

Summary:
Transcriptional enhancers direct precise gene expression patterns during development and harbor the majority of variants associated with phenotypic diversity, evolutionary adaptations, and disease. Pinpointing which enhancer variants contribute to changes in gene expression and phenotypes is a major challenge. Here we find that suboptimal or low-affinity binding sites are necessary for precise gene expression during heart development. Single nucleotide variants can optimize the affinity of ETS binding sites, causing gain-of-function gene expression, cell migration defects, and phenotypes as severe as extra beating hearts in the marine chordate Ciona robusta. In human iPSC-derived cardiomyocytes, a single nucleotide variant within a human GATA4 enhancer increases ETS binding affinity and causes gain-of-function enhancer activity. The prevalence of suboptimal-affinity sites within enhancers creates a vulnerability whereby affinity-optimizing SNVs can lead to gain-of-function gene expression, changes in cellular identity, and organismal-level phenotypes that could contribute to the evolution of novel traits or diseases.
In Brief:
Jindal et al. show that low-affinity ETS sites are critical for tissue-specific enhancer activity within the developing heart, and that single nucleotide variants can increase binding affinity, causing gain-of-function enhancer activity disrupting heart development. These findings have implications for understanding how enhancer variants alter phenotypes in health, evolution, and disease.
Graphical Abstract
Introduction
Enhancers are genomic elements that act as switches to control the location, level, and timing of gene expression to ensure the successful development and integrity of an organism1,2. Sequence changes within enhancers can lead to changes in gene expression, phenotypic diversity, novel traits, and disease3–5. Indeed, most disease-associated variants within the genome lie within enhancers6–8. Yet, pinpointing causal base-pair changes within enhancers is challenging as they are typically associated with many inert variants due to linkage disequilibrium furthermore it is hard to functionally validate variants at scale in the relevant biological context9. Our inability to predict causal enhancer variants is stalling efforts to harness the full potential of genomic data to understand the genetic basis of health and disease. Here we use a mechanistic understanding of the regulatory principles governing enhancers to pinpoint causal enhancer variants that alter gene expression and heart development.
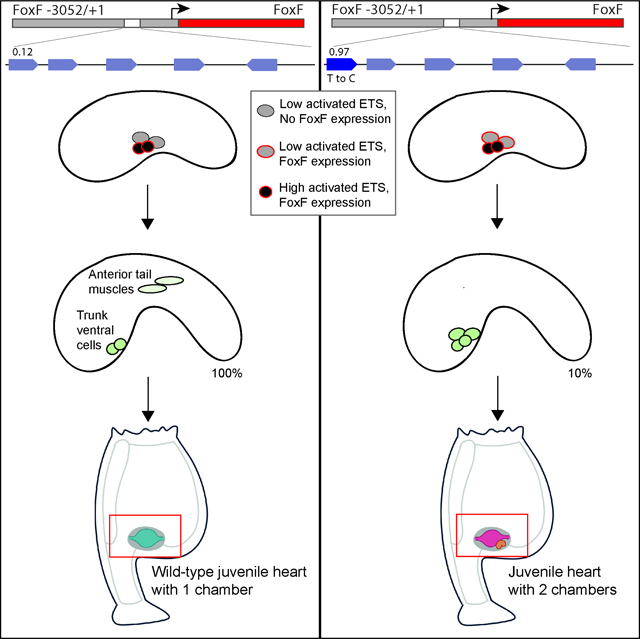
FGF (Fibroblast Growth Factor) signaling plays a critical and conserved role in heart development across bilaterians10,11. FGF signaling mediates changes in gene expression through binding of activated ETS transcription factors (TFs) to enhancers12. Loss and gain of FGF signaling pathway components cause heart defects in organisms as diverse as Drosophila and human13–15. For example, gain of FGF is implicated in cardiac hypertrophy16 while manipulation of ETS-1 in mice recapitulates some of the most common congenital heart defects in Jacobsen syndrome17. In Ciona, expression of a constitutively active form of ETS (ETS-VP16) in non-heart muscle cells leads to development of a multi-chambered heart by recruitment of more cardiac progenitor cells to the ventral midline18.
Ciona robusta (Ciona) is a marine chordate and member of the urochordates, the sister group to vertebrates19. Like vertebrates, Ciona have a pumping heart within a pericardium, and early heart specification involves the TFs ETS and MesP20. Across bilaterians, migration of the heart cells to the ventral midline is dependent on FGF signaling. In Ciona, the Trunk Ventral Cells (TVCs) give rise to the heart and pharyngeal cells21,22. FGF signaling and MesP activate expression of FoxF in the TVCs, this triggers migration of the TVCs to the ventral midline. Ciona is experimentally tractable, allowing us to investigate enhancer activity in hundreds of developing embryos and easily visualize the migration of the TVCs to the ventral midline20. Thus, Ciona heart development and the FoxF enhancer provide an ideal system to investigate the relationship between enhancer sequence, tissue-specific gene expression and organismal-level phenotypes.
Developmental enhancers contain suboptimal-affinity sites (also referred to as submaximal and low-affinity sites), which ensure tissue-specific gene expression23–28. While the use of suboptimal-affinity binding sites to encode precise gene expression has been seen in a variety of contexts, it has not been explored within heart development. Additionally, in the previous studies, many nucleotide changes are made to the sequences to turn low-affinity sites into high-affinity sites, but these large numbers of changes do not reflect the sequence changes naturally arising within genomic enhancers. Here we seek to determine if low-affinity binding sites are important in heart development, and if single nucleotide variants (SNVs) that increase binding affinity (affinity-optimizing SNVs) could cause gain-of-function gene expression that contributes to phenotypic changes in heart development.
Results
The FoxF enhancer drives expression in TVCs and contains low-affinity ETS binding sites
FoxF is expressed within the TVCs and triggers migration of the TVCs to the ventral midline. A 295bp FoxF enhancer upstream of a minimal promoter (bpFog) driving expression of nuclear mCherry recapitulates this TVC specific expression pattern (Figures 1A,E,S1A)29,30. An E-box site, ATTA sites and three ETS sites are important for activity of the FoxF enhancer (Figure S1B)21,31. To identify additional ETS sites within the FoxF enhancer, we search for ETS cores (GGAW) within the enhancer and identify three additional putative ETS sites. We assign affinities of all six ETS sites from Protein Binding Microarray (PBM) data, which measures the interaction of ETS1 with all possible 8-mers32,33. We use PBM data for mouse ETS1 as a proxy for Ciona ETS1 binding, as the DNA-binding domain and binding specificity of ETS1 is highly conserved25,34. We calculate the relative affinity as the ratio of each 8-mer’s fluorescence to the fluorescence of CCGGAAGT, the 8-mer with the highest affinity for ETS1 in Drosophila, Ciona, mouse, and human25,33,34. The putative ETS sites within the FoxF enhancer bind ETS with low relative affinities ranging from 0.09 to 0.24 (Figure S1C).
Figure 1. Low-affinity ETS sites are necessary for tissue-specific expression.
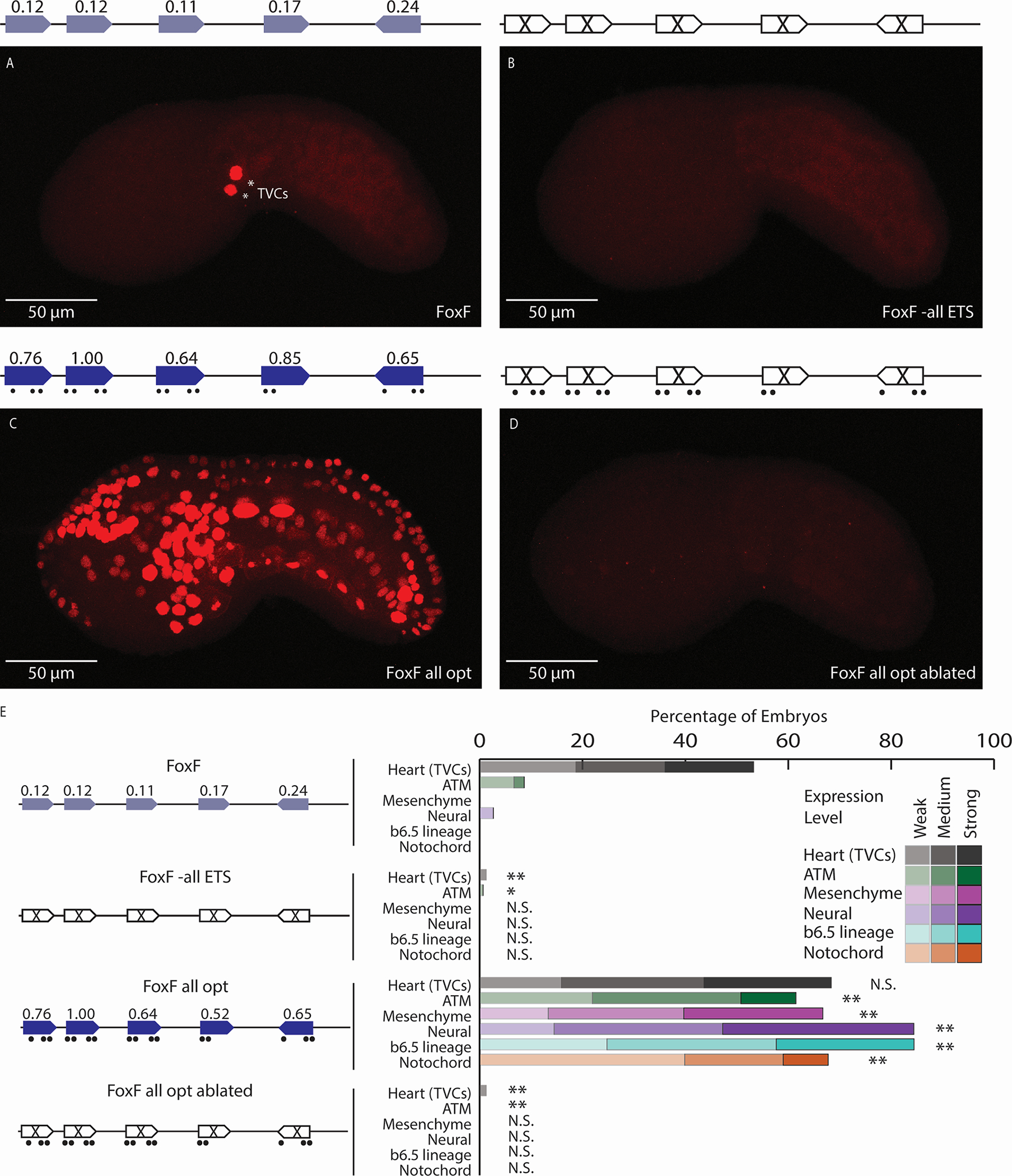
A. The FoxF enhancer drives expression in the TVCs, marked by asterisks. B. An enhancer with all six ETS sites ablated (by changing each site from GGAW to GCAW) drives no enhancer activity. C. Optimizing the affinity of all ETS sites leads to ectopic expression in ATMs, mesenchyme, nervous system, and notochord. D. Ablating the ETS sites within the context of the FoxF all opt (FoxF all opt ablated) drives no enhancer activity. E. Embryo scoring for above constructs (3 replicates, n ≥ 32 embryos per replicate). P-values are obtained using Chi-square test comparing each condition to WT with Bonferroni correction: ** P<0.0017, * P<0.017, N.S. not significant. Schematics show electroporated enhancers, values are affinities, dots show nucleotide changes. ETS6 is not shown in the schematic as ablation of this site did not impact expression (see Figure S1). Scale bars, 50 μm. Data in stacked bar chart are represented as the mean. See also Figures S1,S2 and Tables S1,S2.
Low-affinity ETS sites are necessary for tissue-specific expression
To determine if these six low-affinity ETS sites are necessary for expression within the TVCs, we ablate these ETS sites using point mutations that disrupt hydrogen bonding between the ETS TF and the binding site (GGAW to GCAW)35. Ablating all of the ETS sites in parallel leads to complete loss of enhancer activity (Figures 1B,E,S1C). To investigate the contribution of each ETS site to FoxF enhancer activity, we ablate individual ETS sites with point mutations (GGAW to GCAW) and test the effect of these changes in reporter assays by scoring mCherry expression levels in embryos. To the best of our knowledge, these ablations do not create or ablate motifs for TFs present within the TVCs (Table S1). Ablating individual sites in the enhancer leads to a significant reduction in enhancer activity for five ETS sites (Figures S1C,S2). The only site without a reduction in expression upon ablation is ETS6, which has the lowest affinity at 0.09. While loss of the ETS6 site doesn’t alter expression, we cannot rule out that this site is redundantly involved in enhancer activity36–38. These results demonstrate that five low-affinity ETS sites are necessary for FoxF enhancer activity in the TVCs and illustrate the functional role of low-affinity ETS sites within a developmental heart enhancer.
To determine if the ETS sites within the FoxF enhancer need to be low-affinity for tissue-specific expression, we increase the affinity of the five ETS sites necessary for activity, keeping the GGAW core constant. Optimizing the affinity of all five ETS sites within the enhancer requires 16 nucleotide changes and leads to ectopic expression in tissues receiving FGF signaling, including the anterior tail muscle cells (ATMs), mesenchyme, nervous system, and notochord (Figure 1C,E). To demonstrate that it is the increase of ETS affinity driving the expression change, we made a FoxF enhancer with the 16 nucleotides that optimize ETS affinity in combination with mutations which ablate ETS binding (GGAW to GCAW) (FoxF all opt ablated). This enhancer has no activity (Figures 1D,E). Thus, low-affinity sites are important for maintaining TVC-specific expression of the FoxF enhancer. Similar results have been seen upon optimizing the affinity of sites within neural and notochord enhancers, indicating that the role of low-affinity sites to encode tissue-specific expression is a general regulatory principle governing developmental enhancers25,26.
Affinity-optimizing SNVs lead to loss of tissue-specific expression
To determine if any Single Nucleotide Variants (SNVs) could optimize ETS binding affinity and alter gene expression, we make every possible SNV in the flanking region of the ETS sites of the FoxF enhancer in silico and calculate the resulting affinity change. Within this manuscript, we use SNVs to denote single base-pair changes, regardless of whether they are synthetic or naturally found within Ciona or human populations. We find that SNVs in three of the five ETS sites cause a ≥3-fold increase in ETS affinity (Figure 2A).
Figure 2. Affinity-optimizing SNVs lead to loss of tissue-specific expression.
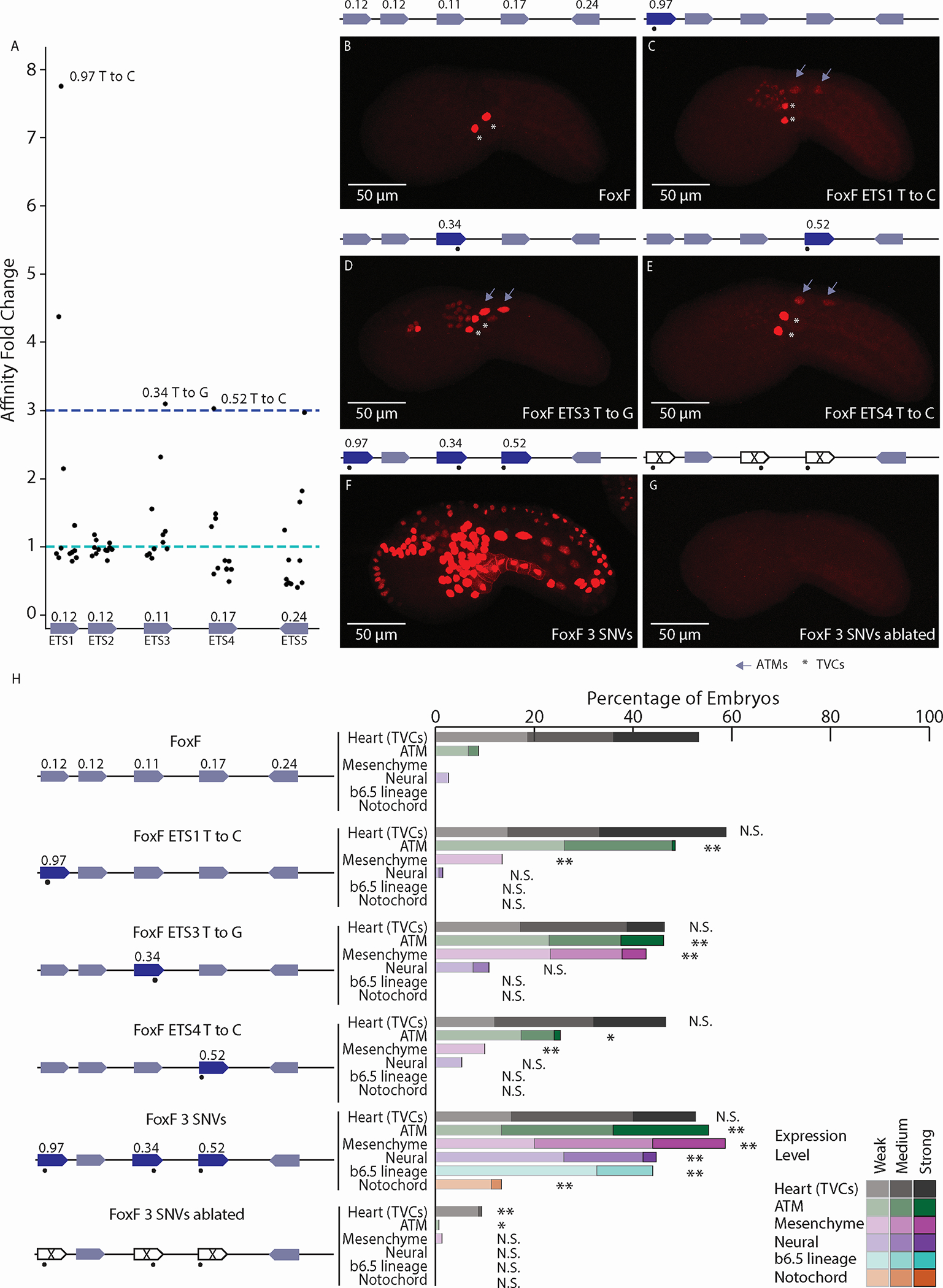
A. Affinity fold change for SNVs within functional ETS sites of the FoxF enhancer. B. The WT FoxF enhancer drives expression in the TVCs. C. The FoxF-ETS1-T-to-C enhancer drives expression in the TVCs, ATMs, and mesenchyme. D. The FoxF-ETS3-T-to-G enhancer drives expression in the TVCs, ATMs, mesenchyme, and neural tissues. E. The FoxF-ETS4-T-to-C enhancer drives expression in the TVCs and ATMs. F. Optimizing the affinity of three ETS sites with SNVs leads to ectopic expression in ATMs, endoderm, mesenchyme, nervous system, and notochord. G. Ablating the ETS sites (GGAW to GCAW) within the context of the 3 SNVs no longer drives ectopic expression, only very weak TVC enhancer activity. H. Embryo scoring for above constructs (3 replicates, n ≥ 17 embryos per replicate). P-values are obtained by using Chi-square test comparing each condition to WT with Bonferroni correction: ** P < 0.001, * P <0.01, N.S. not significant. Asterisks mark TVCs and arrows mark ATMs. Schematics show electroporated enhancers, values are affinities, dots mark SNVs. Scale bars, 50 μm. Data in stacked bar chart are represented as the mean. See also Figures S2,S3 and Tables S1,S2.
The greatest affinity change occurs within the ETS1 site which has a starting affinity of 0.12, a SNV increases affinity 8-fold creating an almost consensus site, with 0.97 affinity. We demonstrate the affinity change using electrophoretic mobility shift assay (EMSA) to detect differential binding of Ciona ETS1 to the WT and affinity-optimized ETS1 sequences (Figure S3). When we make this SNV within the FoxF enhancer (FoxF-ETS1-T-to-C), the enhancer drives ectopic mCherry expression in the ATMs and mesenchyme, both cell types which receive FGF signaling (Figures 2C,H).
We also find affinity-optimizing SNVs within the ETS3 and ETS4 sites. A SNV within the ETS3 site causes a 3-fold increase in affinity, from 0.11 to 0.34. This affinity-optimizing SNV drives ectopic expression in ATMs, mesenchyme, and neural tissues (Figures 2D,H). The affinity-optimizing SNV within the ETS4 site increases affinity from 0.17 to 0.52, this 3-fold affinity increase causes ectopic expression in the ATMs and mesenchyme (Figures 2E,H). These results suggest that affinity-optimizing SNVs are sufficient to cause loss of tissue-specific gene expression.
A FoxF enhancer containing all three affinity-optimizing SNVs (FoxF 3 SNVs) drives strong activity in tissues responding to FGF signaling, similar to the FoxF all opt enhancer and the effect of these three SNVs appears to be synergistic (Figures 2F,H). A FoxF enhancer with all three affinity-optimizing SNVs and ablated ETS cores (GGAW to GCAW) drives no expression, demonstrating that the effect of these SNVs is ETS-dependent (Figures 2G,H).
Affinity-optimizing SNVs cause migration defects and disrupt heart development
All three enhancers containing a single affinity-optimizing SNV show ectopic reporter expression in the ATMs (Figure 2). The ATMs and TVCs are related cell types that both receive FGF9 ligand; however, the TVCs contain higher levels of activated ETS due to FGF receptor enrichment39,40. The high levels of activated ETS within the TVCs causes TVC-specific expression of FoxF and migration of the TVCs to the ventral midline18. As the ATMs have low levels of activated ETS, they do not express FoxF and do not migrate (Figure 3A; Movie S1)18. Overexpression of constitutively active ETS or FoxF in the ATMs causes the ATMs to migrate with the TVCs18,21. We therefore hypothesize that the ectopic ATM expression driven by FoxF enhancers containing affinity-optimizing SNVs could cause migration of the ATMs to the ventral midline.
Figure 3. Affinity-optimizing SNVs cause migration defects and disrupt heart development.

A. Schematic showing a tailbud wild-type embryo, the concentration of activated ETS in the TVCs and ATMs and the role of FoxF in migration of the TVCs to the ventral midline. The TVCs have high levels of activated ETS while the ATMs have lower levels of activated ETS. FoxF enhancer drives expression of FoxF in the TVCs, causing them to migrate. B. Schematic showing a tailbud embryo containing the FoxF-ETS1-T-to-C enhancer driving FoxF. TVC cells have high levels of activated ETS, while ATMs have low levels of activated ETS. Embryos with the FoxF-ETS1-T-to-C enhancer drive expression of FoxF in the TVCs and ATMs causing the ATMs to express FoxF and migrate with the TVCs. C. Embryo with WT FoxF enhancer driving FoxF, co-electroporated with a MesP enhancer driving GFP marking the ATMs and TVCs at 16 hpf. D. Embryo with FoxF-ETS1-T-to-C enhancer driving FoxF, co-electroporated with a MesP enhancer driving GFP in the TVCs and ATMs at 16 hpf; all cells migrate to the ventral midline. Black cells in C and D are the otolith and ocellus. E. Juvenile with WT FoxF enhancer driving FoxF with heart colored in blue; normal heart morphology is seen. F. Juvenile with FoxF-ETS1-T-to-C enhancer driving FoxF has two hearts with main heart colored in pink, auxiliary heart colored in orange; abnormal heart morphology is seen in 79% (41/52) of embryos with migration defects. Hearts were false-colored digitally. G. Schematic of juvenile Ciona with normal heart morphology. H. Schematic of juvenile Ciona with auxiliary heart. Scale bars, 50 μm. See also Figure S4 and Movies S1,S2.
To test this hypothesis, we co-electroporate a Mesp-2kb>GFP reporter to mark both the TVCs and ATMs and a construct containing the FoxF 3kb regulatory element, including the endogenous promoter, driving FoxF mRNA (Figure S4A). We use the 3Kb regulatory element rather than a minimal enhancer to better recapitulate the regulatory region. When the wild-type FoxF enhancer drives FoxF, we observe no migration of ATMs to the ventral midline (Figures 3A,C,S4; Movie S1) and juvenile Ciona have normal hearts (Figure 3E,G; Movie S2). When we electroporate the FoxF-ETS1-T-to-C enhancer driving FoxF, we see migration defects in 9.3% of embryos (Figures 3B,D,S4; Movie S1). The FoxF-ETS3-T-to-G enhancer driving FoxF causes migration defects in 9.9% of embryos, despite the fact this SNV only increases the affinity of the site to 0.34 (Figure S4). The FoxF-ETS4-T-to-C driving FoxF causes migration defects in 8.0% of embryos (Figure S4). Thus, SNVs that lead to ≥3-fold increase in affinity, even those that result in relatively low-affinity sites, have the potential to contribute to migration defects during heart development. The strong impact of the optimized ETS3 site on migration could be due to nearby E-box and ATTA sites21,31, suggesting that enhancer grammar41 may influence the functional consequences of affinity-optimizing SNVs within genomes.
To determine the impact of migration defects on heart development, we followed 52 FoxF-ETS1-T-to-C embryos with migration defects from the larval to juvenile stages. Strikingly, 79% of embryos with migration defects have abnormally developed hearts (Figures 3F,H). Phenotypes range from enlarged hearts to two distinct beating hearts within the same pericardium (Movie S2). As the Ciona age, hearts from animals with the FoxF-ETS1-T-to-C SNV could not pump blood and these animals did not survive beyond twelve days post metamorphosis.
Low-affinity ETS sites are prevalent in putative developmental heart enhancers across chordates and these enhancers are vulnerable to affinity-optimizing SNVs.
The role of FGF signaling in specification and migration of heart cells is conserved from flies to vertebrates10,11. To determine if low-affinity sites are common within developmental heart enhancers, we used epigenomic datasets to define putative developmental heart enhancers (DHEs) in both Ciona and human. Using ATAC-seq data42 for Ciona heart cells at 6.5hr post fertilization, a time at which TVCs receive FGF signaling, we identify 15,174 putative Ciona DHEs. Using ATAC-seq and ChIP-seq for p300 and H3K27Ac from primary fetal human hearts and iPSC-derived embryonic-like cardiomyocytes at time points when FGF signaling is important for heart development, we find 252,931 putative human DHEs43–46. In both Ciona and human DHEs, the median affinity of putative ETS sites is 0.12, suggesting that low-affinity sites are prevalent within chordate putative DHEs. Within 6,618 (44%) of Ciona DHEs and 154,685 (61%) of human putative DHEs, a SNV can increase the affinity of ETS sites by ≥3-fold with the average affinity after optimization being 0.52 (Figure 4A). Thus, DHEs are vulnerable to affinity-optimizing SNVs, some of which may lead to GOF gene activity and changes in cellular identity.
Figure 4. Low-affinity ETS binding sites are prevalent within developmental heart enhancers and affinity-optimizing SNVs lead to gain-of-function expression in human iPSC-CMs.
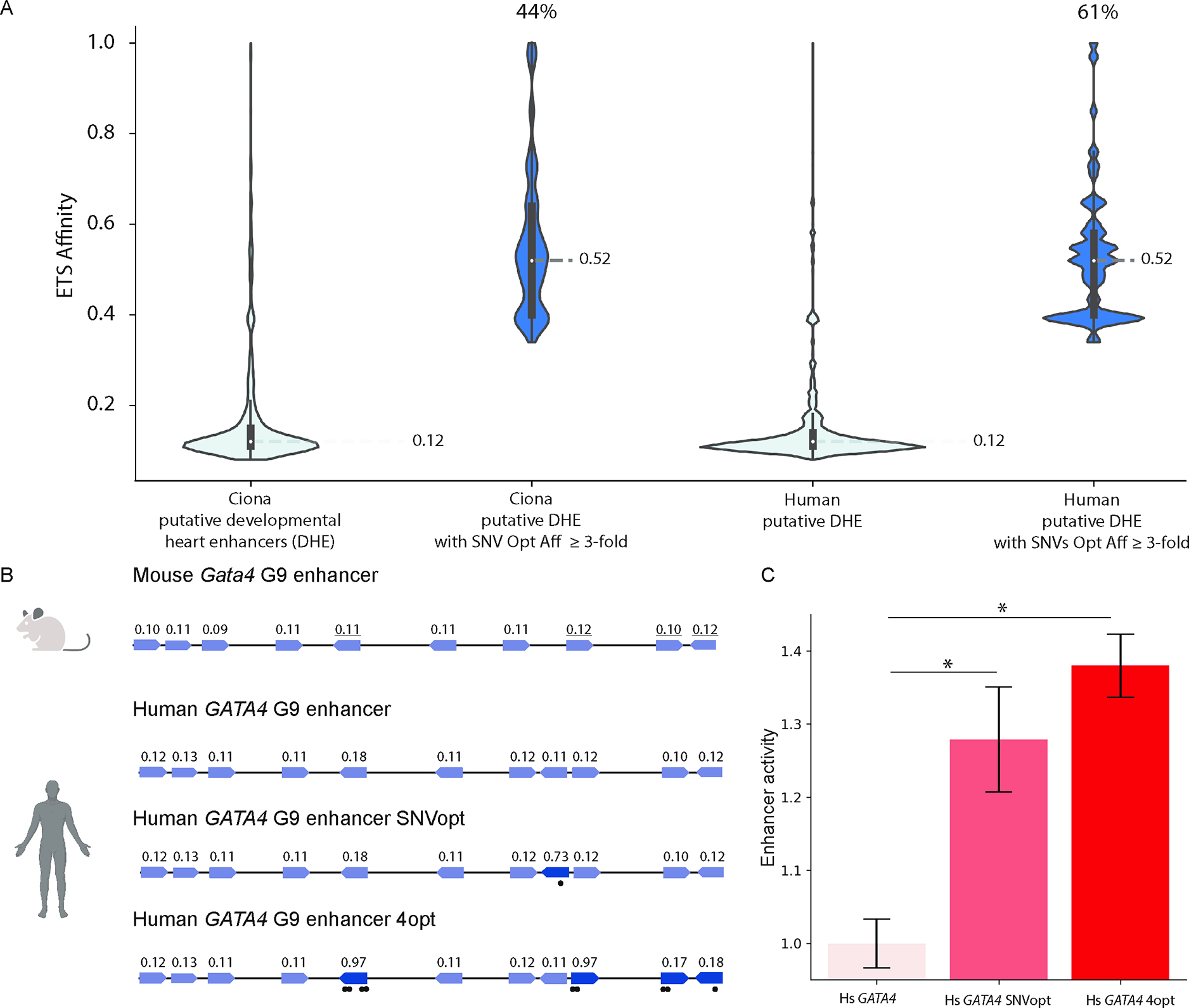
A. Violin plots in light blue of ETS site affinities within Ciona and human putative DHEs. 44% of Ciona DHEs and 61% of human DHEs contain an ETS site in which a SNV can increase ETS affinity ≥3-fold. Dark blue violin plots show distributions of the affinity-optimizing SNVs. B. Schematics of human and mouse Gata4 enhancers show high conservation of low-affinity ETS sites. Sites with underlined affinities mark functionally validated ETS sites in mouse35. GATA4 G9 enhancer SNVopt harbors a SNV that increases relative affinity from 0.11 to 0.73. GATA4 G9 enhancer 4opt has four ETS sites with increased affinity. C. Hs GATA4, Hs GATA4 SNVopt and Hs GATA4 4opt enhancer activity as measured by reporter assay in human iPSC-CMs. Data are represented as mean ± SEM. * p<1e-5. See also Table S2.
Human and mouse Gata4 heart enhancers contain low-affinity ETS sites
GATA4 is a key TF within the developing heart. One of the human DHEs is the ortholog of the mouse enhancer for Gata4 (Figure 4B). The mouse Gata4 G9 enhancer is ETS-dependent and drives expression in the endocardial and myocardial heart layers35. Ablating four ETS sites within the mouse Gata4 G9 enhancer leads to loss of expression, suggesting that these sites are functional and required for enhancer activity35. However, the affinity of these ETS sites has not been studied. We assign the affinity of all ETS sites within the Gata4 G9 enhancer using mouse ETS1 PBM data. All of the ETS sites within the mouse Gata4 G9 enhancer have affinities ranging from 0.09 and 0.12. The four sites previously shown to contribute to enhancer activity and bind ETS-1 have affinities of 0.11, 0.12, 0.10, and 0.12, respectively. These findings demonstrate that low-affinity ETS sites are necessary for heart-specific expression. The human GATA4 G9 enhancer contains low-affinity binding sites that are almost identical to those within the mouse Gata4 G9 enhancer. A reporter construct containing the human GATA4 G9 enhancer, basal SCP promoter and reporter (GFP and barcode) drives expression in human iPSC-derived fetal-like cardiomyocytes, which are the primary cell type found within the myocardial layers of the heart (Figure 4C).
Affinity-optimizing SNVs within the human GATA4 G9 enhancer cause gain-of-function expression in human iPSC-derived cardiomyocytes
To determine if optimizing the affinity of the ETS sites within the GATA4 G9 enhancer leads to GOF expression, in this case an increase in expression within cardiomyocytes, we conducted a reporter assay in human iPSC-CMs to compare the expression of the reference human GATA4 G9 enhancer (Hs GATA4) to the same enhancer with a single (Hs GATA4 SNVOpt) or four affinity-optimizing mutations (Hs GATA4 4Opt)47,48. Increasing the affinity of a single ETS site from 0.11 to 0.73 (Hs GATA4 SNVopt) leads to a significant increase in expression. Increasing the affinity of four ETS sites (Hs GATA4 4opt) leads to an even larger increase in expression (Figure 4C). Thus, in the human GATA4 G9 heart enhancer, affinity-optimizing variants drive gain-of-function gene expression, consistent with our findings in Ciona.
Discussion
Low-affinity ETS sites are a prevalent feature of developmental heart enhancers
Our genome-wide analysis finds low-affinity sites are a common feature of developmental heart enhancers across Ciona, mouse and human. Ablation of low-affinity sites both within the Ciona FoxF enhancer and mouse Gata4 enhancer cause a significant reduction in enhancer activity, demonstrating that these low-affinity sites are functional. Low-affinity sites are also important within enhancers activated by other TFs downstream of signaling pathways and pleiotropic factors23,25,49–51. Our findings highlight the importance and vastly underappreciated contribution of very low-affinity binding sites to regulation of gene expression and the advantages of scoring the affinity of sites.
Affinity-optimizing SNVs cause gain-of-function enhancer activity, which can contribute to phenotypes
In Ciona, affinity-optimizing SNVs lead to loss of tissue-specific expression and migration defects that alter heart development, in the most extreme cases we see two beating hearts. We propose that ETS affinity-optimizing SNVs likely contribute to congenital heart disease and cardiac traits. The ubiquitous role of FGF in other developmental programs52,53 and cancer54 indicates that ETS affinity-optimizing SNVs are likely involved in other enhanceropathies. Searching for affinity increases as a method to filter for causal enhancer variants is not common, yet from our work and a few other studies it appears this could be a powerful approach. To our knowledge, only three studies search for an affinity increase and measure the impact on gene expression, yet do not analyze phenotype55–57. In a handful of examples, causal enhancer variants have been functionally validated via reporter assays. After the validation of the impact of these variants on gene expression, these sequence changes have been identified as increasing affinity. Although using affinity to filter for causal variants is in its infancy, this approach appears to generalize beyond ETS as these other aforementioned studies have found sequence changes that increase affinity in TFs as diverse as Wnt effectors and Hox members that can alter gene expression and are associated with phenotypes58–61.
Affinity-optimizing SNVs may contribute to evolution of novel traits
There is a fine line between creation of novel phenotypes that are beneficial or detrimental5. The Ciona heart, like all invertebrate chordates, is single-chambered, while all extant vertebrates have at least two chambers. The evolution of a dual-chambered heart in vertebrates is thought to involve recruitment of additional precursor cells to the ventral midline18. Our study indicates that mutations within enhancers that increase binding affinity for ETS result in more cells migrating to the ventral midline and creation of another compartment within the pericardium. Although the animals in our study with two hearts did not survive, it is possible that some animals with multi-chambered hearts could survive and have a selective advantage. More generally, affinity-optimizing SNVs could contribute to the evolution of traits by affecting gene expression patterns and cell behavior.
Violations in regulatory principles governing enhancers can pinpoint causal enhancer variants
How enhancer variants contribute to phenotypes is a pressing challenge we need to solve if we want to causally link genomic variation to phenotypic diversity in health and disease. To date, only a handful of enhancer variants have been functionally validated as altering gene expression and phenotype. Yet thousands of non-coding variants are associated with changes in gene expression and phenotypic variation. We cannot possibly test all non-coding variants in the relevant cell types and time points to pinpoint causal enhancer variants. Finding rules that generalize across different enhancers relies on understanding the regulatory principles that govern enhancer function. One such regulatory principle is the use of low-affinity binding sites to encode tissue-specific expression and levels of expression. Here we show that SNVs that violate this regulatory principle can cause gain-of-function gene expression and organismal-level phenotypic changes. Identification of other regulatory principles and subsequently variants that violate these principles could provide further mechanistic approaches to pinpoint causal enhancer variants at scale.
Limitations of the Study
While many studies use position weight matrices (PWM) to find matches to binding sites, here we use PBM data, an in vitro measurement of relative binding affinity that allows us to give a relative affinity score to every 8mer sequence. In vitro PBM measurements do not consider chromatin structure, protein-protein interactions, cooperative binding between multiple sites and phase separation. While this is a limitation, it also has some advantages as it enables us to have a fixed measurement of relative binding affinity that can be used across genomes, cell types and species to annotate putative binding sites. As illustrated in our study, this approach can predict binding and the impact of SNVs in different cell types and organisms (e.g FoxF Ciona TVC enhancer and human GATA4 cardiomyocyte enhancer). As such, it is a score that can be used to analyze genomic sequences and predict the impact of enhancer SNVs without the need to measure specific binding affinity of every site at every time point and cell type of an organism.
Our studies take advantage of disparate biological systems Ciona and human cardiomyocytes to detect gain-of-function gene expression. In Ciona, we are able to assay expression in all cells of the embryo, but it is difficult to quantify expression levels within these cell types. While in human cells we see that the affinity-optimizing SNV increase levels of expression, however, we cannot assay the impact of this SNV on specificity of gene expression within the entire human heart. Together, these complementary assays illustrate that affinity-optimizing SNVs can increase the levels of expression within the endogenous location and also lead to expression in other cell types, both types of GOF expression could contribute to phenotypes.
Another limitation relates to how we study the role of enhancer variants on cell behavior. We characterize Ciona heart phenotypes by electroporation of a plasmid carrying a large 3kb regulatory region driving FoxF cDNA into fertilized eggs. While the WT 3kb region driving FoxF mRNA does not alter migration, the same regulatory element with only a single SNV leads to migration defects. Indeed, we see consistent results for three different SNVs tested in this way. Ideally, we would show that a SNV within the endogenous locus has the same effect. However, targeted genome editing with HDR, or base editing is not possible within Ciona. Once these techniques are established, testing the impact of the FoxF SNVs within the genome will be important. While we have not tested the FoxF SNVs in the endogenous locus, we have done similar studies in mice and find that affinity-optimizing SNVs within the endogenous locus in mice lead to GOF expression and phenotypes62.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Emma Farley (efarley@ucsd.edu).
Materials availability
Plasmids generated in this study will be made available from lead contact upon request.
Data and code availability
Sequencing data have been deposited at SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Microscopy and scoring data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOI is listed in the key resources table.
Bed files of putative Ciona developmental heart enhancers and putative human developmental heart enhancers have been deposited at GitHub and DOI is listed in the key resources table
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resource table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| mouse ETS1 universal PBM data | Wei et al.33 | thebrain.bwh.harvard.edu/uniprobe/index.php |
| Ciona robusta genome | Satou et al.69 | N/A |
| LacZ_6hpf_1–3 | Racioppi et al.42 | GEO: GSE126691 |
| Human reference genome NCBI build 38, GRCh38 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human |
| Human fetal heart putative enhancers predicted from H3K27ac ChIP and ATAC-seq | Richter et al.43 | Supplementary Table 11 in source |
| iPSC-cardiomyocyte putative enhancers predicted from ATAC- seq and H3K27ac ChIP | Benaglio et al.44 | GEO: GSE133833 |
| Human fetal heart putative enhancers predicted from p300/CBP | May et al.45 | GEO: GSE32587 |
| Sequencing data for MPRA | This paper | SRA: PRJNA999706 |
| Experimental Models: Organisms/Strains | ||
| Ciona intestinalis type A (Ciona robusta) | M-REP | N/A |
| iPSC-CMs | Frazer Lab | UDID_10948 |
| iPSC-CMs | Frazer Lab | UDID_13948 |
| Oligonucleotides | ||
| Oligonucleotides for mutagenesis, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: foxf295-bpfog>H2B-mCherry | Levine & Christiaen Labs | N/A |
| Plasmid: foxf295-ets1mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets2mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets3mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets4mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets5mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets6mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets1optT2C-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets3optT2G-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets4optT2C-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets134opt-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets134optAbl-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets12345opt-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets12345optAbl-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets123456mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf3kb>foxfcDNA | This paper | N/A |
| Plasmid: foxf3kb-ets1OptT2C>foxfcDNA | This paper | N/A |
| Plasmid: foxf3kb-ets3OptT2G>foxfcDNA | This paper | N/A |
| Plasmid: foxf3kb-ets4OptT2C>foxfcDNA | This paper | N/A |
| Plasmid: gata4-g9-scp>unc76-GFP | This paper | N/A |
| Plasmid: gata4-g9-SNVopt-scp>unc76-GFP | This paper | N/A |
| Plasmid: gata4-g9–4opt-scp>unc76-GFP | This paper | N/A |
| Plasmid: pTNT-ciETS1 | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | Schneider et al.76 | https://imagej.nih.gov/ij/ |
| FastQC (version 0.11.2) | N/A | http://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| Trim Galore (version 0.4.4) | N/A | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore |
| Bowtie2 (version 2.3.2) | Langmead et al.70 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools (version 1.2) | Li et al.71 | https://samtools.sourceforge.net |
| MACS2 (version 2.7.9) | Zhang et al.72 | N/A |
| BEDTools | Quinlan et al.74 | N/A |
| pyliftover 0.4 | N/A | https://pypi.org/project/pyliftover |
| Adobe Premiere Pro | Adobe | N/A |
| Adobe Animate | Adobe | N/A |
| Code to generate Fig 2A, 4A, 4C and putative Ciona developmental heart enhancers and putative human developmental heart enhancers | This paper | https://doi.org/10.5281/zenodo.8215976 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Tunicates
Adult C. intestinalis type A aka Ciona robusta (obtained from M-Rep) were maintained under constant illumination in seawater (obtained from Reliant Aquariums) at 18°C. Ciona filter feed on material in seawater and we do not add any food for the week we keep them for. Ciona are hermaphroditic, therefore there is only one possible sex for individuals. Age or developmental stage of the embryos studied are indicated in the main text.
iPSC-derived cardiomyocytes
iPSC-CMs from line UDID_109 and from line UDID_139 47 were thawed and seeded onto gelatin-coated 48-well plates at a density of 1.3 × 105/cm2. UDID_109 cell line is from an Asian male and UDID_139 cell line is from an Asian female. Cells were cultured using previously described culture conditions63.
METHOD DETAILS
Mutagenesis and cloning
The FoxF enhancer construct was a gift from the Levine and Christiaen labs. The construct consists of the following elements in order: AscI, FoxF enhancer, XbaI, bpfog promoter, NotI, H2B-mCherry, EcoRI. Point mutations were introduced into the construct using mutagenesis with partially overlapping primers with 3’-overhangs. To clone the construct with all 6 ETS sites ablated from GGAW to GCAW, we ordered the FoxF enhancer synthesized from IDT. For experiments using the 3kb FoxF enhancer and FoxF cDNA, the 3kb FoxF regulatory region was amplified from the genome and cloned into our electroporation vector, the FoxF cDNA was also cloned into this vector The FoxF cDNA sequence can be found in Aniseed with the Gene Model ID KH2012:KH.C3.170. The GATA4 G9 reporter plasmid for testing in iPSC-CMs consists of the following elements in order: GATA4 fragment, supercore promoter, unc76-EGFP, 30-nucleotide barcode and polyA.
Electroporation
Constructs to be compared to each other are midiprepped in the same batch using the NucleoBond Xtra Midi kit (Macherey-Nagel). For each construct, 70 μg of DNA in 100 μL is added to 400 μL of 0.96 M D-mannitol. Dechorionation, in vitro fertilization and electroporation were performed as described previously 64. Typically for each electroporation, eggs and sperm were collected from 6 adults. Embryos were fixed at the appropriate developmental stage for 15 minutes in 3.7% formaldehyde. The tissue was then cleared in a series of washes of 0.3% Triton-X in PBS and then of 0.01% Triton-X in PBS. Samples were mounted in Prolong Gold. Differential interference contrast microscopy was used to obtain transmitted light micrographs with an Olympus FV3000, using the 40X objective. The same microscope was used to obtain all fluorescent images. Three biological replicates were analyzed for each construct unless otherwise noted.
Counting embryos
In Ciona, as there are defined cell lineages, we can determine cell identity based on visually inspecting the nuclear signal and the region of the embryo in which the signal occurs. Strong, moderate, and weak expression levels through the eyepiece were determined as strong being visible at 25x of light source, moderate being visible at 50x of light source, and weak being visible at 100x of light source. Fifty embryos were counted for each biological replicate, unless otherwise noted. For each experiment, slides were counted blind.
Acquisition of Images
For enhancers being compared, images were taken from electroporations performed on the same day using identical settings. For representative images, embryos were chosen that represented the average from counting data. Representative embryos on the slide were chosen and imaged prior to unblinding the slides. Two exposure times were taken for each construct. In each figure, the same exposure time for each image is shown to allow direct comparison, which occasionally leads to overexposed images being used for stronger constructs (e.g., Fig 1C).
Electrophoretic mobility shift assay
EMSAs were performed using the LightShift™ Chemiluminescent EMSA Kit (ThermoFisher Scientific) with biotinylated and non-biotinylated double-stranded oligonucleotides corresponding to the ETS1 region of the FoxF enhancer and mutations that alter affinity to 0.97 and 1.00. Oligonucleotides were annealed according to the advanced protocol at this link (https://tools.thermofisher.com/content/sfs/brochures/TR0045-Anneal-oligos.pdf). Ciona ETS1 DBD protein was synthesized using the TNT Quick Coupled Transcription/Translation System (Promega) from the pTNT plasmid containing Ciona ETS1 DBD. Ciona ETS1 DBD was synthesized with flanking XhoI and NotI sites using Twist Bioscience and cloned into the pTNT-B18R vector (Addgene #58978). The Ciona ETS1 DBD sequence was determined as the sequence of Ciona ETS1 that aligns to Mouse ETS1 DBD65,66. The binding reaction was carried out in a 20 μL volume containing 2 μL of 10X Binding Buffer (100 mM Tris, 500 mM KCl, 10 mM DTT; pH 7.5), 50 ng Poly(dI:dC), 4 μL of protein extract and 20 femtomol biotin-labeled probe. For competition experiments, a 200-fold molar excess of unlabeled probe was added. Binding reactions were pre-incubated for 10 minutes before adding the biotin-labeled probe. Binding reactions were then incubated at room temperature for 20 min and loaded onto a DNA retardation gel (6%). After sample electrophoresis with 0.5X TBE on ice and transfer to a 0.45-μm BiodyneTM B Nylon membrane (ThermoFisher Scientific) in the cold room, DNA was crosslinked for 15 minutes using 312 nm light, and the membrane was put between blank sheets of paper overnight. The next day, the biotinylated probes were detected using the Chemiluminescent Nucleic Acid Detection Module (ThermoFisher Scientific). Images of the resulting membrane were acquired using a Chemidoc MP imaging system (Bio-Rad).
Counting Larvae for Migration Defects
Embryos were transferred from gelatin-coated electroporation plates to 15cm uncoated plastic plates filled with filtered seawater at 7 hpf. All well-developed larvae on the plate were counted from 16–20 hpf and scored for presence or absence of normal ATM development. All larvae with migration defects were marked for heart morphology analysis in the juvenile stage. For each replicate, 10–20 larva with normal ATM development were selected for heart morphology analysis in the juvenile stage.
Juvenile Heart Morphology Analysis and Imaging
Juvenile hearts were analyzed for morphological defects starting at 48hrs after metamorphosis. Hearts with abnormal structure or function were scored as “deformed.” Images and video of hearts were taken on an Axiozoom microscope with a mounted iPhone 7. Video editing was done in Adobe Premiere Pro. Heartbeat animations were made in Adobe Animate.
TVC Migration Time-lapse Imaging
Embryos were electroporated with MesP 2kb>GFP as previously described and checked for GFP fluorescence at 6 hpf. Well-developed embryos with strong GFP expression were transferred to a glass-bottom microscopy plate in seawater. Z-stacks were taken of each embryo every 30 minutes for 8 hours with an Olympus FV3000 microscope with a 20x objective lens. Resulting time-lapses were analyzed in Fiji. A max projection was taken of the GFP channel and merged with the DIC. Time-lapses were cropped and rotated in Adobe Photoshop.
Scoring Relative Affinities
Relative affinities were calculated using high-throughput binding datasets 67 (thebrain.bwh.harvard.edu/uniprobe/index.php). They were calculated using median signal intensities of mouse ETS1 universal protein binding microarray data from the UniProbe database 33. It has previously been shown that the binding specificity of ETS1 across 600 million years from flies to humans is conserved and thus the use of mouse ETS1 to calculate ETS affinity is a valid approach 34. The relative affinity represents the ratio of the median signal intensity of the native 8-mer motif to the optimal 8-mer motif for ETS.
Computational Search for Motifs Altered by each SNV
For each SNV, a 30 base-pair region centered on the ETS site was put into JASPAR and scanned for all 1205 vertebrate profiles with 80% cutoff. Then, the same 30 base-pair region centered on the ETS site with the SNV was put into JASPAR 68 and scanned for all 1205 vertebrate profiles with 80% cutoff. The list of sites destroyed and created by the SNV were found, we further filtered this list to focus on individual transcription factor sites with mutations that affect nucleotides critical for transcription factor binding. Of the ETS-family JASPAR profiles, we used MA0098.1.ETS1. For the GGAW to GCAW ablations, no TFBS for repressors expressed in the TVCs (ANISEED database) were created and no TFBS for activators expressed in the TVCs (ANISEED database) were destroyed. For the SNV opt mutations, no TFBS for repressors expressed in the ATMs (ANISEED database) were destroyed and no TFBS for activators expressed in the ATMs (ANISEED database) were created.
ATAC-seq Data Analysis
Alignment of ATAC-seq reads.
ATAC-seq alignment was performed following the methods in 42. Raw reads from 6 hours post fertilization (hpf) (GEO accession GSE126691, LacZ_6hpf_1–3, LacZ_10hpf_1–4) were first preprocessed by FastQC (version 0.11.2, http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Adaptors were trimmed using Trim Galore (version 0.4.4, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore) and trimmed reads were aligned to the Ciona robusta genome 69 using Bowtie2 (version 2.3.2, 70) with the parameters –very-sensitive -p 16 -X 1000. Reads with mapping quality score > 30 were kept for downstream analysis using SAMtools (version 1.2, 71). Mitochondrial reads were removed using bash command egrep -v. Following examination of read quality of the 6hpf data, replicate 2 was removed. Read pileup (BAM) files for replicates 1 and 3 at 6hpf were merged using the SAMtools merge function and peaks were called using MACS2 (version 2.7.9) 72 (--nomodel --bdg --g 99000000 -f BAMPE -q 0.01). To correct for nonspecific sequencing biases, we subtracted gDNA from these libraries 73.
Calculating top ATAC peaks.
To find the highest confidence peaks in our dataset, we calculated the area under the curve (AUC) as the sum of the read depth at each position in the peak. Bedtools genomecov (-bga) and intersect functions were used to calculate the read depth at every genomic position within all the called peaks 74. Peaks with AUCs in the top 90% were kept and the rest were discarded.
Downloading putative enhancers.
Human fetal heart putative enhancers were identified from epigenomic datasets from the key resource table. Any coordinates for hg18 or hg19 were lifted over to hg38 using pyliftover 0.4 (https://pypi.org/project/pyliftover).
Identifying affinity optimizing SNVs in putative developmental heart enhancers.
We first determined the distribution of ETS affinities in putative developmental heart enhancers. We collected affinities for all ETS sites defined as NNGGAWNN. Once all of the ETS binding site affinities were determined, we then performed all possible point mutations to each binding site and plotted those which resulted in ≥3-fold change. We reported the number of putative heart enhancers which contained ≥1 ETS which could be affinity-optimized ≥3 fold by a SNV.
Library Construction
A library containing Hs GATA4, Hs GATA4 SNVopt and Hs GATA4 4opt enhancers upstream of the supercore promoter, GFP and barcode, was constructed as previously described in 75.
iPSC-CM transfection and collection
iPSC-CMs from line UDID_109 and from line UDID_13947 were thawed and seeded onto gelatin-coated 48-well plates at a density of 1.3 × 105/cm2. The iPSC-CMs were transfected with 325nM/well of plasmid containing the enhancer library driving GFP using ViaFect™ Transfection Reagent (Promega) at a 6:1 ratio in a medium containing 10% FBS and 5uM (Y27632) ROCK Inhibitor (Sigma). 16hrs post transfection and every other day, medium was changed. Cells were then collected 96hrs post transfection and stored in TriZol reagent frozen at −80°C. Library transfection was performed in duplicate for line 109 and triplicate for line 139.
RNA extraction, cDNA synthesis, DNA extraction, barcode extraction, sequencing
Total RNA was extracted using TriZol extraction and treated to remove contaminating DNA (TURBO DNA-free DNase digestion kit, Ambion). Subsequent mRNA was isolated (Dynabeads mRNA Isolation kit, Invitrogen) and was used for cDNA synthesis (Transcriptor High Fidelity cDNA Synthesis kit, Sigma-Aldrich). Subsequent DNA extraction, barcode extraction, and sequencing was performed as previously described 75.
Enhancer to Barcode Assignment, Dictionary Analysis, and SEL-Seq Data Analysis
Assignment of barcodes to enhancers, dictionary setup and SEL-Seq data analysis was performed as previously described75, with the only difference being that assignment of barcodes to enhancers was done using PacBio sequencing. Active enhancers will transcribe the mRNA barcode, thus the mRNA barcodes were measured to calculate enhancer activity. The library was transfected into human iPSC-CMs, samples were collected 96 hours after transfection, and we isolated barcode mRNA and DNA. To normalize the enhancer activity to differences in the amount of plasmid, we calculated log2(mRNA RPM / DNA RPM). This was done in 6 biological replicates across 2 cell lines, with an average Pearson correlation coefficient between replicates of 0.86.
QUANTIFICATION AND STATISTICAL ANALYSIS
For comparisons of counting data, the chi-squared test was used in Excel with the CHISQ.TEST function. For the post hoc test to avoid false positives, Bonferroni correction was used to adjust thresholds for significance. With N comparisons, the Bonferroni-adjusted P value needed for significance is 0.05/N. Enhancer activity scores in Figure 4 were statistically analyzed using a standard two-sided T-test with Benjamini-Hochberg adjustment.
Supplementary Material
Movie S1. Ectopic FoxF expression in ATMs drives cell migration. Related to Figure 3. (Top) Embryo with WT FoxF 3Kb regulatory region driving the FoxF gene shows normal TVC migration to the ventral midline while ATMs remain in the tail and begin to elongate. (Bottom) Embryo electroporated with FoxF-ETS1-T-to-C 3Kb regulatory region driving FoxF gene shows migration of TVCs and ATMs together to the ventral midline. Timelapses taken from late neurula to late tailbud stages (St. 16–23).
Movie S2. Juvenile Ciona with ATM migration defect develops an extra heart. Related to Figure 3. (Left) The wild-type juvenile Ciona heart consists of a single linear chamber within the heart cavity in which peristaltic motion cycles blood through the body. (Right) Embryos with FoxF-ETS1-T-to-C driving FoxF with migrated ATMs were tracked through metamorphosis. This juvenile developed extra heart tissue, which contracts independently of the main heart.
Table S1: Table containing the wild-type and mutated ETS binding sites in FoxF enhancer. Related to Figures 1 and 2. The first sheet lists all of the FoxF enhancer variants and the sequence of each ETS transcription factor binding site. The subsequent sheets of the table contain a list of putative transcription factor binding sites created or deleted in the following constructs: FoxF -ETS1, FoxF -ETS2, FoxF -ETS3, FoxF -ETS4, FoxF -ETS5, FoxF -ETS6, FoxF ETS1T2C, FoxF ETS3T2G and FoxF ETS4T2C.
Highlights:
Low-affinity binding sites are prevalent within developmental heart enhancers
Single nucleotide variants that increase ETS affinity drive excess enhancer activity
Affinity-optimizing enhancer variants disrupt heart development.
Acknowledgements:
We thank the Farley and Frazer labs, especially Matteo D’Antonio for helpful discussions. We thank Jim Posakony, Mike Levine, and Krissie Tellez for a critical reading of the manuscript. We thank Swetha Mahesula, and Samara Reck-Peterson for assistance with the EMSA experiments. We thank Alta Fang for data analysis assistance. We thank the San Diego Supercomputer Center for providing computational resources through the Triton Shared Computer Cluster. We thank Steve LePage at M-REP for collection of Ciona used in this study. Figure 4 created with Biorender.com. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a NIH SIG grant (#S10 OD026929). This publication also includes data generated at the UC San Diego IGM Genomics Center utilizing a PacBio sequencer that was purchased with funding from NIH R01MH113715, NIH U01DA051234, and NIH P50DA037844.
Funding:
G.A.J. is supported by NIH R01HG011485 and has past support from a Hartwell fellowship, American Heart Association Grant 18POST34030077, NIH T32HL007444, and UC San Diego Chancellor’s Research Excellence Scholars Program. A.T.B. is supported by NIH T32GM133351. J.J.S. is supported by NIH T32GM127235. B.P.S. was supported by NIH T32GM133351. M.F.R. was supported by NIH T32GM008666. E.K.F., G.A.J., A.T.B., J.J.S., J.L.G., F.L., S.H.L., B.P.S., M.F.R., A.K., R.O.L. were supported by NIH DP2HG010013. K.A.F. and A.D-C. were supported by CIRM GC1R-06673-B, NSF-CMMI1728497, NIH U01HL107442.
Inclusion and Diversity:
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as living with a disability.
Footnotes
Declaration of Interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Levine M (2010). Transcriptional Enhancers in Animal Development and Evolution. Curr. Biol. 20, R754–R763. 10.1016/j.cub.2010.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryan GE, and Farley EK (2020). Functional genomic approaches to elucidate the role of enhancers during development. Wiley Interdiscip. Rev. Syst. Biol. Med. 12, e1467. 10.1002/wsbm.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lettice LA, Hill AE, Devenney PS, and Hill RE (2008). Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum. Mol. Genet. 17, 978–985. 10.1093/hmg/ddm370. [DOI] [PubMed] [Google Scholar]
- 4.Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, et al. (2014). Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375. 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tournamille C, Colin Y, Cartron JP, and Kim CLV (1995). Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat. Genet. 10, 224–228. [DOI] [PubMed] [Google Scholar]
- 6.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. (2012). Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 337, 1190–1195. 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tak YG, and Farnham PJ (2015). Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin 8, 57. 10.1186/s13072-015-0050-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Visel A, Rubin EM, and Pennacchio LA (2009). Genomic views of distant-acting enhancers. Nature 461, 199–205. 10.1038/nature08451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gallagher MD, and Chen-Plotkin AS (2018). The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 102, 717–730. 10.1016/j.ajhg.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harvey RP (2002). Patterning the vertebrate heart. Nat. Rev. Genet. 3, 544–556. 10.1038/nrg843. [DOI] [PubMed] [Google Scholar]
- 11.Zaffran S, and Frasch M (2002). Early Signals in Cardiac Development. Circ. Res. 91, 457–469. 10.1161/01.RES.0000034152.74523.A8. [DOI] [PubMed] [Google Scholar]
- 12.Sharrocks AD (2001). The ETS-domain transcription factor family. Nat. Rev. Mol. Cell Biol. 2, 827–837. 10.1038/35099076. [DOI] [PubMed] [Google Scholar]
- 13.Beiman M, Shilo BZ, and Volk T (1996). Heartless, a Drosophila FGF receptor homolog, is essential for cell migration and establishment of several mesodermal lineages. Genes Dev. 10, 2993–3002. 10.1101/gad.10.23.2993. [DOI] [PubMed] [Google Scholar]
- 14.Buckingham M, Meilhac S, and Zaffran S (2005). Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 6, 826–835. 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 15.Reifers F, Walsh EC, Leger S, Stainier DYR, and Brand M (2000). Induction and differentiation of the zebrafish heart requires fibroblast growth factor 8 (fgf8/acerebellar). Development 127, 225–235. [DOI] [PubMed] [Google Scholar]
- 16.House SL, House BE, Glascock B, Kimball T, Nusayr E, Schultz EJ, and Doetschman T (2010). Fibroblast Growth Factor 2 Mediates Isoproterenol-induced Cardiac Hypertrophy through Activation of the Extracellular Regulated Kinase. Mol Cell Pharmacol 2, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye M, Coldren C, Liang X, Mattina T, Goldmuntz E, Benson DW, Ivy D, Perryman MB, Garrett-Sinha LA, and Grossfeld P (2010). Deletion of ETS-1, a gene in the Jacobsen syndrome critical region, causes ventricular septal defects and abnormal ventricular morphology in mice. Hum. Mol. Genet. 19, 648–656. 10.1093/hmg/ddp532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davidson B, Shi W, Beh J, Christiaen L, and Levine M (2006). FGF signaling delineates the cardiac progenitor field in the simple chordate, Ciona intestinalis. Genes Dev. 20, 2728–2738. 10.1101/gad.1467706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delsuc F, Brinkmann H, Chourrout D, and Philippe H (2006). Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 439, 965–968. 10.1038/nature04336. [DOI] [PubMed] [Google Scholar]
- 20.Davidson B (2007). Ciona intestinalis as a model for cardiac development. Semin. Cell Dev. Biol. 18, 16–26. 10.1016/j.semcdb.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beh J, Shi W, Levine M, Davidson B, and Christiaen L (2007). FoxF is essential for FGF-induced migration of heart progenitor cells in the ascidian Ciona intestinalis. Development 134, 3297–3305. 10.1242/dev.010140. [DOI] [PubMed] [Google Scholar]
- 22.Christiaen L, Davidson B, Kawashima T, Powell W, Nolla H, Vranizan K, and Levine M (2008). The Transcription/Migration Interface in Heart Precursors of Ciona intestinalis. Science 320, 1349–1352. 10.1126/science.1158170. [DOI] [PubMed] [Google Scholar]
- 23.Crocker J, Abe N, Rinaldi L, McGregor AP, Frankel N, Wang S, Alsawadi A, Valenti P, Plaza S, Payre F, et al. (2015). Low Affinity Binding Site Clusters Confer Hox Specificity and Regulatory Robustness. Cell 160, 191–203. 10.1016/j.cell.2014.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delker RK, Ranade V, Loker R, Voutev R, and Mann RS (2019). Low affinity binding sites in an activating CRM mediate negative autoregulation of the Drosophila Hox gene Ultrabithorax. PLoS Genet. 15, e1008444. 10.1371/journal.pgen.1008444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farley EK, Olson KM, Zhang W, Brandt AJ, Rokhsar DS, and Levine MS (2015). Suboptimization of developmental enhancers. Science 350, 325–328. 10.1126/science.aac6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farley EK, Olson KM, Zhang W, Rokhsar DS, and Levine MS (2016). Syntax compensates for poor binding sites to encode tissue specificity of developmental enhancers. Proc. Natl. Acad. Sci. 113, 6508–6513. 10.1073/pnas.1605085113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kribelbauer JF, Rastogi C, Bussemaker HJ, and Mann RS (2019). Low-Affinity Binding Sites and the Transcription Factor Specificity Paradox in Eukaryotes. Annu. Rev. Cell Dev. Biol. 35, 357–379. 10.1146/annurev-cellbio-100617-062719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swanson CI, Schwimmer DB, and Barolo S (2011). Rapid Evolutionary Rewiring of a Structurally Constrained Eye Enhancer. Curr. Biol. 21, 1186–1196. 10.1016/j.cub.2011.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Razy-Krajka F, Gravez B, Kaplan N, Racioppi C, Wang W, and Christiaen L (2018). An FGF-driven feed-forward circuit patterns the cardiopharyngeal mesoderm in space and time. eLife 7, e29656. 10.7554/eLife.29656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothbächer U, Bertrand V, Lamy C, and Lemaire P (2007). A combinatorial code of maternal GATA, Ets and beta-catenin-TCF transcription factors specifies and patterns the early ascidian ectoderm. Dev. Camb. Engl. 134, 4023–4032. 10.1242/dev.010850. [DOI] [PubMed] [Google Scholar]
- 31.Woznica A, Haeussler M, Starobinska E, Jemmett J, Li Y, Mount D, and Davidson B (2012). Initial deployment of the cardiogenic gene regulatory network in the basal chordate, Ciona intestinalis. Dev. Biol. 368, 127–139. 10.1016/j.ydbio.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berger MF, and Bulyk ML (2009). Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat. Protoc. 4, 393–411. 10.1038/nprot.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei G-H, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, et al. (2010). Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 29, 2147–2160. 10.1038/emboj.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nitta KR, Jolma A, Yin Y, Morgunova E, Kivioja T, Akhtar J, Hens K, Toivonen J, Deplancke B, Furlong EEM, et al. (2015). Conservation of transcription factor binding specificities across 600 million years of bilateria evolution. eLife 4, e04837. 10.7554/eLife.04837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schachterle W, Rojas A, Xu S-M, and Black BL (2012). ETS-dependent regulation of a distal Gata4 cardiac enhancer. Dev. Biol. 361, 439–449. 10.1016/j.ydbio.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuqua T, Jordan J, van Breugel ME, Halavatyi A, Tischer C, Polidoro P, Abe N, Tsai A, Mann RS, Stern DL, et al. (2020). Dense and pleiotropic regulatory information in a developmental enhancer. Nature 587, 235–239. 10.1038/s41586-020-2816-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lettice LA, Williamson I, Wiltshire JH, Peluso S, Devenney PS, Hill AE, Essafi A, Hagman J, Mort R, and Grimes G (2012). Opposing functions of the ETS factor family define Shh spatial expression in limb buds and underlie polydactyly. Dev. Cell 22, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spivakov M (2014). Spurious transcription factor binding: non-functional or genetically redundant? BioEssays News Rev. Mol. Cell. Dev. Biol. 36, 798–806. 10.1002/bies.201400036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cooley J, Whitaker S, Sweeney S, Fraser S, and Davidson B (2011). Cytoskeletal polarity mediates localized induction of the heart progenitor lineage. Nat. Cell Biol. 13, 952–957. 10.1038/ncb2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cota CD, and Davidson B (2015). Mitotic Membrane Turnover Coordinates Differential Induction of the Heart Progenitor Lineage. Dev. Cell 34, 505–519. 10.1016/j.devcel.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 41.Jindal GA, and Farley EK (2021). Enhancer grammar in development, evolution, and disease: dependencies and interplay. Dev. Cell 56, 575–587. 10.1016/j.devcel.2021.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Racioppi C, Wiechecki KA, and Christiaen L (2019). Combinatorial chromatin dynamics foster accurate cardiopharyngeal fate choices. eLife 8, e49921. 10.7554/eLife.49921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richter F, Morton SU, Kim SW, Kitaygorodsky A, Wasson LK, Chen KM, Zhou J, Qi H, Patel N, DePalma SR, et al. (2020). Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat. Genet. 52, 769–777. 10.1038/s41588-020-0652-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benaglio P, D’Antonio-Chronowska A, Ma W, Yang F, Young Greenwald WW, Donovan MKR, DeBoever C, Li H, Drees F, Singhal S, et al. (2019). Allele-specific NKX2–5 binding underlies multiple genetic associations with human electrocardiographic traits. Nat. Genet. 51, 1506–1517. 10.1038/s41588-019-0499-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.May D, Blow MJ, Kaplan T, McCulley DJ, Jensen BC, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, et al. (2012). Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 44, 89–93. 10.1038/ng.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sylva M, van den Hoff MJB, and Moorman AFM (2014). Development of the human heart. Am. J. Med. Genet. A. 164A, 1347–1371. 10.1002/ajmg.a.35896. [DOI] [PubMed] [Google Scholar]
- 47.D’Antonio-Chronowska A, Donovan MKR, Young Greenwald WW, Nguyen JP, Fujita K, Hashem S, Matsui H, Soncin F, Parast M, Ward MC, et al. (2019). Association of Human iPSC Gene Signatures and X Chromosome Dosage with Two Distinct Cardiac Differentiation Trajectories. Stem Cell Rep. 13, 924–938. 10.1016/j.stemcr.2019.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Panopoulos AD, D’Antonio M, Benaglio P, Williams R, Hashem SI, Schuldt BM, DeBoever C, Arias AD, Garcia M, Nelson BC, et al. (2017). iPSCORE: A Resource of 222 iPSC Lines Enabling Functional Characterization of Genetic Variation across a Variety of Cell Types. Stem Cell Rep. 8, 1086–1100. 10.1016/j.stemcr.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crocker J, Preger-Ben Noon E, and Stern DL (2016). Chapter Twenty-Seven - The Soft Touch: Low-Affinity Transcription Factor Binding Sites in Development and Evolution. In Current Topics in Developmental Biology Essays on Developmental Biology, Part B, Wassarman PM, ed. (Academic Press; ), pp. 455–469. 10.1016/bs.ctdb.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 50.Rowan S, Siggers T, Lachke SA, Yue Y, Bulyk ML, and Maas RL (2010). Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes Dev. 24, 980–985. 10.1101/gad.1890410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai A, Muthusamy AK, Alves MR, Lavis LD, Singer RH, Stern DL, and Crocker J (2017). Nuclear microenvironments modulate transcription from low-affinity enhancers. eLife 6, e28975. 10.7554/eLife.28975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jindal GA, Goyal Y, Burdine RD, Rauen KA, and Shvartsman SY (2015). RASopathies: unraveling mechanisms with animal models. Dis. Model. Mech. 8, 769–782. 10.1242/dmm.020339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie Y, Su N, Yang J, Tan Q, Huang S, Jin M, Ni Z, Zhang B, Zhang D, Luo F, et al. (2020). FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 5, 181. 10.1038/s41392-020-00222-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grose R, and Dickson C (2005). Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev. 16, 179–186. 10.1016/j.cytogfr.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, et al. (2004). A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 119, 591–602. 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 56.Kheradpour P, Ernst J, Melnikov A, Rogov P, Wang L, Zhang X, Alston J, Mikkelsen TS, and Kellis M (2013). Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 23, 800–811. 10.1101/gr.144899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arnosti DN, Barolo S, Levine M, and Small S (1996). The eve stripe 2 enhancer employs multiple modes of transcriptional synergy. Dev. Camb. Engl. 122, 205–214. 10.1242/dev.122.1.205. [DOI] [PubMed] [Google Scholar]
- 58.Jia L, Landan G, Pomerantz M, Jaschek R, Herman P, Reich D, Yan C, Khalid O, Kantoff P, Oh W, et al. (2009). Functional enhancers at the gene-poor 8q24 cancer-linked locus. PLoS Genet. 5, e1000597. 10.1371/journal.pgen.1000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tuupanen S, Turunen M, Lehtonen R, Hallikas O, Vanharanta S, Kivioja T, Björklund M, Wei G, Yan J, Niittymäki I, et al. (2009). The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 41, 885–890. 10.1038/ng.406. [DOI] [PubMed] [Google Scholar]
- 60.French JD, Ghoussaini M, Edwards SL, Meyer KB, Michailidou K, Ahmed S, Khan S, Maranian MJ, O’Reilly M, Hillman KM, et al. (2013). Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am. J. Hum. Genet. 92, 489–503. 10.1016/j.ajhg.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang Q, Whitington T, Gao P, Lindberg JF, Yang Y, Sun J, Väisänen M-R, Szulkin R, Annala M, Yan J, et al. (2014). A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat. Genet. 46, 126–135. 10.1038/ng.2862. [DOI] [PubMed] [Google Scholar]
- 62.Lim F, Ryan GE, Le SH, Solvason JJ, Steffen P, and Farley EK Affinity-optimizing variants within the ZRS enhancer disrupt limb development. bioRxiv. 10.1101/2022.05.27.493789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.D’Antonio-Chronowska A, D’Antonio M, and Frazer KA (2020). In vitro Differentiation of Human iPSC-derived Cardiovascular Progenitor Cells (iPSC-CVPCs). Bio-Protoc. 10, e3755. 10.21769/BioProtoc.3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Christiaen L, Wagner E, Shi W, and Levine M (2009). Electroporation of Transgenic DNAs in the Sea Squirt Ciona. Cold Spring Harb. Protoc. 2009, pdb.prot5345. 10.1101/pdb.prot5345. [DOI] [PubMed] [Google Scholar]
- 65.De Val S, Anderson JP, Heidt AB, Khiem D, Xu S-M, and Black BL (2004). Mef2c is activated directly by Ets transcription factors through an evolutionarily conserved endothelial cell-specific enhancer. Dev. Biol. 275, 424–434. 10.1016/j.ydbio.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 66.Nye JA, Petersen JM, Gunther CV, Jonsen MD, and Graves BJ (1992). Interaction of murine ets-1 with GGA-binding sites establishes the ETS domain as a new DNA-binding motif. Genes Dev. 6, 975–990. 10.1101/gad.6.6.975. [DOI] [PubMed] [Google Scholar]
- 67.Hume MA, Barrera LA, Gisselbrecht SS, and Bulyk ML (2015). UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein–DNA interactions. Nucleic Acids Res. 43, D117–D122. 10.1093/nar/gku1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Pérez N, et al. (2022). JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173. 10.1093/nar/gkab1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Satou Y, Nakamura R, Yu D, Yoshida R, Hamada M, Fujie M, Hisata K, Takeda H, and Satoh N (2019). A Nearly Complete Genome of Ciona intestinalis Type A (C. robusta) Reveals the Contribution of Inversion to Chromosomal Evolution in the Genus Ciona. Genome Biol. Evol. 11, 3144–3157. 10.1093/gbe/evz228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Toenhake CG, Fraschka SA-K, Vijayabaskar MS, Westhead DR, van Heeringen SJ, and Bártfai R (2018). Chromatin Accessibility-Based Characterization of the Gene Regulatory Network Underlying Plasmodium falciparum Blood-Stage Development. Cell Host Microbe 23, 557–569.e9. 10.1016/j.chom.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Song BP, Ragsac MF, Tellez K, Jindal GA, Grudzien JL, Le SH, and Farley EK (2022). Diverse logics and grammar encode notochord enhancers. bioRxiv. 10.1101/2022.07.25.501440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Ectopic FoxF expression in ATMs drives cell migration. Related to Figure 3. (Top) Embryo with WT FoxF 3Kb regulatory region driving the FoxF gene shows normal TVC migration to the ventral midline while ATMs remain in the tail and begin to elongate. (Bottom) Embryo electroporated with FoxF-ETS1-T-to-C 3Kb regulatory region driving FoxF gene shows migration of TVCs and ATMs together to the ventral midline. Timelapses taken from late neurula to late tailbud stages (St. 16–23).
Movie S2. Juvenile Ciona with ATM migration defect develops an extra heart. Related to Figure 3. (Left) The wild-type juvenile Ciona heart consists of a single linear chamber within the heart cavity in which peristaltic motion cycles blood through the body. (Right) Embryos with FoxF-ETS1-T-to-C driving FoxF with migrated ATMs were tracked through metamorphosis. This juvenile developed extra heart tissue, which contracts independently of the main heart.
Table S1: Table containing the wild-type and mutated ETS binding sites in FoxF enhancer. Related to Figures 1 and 2. The first sheet lists all of the FoxF enhancer variants and the sequence of each ETS transcription factor binding site. The subsequent sheets of the table contain a list of putative transcription factor binding sites created or deleted in the following constructs: FoxF -ETS1, FoxF -ETS2, FoxF -ETS3, FoxF -ETS4, FoxF -ETS5, FoxF -ETS6, FoxF ETS1T2C, FoxF ETS3T2G and FoxF ETS4T2C.
Data Availability Statement
Sequencing data have been deposited at SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Microscopy and scoring data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOI is listed in the key resources table.
Bed files of putative Ciona developmental heart enhancers and putative human developmental heart enhancers have been deposited at GitHub and DOI is listed in the key resources table
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resource table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| mouse ETS1 universal PBM data | Wei et al.33 | thebrain.bwh.harvard.edu/uniprobe/index.php |
| Ciona robusta genome | Satou et al.69 | N/A |
| LacZ_6hpf_1–3 | Racioppi et al.42 | GEO: GSE126691 |
| Human reference genome NCBI build 38, GRCh38 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human |
| Human fetal heart putative enhancers predicted from H3K27ac ChIP and ATAC-seq | Richter et al.43 | Supplementary Table 11 in source |
| iPSC-cardiomyocyte putative enhancers predicted from ATAC- seq and H3K27ac ChIP | Benaglio et al.44 | GEO: GSE133833 |
| Human fetal heart putative enhancers predicted from p300/CBP | May et al.45 | GEO: GSE32587 |
| Sequencing data for MPRA | This paper | SRA: PRJNA999706 |
| Experimental Models: Organisms/Strains | ||
| Ciona intestinalis type A (Ciona robusta) | M-REP | N/A |
| iPSC-CMs | Frazer Lab | UDID_10948 |
| iPSC-CMs | Frazer Lab | UDID_13948 |
| Oligonucleotides | ||
| Oligonucleotides for mutagenesis, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: foxf295-bpfog>H2B-mCherry | Levine & Christiaen Labs | N/A |
| Plasmid: foxf295-ets1mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets2mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets3mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets4mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets5mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets6mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets1optT2C-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets3optT2G-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets4optT2C-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets134opt-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets134optAbl-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets12345opt-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets12345optAbl-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf295-ets123456mut-bpfog>H2B-mCherry | This paper | N/A |
| Plasmid: foxf3kb>foxfcDNA | This paper | N/A |
| Plasmid: foxf3kb-ets1OptT2C>foxfcDNA | This paper | N/A |
| Plasmid: foxf3kb-ets3OptT2G>foxfcDNA | This paper | N/A |
| Plasmid: foxf3kb-ets4OptT2C>foxfcDNA | This paper | N/A |
| Plasmid: gata4-g9-scp>unc76-GFP | This paper | N/A |
| Plasmid: gata4-g9-SNVopt-scp>unc76-GFP | This paper | N/A |
| Plasmid: gata4-g9–4opt-scp>unc76-GFP | This paper | N/A |
| Plasmid: pTNT-ciETS1 | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | Schneider et al.76 | https://imagej.nih.gov/ij/ |
| FastQC (version 0.11.2) | N/A | http://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| Trim Galore (version 0.4.4) | N/A | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore |
| Bowtie2 (version 2.3.2) | Langmead et al.70 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools (version 1.2) | Li et al.71 | https://samtools.sourceforge.net |
| MACS2 (version 2.7.9) | Zhang et al.72 | N/A |
| BEDTools | Quinlan et al.74 | N/A |
| pyliftover 0.4 | N/A | https://pypi.org/project/pyliftover |
| Adobe Premiere Pro | Adobe | N/A |
| Adobe Animate | Adobe | N/A |
| Code to generate Fig 2A, 4A, 4C and putative Ciona developmental heart enhancers and putative human developmental heart enhancers | This paper | https://doi.org/10.5281/zenodo.8215976 |