
Fig. 2.
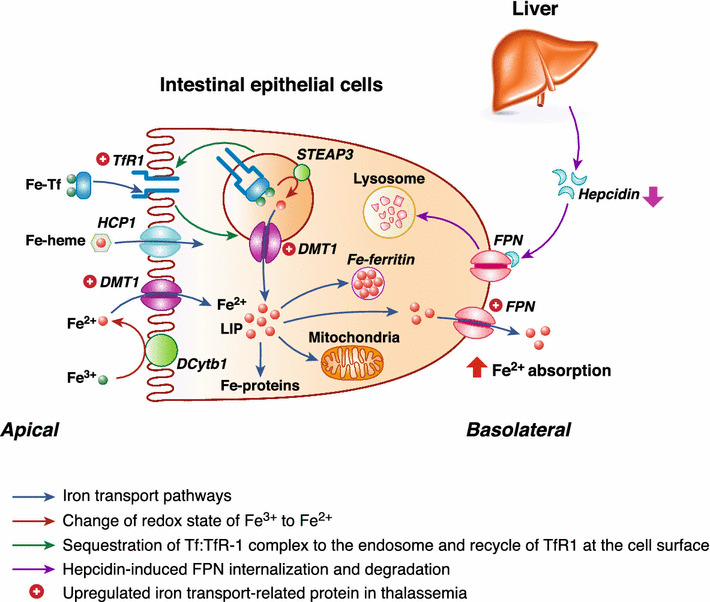
Cellular mechanisms of iron transport in thalassemia. Under normal conditions, dietary iron in the intestinal lumen can traverse the apical membrane by several pathways, such as transferrin receptor 1 (TfR1)-mediated uptake of iron-bound transferrin (Tf), divalent metal transporter 1 (DMT1)-mediated uptake of Fe2+, and Fe-heme uptake by heme carrier protein (HCP)-1. Although Fe3+ (non-heme iron) is more abundant than Fe2+, DMT1 predominantly transports Fe2+ in the presence of H+ in the lumen; therefore, it needs DCytb1 to change the iron redox state. Meanwhile, after endocytosis, iron ions are liberated from Fe-Tf-TfR1 by a metalloreductase, six-transmembrane epithelial antigen of the prostate (STEAP)-3, before being transported into the cytoplasm via DMT1 to join the cellular labile iron pool (LIP). Iron in LIP can be distributed into mitochondria and ferritin, or extruded across the basolateral membrane by ferroportin-1 (FPN). FPN is also a receptor for hepcidin, which can induce FPN internalization and degradation. In thalassemia, the upregulated expressions of DMT1 and TfR1 enhance apical iron uptake into the intestinal epithelial cells. In addition, a reduction in the hepcidin levels decreases FPN internalization and degradation, which can, in turn, increase the number of FPN proteins in the basolateral membrane, and then enhances the intestinal iron absorption