

Abstract
Enzymatically dissociated mouse FDB muscle fibers, loaded with Fura-2 AM, were used to study the effect of mitochondrial uncoupling on the capacitative Ca2+ entry, SOCE. Sarcoplasmic reticulum (SR) Ca2+ stores were depleted by repetitive exposures to high K+ or 4-chloro-m-Cresol (4-CmC) in the absence of extracellular Ca2+. SR Ca2+ store replenishment was substantially reduced using 5 μM cyclopiazonic acid (CPA). Readmission of external Ca2+ (5 mM) increased basal [Ca2+]i under two modalities. In mode 1 [Ca2+]i initially increased at a rate of 0.8 ± 0.1 nM/s and later at a rate of 12.3 ± 2.6 nM/s, reaching a final value of 477.8 ± 36.8 nM in 215.7 ± 25.9 s. In mode 2, [Ca2+]i increased at a rate of 0.8 ± 0.1 nM/s to a value of 204.9 ± 20.6 nM in 185.4 ± 21.1 s. FCCP, 2 μM, reduced this Ca2+ entry. In nine FCCP-poisoned fibers, the initial rate of Ca2+ increase was 0.34 ± 0.1 nM/s (mean ± SEM), reaching a plateau of 149.2 ± 14.1 nM in 217 ± 19 s. The results may likely be explained by the hypothesis that SOCE is inhibited by mitochondrial uncouplers, pointing to a possible mitochondrial role in its activation. Using time-scan confocal microscopy and the dyes CaOr-5N AM or Rhod-2 AM to label mitochondrial Ca2+, we show that during depletion [Ca2+]mito initially increases and later diminishes. Finally, we show that the increase in basal [Ca2+]i, associated with SOCE activation, diminishes upon external Na+ withdrawal. Na+ entry through the SOCE pathway and activation of the reversal of Na+/Ca2+ exchanger could explain this SOCE modulation by Na+.
Keywords: Skeletal muscle, Mitochondria, SOCE, Ca2+ imaging, Na+/Ca2+ exchange
Introduction
In many cells Ca2+ entry occurs through a pathway whose activation is dependent on the Ca2+ content of intracellular stores. This Ca2+ influx, known as capacitative Ca2+ entry or store operated Ca2+ entry (SOCE), was first described and well characterized in non-excitable cells, where it constitutes a major pathway for Ca2+ influx [1, 2]. The electrical manifestation of this Ca2+ entry in the continuous presence of extracellular Ca2+ is represented by a Ca2+ current, Icrac, that flows through channels activated by substantial depletion of endoplasmic reticulum (ER) Ca2+ stores, normally operated by IP3 receptors [2–6].
This mode of Ca2+ entry has also been described in excitable cells, as is the case for adult skeletal muscle fibers, where the sarcoplasmic reticulum (SR) calcium stores are operated by ryanodine receptors. The presence of SOCE in adult skeletal muscle fibers has been reported by different authors using different techniques [7–12]. However, attempts to measure SOCE single channel currents during SR Ca2+ release and/or SR Ca2+ depletion of muscle fibers under cell attached patch conditions have produced conflicting results [10, 13]. The reason for this could lie in the small magnitude of the currents with Ca2+ as the charge carrier and their preferential localization at level of the t-tubules membrane [11]. The physiological role of SOCE in skeletal muscle is not well understood, although it has been suggested that it could be relevant for replenishing exhausted SR Ca2+ stores during prolonged contractile activity. Furthermore, there is much less information on the regulatory mechanisms involved in SOCE activation in excitable cells, in particular adult skeletal muscle fibers, than in non-excitable cells. The main aim of this work was to study the effect of mitochondrial poisoning with FCCP on SOCE in mouse skeletal muscle fibers. We have measured SOCE in terms of cytoplasmic Ca2+ increases in the presence of CPA, a potent inhibitor of the Ca2+ SR pump. We show, for the first time that, in adult mouse skeletal muscle fibers, mitochondria poisoning by FCCP reduces the cytoplasmic Ca2+ increase likely to be associated with SOCE. Furthermore, we present some evidence that mitochondria may take up Ca2+ during the early stages of the store depletion procedure, but release it when depletion is completed, thus suggesting the possibility of cross-talking between SR and mitochondria. Finally, we have tested the possibility that also in mice muscle fibers, as occurs in human platelets [14], Na+ entry might occur simultaneously with SOCE, promoting additional Ca2+ entry through the operation of the Na+/Ca2+ exchanger in its reverse mode.
Methods
Fiber preparation
The enzymatic dissociation method was similar to the one previously published [15, 16]. Briefly, flexor digitorum brevis (FDB) muscles were dissected from adult (42 days) mice (NMR IVIC). The mice were killed by rapid cervical dislocation. Procedures were approved by the local animal care committee. Muscles were incubated in a modified mammalian Ringer solution (see below) containing 1 mM Ca2+ and 4 mg/ml collagenase (Worthington CLS2) for 1 h at 36°C. After incubation with collagenase, the muscles were washed three times with the 1 mM Ca2+ Ringer's solution and gently separated from tendons and remaining tissue with a fire-polished Pasteur pipette. All the experiments were carried out at room temperature (20–22°C).
Experimental solutions and chemicals
The composition of the basic experimental solution (mammalian normal Ringer's solution, NR) was as follows (in mM): 145 NaCl, 2.5 KCl, 1.0 MgSO4, 2.5 CaCl2, 10 d-glucose, 10 HEPES, pH 7.4. The 5 Ca2+ solution was the same as NR but with 5 mM CaCl2. Ca2+-free (0Ca) solution had the following composition (in mM): 145 NaCl, 2.8 KCl, 2 MgSO4, 10 d-glucose, 10 HEPES, 0.5 EGTA, pH 7.4, 290 mOsm. The high K+–0Ca2+ solution had the following composition (in mM): 75 K2SO4, 5 NaCl, 2 MgSO4, 10 HEPES, 10 glucose, pH 7.4.
Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), cyclopiazonic acid (CPA), N-benzyl-ptoluene sulphonamide (BTS) (Sigma St Louis, MO), 4-chloro-m-Cresol (4-CmC) (Fluka Chemical Corp, RonKonKoma, NY), 2-aminoethoxydiphenyl borate (2-APB)(Calbiochem, La Jolla, CA) and KB-R7943 (EMD Biosciences, Inc. San Diego, CA) were added from stock solutions to the concentrations indicated. All fluorescent dyes, Fura-2 AM, Rhod-2 AM, Calcium Orange-5N AM, MitoTracker® Green FM and SBFI AM (benzofuran isophthalate), were from Molecular Probes (Eugene, OR). They were dissolved in DMSO at a final concentration of 0.1%.
Depletion protocol
The fiber viability was verified by electrical stimulation followed by exposures to high K+ solution prior to depleting the SR Ca2+ stores of fibers poisoned with cyclopiazonic acid. This procedure consisted of a series of exposures either individually or in combination with high K+ for 1–3 s, or with the ryanodine receptor agonist 4-CmC (500 μM) for 10–40 s, in the absence of external Ca2+. The criterion for depletion was based on the drastic reduction of the Ca2+ transient responses to the high K+ or the 4-CmC challenges as illustrated in Fig. 1. In 29 different fibers, the number of exposures to the high K+ and/or 4-CmC was 9.8 ± 0.5, (mean ± SEM, n = 29).
Fig. 1.
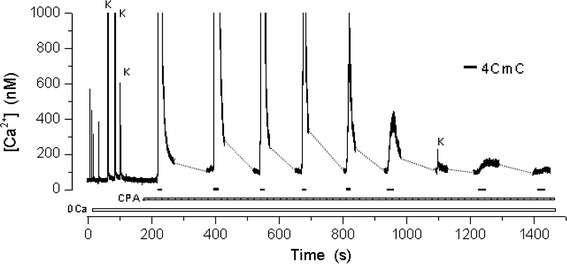
Depletion procedure. One fiber, bathed in a 0 Ca2+ medium, was electrically stimulated four times and challenged three times with the high K+ solution to verify its viability before treating it with CPA. In the presence of this agent, the fiber was exposed several times to 4-CmC until the responses to high K+ and to 4-CmC were practically abolished. In this and following figures, the dashed lines represent acquisition interruptions. A similar protocol, not entirely shown, preceded the traces of the experiments of Figs. 2, 3, 4, 5 and 8
Movement artifacts
As previously shown [16], Ca2+ transients can be recorded with few movement artifacts by selecting fibers that spontaneously adhered to the glass cover slide bottom of the experimental chamber. To further reduce movement artifacts, all experimental solutions contained 40 μM BTS to diminish contraction. This was particularly important in confocal microscopy experiments, since we intended to carry out measurements of changes in mitochondrial Ca2+-dependent fluorescence in the same fiber region during the depletion procedure. In this case, we also coated the glass cover slides with 1 mg/ml laminin (Sigma) to improve attachment of the fibers and allowed them to rest between 2 and 3 h before starting the experiment.
Fluorescence recording
Fura-2 basal Ca2+ measurements
Dissociated fibers were loaded with the Ca2+-sensitive fluorescent indicator Fura-2 AM 5 μM (Molecular Probes) for 30 min at room temperature in the dark and then washed with standard solution. Once placed in the experimental chamber, the loaded cells were allowed to rest an additional 15 min to allow further de-esterification.
Fura-2 fluorescence was measured with a fluorescence imaging apparatus (Ionoptix Co., Milton, MA) mounted on an inverted Nikon Diaphot TMD microscope. For fura-2 measurements the light from a 100-W xenon lamp was filtered alternating 340/20- and 380/18-nm interference filters (Chroma Technology Corp., Rockingham, VT). The resultant fluorescence was passed through a 400-nm dichroic mirror, filtered at 510/40 nm (Chroma Technology Corp., Rockingham, VT) and collected using an intensified CCD camera. Fluorescence images were taken at a rate of 33 ms/frame, digitalized and analyzed using the IonOptix software. The Ca2+ concentration was calculated according to the formula [17]:
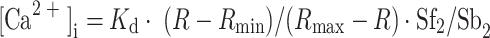 |
where R is the measured fluorescence ratio. The values of R max and R min and the constant Sf2/Sb2 (fluorescence of free and Ca2+-bound fura-2 at 380 nm) were calculated in vitro using variable CaEGTA/EGTA ratios to give different [Ca2+] (Kit no. 1, Molecular Probes). The dissociation constant K d, for the fura-Ca2+ complex, was taken as 89 nM, which is a value obtained from the on and off rate constants for calcium binding to fura-2 in muscle fibers [18].
Since with our configuration, the equipment allowed the acquisition of images at video rate, the Ca2+ transients elicited by electrical stimulation were heavily filtered and are only shown as proof of fiber excitability.
SBFI sodium measurements
The dissociated muscle fibers were loaded with the membrane-permeable acetoxy methyl ester of the fluorescent sodium-binding benzofuran isophthalate, SBFI-AM (Molecular Probes). Stock solution was prepared by dissolving SBFI-AM in dimethyl sulphoxide (DMSO) to a concentration of 2 mM. The loading solution was made by adding 2 μl of 25% wt/wt Pluronic F-127 (Molecular Probes) in DMSO to 3 μl of SBFI-AM stock solution, sonicated and then diluted in 1 ml of NR. The final concentration of SBFI-AM in the solution was 6 μM [19]. The dye was excited at 340/20 and 383/18 nm wavelengths, and emitted light was recorded at 510/40 nm. Only fibers that responded to electrical stimulation were selected.
Laser scanning confocal microscopy
Cells loaded with Calcium Orange-5N AM (CaOr-5N) or with Rhod-2 AM fluorescent dyes were plated on cover slides coated with laminine as described above. The cover slide was placed as the bottom of the superfusion open chamber mounted on the stage of an inverted Nikon Eclipse TE2000 microscope equipped with a Plan Fluor 100× 1.3 NA oil-immersion objective and coupled to a laser scanning confocal system Nikon C1, with Neon (543 nm) and air-cooled Argon (488) lasers, 515/530 (BP) and 570 (LP) emission filters.
Mitochondria were observed in cells loaded with 200 nM MitoTracker Green FM, a cell permeant mitochondrion selective dye (490 nm excitation/516 nm emission) that passively diffuses across the plasma membrane and accumulates in the lipid environment of mitochondria (Molecular Probes) [20].
Time-scan confocal microscopy was used in individual cells to follow mitochondrial calcium ([Ca2+]mito) changes during the depletion procedure. To follow [Ca2+]mito changes, dissociated fibers were loaded with 5 μM CaOr-5N AM, K d 87 μM [21] for 60 min at 37°C or 7 μM Rhod-2 AM, K d 570 nM (Molecular Probes) for 120 min at 37°C in mammalian Ringer's solution, washed and then incubated for an additional 10 min at 37°C. Data were acquired with the Nikon EZ-C1 software and processed with the Nikon Viewer 1.00. Mitochondrial fluorescence intensity was obtained using the particle analysis facility of the ImageJ 1.41 g program (Free Software, National Institutes of Health). This program allows recognition and measurement of the area of the selected objects for analysis, giving the brightness value for each pixel. As shown in Fig. 7b, basically all the mitochondria present in the confocal image could be selected for analysis.
Fig. 7.
Variations in [Ca2+]mit during SR Ca2+ depletion. a A confocal image of a fiber loaded with Rhod-2; b A “mask” of the image in a, obtained using the Particle Analysis module of the ImageJ program, as explained in the Methods section. c The time course of the mean Rhod-2 fluorescence variation of the mitochondria during the depletion procedure. The points represent the mitochondrial fluorescence expressed as arbitrary units (mean ± SEM, n = 354). Calibration bar 10 μm
Results
Ca2+-dependent [Ca2+]myo increase in SR Ca2+ depleted fibers
Figure 1 shows that following depletion, the basal myoplasmic Ca2+ concentration [Ca2+]myo was somewhat higher than before depletion. In all figures, the dashed lines represent breaks in acquisition in order to diminish illumination periods and prevent fiber photo-damage. Table 1 shows that after depleting the SR Ca2+ stores, [Ca2+]myo increased from an initial value of 61 ± 2 to 86 ± 3 nM (mean ± SEM, n = 29).
Table 1.
Effect of FCCP on SOCE
| [Ca2+]bsl | [Ca2+]myo increase | |||||
|---|---|---|---|---|---|---|
| Pre-deplet (nM) | Post-deplet (nM) | Initial rate (nM/s) | Final rate (nM/s) | Final level (nM) | ||
| Modes 1 and 2 | Mode 1 | Mode 1 | Mode 2 | |||
| Control | 61.1* ± 2.4 (29)* | 85.9 ± 2.7 (29) | 0.8 ± 0.1 (29) | 12.3 ± 2.6 (14) | 477.8 ± 36.8 (14) | 204.9 ± 20.6 (15) |
| FCCP | 67.1 ± 5.1 (11) | 85.5 ± 4.1 (11) | 0.3 ± 0.1 (11) | – | – | 149.2 ± 14.0 (11) |
* Mean ± SEM (N)
Following depletion of the SR Ca2+ stores in the absence of external Ca2+, exposure of the fibers to a solution containing 5 mM Ca2+ caused an increase in the [Ca2+]myo. This increase occurred mainly under two different modalities as illustrated in Fig. 2. In this and the following figures, the experimental record started once depletion was completed. Figure 2a shows the result of a run in which, following exposure to 5 mM Ca2+, the [Ca2+]myo started to rise at first slowly and then increasingly faster from an initial level of about 80 nM to a value of about 500 nM in 280 s. At this point the fiber was exposed to the normal Ringer's solution (NR) containing 2 mM Ca2+ and no CPA, which caused a relatively fast decay of the [Ca2+]myo to a quasi-basal condition. The interruption of the [Ca2+]myo increase in response to 5 mM Ca2+ was necessary to avoid reaching higher [Ca2+]myo that would have resulted in fiber contraction and the end of the experiment. We define this type of response as mode 1. Figure 2b shows the result of another experiment in which following exposure to the 5 mM external Ca2+ solution, the [Ca2+]myo increased slowly from an initial level of about 75 nM to a plateau level of about 170 nM in 156 s, which was reversed upon exposure of the fiber to NR. This is defined as a mode 2 response. In both cases, the fibers fully reacquired their capacity to respond to high K+ solution after a recovery period of about 180 s. Table 1 shows that in 29 fibers tested, 14 showed a mode 1 response, while 15 responded in mode 2. The table also shows that for the fibers responding in mode 1, the initial and final rates of [Ca2+]myo increase were respectively 0.8 ± 0.1 and 12.3 ± 3 nM/s, while the [Ca2+]myo attained before the response interruption was 477.8 ± 36.8 nM. The time to reach this value was 215.7 ± 25.9 s. In the case of fibers responding in mode 2, the initial rate of [Ca2+]myo increase was 0.8 nM/s, while the plateau [Ca2+]myo value was 204.9 ± 20.6 nM, attaining this value in 185.4 ± 21.1 s.
Fig. 2.
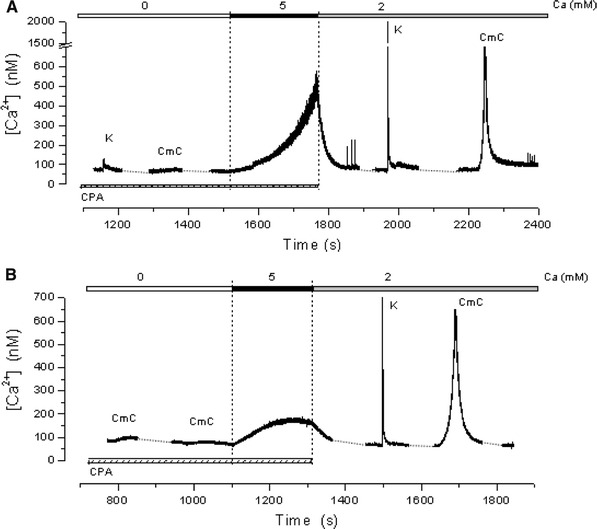
Two modalities of SOCE activation. Following depletion of the SR Ca2+ stores, achieved in the absence of external Ca2+, exposing the fibers to a solution containing 5 mM Ca2+, causes an increase in the [Ca2+]myo that basically follows two different modalities. Trace in a shows an experiment in which [Ca2+]myo, after a slow start, increases rapidly to a value higher than 500 nM, rendering it necessary to reverse it to avoid fiber contracture and damage, mode 1. Trace in b shows that in another fiber, the [Ca2+]myo increase is slower and saturates at a lower value, mode 2. The characteristics parameters of the two modes are resumed in Table 1. Notice the recovery of the responses to K+ and 4-CmC at the end of the runs
Figure 3 shows three runs carried out with different fibers in which CPA was washed out at varying times during the experiment. Figure 3a shows that after completing the depletion procedure, the fiber was exposed to 5 mM Ca2+, which caused the basal [Ca2+]myo to increase from 110 to 231 nM. CPA was washed out 176 s after exposure to 5 mM Ca2+ and, after a delay of about 10 s, [Ca2+]myo began to decrease, reaching the baseline level in about 110 s. In Fig. 3b, CPA was withdrawn simultaneously with the exposure to 5 mM Ca2+, resulting in a small and transient increase of [Ca2+]myo from 103 to 138 nM followed by a spontaneous decay to the initial value. This decay could be due to Ca2+ uptake by the SR, and soon after washing out CPA competes with SOCE. This would explain the recovery of the K+ contracture elicited after switching to the 0 Ca2+ condition. In Fig. 3c, CPA was washed out 74 s before exposure to the 5 mM Ca2+ solution; in this case the [Ca2+]myo increase was almost abolished. In addition to showing the reversibility of the CPA effect, these results demonstrate that in the absence of CPA, the SR Ca2+ pump is an effective buffer that successfully competes with the mechanism responsible for the described Ca2+ increase.
Fig. 3.
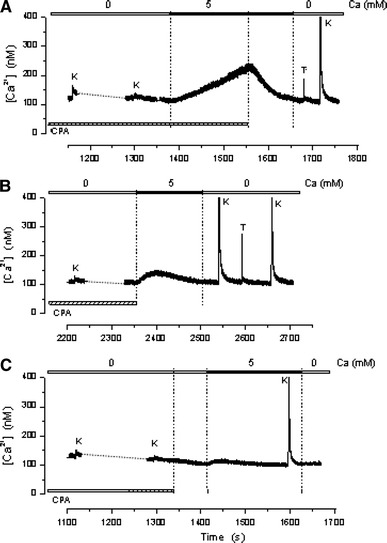
Reversibility of the CPA effect on the SR Ca2+ pump. The three runs in the figure, carried out with different fibers, show that following the depletion procedure, washing out CPA at different times: a after, b simultaneously or c before activating SOCE with 5 mM Ca2+, rapidly reverses or reduces the [Ca2+]myo. The results demonstrate that the unpoisoned SR Ca2+ pump effectively competes with SOCE
Mechanism of [Ca2+]myo increase
In order to identify the mechanism responsible for the [Ca2+]myo increase following depletion of intracellular Ca2+ stores, we used 2-APB at high concentration and Gd3+, known inhibitors of SOCE.
Figure 4 shows the effect of 2-APB in two different fibers. Trace A shows that exposure to 80 μM 2-APB, partially reversed the [Ca2+]myo increase induced by 2 mM Ca2+ in a depleted fiber. Trace B shows that after completing the depletion procedure, simultaneous exposure of the fiber to 5 mM Ca2+ and 2-APB failed to cause the [Ca2+]myo increase. A small delayed increase could be observed after washing out the drug.
Fig. 4.
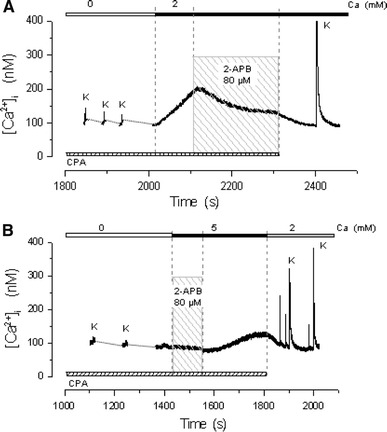
SOCE blocking by 2-APB. a Reversion of the [Ca2+]myo increase by exposure to 80 μM 2-APB. b Inhibition of the [Ca2+]myo increase by simultaneous exposure of the fiber to 5 mM Ca2+ and 2-APB. A small delayed increase could be observed after washing out the drug
Similar results were obtained with 10 μM Gd3+ that effectively reverted the [Ca2+]myo increase induced by 5 mM Ca2+ in a depleted fiber. In other experiments, addition of Gd3+ completely prevented the [Ca2+]myo increase when it was applied before the addition of 5 mM Ca2+. We also used Mn2+, which was effective in reducing fura-2 fluorescence, as indicated by the reversal of the 340/380 ratio increase after stimulation with 5 mM Ca2+ (experiments not shown). Mn2+ is used to reveal ICRAC in non-excitable cells where there are no other Ca2+ entry pathways [22]. The observed [Ca2+]myo increase was not affected by 10 μM nifedipine or 10 μM D-600, well known blockers of the L-type Ca2+ channels. The above experiments suggest that the [Ca2+]myo increase observed in depleted fibers exposed to 2.5 or 5 mM Ca2+ is mediated by Ca2+ entry through SOCE.
Effects of FCCP on [Ca2+]myo increase mediated by SOCE
Figure 5a shows an experiment in which treatment of a fiber with 2 μM FCCP for 2 min, after Ca2+ store depletion under 0Ca2+ conditions, caused a small increase in [Ca2+]myo. Exposure to 5 mM external Ca2+ caused a further increase, which was however considerably smaller than those obtained in the absence of FCCP (see Fig. 2b). The last portion of the record shows that after a recovery period, the fiber responded to high K+. Table 1 summarizes the results obtained in several experiments of this type. The results indicate that mitochondrial poisoning effectively diminishes SOCE activation. Experiments were carried out to test whether FCCP could affect SOCE after it had been activated. Figure 5b shows an experiment in which, after completing the depletion procedure, the fiber was exposed first to 5 mM Ca2+ and after 70 s to 2 μM FCCP. Exposure to Ca2+ caused an increase in [Ca2+]myo, which reached 300 nM in about 170 s. Exposure to FCCP not only blocked, with a 100 s delay, the increase in [Ca2+]myo, but also caused a small decrease in its value. The [Ca2+]myo decay was accelerated by washing out CPA, without, however, reaching baseline levels, as has previously been described in the experiments carried out in the absence of FCCP (Fig. 3). Similar results were obtained in three other fibers. The results show that FCCP is still capable of affecting SOCE, even after its activation. Interestingly, it also suggests that FCCP treatment may affect the fiber Ca2+ clearance capacity after CPA is removed.
Fig. 5.
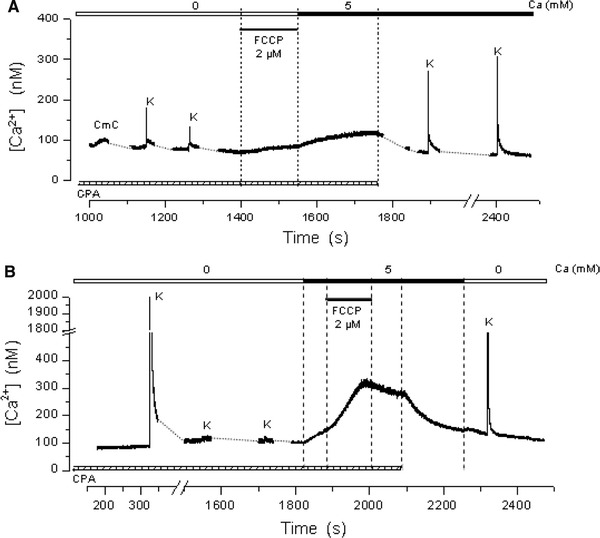
Inhibition of SOCE by mitochondrial uncoupling. a Treatment with FCCP 2 μM effectively reduces SOCE activation. FCCP by itself causes a small increase in the [Ca2+]myo. b FCCP affects SOCE even after its activation. The experiment shows that the increase in [Ca2+]myo is halted and even reversed by FCCP applied after SOCE activation
Mitochondrial Ca2+ following depletion of Ca2+ stores
An important observation resulting from the experiment in Fig. 5a is that after depletion of the SR Ca2+ stores, exposure of the fibers to FCCP causes a much smaller increase in the basal [Ca2+]myo than that recently described in rested, non-depleted fibers [23]. The experiment of Fig. 6a shows the effect of FCCP on the basal [Ca2+]myo in a fiber not subjected to the depletion procedure. The experiment shows that myoplasmic Ca2+ starts to increase 45 s after exposure to 2 μM FCCP and reaches a level of 234 nM from the 108 nM initial value, whereas, in non-depleted fibers, the mean increase in [Ca2+]myo was 233 ± 14.9 nM, n = 10. Figure 6b compares the reduction of Ca2+ responses to high K+, 4-CmC and FCCP observed before and after store depletion. A comparison of the FCCP effect is made between the mean values from two sets of experiments: the value before depletion, taken from a previous work [23], and the value after depletion, taken from this work. In the case of the high K+ and 4-CmC responses, the values obtained under normal conditions represent a low estimate since in many cases the response peak exceeded the saturation level of our acquisition system. Figure 6b shows that after depletion, the responses to high K+, 4-CmC and FCCP were reduced by 98.6, 99.4 and 93.5%, respectively. It should be noted that the absolute value of the residual response is rather similar in the three cases: 26.2, 13.1 and 15.2 nM, respectively. This could indicate that following the SR Ca2+ depletion procedure, the content of the mitochondria is also reduced. This possibility was explored by measuring the fluorescence of the Ca2+-sensitive dyes CaOr-5N and Rhod-2, before, during and after the depletion procedure. Control experiments (not shown) confirmed that these dyes are highly co-localized with MitoTracker Green, which selectively stains mitochondria and therefore selectively labels mitochondrial Ca2+ [20]. The image in Fig. 7a, obtained with a Nikon C1 Confocal Laser Scanning Microscope, shows that most of the Rhod-2 fluorescence originated at the mitochondria level; it is also present in the myoplasm, albeit in a diffuse way. The image in Fig. 7b represents the mask created by the ImageJ 1.41 g Particle Analysis program and shows the mitochondria selected for analysis. Images from the same fiber were acquired at different times during the depletion procedure. The graph in Fig. 7c shows the change of mitochondrial fluorescence value occurring during the depletion procedure. The experimental points in the graph represent the mean (±SEM) value of mitochondria fluorescence, expressed in arbitrary units. The results indicate an increase in mitochondrial Ca2+ during the early stages of Ca2+ store depletion that later diminishes; equal results were obtained in two additional experiments carried out with CaOr-5N.
Fig. 6.
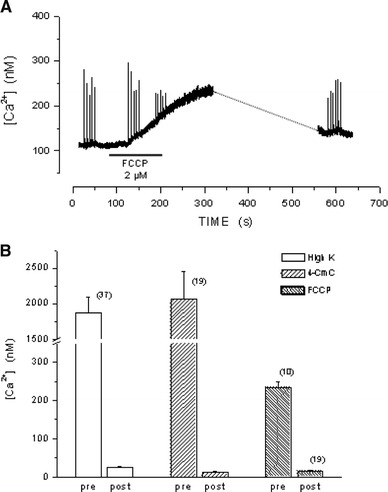
a Effect of FCCP 2 μM on the basal [Ca2+]myo in a non-depleted fiber. b Reduction of the [Ca2+]myo increases in response to high K+, 4-CmC and FCCP after store depletion (column labeled post)
A role for the reverse Na+/Ca2+ exchanger during SOCE activation
In view of recent results obtained with human platelets [14], we thought it of interest to explore whether the Na+/Ca2+ exchanger, operating in its reverse mode (Na+ out/Ca2+ in) could have a role in the [Ca2+]myo increase due to SOCE activation in mammalian muscle. Figure 8a shows an experiment in which, following the depletion procedure, SOCE was activated by exposure to 2.5 mM Ca2+, reaching a [Ca2+]myo 250 nM in about 175 s. This increase was interrupted by exposure to 20 μM KB-R7943, a potent but not very specific inhibitor of the reversal mode of Na+/Ca2+ exchanger, causing a partial reverse of the [Ca2+]myo increase. The removal of the inhibitor caused resumption of the [Ca2+]myo increase, reaching a value of approximately 370 nM, at which time the fiber was exposed to a solution in which Na+ was substituted by Li+, causing a rapid decrease in the myoplasmic Ca2+ concentration. The effect was repeated by again exposing the fiber to a Na+–0Na+ cycle. It is interesting to note that, when both external Na+ and Ca2+ were washed out, the decay to the initial baseline was not complete and only occurred when the SR Ca2+ pump inhibitor CPA was also removed. This protocol was repeated with two other fibers. The effect of Na+ withdrawal (substituted by Li+ or Tris+) on the [Ca2+]myo levels was consistently observed in several fibers as shown in Table 2. This result indicates that the myoplasmic Ca2+ increase, associated with SOCE activation, depends at least in part on the presence of external Na+. The possibility that muscle fibers could be loaded with Na+ during SOCE activation was tested. The compound SBFI was used to report intracellular Na+ increase in one fiber subjected to the same protocol of the experiment in Fig. 8a. In the run shown in Fig. 8b, the changes in myoplasmic Na+ levels are reported in terms of the 340/380 fluorescence ratio for this dye. It can be observed that following the depletion procedure, when the fiber is exposed to external Ca2+, the internal Na+ level increases. Exposure of the fiber to the KB-R7943 inhibitor discontinues this increase and perhaps begins its reversal, while external Na+ withdrawal completely reverses it. Re-adding Na+ in the external medium causes a new increase in myoplasmic Na+.
Fig. 8.
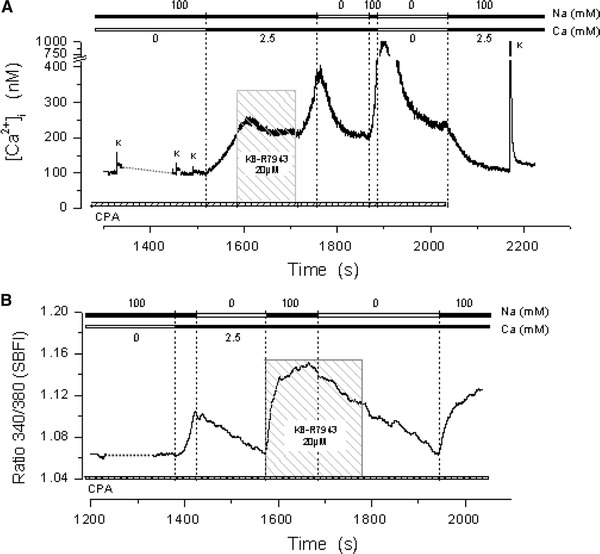
SOCE dependence on extracellular Na+. a The activation-inhibition cycle on Ca2+ entry by SOCE when external Na+ is substituted by Tris or exposed to the Na+/Ca2+ exchange inhibitor KB-R7943. b Changes in cytoplasmic Na+ in a similar experiment carried out in a different fiber loaded with Na+ indicator SBFI. The figure demonstrates that following depletion, SOCE activation causes simultaneous increases in cytoplasmic Ca2+ and Na+
Table 2.
External sodium effect on SOCE
| Δ[Ca2+] (nM) | Δ[Ca2+]/[Ca2+]max | N | |
|---|---|---|---|
| 0Na (Li) | −33.8 ± 21.4 | 0.5 ± 0.10 | 5 |
| 0Na (Tris) | −76.6 ± 6.4 | 0.6 ± 0.04 | 16 |
| 25Na (Tris) | −62.3 ± 14.2 | 0.6 ± 0.07 | 4 |
| 50Na (Tris) | −45.0 ± 11.4 | 0.4 ± 0.06 | 5 |
Mean ± SEM, [Ca2+]max value before the withdrawal of external Na+
Discussion
SOCE in skeletal muscle fibers
In non-excitable cells, SOCE constitutes a major pathway for Ca2+ entry that by interaction with different intracellular Ca2+ stores, such as the endoplasmic reticulum and mitochondria, may modulate Ca2+ signals [6, 24–27]. SOCE has also been demonstrated in adult skeletal muscle cells [7, 8, 10–12]. In muscle fibers Ca2+ transients elicited by electrical stimulation are the most stereotyped signals; the sarcoplasmic reticulum has a protagonist role in the release of the Ca2+ necessary for contractile activation and in its sequestration during relaxation. In this scenario, the role of SOCE has yet to be well established. It has recently been proposed that this mechanism could in principle provide a source of Ca2+ to replenish intracellular Ca2+ stores and avoid their depletion during repetitive activity [11].
In this work SOCE was studied by following the changes in myoplasmic Ca2+ concentration by measuring fura-2 fluorescence in mouse muscle fibers whose SR Ca2+ stores had been severely depleted and its replenishment blocked by the use of CPA, a potent and reversible inhibitor of the SR Ca2+ ATPase. Under these conditions, the maximum rate of [Ca2+]myo increase was 12 nM s−1 when extracellular Ca2+ was 5 mM. This value is much smaller than that of the directly measured Ca2+ flux into the fiber, 16 μM s−1, which was recently determined [11]. Apart from the different techniques and preparations, an additional interesting possibility for this difference could be the presence of undetermined Ca2+ sinks in intact fibers affecting our measurements.
Effect of FCCP
An important result of this work is the demonstration that, in adult mouse skeletal muscle fibers, the mitochondrial uncoupler FCCP diminishes SOCE significantly.
The importance of mitochondria in the regulation of SOCE has been demonstrated by the work of various authors [6, 24–27]. In these preparations, mitochondrial poisoning causes inactivation or reduction of Icrac [6, 26, 28]. The work of Parekh and colleagues has shown that Ca2+ uptake by fully energized mitochondria is required for Icrac activation by InsP3, under physiological conditions. Mitochondrial poisoning has two simultaneous effects mediated by the Ca2+ buffering action: (1) removal of mitochondrial competition with SERCA pumps that facilitates store replenishment, stopping SOCE [29]; (2) reduction of Ca2+ uptake by mitochondria increases cytoplasmic Ca2+, thus favoring Ca2+-dependent inactivation of the SOC channels [24]. In our work, the effect of mitochondrial poisoning by FCCP cannot be explained in a similar way because of the widely different experimental conditions. Under these conditions, mitochondrial poisoning may have caused the loss of some signal necessary for SOCE activation or maintenance, in agreement with the proposal [26] that mitochondria may also contribute to SOCE activation by a Ca2+ buffering independent mechanism, involving some hitherto unidentified factor produced by functional mitochondria.
The complex mechanisms involved in SOCE activation render it difficult to speculate on the mode of action of unidentified compounds that might interfere with it. However, it has been recently shown that in mice skeletal muscle, a STIM1-specific antibody highly co-localizes with a RyR1 antibody, indicating the presence of STIM1 molecules in the junctional region of the SR where also mitochondria are closely localized [30]. This proximity should favor an active interaction between mitochondria and the STIM1 and ORAI components of the Icrac.
We also report that in some fibers the responses to high K+ or to 4-CmC at the end of the experiment were larger than those obtained at its beginning, indicating that in resting state, the SR is not completely filled, in agreement with a previous report showing that the SR of fast twitch muscle is only 1/3 full [31]. This observation, however, is apparently not related to the fact that, under our experimental conditions, there were two modalities of SOCE activation.
Mitochondrial Ca2+ during the depletion protocol
Another observation pertains to the increase in myoplasmic Ca2+ observed in response to exposure of depleted fibers to FCCP. This increase is smaller than that previously reported [23] for the case of non-depleted fibers, and its magnitude is similar to that of the Ca2+ responses to high K+ or 4-CmC obtained in depleted fibers (see Fig. 6). This could mean either that the [Ca2+]myo increase caused by FCCP originates from the SR and not from the mitochondria, or that the mitochondria content also diminishes following the depletion procedure. Although, the first possibility cannot be totally ruled out, we have presented evidence in favor of the second possibility.
In fact, the confocal experiments suggest that during the first phase of store depletion, the Ca2+ content of the mitochondria measured with CaOr-5N or with Rhod-2 increases, but in the final phases of depletion the mitochondrial Ca2+ concentration diminishes. A possible explanation for the biphasic change in the Ca2+ content of the mitochondrial compartment during acute SR Ca2+ depletion is that during the first phase (increased Ca2+ content), Ca2+ released from the SR by high K+ or 4-CmC is partially taken into the mitochondria through the mitochondrial calcium uniporter (MCU). The decreased mitochondrial content that occurs when the SR is partially depleted could indicate that it may be determined by the SR Ca2+ content, as previously suggested from results obtained under different experimental conditions [32, 33]. In striated muscle, a coupling between SR and mitochondria would be facilitated by the physical proximity of the two structures. Recently, small bridges or tethers have been described in skeletal muscle, providing a rather strong link between the outer mitochondrial membrane and SR [34], possibly acting not only as anchors but also as sensors.
A possible role of the Na+/Ca2+ exchange
Finally, we also show that the increase in myoplasmic Ca2+ observed after SOCE activation can be partially reversed by external Na+ withdrawal. This effect could be explained assuming that in muscle fibers, Ca2+ entry following SOCE activation is accompanied by an entry of Na+. In human platelets, it has been shown that Na+ may enter the cell through the SOCE pathway [14]. However, in rat peritoneal mast cells [5] and in Jurkat T lymphocytes [35], Icrac has been shown to be impermeant to Na+ when external Ca2+ is present. Therefore, we may tentatively assume that Na+ enters muscle fibers through an unidentified pathway.
The increase in intracellular Na+, which in principle could reach very high levels in microdomains near the membrane, possibly increases Ca2+ entry by activation of the Na+/Ca2+ exchanger in its reverse mode. This could explain why Na+ withdrawal causes a decrease in the Ca2+ levels attained during SOCE. Preliminary experiments carried out with the Na+/Ca2+ exchanger inhibitor KB-R7943 and with the Na+-sensitive fluorescent dye SBFI appear to support this possibility. An increase in intracellular Na+ could also facilitate Ca2+ efflux from the mitochondria through the activation of the mitochondrial Na+/Ca2+ exchanger, as suggested above.
Apart from the FCCP effect on SOCE, for which we do not have a plausible explanation, the other major finding obtained in this work is summarized in the diagram in Fig. 9. Depletion of SR Ca2+ content by inhibitors of the SR Ca2+-ATPase, agonists of RYR and/or repetitive plasma membrane depolarization with high external potassium solutions (1) induces the opening of SOCE (2), thus allowing Ca2+ entry into the myoplasm, which in part is taken up by the mitochondrial compartment via the mitochondrial calcium uniporter, MCU (3), as clearly shown in our confocal results. Although the link between the state of the mitochondrial compartment, SOCE, SR Ca2+ content and release is unknown (4), our data clearly show an interrelationship among them. The increase of [Na+] in the myoplasm and the sodium dependency of the intracellular Ca2+ signal indicates that activation of the plasma membrane electrogenic Na+/Ca2+ exchange could contribute to the overall Ca2+ increase in the myoplasm when SOCE is activated [5]. Finally, Ca2+ release from the mitochondria through its electro neutral Na+/Ca2+ exchange might also contribute to the overall Ca2+ signal. Further work is in progress in our laboratory to characterize the relationship between these different mechanisms.
Fig. 9.
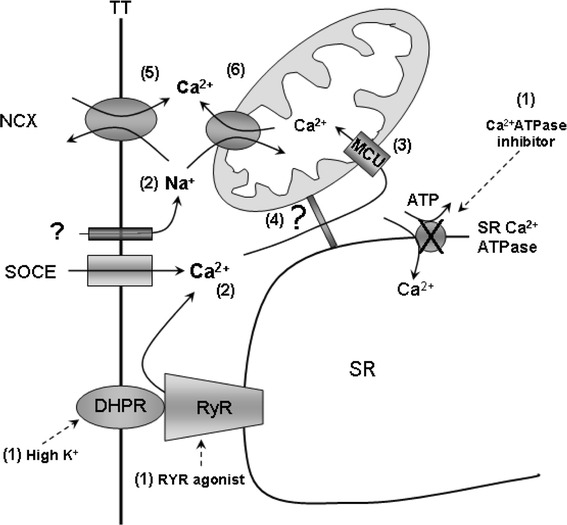
Schematic diagram of the interrelationships among acute Ca2+ depletion of the SR, opening of the SOCE, Ca2+ uptake and release from the mitochondrial compartment and Ca2+ entry through the reverse plasma membrane Na+/Ca2+ exchange. 1 SR Ca2+ depletion procedure. 2 Ca2+ entry through SOC and Na+ entry by an unidentified pathway. 3 Ca2+ uptake into the mitochondria through its uniporter following the electrochemical gradient. 4 An unknown step linking mitochondria, SOCE and SR-Ca2+ content. 5 Ca2+ entry through the electrogenic plasma membrane Na+/Ca2+ exchange and 6 Ca2+ release from the mitochondria through its electroneutral Na+/Ca2+ exchange as a consequence of the rise in [Na+]i (see text for explanation)
Acknowledgments
We acknowledge the kind gift of CaOr-5 N by Dr. Ariel Escobar. This work was supported by FONACIT grant G-2001000637.
References
- 1.Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 2.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 3.Parekh AB, Penner R. Activation of store-operated calcium influx at resting InsP3 levels by sensitization of the InsP3 receptor in rat basophilic leukaemia cells. J Physiol. 1995;489(Pt 2):377–382. doi: 10.1113/jphysiol.1995.sp021058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fierro L, Parekh AB. Substantial depletion of the intracellular Ca2+ stores is required for macroscopic activation of the Ca2+ release-activated Ca2+ current in rat basophilic leukaemia cells. J Physiol. 2000;522:247–257. doi: 10.1111/j.1469-7793.2000.t01-1-00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilabert JA, Parekh AB. Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca2+ current Icrac. EMBO J. 2000;19:6401–6407. doi: 10.1093/emboj/19.23.6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurebayashi N, Ogawa Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J Physiol. 2001;533:185–199. doi: 10.1111/j.1469-7793.2001.0185b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez Narvaez AA, Castillo A. Ca2+ store determines gating of store operated calcium entry in mammalian skeletal muscle. J Muscle Res Cell Motil. 2007;28:105–113. doi: 10.1007/s10974-007-9105-x. [DOI] [PubMed] [Google Scholar]
- 9.Ma J, Pan Z. Retrograde activation of store-operated calcium channel. Cell Calcium. 2003;33:375–384. doi: 10.1016/S0143-4160(03)00050-2. [DOI] [PubMed] [Google Scholar]
- 10.Ducret T, Vandebrouck C, Cao ML, Lebacq J, Gailly P. Functional role of store-operated and stretch-activated channels in murine adult skeletal muscle fibres. J Physiol. 2006;575:913–924. doi: 10.1113/jphysiol.2006.115154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Launikonis BS, Rios E. Store-operated Ca2+ entry during intracellular Ca2+ release in mammalian skeletal muscle. J Physiol. 2007;583:81–97. doi: 10.1113/jphysiol.2007.135046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma J, Pan Z. Junctional membrane structure and store operated calcium entry in muscle cells. Front Biosci. 2003;8:d242–d255. doi: 10.2741/977. [DOI] [PubMed] [Google Scholar]
- 13.Allard B, Couchoux H, Pouvreau S, Jacquemond V. Sarcoplasmic reticulum Ca2+ release and depletion fail to affect sarcolemmal ion channel activity in mouse skeletal muscle. J Physiol. 2006;575:68–81. doi: 10.1113/jphysiol.2006.112367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harper AG, Sage SO. A key role for reverse Na+/Ca2+ exchange influenced by the actin cytoskeleton in store-operated Ca2+ entry in human platelets: evidence against the de novo conformational coupling hypothesis. Cell Calcium. 2007;42:606–617. doi: 10.1016/j.ceca.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Carroll SL, Klein M, Schneider MF. Calcium transients in intact rat skeletal muscle fibers in agarose gel. Am J Physiol. 1995;269:C28–C34. doi: 10.1152/ajpcell.1995.269.1.C28. [DOI] [PubMed] [Google Scholar]
- 16.Capote J, Bolaños P, Schuhmeier RP, Melzer W, Caputo C. Calcium transients in developing mouse skeletal muscle fibres. J Physiol. 2005;564:451–464. doi: 10.1113/jphysiol.2004.081034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 18.Klein MG, Simon BJ, Szucs G, Schneider MF. Simultaneous recording of calcium transients in skeletal muscle using high- and low-affinity calcium indicators. Biophys J. 1988;53:971–988. doi: 10.1016/S0006-3495(88)83178-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeung EW, Ballard HJ, Bourreau JP, Allen DG. Intracellular sodium in mammalian muscle fibers after eccentric contractions. J Appl Physiol. 2003;94:2475–2482. doi: 10.1152/japplphysiol.01128.2002. [DOI] [PubMed] [Google Scholar]
- 20.Bolaños P, Guillen A, Rojas H, Boncompagni S, Caputo C. The use of Calcium Orange-5N as a specific marker of mitochondrial Ca2+ in mouse skeletal muscle fibers. Pflugers Arch. 2008;455:721–731. doi: 10.1007/s00424-007-0312-5. [DOI] [PubMed] [Google Scholar]
- 21.Zhao M, Hollingworth S, Baylor SM. Properties of tri- and tetracarboxylate Ca2+ indicators in frog skeletal muscle fibers. Biophys J. 1996;70:896–916. doi: 10.1016/S0006-3495(96)79633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fasolato C, Hoth M, Penner R. Multiple mechanisms of manganese-induced quenching of fura-2 fluorescence in rat mast cells. Pflugers Arch. 1993;423:225–231. doi: 10.1007/BF00374399. [DOI] [PubMed] [Google Scholar]
- 23.Caputo C, Bolaños P. Effect of mitochondria poisoning by FCCP on Ca2+ signaling in mouse skeletal muscle fibers. Pflugers Arch. 2008;455:733–743. doi: 10.1007/s00424-007-0317-0. [DOI] [PubMed] [Google Scholar]
- 24.Hoth M, Button DC, Lewis RS. Mitochondrial control of calcium-channel gating: a mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc Natl Acad Sci USA. 2000;97:10607–10612. doi: 10.1073/pnas.180143997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilabert JA, Bakowski D, Parekh AB. Energized mitochondria increase the dynamic range over which inositol 1, 4, 5-trisphosphate activates store-operated calcium influx. EMBO J. 2001;10:2672–2679. doi: 10.1093/emboj/20.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glitsch MD, Bakowski D, Parekh AB. Store-operated Ca2+ entry depends on mitochondrial Ca2+ uptake. EMBO J. 2002;21:6744–6754. doi: 10.1093/emboj/cdf675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parekh AB. Mitochondrial regulation of intracellular Ca2+ signaling: more than just simple Ca2+ buffers. News Physiol Sci. 2003;18:252–256. doi: 10.1152/nips.01458.2003. [DOI] [PubMed] [Google Scholar]
- 28.Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parekh AB. Store-operated Ca2+ entry: dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane. J Physiol. 2003;547:333–348. doi: 10.1113/jphysiol.2002.034140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stiber J, Hawkins A, Zhang ZS, Wang S, Burch J, Graham V, Ward CC, Seth M, Finch E, Malouf N, Williams RS, Eu JP, Rosenberg P. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10:688–697. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fryer MW, Stephenson DG. Total and sarcoplasmic reticulum calcium contents of skinned fibres from rat skeletal muscle. J Physiol. 1996;493(Pt 2):357–370. doi: 10.1113/jphysiol.1996.sp021388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudolf R, Mongillo M, Magalhaes PJ, Pozzan T. In vivo monitoring of Ca2+ uptake into mitochondria of mouse skeletal muscle during contraction. J Cell Biol. 2004;166:527–536. doi: 10.1083/jcb.200403102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vandebrouck A, Ducret T, Basset O, Sebille S, Raymond G, Ruegg U, Gailly P, Cognard C, Constantin B. Regulation of store-operated calcium entries and mitochondrial uptake by minidystrophin expression in cultured myotubes. Faseb J. 2006;20:136–138. doi: 10.1096/fj.04-3633fje. [DOI] [PubMed] [Google Scholar]
- 34.Boncompagni S, Rossi AE, Micaroni M, Beznoussenko GV, Polishchuk RS, Dirksen RT, Protasi F. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol Biol Cell. 2009;20:1058–1067. doi: 10.1091/mbc.E08-07-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lepple-Wienhues A, Cahalan MD. Conductance and permeation of monovalent cations through depletion-activated Ca2+ channels (ICRAC) in Jurkat T cells. Biophys J. 1996;71:787–794. doi: 10.1016/S0006-3495(96)79278-0. [DOI] [PMC free article] [PubMed] [Google Scholar]