.0125033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Whitehurst RM, Zhang M, Bhattacharjee A, Li M. Dexamethasone-induced hypertrophy in rat neonatal cardiac myocytes involves an elevated L-type Ca(2+)current. J Mol Cell Cardiol 1999;31:1551–8. 10.1006/jmcc.1999.0990. [DOI] [PubMed] [Google Scholar]
- [83].Gan L, Li Q, Pan J, Chen L. Glucocorticoids rapidly promote YAP phosphorylation via the cAMP-PKA pathway to repress mouse cardiomyocyte proliferative potential. Mol Cell Endocrinol 2022;548:111615. 10.1016/j.mce.2022.111615. [DOI] [PubMed] [Google Scholar]
- [84].Huang W-C, Yang C-C, Chen I-H, Liu Y-ML, Chang S-J, Chuang Y-J. Treatment of glucocorticoids inhibited early immune responses and impaired cardiac repair in adult zebrafish. PLoS ONE 2013;8:e66613. 10.1371/journal.pone.0066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Jaźwińska A, Sallin P. Regeneration versus scarring in vertebrate appendages and heart. J Pathol 2016;238:233–46. 10.1002/path.4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Pianca N, Sacchi F, Umansky KB, Chirivì M, Iommarini L, Da Pra S, et al. Glucocorticoid receptor antagonization propels endogenous cardiomyocyte proliferation and cardiac regeneration. Nat Cardiovasc Res 2022. 10.1038/s44161-022-00090-0. [DOI] [PubMed] [Google Scholar]
- [87].Lamers WH, Mooren PG, Griep H, Endert E, Degenhart HJ, Charles R. Hormones in perinatal rat and spiny mouse: relation to altricial and precocial timing of birth. Am J Physiol 1986;251:E78–85. 10.1152/ajpendo.1986.251.1.E78. [DOI] [PubMed] [Google Scholar]
- [88].Diez JA, Sze PY, Ginsburg BE. Postnatal development of mouse plasma and brain corticosterone levels: new findings contingent upon the use of a competitive protein-binding assay. Endocrinology 1976;98:1434–42. 10.1210/endo-98-6-1434. [DOI] [PubMed] [Google Scholar]
- [89].Sirajudeen S, Shah I, Al Menhali A. A narrative role of vitamin D and its receptor: with current evidence on the gastric tissues. Int J Mol Sci 2019;20. 10.3390/ijms20153832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Chen S, Law CS, Grigsby CL, Olsen K, Hong T-T, Zhang Y, et al. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation 2011;124:1838–47. 10.1161/CIRCULATIONAHA.111.032680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Cutie S, Payumo AY, Lunn D, Huang GN. In vitro and in vivo roles of glucocorticoid and vitamin D receptors in the control of neonatal cardiomyocyte proliferative potential. J Mol Cell Cardiol 2020;142:126–34. 10.1016/j.yjmcc.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Hlaing SM, Garcia LA, Contreras JR, Norris KC, Ferrini MG, Artaza JN. 1,25-Vitamin D3 promotes cardiac differentiation through modulation of the WNT signaling pathway. J Mol Endocrinol 2014;53:303–17. 10.1530/JME-14-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007;103:533–7. 10.1016/j.jsbmb.2006.12.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Han Y, Chen A, Umansky K-B, Oonk KA, Choi W-Y, Dickson AL, et al. Vitamin D stimulates cardiomyocyte proliferation and controls organ size and regeneration in zebrafish. Dev Cell 2019;48:853–863.e5. 10.1016/j.devcel.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Alshak MN, Das JM. Neuroanatomy, sympathetic nervous system. StatPearls, Treasure Island (FL): StatPearls Publishing; 2022. [PubMed] [Google Scholar]
- [96].McCorry LK. Physiology of the autonomic nervous system. Am J Pharm Educ 2007;71:78. 10.5688/aj710478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wehrwein EA, Orer HS, Barman SM. Overview of the anatomy, physiology, and pharmacology of the autonomic nervous system. Compr Physiol 2016;6:1239–78. 10.1002/cphy.c150037. [DOI] [PubMed] [Google Scholar]
- [98].Shibasaki M, Crandall CG. Mechanisms and controllers of eccrine sweating in humans. Front Biosci (Schol Ed) 2010;2:685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Manousiouthakis E, Mendez M, Garner MC, Exertier P, Makita T. Venous endothelin guides sympathetic innervation of the developing mouse heart. Nat Commun 2014;5:3918. 10.1038/ncomms4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Bartolomé J, Lau C, Slotkin TA. Ornithine decarboxylase in developing rat heart and brain: role of sympathetic development for responses to autonomic stimulants and the effects of reserpine on maturation. J Pharmacol Exp Ther 1977;202:510–8. [PubMed] [Google Scholar]
- [101].Kreipke RE, Birren SJ. Innervating sympathetic neurons regulate heart size and the timing of cardiomyocyte cell cycle withdrawal. J Physiol (Lond) 2015;593:5057–73. 10.1113/JP270917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Tampakakis E, Gangrade H, Glavaris S, Htet M, Murphy S, Lin BL, et al. Heart neurons use clock genes to control myocyte proliferation. Sci Adv 2021;7:eabh4181. 10.1126/sciadv.abh4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sakabe M, Thompson M, Chen N, Verba M, Hassan A, Lu R, et al. Inhibition of adrenergic β1-AR/Gαs signaling promotes cardiomyocyte proliferation through activation of RhoA-YAP axis. BioRxiv 2021. 10.1101/2021.10.20.465083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Liu H, Zhang C-H, Ammanamanchi N, Suresh S, Lewarchik C, Rao K, et al. Control of cytokinesis by β-adrenergic receptors indicates an approach for regulating cardiomyocyte endowment. Sci Transl Med 2019;11. 10.1126/scitranslmed.aaw6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Yu F-X, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012;150:780–91. 10.1016/j.cell.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Tindle J, Tadi P. Neuroanatomy, parasympathetic nervous system. StatPearls, Treasure Island (FL): StatPearls Publishing; 2022. [PubMed] [Google Scholar]
- [107].Mahmoud AI, O’Meara CC, Gemberling M, Zhao L, Bryant DM, Zheng R, et al. Nerves regulate cardiomyocyte proliferation and heart regeneration. Dev Cell 2015;34:387–99. 10.1016/j.devcel.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Todd T On the process of reproduction of the members of the aquatic salamander. Quart J Sci Arts Lib 1823;16:84–6. [Google Scholar]
- [109].Drachman DB. Atrophy of skeletal muscle in chick embryos treated with botulinum toxin. Science 1964;145:719–21. 10.1126/science.145.3633.719. [DOI] [PubMed] [Google Scholar]
- [110].Singer M, Davis MH, Scheuing MR. The influence of atropine and other neuropharmacological substances on regeneration of the forelimb in the adult urodele, Triturus. J Exp Zool 1960;143:33–45. 10.1002/jez.1401430104. [DOI] [Google Scholar]
- [111].Wang J, Liu S, Heallen T, Martin JF. The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nat Rev Cardiol 2018;15:672–84. 10.1038/s41569-018-0063-3. [DOI] [PubMed] [Google Scholar]
- [112].Zheng A, Chen Q, Zhang L. The Hippo-YAP pathway in various cardiovascular diseases: Focusing on the inflammatory response. Front Immunol 2022;13:971416. 10.3389/fimmu.2022.971416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Del Re DP. Hippo-Yap signaling in cardiac and fibrotic remodeling. Curr Opin Physiol 2022;26:100492. 10.1016/j.cophys.2022.100492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Xiao Y, Leach J, Wang J, Martin JF. Hippo/yap signaling in cardiac development and regeneration. Curr Treat Options Cardiovasc Med 2016;18:38. 10.1007/s11936-016-0461-y. [DOI] [PubMed] [Google Scholar]
- [115].Zheng Y, Pan D. The hippo signaling pathway in development and disease. Dev Cell 2019;50:264–82. 10.1016/j.devcel.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011;332:458–61. 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci USA 2012;109:2394–9. 10.1073/pnas.1116136109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, et al. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal 2011;4:ra70. 10.1126/scisignal.2002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Embryonic expression of an Nkx2–5/Cre gene using ROSA26 reporter mice. Genesis 2001;31:176–80. 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- [120].Del Re DP. Hippo-Yap signaling in cardiac and fibrotic remodeling. Curr Opin Physiol 2022;26:100492. 10.1016/j.cophys.2022.100492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, et al. Hippo signaling impedes adult heart regeneration. Development 2013;140:4683–90. 10.1242/dev.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci USA 2013;110:13839–44. 10.1073/pnas.1313192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Liu S, Li K, Wagner Florencio L, Tang L, Heallen TR, Leach JP, et al. Gene therapy knockdown of Hippo signaling induces cardiomyocyte renewal in pigs after myocardial infarction. Sci Transl Med 2021;13. 10.1126/scitranslmed.abd6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Zhang S, Liu S, Leach JP, Li K, Perin EC, Martin JF. Gene therapy knockdown of hippo signaling resolves arrhythmic events in pigs after myocardial infarction. Circulation 2022;146:1558–60. 10.1161/CIRCULATIONAHA.122.059972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Mehdipour M, Liu Y, Liu C, Kumar B, Kim D, Gathwala R, et al. Key Age-Imposed Signaling Changes That Are Responsible for the Decline of Stem Cell Function. Subcell Biochem 2018;90:119–43. 10.1007/978-981-13-2835-0_5. [DOI] [PubMed] [Google Scholar]
- [126].Wodarz A, Nusse R. MECHANISMS OF WNT SIGNALING IN DEVELOPMENT. Annu Rev Cell Dev Biol 1998;14:59–88. 10.1146/annurev.cellbio.14.1.59. [DOI] [PubMed] [Google Scholar]
- [127].Wright M, Aikawa M, Szeto W, Papkoff J. Identification of a Wnt-responsive signal transduction pathway in primary endothelial cells. Biochem Biophys Res Commun 1999;263:384–8. 10.1006/bbrc.1999.1344. [DOI] [PubMed] [Google Scholar]
- [128].Li F, Chong ZZ, Maiese K. Winding through the WNT pathway during cellular development and demise. Histol Histopathol 2006;21:103–24. 10.14670/HH-21.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Ozhan G, Weidinger G. Wnt/β-catenin signaling in heart regeneration. Cell Regen (Lond) 2015;4:3. 10.1186/s13619-015-0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol 2009;10:468–77. 10.1038/nrm2717. [DOI] [PubMed] [Google Scholar]
- [131].MacDonald BT, He X. Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb Perspect Biol 2012;4. 10.1101/cshperspect.a007880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Li D, Sun J, Zhong TP. Wnt signaling in heart development and regeneration. Curr Cardiol Rep 2022;24:1425–38. 10.1007/s11886-022-01756-8. [DOI] [PubMed] [Google Scholar]
- [133].Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Göke B, et al. Comprehensive analysis of β-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/β-catenin signaling. BMC Genomics 2014;15:74. 10.1186/1471-2164-15-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Valenta T, Hausmann G, Basler K. The many faces and functions of β-catenin. EMBO J 2012;31:2714–36. 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Cadigan KM, Waterman ML. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol 2012;4. 10.1101/cshperspect.a007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Tseng A-S, Engel FB, Keating MT. The GSK-3 inhibitor BIO promotes proliferation in mammalian cardiomyocytes. Chem Biol 2006;13:957–63. 10.1016/j.chembiol.2006.08.004. [DOI] [PubMed] [Google Scholar]
- [137].Quaife-Ryan GA, Mills RJ, Lavers G, Voges HK, Vivien CJ, Elliott DA, et al. β-Catenin drives distinct transcriptional networks in proliferative and nonproliferative cardiomyocytes. Development 2020;147. 10.1242/dev.193417. [DOI] [PubMed] [Google Scholar]
- [138].Li H, Chang C, Li X, Zhang R. The roles and activation of endocardial Notch signaling in heart regeneration. Cell Regen (Lond) 2021;10:3. 10.1186/s13619-020-00060-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Siebel C, Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol Rev 2017;97:1235–94. 10.1152/physrev.00005.2017. [DOI] [PubMed] [Google Scholar]
- [140].Kopan R, Ilagan MXG. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 2009;137:216–33. 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Gordon WR, Zimmerman B, He L, Miles LJ, Huang J, Tiyanont K, et al. Mechanical allostery: evidence for a force requirement in the proteolytic activation of notch. Dev Cell 2015;33:729–36. 10.1016/j.devcel.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Raya A, Koth CM, Büscher D, Kawakami Y, Itoh T, Raya RM, et al. Activation of Notch signaling pathway precedes heart regeneration in zebrafish. Proc Natl Acad Sci USA 2003;100 Suppl 1:11889–95. 10.1073/pnas.1834204100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Zhang P, Zhao Y, Sun X-H. Notch-regulated periphery B cell differentiation involves suppression of E protein function. J Immunol 2013;191:726–36. 10.4049/jimmunol.1202134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Zhao D, Mo Y, Li M-T, Zou S-W, Cheng Z-L, Sun Y-P, et al. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J Clin Invest 2014;124:5453–65. 10.1172/JCI76611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Croquelois A, Domenighetti AA, Nemir M, Lepore M, Rosenblatt-Velin N, Radtke F, et al. Control of the adaptive response of the heart to stress via the Notch1 receptor pathway. J Exp Med 2008;205:3173–85. 10.1084/jem.20081427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].He Y, Pang S, Huang J, Zhu K, Tong J, Tang Y, et al. Blockade of RBP-J-Mediated Notch Signaling Pathway Exacerbates Cardiac Remodeling after Infarction by Increasing Apoptosis in Mice. Biomed Res Int 2018;2018:5207031. 10.1155/2018/5207031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Sorensen DW, van Berlo JH. The Role of TGF-β Signaling in Cardiomyocyte Proliferation. Curr Heart Fail Rep 2020;17:225–33. 10.1007/s11897-020-00470-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Todorovic V, Jurukovski V, Chen Y, Fontana L, Dabovic B, Rifkin DB. Latent TGF-beta binding proteins. Int J Biochem Cell Biol 2005;37:38–41. 10.1016/j.biocel.2004.03.011. [DOI] [PubMed] [Google Scholar]
- [149].Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, et al. Latent TGF-β structure and activation. Nature 2011;474:343–9. 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Zhang YE, Newfeld SJ. Meeting report - TGF-β superfamily: signaling in development and disease. J Cell Sci 2013;126:4809–13. 10.1242/jcs.142398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Heldin C-H, Moustakas A Signaling Receptors for TGF-β Family Members. Cold Spring Harb Perspect Biol 2016;8. 10.1101/cshperspect.a022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Ten Dijke P, Goumans M-J, Itoh F, Itoh S. Regulation of cell proliferation by Smad proteins. J Cell Physiol 2002;191:1–16. 10.1002/jcp.10066. [DOI] [PubMed] [Google Scholar]
- [153].Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992;359:693–9. 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet 1995;11:409–14. 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet 1995;11:415–21. 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- [156].Uribe V, Ramadass R, Dogra D, Rasouli SJ, Gunawan F, Nakajima H, et al. In vivo analysis of cardiomyocyte proliferation during trabeculation. Development 2018;145. 10.1242/dev.164194. [DOI] [PubMed] [Google Scholar]
- [157].McKoy G, Bicknell KA, Patel K, Brooks G. Developmental expression of myostatin in cardiomyocytes and its effect on foetal and neonatal rat cardiomyocyte proliferation. Cardiovasc Res 2007;74:304–12. 10.1016/j.cardiores.2007.02.023. [DOI] [PubMed] [Google Scholar]
- [158].Sridurongrit S, Larsson J, Schwartz R, Ruiz-Lozano P, Kaartinen V. Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev Biol 2008;322:208–18. 10.1016/j.ydbio.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Inman GJ, Nicolás FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol 2002;62:65–74. 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- [160].Wang Y, Wei J, Zhang P, Zhang X, Wang Y, Chen W, et al. Neuregulin-1, a potential therapeutic target for cardiac repair. Front Pharmacol 2022;13:945206. 10.3389/fphar.2022.945206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Rupert CE, Coulombe KL. The roles of neuregulin-1 in cardiac development, homeostasis, and disease. Biomark Insights 2015;10:1–9. 10.4137/BMI.S20061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127–37. 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- [163].Sudol M. Neuregulin 1-activated ERBB4 as a “dedicated” receptor for the Hippo-YAP pathway. Sci Signal 2014;7:pe29. 10.1126/scisignal.aaa2710. [DOI] [PubMed] [Google Scholar]
- [164].Bersell K, Arab S, Haring B, Kühn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 2009;138:257–70. 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- [165].Wadugu B, Kühn B. The role of neuregulin/ErbB2/ErbB4 signaling in the heart with special focus on effects on cardiomyocyte proliferation. Am J Physiol Heart Circ Physiol 2012;302:H2139–47. 10.1152/ajpheart.00063.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Polizzotti BD, Ganapathy B, Walsh S, Choudhury S, Ammanamanchi N, Bennett DG, et al. Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci Transl Med 2015;7:281ra45. 10.1126/scitranslmed.aaa5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol 2015;17:627–38. 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- [168].Aharonov A, Shakked A, Umansky KB, Savidor A, Genzelinakh A, Kain D, et al. ERBB2 drives YAP activation and EMT-like processes during cardiac regeneration. Nat Cell Biol 2020;22:1346–56. 10.1038/s41556-020-00588-4. [DOI] [PubMed] [Google Scholar]
- [169].Haskins JW, Nguyen DX, Stern DF. Neuregulin 1-activated ERBB4 interacts with YAP to induce Hippo pathway target genes and promote cell migration. Sci Signal 2014;7:ra116. 10.1126/scisignal.2005770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Pettinato AM, Ladha FA, Hinson JT. The cardiac sarcomere and cell cycle. Curr Cardiol Rep 2022;24:623–30. 10.1007/s11886-022-01682-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Barnum KJ, O’Connell MJ. Cell cycle regulation by checkpoints. Methods Mol Biol 2014;1170:29–40. 10.1007/978-1-4939-0888-2_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Théry M, Racine V, Pépin A, Piel M, Chen Y, Sibarita J-B, et al. The extracellular matrix guides the orientation of the cell division axis. Nat Cell Biol 2005;7:947–53. 10.1038/ncb1307. [DOI] [PubMed] [Google Scholar]
- [173].Pollard TD, O’Shaughnessy B. Molecular mechanism of cytokinesis. Annu Rev Biochem 2019;88:661–89. 10.1146/annurev-biochem-062917-012530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Jones MC, Zha J, Humphries MJ. Connections between the cell cycle, cell adhesion and the cytoskeleton. Philos Trans R Soc Lond B Biol Sci 2019;374:20180227. 10.1098/rstb.2018.0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009;28:2925–39. 10.1038/onc.2009.170. [DOI] [PubMed] [Google Scholar]
- [176].Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, et al. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science 1992;257:1689–94. 10.1126/science.1388288. [DOI] [PubMed] [Google Scholar]
- [177].Blethrow JD, Glavy JS, Morgan DO, Shokat KM. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc Natl Acad Sci USA 2008;105:1442–7. 10.1073/pnas.0708966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Cukier IH, Li Y, Lee JM. Cyclin B1/Cdk1 binds and phosphorylates Filamin A and regulates its ability to cross-link actin. FEBS Lett 2007;581:1661–72. 10.1016/j.febslet.2007.03.041. [DOI] [PubMed] [Google Scholar]
- [179].Wang W, Chen L, Ding Y, Jin J, Liao K. Centrosome separation driven by actin-microfilaments during mitosis is mediated by centrosome-associated tyrosine-phosphorylated cortactin. J Cell Sci 2008;121:1334–43. 10.1242/jcs.018176. [DOI] [PubMed] [Google Scholar]
- [180].Yoshizumi M, Lee WS, Hsieh CM, Tsai JC, Li J, Perrella MA, et al. Disappearance of cyclin A correlates with permanent withdrawal of cardiomyocytes from the cell cycle in human and rat hearts. J Clin Invest 1995;95:2275–80. 10.1172/JCI117918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Chaudhry HW, Dashoush NH, Tang H, Zhang L, Wang X, Wu EX, et al. Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem 2004;279:35858–66. 10.1074/jbc.M404975200. [DOI] [PubMed] [Google Scholar]
- [182].Cheng RK, Asai T, Tang H, Dashoush NH, Kara RJ, Costa KD, et al. Cyclin A2 induces cardiac regeneration after myocardial infarction and prevents heart failure. Circ Res 2007;100:1741–8. 10.1161/CIRCRESAHA.107.153544. [DOI] [PubMed] [Google Scholar]
- [183].Shapiro SD, Ranjan AK, Kawase Y, Cheng RK, Kara RJ, Bhattacharya R, et al. Cyclin A2 induces cardiac regeneration after myocardial infarction through cytokinesis of adult cardiomyocytes. Sci Transl Med 2014;6:224ra27. 10.1126/scitranslmed.3007668. [DOI] [PubMed] [Google Scholar]
- [184].Woo YJ, Panlilio CM, Cheng RK, Liao GP, Atluri P, Hsu VM, et al. Therapeutic delivery of cyclin A2 induces myocardial regeneration and enhances cardiac function in ischemic heart failure. Circulation 2006;114:I206–13. 10.1161/CIRCULATIONAHA.105.000455. [DOI] [PubMed] [Google Scholar]
- [185].Soonpaa MH, Koh GY, Pajak L, Jing S, Wang H, Franklin MT, et al. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest 1997;99:2644–54. 10.1172/JCI119453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Pasumarthi KBS, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res 2005;96:110–8. 10.1161/01.RES.0000152326.91223.4F. [DOI] [PubMed] [Google Scholar]
- [187].Poolman RA, Li JM, Durand B, Brooks G. Altered expression of cell cycle proteins and prolonged duration of cardiac myocyte hyperplasia in p27KIP1 knockout mice. Circ Res 1999;85:117–27. 10.1161/01.res.85.2.117. [DOI] [PubMed] [Google Scholar]
- [188].Flink IL, Oana S, Maitra N, Bahl JJ, Morkin E. Changes in E2F complexes containing retinoblastoma protein family members and increased cyclin-dependent kinase inhibitor activities during terminal differentiation of cardiomyocytes. J Mol Cell Cardiol 1998;30:563–78. 10.1006/jmcc.1997.0620. [DOI] [PubMed] [Google Scholar]
- [189].Tane S, Ikenishi A, Okayama H, Iwamoto N, Nakayama KI, Takeuchi T. CDK inhibitors, p21(Cip1) and p27(Kip1), participate in cell cycle exit of mammalian cardiomyocytes. Biochem Biophys Res Commun 2014;443:1105–9. 10.1016/j.bbrc.2013.12.109. [DOI] [PubMed] [Google Scholar]