

Abstract
Stimulation of the Na+–H+ exchanger plays an important role in the pathway of myocardial dysfunction and injury following hemorrhagic shock. Inhibition of the Na+–H+ exchanger appears to be a new pharmacological tool for myocardial protection. Despite the extensive research that has been done on the role of the Na+–H+ exchanger in ischemia reperfusion, little is known about the role of the exchanger following hemorrhagic shock. The purpose of this study was to examine the protective effects of blocking the cardiac Na+–H+ exchanger, using 20 μM dimethyl amiloride (DMA), a specific Na+–H+ exchanger blocker, on myocardial contractile function after ex vivo perfusion of isolated rat heart following 1 h of hemorrhagic shock. Sprague–Dawley rats were assigned to hemorrhage + DMA, hemorrhage, sham hemorrhage + DMA and sham hemorrhage groups (n = 6 per group). Hearts were perfused with a balanced salt solution for 60 min. In the DMA treated group, 20 μM DMA was added for the first 5 min of the 60-min ex vivo heart resuscitation. The results showed that inhibition of the Na+–H+ exchanger for 5 min on ex vivo perfusion of the isolated hearts following hemorrhagic shock using 20 μM DMA improved myocardial contractile function. Blocking the Na+–H+ exchanger on ex vivo perfusion of isolated hearts using 20 μM DMA has protective effects on myocardial contractile function.
Keywords: Hemorrhage, Rat, Isolated heart, Contractility, Dimethyl amiloride, Calcium, Sodium, Lactate, pH, Langendorff
Introduction
Hemorrhagic shock is well known to result in myocardial contractile dysfunction and heart failure [1]. The exact mechanism(s) is not clear. However, there are several mechanisms involved in the pathogenesis of myocardial contractile dysfunction following hemorrhagic shock, including energy depletion, intracellular acidosis and inflammatory mediators. One of the major pathways is via stimulation of the Na+–H+ exchanger. There is growing interest in developing safer and more effective resuscitation methods that would protect the heart from injury after hemorrhage and resuscitation.
Hemorrhagic shock results in inadequate perfusion of the tissues that will shift the body metabolism to the anaerobic pathway, which will eventually result in production of large amounts of lactic acid and extracellular acidosis [2]. It is well known that acidosis results in myocardial dysfunction [3, 4].
Extracellular acidosis will cause intracellular acidosis, which will stimulate the Na+–H+ exchanger [5, 6]. The Na+–H+ exchanger is the major transporter responsible for the regulation of myocardial pH [2]. The Na+–H+ exchanger is electro-neutral, exchanging one Na+ for one H+. This will result in an increase in intracellular Na+, which will stimulate another exchanger, the Na+–Ca2+ exchanger [7], which will result in an efflux of 3 Na+ in exchange with the influx of 2 Ca2+ with subsequent elevation of intracellular Ca2+. The increase in intracellular Ca2+ has an important role in the pathological changes that occur and result in myocardial contractile dysfunction [8]. For this reason, there is great interest in the mechanism of increase in the intracellular Ca2+ and the contribution of blocking the Na+–H+ exchanger in preventing this increase and protecting the myocardium following hemorrhagic shock and resuscitation.
Extensive research has been done to demonstrate the protective effects of blocking the Na+–H+ exchanger in case of ischemia-reperfusion using amiloride and its more specific analogues [9, 10, 11, 12]. This field has recently become a novel pharmacological target for myocardial protection against reperfusion injury. During hemorrhagic shock, the Na+–H+ exchanger will be stimulated, as described earlier, and the sequence of events mentioned will occur, causing calcium overload and myocardial injury. During rapid resuscitation, which occurs in clinical life, the exchanger will be stimulated more rapidly and aggressively to correct the acidosis that occurred because of inadequate perfusion, which will cause more increase in the levels of intracellular sodium, and eventually calcium. Our hypothesis is that if we treat the animal with a Na+–H+ exchange inhibitor for a short period of time before resuscitation, this may prevent the post-resuscitation myocardial injury and dysfunction by giving the cell time to correct the intracellular acidosis by other mechanisms than the Na+–H+ exchanger, which will prevent the overload of the cell with calcium.
The protective effects of blocking the Na+–H+ exchanger on myocardial contractile function following resuscitation from hemorrhagic shock has been investigated for the first time in previous work in our laboratory using amiloride [13, 14]. The purpose of this study was to investigate the protective effects of blocking the Na+–H+ exchanger using DMA, a specific Na+–H+ exchanger blocker with no significant effects on Na+–Ca2+ exchanger [15] and sodium channels [16], in rats. For this reason, after 60 min of hemorrhagic shock, we treated the isolated hearts with DMA, before ex vivo resuscitation for 60 min using the Langendorff Apparatus.
Materials and methods
Animal preparation
Male Sprague–Dawley rats were injected intra-peritonealy (i.p.) with heparin sodium 2,000 IU 15 min prior to anesthesia. The rats were then anesthetized using intra-peritoneal urethane (125 mg/kg). The left carotid artery was cannulated using polyethylene tubing size 60 and was connected to an in-line pressure transducer for continuous blood pressure monitoring. Animals were allowed to stabilize for a period of 30 min. The animals were assigned randomly to hemorrhage treated, untreated or similar time matched control groups (n = 6 per group).
Experimental protocol
Rats were hemorrhaged using a reservoir (a 10-ml syringe) that was connected to the carotid artery via a three-way stopcock. Blood was aspirated at a rate of 1 ml/min. Blood was continuously withdrawn or re-infused to the animal to maintain a mean arterial pressure of approximately 40 mmHg. The same surgical procedure was performed as for the sham hemorrhage groups except rats were not hemorrhaged.
Ex vivo perfusion
After 1 h of hemorrhagic shock, hearts were harvested and resuscitated ex vivo by perfusion using the Langendorff apparatus for 60 min; data were collected and analyzed by the BIOPAC data acquisition System and the AcKnowledge software.
The isolated hearts were perfused with Krebs-Henseleit bicarbonate (KHB) buffer consisting of the following (in mM): sodium chloride, 118; calcium chloride, 1.25; potassium chloride, 4.7; sodium bicarbonate, 21; magnesium sulphate, 1.2; glucose, 11; potassium biphosphate, 1.2; EDTA, 0.5. In the treatment group, hearts were perfused with 20 μM DMA for 5 min, then shifted to perfusion with KHB for 55 min. The dose of DMA has been chosen with reference to the work that has been done before using DMA in case of ischemia-reperfusion and proved the cardioprotective effects of inhibiting the Na+–H+ exchanger using 20 μM DMA [23]. The aim of our study was to examine the cardioprotective effect of using the same dose of DMA in case of hemorrhage–resuscitation.
A saline-filled cellophane balloon-tipped catheter was placed into the left ventricle (LV) via the mitral valve and was used to measure LV pressure. LV end diastolic pressure (LVEDP) was adjusted at 5 mmHg by inflating the intraventricular balloon with 0.4–0.5 ml saline at the start of the perfusion. After that, the LVEDP was recorded, and no more adjustments were made in the balloon volume. The rate of perfusion was maintained at 10 ml/min by using a peristaltic pump purchased from Harvard Apparatus Inc. (model no. ‘1210’ series). The perfusate then passes to the aortic cannula through which it will enter the aorta to perfuse the heart. The coronary perfusion pressure was measured using a pressure transducer that was connected to the side arm of the aortic perfusion catheter and positioned at the level of the aorta. Hearts were stimulated electrically at 5 Hz, the pulse width was 1 mS, and the pulse amplitude was 5–7 volts, using an electrical stimulator (6020 Stimulator from Harvard Apparatus). The stimulator was connected to the heart via a double-stranded copper wire that was embedded into the tissue of the right atrial appendage. A reference electrode was attached to a stainless steel aortic cannula as a ground. Perfusate temperature was maintained at 37°C by the surrounding heated water that came from the thermocirculator purchased from Harvard Apparatus (serial no. K12248, item no. 50-1940). The perfusate was gassed with a mixture of 95% O2 + 5% CO2 via the oxygenator bubbler connected to a gas cylinder. The pH was checked using a pH meter from Harvard Apparatus.
Statistical analysis
All data were initially analyzed with Bartlett’s test for homogeneity. Data were analyzed with multivariate analysis of variance (ANOVA). Means were analyzed using Duncan’s test and were considered significant when yielding a P value less than 0.05. Data are expressed as means ± SEM.
Results
Figure 1 shows the changes in the LVEDP during the 1 h experimental protocol. LVEDP was elevated in the hemorrhage group compared to the sham hemorrhage group and increased more at 45 min of the experimental time. Left ventricular peak systolic pressure was significantly higher in the hemorrhage treated group as compared to the untreated hemorrhage group (P < 0.05) (Fig. 2). Left ventricular generated pressure was significantly low in the hemorrhage group compared to the sham hemorrhage group, while the hemorrhage treated group showed a significant increase in the left ventricular generated pressure compared to the hemorrhage group (Fig. 3). Left ventricular maximum +dP/dt (Fig. 4) was significantly lower in the hemorrhage group as compared to the sham hemorrhage group and significantly higher in the hemorrhage treated compared to the hemorrhage group; maximum −dP/dt was significantly higher in the hemorrhage group than in the sham hemorrhage group (P < 0.05) (Figs. 5, 6), while −dP/dt was significantly lower in the hemorrhage treated compared to the hemorrhage group (P < 0.05). There was no significant difference between the sham and the sham treated with DMA, indicating that with this dose of DMA, there was no effect of DMA on the sham hemorrhage hearts. Figure 6 shows an example of the raw data collected by the BIOPAC system and the AcqKnowledge software.
Fig. 1.
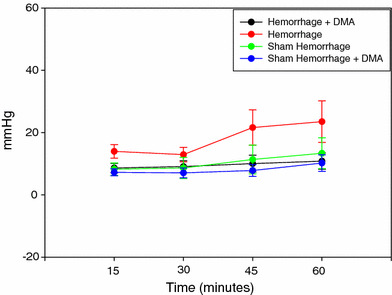
Left ventricular end-diastolic pressure of the sham hemorrhage treated and untreated, hemorrhage treated and untreated group over 1 h of ex vivo perfusion (n = 6)
Fig. 2.
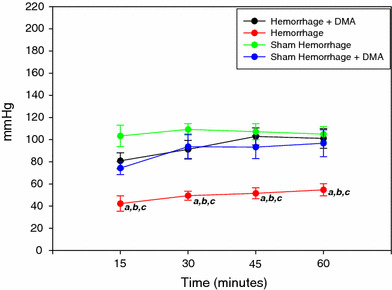
Left ventricular peak systolic pressure of the sham hemorrhage treated and untreated, hemorrhage treated and untreated group over 1 h ex vivo perfusion (n = 6). a = P < 0.05 compared to hemorrhage + DMA; b = P < 0.05 compared to sham hemorrhage; c = P < 0.05 compared to sham hemorrhage + DMA
Fig. 3.
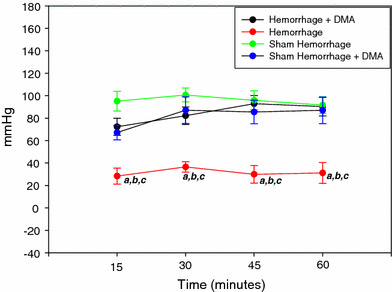
Left ventricular generated pressure of the sham hemorrhage treated and untreated, hemorrhage treated and untreated group over 1 h ex vivo perfusion (n = 6). a = P < 0.05 compared to hemorrhage + DMA, b = P < 0.05 compared to sham hemorrhage, c = P < 0.05 compared to sham hemorrhage + DMA
Fig. 4.
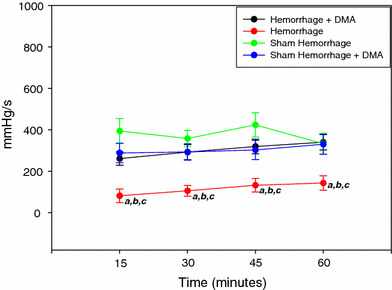
Left ventricular +dP/dt of the sham-hemorrhage treated and untreated, hemorrhage treated and untreated group over 1 h ex vivo perfusion (n = 6). a = P < 0.05 compared to hemorrhage + DMA, b = P < 0.05 compared to sham hemorrhage, c = P < 0.05 compared to sham hemorrhage + DMA
Fig. 5.
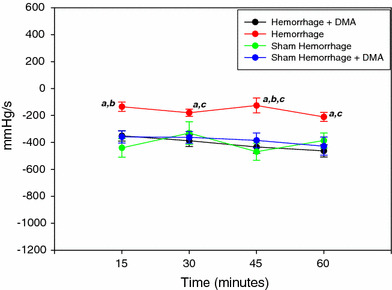
Left ventricular −dP/dt of the sham-hemorrhage treated and untreated, hemorrhage treated and untreated group over 1 h ex vivo perfusion (n = 6). a = P < 0.05 compared to hemorrhage + DMA, b = P < 0.05 compared to sham hemorrhage, c = P < 0.05 compared to sham hemorrhage + DMA
Fig. 6.
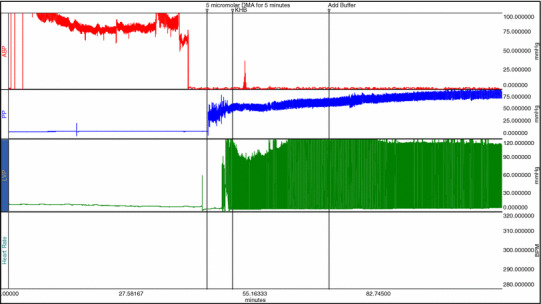
Example of the raw data in the AcqKnowledge software showing the measurements of arterial blood pressure in channel 1, coronary perfusion pressure in channel 2 and LV pressure in channel 3
Discussion
This study showed that treatment with DMA for 5 min on ex vivo perfusion, before 60 min of ex vivo resuscitation, following 1 h of hemorrhage, improved LV function, prevented the myocardial dysfunction and improved the recovery.
These results further reinforce the previous studies that have been done in case of ischemia-reperfusion and demonstrated that blocking the Na+–H+ exchanger has beneficial effects on myocardial function.
Activation of the Na+–H+ exchanger appears to play an important role in the pathophysiology of contractile dysfunction and heart failure following hemorrhagic shock. Inhibition of the Na+–H+ exchanger has been shown to protect the heart in a variety of ischemia reperfusion models [17–19]. However, little is known about the protective effects of blocking the Na+–H+ exchanger in case of hemorrhagic shock.
The Na+–H+ exchanger is one of the main regulators of the intracellular pH [20]. Hemorrhagic shock has been shown to result in extracellular acidosis that will eventually cause intracellular acidosis [5]. Extensive pre-clinical studies indicated that Na+–H+ exchanger inhibitors afford protection in animal models of myocardial ischemia (MI) and reperfusion [9].
Moreover, clinical investigations have been reported in patients with myocardial infarction [21]. In 1991, Meng et al. [22] studied the effect of blocking the Na+–H+ exchanger using DMA on the post-ischemic recovery of cardiac contractile function in isolated perfused rat hearts in the Langendorff. Treating the hearts with 20 μM DMA during reperfusion significantly reduced cellular Na+ and Ca2+, which supports the important role of Na+–H+ exchanger in the development of ischemia reperfusion injury. In 2000, Hurtado and Pierce [23] demonstrated the cardioprotective effects of inhibition of Na+–H+ exchanger with DMA at the beginning of reperfusion in isolated, beating adult cardiomyocytes. The role of the Na+–H+ exchanger following hemorrhage and resuscitation investigated in our laboratory exchanger using amiloride, non-specific blocker, was found to provide protection to the myocardium against contractile dysfunction following resuscitation from hemorrhagic shock [14, 24]. Wu et al. [25] demonstrated that inhibition of the Na+–H+ exchanger delays the onset of hypovolemic circulatory shock in pigs.
This study demonstrates a significant beneficial effect of blocking the Na+–H+ exchanger using a specific blocker, DMA, on myocardial contractile function following 60 min of ex vivo and was found to be activated during hemorrhagic shock [13], in addition, blocking the resuscitation of hemorrhagic shock. Future research is needed to prove the effectiveness of myocardial protection by treatment with DMA before resuscitation and the duration of recovery time.
The main novel contribution of this study is that it is the first to test the cardioprotective effects of using a specific Na+–H+ exchange inhibitor, DMA, in case of hemorrhagic shock and resuscitation. Despite the extensive research that has been done on the cardioprotective effects of using Na+–H+ exchange inhibitors in case of ischemia-reperfusion injury, the role of the Na+–H+ exchange inhibitors in case of hemorrhagic shock has not been investigated. Previously, our laboratory only showed the protective effects of using amiloride, a non-specific inhibitor.
The present study will add to our knowledge regarding the beneficial effects of using specific Na+–H+ exchange inhibitors as a therapeutic intervention before resuscitation following hemorrhagic shock. If this proves to be beneficial, it will open a new window for cardio protection before resuscitation of trauma patients. This has a very important clinical implication, since the death rate following resuscitation of trauma patients is among the highest in the world, due to myocardial dysfunction and multiple organ failure, despite the development in resuscitation strategies.
Further studies are needed to investigate the protective effects of more specific blockers and to investigate the exact mechanism of protection.
Acknowledgments
This research was supported by a grant from the College of Medicine Research Center, King Saud University. The author thanks Mr. Sabirine Sahipa for his excellent technical assistance.
References
- 1.Horton JW. Hemorrhagic shock depresses myocardial contractile function in the guinea pig. Circ Shock. 1989;28(1):23–35. [PubMed] [Google Scholar]
- 2.Lazdunski M, Frelin C, Vigne P. The sodium/hydrogen exchange system in cardiac cells: its biochemical and pharmacological properties and its role in regulating internal concentrations of sodium and internal pH. J Mol Cell Cardiol. 1985;17:1029–1042. doi: 10.1016/S0022-2828(85)80119-X. [DOI] [PubMed] [Google Scholar]
- 3.Cingolani H, Koretsune Y, Marban E. Recovery of contractility and pH during respiratory acidosis in ferret hearts: role of Na-H exchange. Am J Physiol. 1990;259:H843–H848. doi: 10.1152/ajpheart.1990.259.3.H843. [DOI] [PubMed] [Google Scholar]
- 4.Mattiazzi AR, Cingolani H. Biphasic effect of hypercapnia on myocardial contrctility. Arch Int Phys Biochem. 1977;85:11–25. [PubMed] [Google Scholar]
- 5.Horton J, McDonald G. Heart and brain nucleotide pools during hemorrhage and resuscitation. Am J Physiol. 1990;259(6):H1788. doi: 10.1152/ajpheart.1990.259.6.H1781. [DOI] [PubMed] [Google Scholar]
- 6.Turvey S, Allen DJ. Changes in myoplasmic sodium concentration during exposure to lactate in perfused rat heart. Cardiovasc Res. 1994;28:987–993. doi: 10.1093/cvr/28.7.987. [DOI] [PubMed] [Google Scholar]
- 7.Renlund DG, et al. Perfusate sodium during ischemia modifies post-ischemic functional and metabolic recovery in the rabbit heart. J Mol Cell Cardiol. 1984;16:795–801. doi: 10.1016/S0022-2828(84)80003-6. [DOI] [PubMed] [Google Scholar]
- 8.Lee JA, Allen DJ. Changes in intracellular free calcium concentration during long exposures to simulated ischemia in isolated mammalian ventricular muscle. Circ Res. 1992;71:58–69. doi: 10.1161/01.res.71.1.58. [DOI] [PubMed] [Google Scholar]
- 9.Tani M, Neely JR. Role of intracellular Na in Ca overload and depressed recovery of ventricular function of reperfused ischemic rat hearts. Circ Res. 1989;65:1045–1056. doi: 10.1161/01.res.65.4.1045. [DOI] [PubMed] [Google Scholar]
- 10.Murphy E, et al. Amiloride delays the ischemia-induced rise in cytosolic free calcium. Circ Res. 1991;68:1250–1258. doi: 10.1161/01.res.68.5.1250. [DOI] [PubMed] [Google Scholar]
- 11.Karmazyn M. Amiloride enhance post-ischemic ventricular recovery: possible role of Na/H exchange. Am J Physiol. 1988;255:H608–H615. doi: 10.1152/ajpheart.1988.255.3.H608. [DOI] [PubMed] [Google Scholar]
- 12.Karmazyn M, Ray M, Haist J. Comparative effects of Na/H exchange inhibitors against cardiac injury produced by ischemia/reperfusion, hypoxia/reoxygenation, and the calcium paradox. J Cardiovasc Pharmacol. 1993;21::172–178. doi: 10.1097/00005344-199301000-00025. [DOI] [PubMed] [Google Scholar]
- 13.Raymond R. Myocardial dysfunction during acute hemorrhagic hypotension in the rat. J Saudi Heart Assoc. 2002;14(4):204–212. [Google Scholar]
- 14.Soliman M, Raymond R. Effects of cardiac Na–H exchange blockade on myocardial contractile dysfunction during hemorrhagic shock. J Saudi Heart Assoc. 2005;17(1):33–42. [Google Scholar]
- 15.Kleyman TR, Cragoe JE. Amiloride and its analogues as tools in the study of ion transport. J Membr Biol. 1988;105:1–21. doi: 10.1007/BF01871102. [DOI] [PubMed] [Google Scholar]
- 16.Dennis S, et al. Effects of proton buffering and of amiloride derivatives on reperfusion arrhythmias in isolated rat heart. Circ Res. 1990;66:1156–1159. doi: 10.1161/01.res.66.4.1156. [DOI] [PubMed] [Google Scholar]
- 17.Scholz W, et al. Protective effects of HOE 642, a selective sodium-hydrogen exchange subtype 1 inhibitor, on cardiac ischemia and reperfusion. Cardiovasc Res. 1995;29:260–268. [PubMed] [Google Scholar]
- 18.Frohlich O, Karmazyn M. The Na–H exchanger revisited: an update on Na-H exchange regulation and the role of the exchanger in hypertention and cardiac function in health and disease. Cardiovasc Res. 1997;36:138–148. doi: 10.1016/S0008-6363(97)00200-9. [DOI] [PubMed] [Google Scholar]
- 19.Karmazyn M. The sodium–hydrogen exchange system in the heart: its role in ischemic and reperfusion injury and therapeutic implications. Can J Cardiol 12:1074–1082. 1996;12:1074–1082. [PubMed] [Google Scholar]
- 20.Pierce GN, Czubryt MP. The contribution of ionic imbalance to ischemia–reperfusion induced injury. J Mol Cell Cardiol. 1995;27:53–63. doi: 10.1016/S0022-2828(08)80007-7. [DOI] [PubMed] [Google Scholar]
- 21.Theroux P. Myocardial call protection. Circulation 101:2874–2876. 2000;101:2874–2876. doi: 10.1161/01.cir.101.25.2874. [DOI] [PubMed] [Google Scholar]
- 22.Meng HP, Lonsberry BB, Pierce GN. Influence of perfusate pH on the postischemic recovery of cardiac contractile function: involvement of sodium–hydrogen exchange. J Pharmacol Exp Ther. 1991;258(3):772–777. [PubMed] [Google Scholar]
- 23.Hurtado C, Pierce N. Inhibition of Na/H exchange at the begining of reperfusion is cardioprotective in isolated, beating adult cardiomyocytes. J Mol Cell Cardiol. 2000;32:1897–1907. doi: 10.1006/jmcc.2000.1222. [DOI] [PubMed] [Google Scholar]
- 24.Wu D, Bassuk J, Arias J, et al. Cardiovascular effects of NA+/H+ exchanger inhibition with BIIB513 following hypovolemic circulatory shock. Shock. 2005;23(3):269–274. [PubMed] [Google Scholar]
- 25.Wu D, Arias J, Bassuk J, et al. Na+/H+ exchange inhibition delays the onset of hypovolemic circulatory shock in pigs. Shock. 2008;29(4):519–525. doi: 10.1097/SHK.0b013e318150757a. [DOI] [PubMed] [Google Scholar]