

Abstract
Precise delivery of genes to therapy-relevant cells is crucial for in vivo gene therapy. Receptor-targeting as prime strategy for this purpose is limited to cell types defined by a single cell-surface marker. Many target cells are characterized by combinations of more than one marker, such as the HIV reservoir cells. Here, we explored the tropism of adeno-associated viral vectors (AAV2) displaying designed ankyrin repeat proteins (DARPins) mono- and bispecific for CD4 and CD32a. Cryo-electron tomography revealed an unaltered capsid structure in the presence of DARPins. Surprisingly, bispecific AAVs transduced CD4/CD32a double-positive cells at much higher efficiencies than single-positive cells, even if present in low amounts in cell mixtures or human blood. This preference was confirmed when vector particles were systemically administered into mice. Cell trafficking studies revealed an increased cell entry rate for bispecific over monospecific AAVs. When equipped with an HIV genome-targeting CRISPR/Cas cassette, the vectors prevented HIV replication in T cell cultures. The data provide proof-of-concept for high-precision gene delivery through tandem-binding regions on AAV. Reminiscent of biological products following Boolean logic AND gating, the data suggest a new option for receptor-targeted vectors to improve the specificity and safety of in vivo gene therapy.
Keywords: DART-AAV, receptor-targeting, designed ankyrin repeat protein, HIV reservoir, CRISPR-Cas, FcγRIIA
Graphical Abstract
Introduction
AAV vectors are the prime choice for in vivo gene therapy to treat a wide range of inherited and acquired diseases [1]. Several AAV-based medicinal products have reached the market addressing diseases such as hemophilia [2], muscular atrophies [3] and retinal dystrophies [4]. The non-enveloped icosahedral capsid is composed of the three viral proteins VP1, VP2 and VP3, which are encoded in overlapping reading frames and differ in the lengths of their N-termini. When configured as a recombinant gene transfer vector, only the flanking ITR (inverted terminal repeat) regions are retained for packaging of the gene of interest, making AAV vectors replication-incompetent [5]. Because of their excellent safety profile and low immunogenicity, AAVs are currently the most prominent vector system for in vivo gene delivery.
Nonetheless, the broad tropism of all naturally occurring serotypes as well as pre-existing immunity can result in severe toxicities, such as hepatotoxicity, and limit delivery efficiencies to therapy-relevant cell types [6–8]. Capsid engineering offers the potential for targeting AAVs to particular cell types while reducing required vector doses [5]. Library-based screening approaches for capsid variants and rational capsid engineering strategies can be distinguished [9]. For rational design, ligands with high-affinity for a receptor expressed on the surface of the targeted cell type are displayed on capsids whose natural receptor recognition sites have been blinded [10]. In the case of AAV2, binding to heparan sulfate proteoglycan (HSPG) is ablated through two point mutations, R585A and R588A [11]. Initially, the N-terminus of VP2 was used to genetically fuse designed ankyrin repeat proteins (DARPins) specific for tumor antigens such as Her2/neu [12], or the lymphocyte marker CD4 [13]. More recently, the AAV2 GH2/3 loop of VP1 was shown to tolerate insertions of nanobodies [14,15] and DARPins [16]. Data available so far indicate that this insertion site improves production yields and gene transfer rates. The corresponding DART (designed ankyrin repeat protein targeted) AAVs displaying DARPins specific for mouse CD8 or glutamate receptor subunit 4 (GluA4), were highly selective for the respective target cells while gene transfer rates were much higher than those achieved with unmodified AAV2 [16,17].
While these monospecific AAVs efficiently transfer genes into their target-receptor-positive cell populations, success with this system relies on the definition of the therapy-relevant target cells by a single cell-surface marker. Often, however, target cells are defined by a specific combination of more than one surface marker. An example is the latently infected HIV reservoir [18]. While more and more surface markers are being identified, none of them defines the reservoir cells by its own. Among them is the FcγRIIa (CD32a), which is overexpressed on HIV latent reservoir cells [19]. Although not uncontroversial [20], CD32a is currently among the most promising markers for identifying reservoir cells [21,22] since it is expressed during the latent and transcriptionally active HIV infection [21,23], localized in infected cells in main tissue reservoirs such as the lymph nodes, the gastrointestinal tract [24], and the cervical tissue [25]. Moreover, a significant fraction of latently infected cells that become reactivated, either naturally or using latency reversal agents (LRA), express this molecule [26]. Confirmation for the expression of CD32a on a small population of CD4+ T lymphocytes (up to 1.2% of CD4 T cells) has recently been demonstrated with DARPin F11, which, in contrast to available antibodies, discriminates between cells expressing CD32a and cells expressing CD32b, a closely related Fcγ receptor [27]. Since CD32a is also expressed by other cells of the hematopoietic system, most prominently platelets [28], additional markers, like CD4, are required to define these cells more precisely and uniquely. However, a vector system addressing two cell surface receptors simultaneously, has not yet been designed and evaluated.
Due to their high stability and aggregation resistance, DARPins are especially well suited to be linked in tandem for multivalent recognition of surface receptors [29,30]. Here, we report the generation and characterization of an engineered AAV2 displaying DARPins specific for CD4 and CD32a either individually or in tandem in the GH2/3 loop of VP1. Much to our surprise, these particles were not only highly specific and efficient in delivering genes into target receptor positive cells, but the bispecific AAVs preferentially targeted cells expressing both markers. The data demonstrate that such particles are structurally unaltered from unmodified AAV2, can be produced with similar efficiency and deliver reporter as well as therapeutic gene cassettes ex vivo as well as in vivo.
Material and methods
Plasmids and molecular cloning
Plasmids encoding DARPins F11 or 55.2 within the GH2/3 loop of VP1 were generated according to [16]. To construct these plasmids, a (G4S)4-linker followed by His- and my-tag was added to the N-terminus of the DARPin sequence, while a G4A-linker was added to the C-terminus. Using the restriction sites SfiI and SpeI the DARPin coding sequence was inserted into the pRC_RR_VP1_r1c3 (Addgene: 65724) [16]. For bispecific AAVs, the N-terminal linker was shortened to a (G4S)2-linker, and both DARPins were separated by a (G4S)3-linker. All targeting plasmids contained point mutations in the splice acceptor site of the common VP2/VP3 coding sequence to prevent VP2- and VP3-DARPin expression. In all productions, VP2 and VP3 expression were complemented from the pRC-VP1-KO plasmid containing a mutated start codon of VP1 [16]. Additionally, all plasmids were mutated to ablate the binding site of the AAV2 capsid proteins for HSPG by introducing R585A and R588A [12].
The EF1α-promoter in pssAAV-EF1α-spCas9 was replaced by the miniature H1 promoter [31] followed by the coding sequence of the gRNA (GTCTCCGCTTCTTCCTGCCAT) [32] resulting in plasmid pssAAV-H1-gRNA-spCas9. This plasmid was used for the production of the ‘all-in-one’ F11-55.2-AAV-Cas9-gRNA vector.
Generation and cultivation of cell lines
HEK-293T (ATCC CRL-11268) cells were cultured in Dulbecco’s modified Eagle’s medium high glucose (Sigma-Aldrich, D6546) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich, F7524) and 1% L-glutamine (Sigma-Aldrich, G7513). SupT1 CD4+ cells (SupT1, DSMZ, ACC 140) were kept in advanced RPMI (Gibco, 12633-012) supplemented with 4% FBS and 1% L-glutamine (advanced RPMI complete). The SupT1 CD4+CD32a+ cell line was generated by transduction with VSVG pseudotyped lentiviral vector transferring the coding sequence of CD32a driven by SFFV promotor. In addition to the CD32a modification, SupT1 CD32a+ cells were treated with a VSVG pseudotyped lentiviral vector encoding a saCas9-gRNA cassette (gRNA ATCTTGCAGAACCAGAAGAAGG) to abolish CD4 expression. SupT1 CD32a+ and SupT1 CD4+CD32a+ were cultured in advanced RPMI complete supplemented with 4 μg/ml puromycin (Gibco, A11138-03).
Primary cells
Human PBMC were isolated from healthy donors or from buffy coats acquired from DRK Blutspendedienst (Frankfurt, Germany) under ethics approval by the committee of Frankfurt University Hospital. PBMC were separated from plasma and red blood cells using Histopaque (Histopaque-1077, Sigma-Aldrich, 10771) gradient density centrifugation. For T cell activation, cells were activated with anti-CD3 (1 μg/ml, Miltenyi Biotec, 130-093-387) antibody coated on a 48-well plate and anti-CD28 (3 μg/ml, Miltenyi Biotec, 130-093-375) added to NutriT-media (4Cell Nutri-T media; Sartorius, 05-11F2001; containing 0.5% penicillin/streptomycin) supplemented with IL-7 (25 U/ml, Miltenyi Biotec, 130-095-361) and IL-15 (50 U/ml, Miltenyi Biotec, 130-095-764).
AAV production
The production and purification of AAV2 via iodixanol gradient centrifugation was described before [16,33]. Optionally, the buffer of AAV recovered from the 40% iodixanol (OptiPrep, Serumwerk, 1893) phase was exchanged to PBS + 0.001% Pluronic (Poloxamer 188 solution, Sigma-Aldrich, P5556) using Amicons (Amicon Ultra-4, Merck Millipore, UFC805024). For Western blot analysis, the cell culture supernatant of transfected HEK293T cells containing AAV particles was filtered (0.45 μm, minisart NML plus, Sartorius, 17829) and digested with 50 U/ml Benzonase (Merck Millipore, E1014) for 30 min at 37°C. Volume was adjusted to 27 ml with PBS and transferred to ultracentrifugation tubes (Quick-seal ultra-clear, Beckman coulter, 344322). Subsequently, 5 ml of 40% sucrose (Sigma-Aldrich, 84097) in TNE buffer (TBS [pH 7.4], EDTA 0.1 mM) was underlaid and centrifuged at 100,000 × g for 16 h. The pellet was resuspended in PBS and stored in aliquots at −80°C. For determining vector genome copies, DNA was isolated from 3 μl vector stock following the kit’s protocol (DNeasy Blood and Tissue kit, Qiagen, 69506). DNA was eluted in 200 μl elution buffer and 5 μl were used to determine signal amplification by qPCR using ITR-specific primers (5’GGAACCCCTAGTGATGGAGTT3’, 5’CGGCCTCAGTGAGCGA3’ and probe 5’6FAM-CACTCCCTCTCTGCGCGCTCG-TAMRA3’. Template DNA was mixed with 15 μl LightCycler 480 Probes Master mix (Roche Diagnostics, 04707494001) and signal amplification was measured on LightCycler 480 II Real-Time PCR System (Roche).
Subcellular fractionation
1×1010 SupT1-CD4/CD32a cells were pre-chilled for 10 min at 4°C followed by incubation with 3×1010 GC of the respective AAV stock for 10 min at 4°C. To remove unbound particles, cells were washed with PBS and resuspended in 1 ml medium. Then, cells were transferred to 48-well plates and incubated for 2 h at 37°C to allow vector endocytosis. Afterwards, cells were washed again in PBS and processed with the subcellular fractionation kit (Thermo Scientific, 78840). From all fractions DNA was isolated using the DNeasy 96 Blood & Tissue Kit (Qiagen, 69581) and eluted in 200 μl elution buffer. 5 μl eluate was used to determine the amount of GFP encoding vector genomes by qPCR using primers 5’ATCATGGCCGACAAGCAGAA3’, 5’TCTCGTTGGGGTCTTTGCTC3’ and probe 5’Cy5-CGGCCCCGTGCTGCTGCCCGA-BHQ3-3’.
Whole blood assay
400 μl human whole blood was mixed with 1×104 SupT1-CD4+/CD32a+ cells and 8×108 GCs of the respective AAV in a 48-well plate at 400 rpm for 6 h in an incubator. Subsequently, blood was diluted with 600 μl PBS and overlaid on 800 μl Histopaque. After centrifugation for 30 min at 1800 × g at room temperature, mononuclear cells were isolated and washed to remove platelets. For cell activation, cells were suspended in NutriT supplemented with cytokines + anti-CD28 and transferred to anti-CD3 coated 48-well plates.
Animal experiments
All animal experiments were performed in accordance with the regulations of the German animal protection law and the respective European Union guidelines under authorization of the local authority (F107/2007). To evaluate the biodistribution of F11-AAV in vivo, NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl) were intravenously injected with a mix of 5×105 SupT1-CD4/CD32a cells and 5×105 HUT87 cells followed by i.v. injection of 2×1011 F11-AAV. Three and seven days after vector application mice were sacrificed. Single cells were isolated from the liver using the liver dissociation kit (Miltenyi Biotec, 130-105-807) and cells from bone marrow were processed as described before [34]. GFP expression was determined by flow cytometry. For in vivo gene delivery by bispecific AAVs, NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl) were i.v. injected with a mixture of 1×106 SupT1-CD4, 1×106 SupT1-CD32a and 1×106 SupT1-CD4/CD32a cells. Thirteen days after cell application 2×1011 GC of the respective AAV were injected i.v. and mice were sacrificed three days after vector application. Cells were isolated from bone marrow and GFP expression was determined by flow cytometry.
HIV production and infections
To generate stocks of the LAI strain of HIV-1, HEK293T cells were transfected with 5 μg of pLAI using Lipofectamine 2000. After two days, the culture supernatant was collected, filtered through a 0.45 μm filter, and then divided into aliquots. The multiplicity of infection (MOI) was determined using previously established methods [35]. Transduced SupT1 cells (2.5e5 cells in 1 ml of medium) were exposed to HIV LAI (MOI 0.02) and cultivated for 12 days with bi-weekly passages. To follow virus replication cell supernatant and cells were collected from each passage and used to determine the levels of CA-p24 antigen using enzyme-linked immunosorbent assay (in-house assay) and flow cytometry. For the determination of HIV-1 CA-p24 protein in the supernatant, the HIV-1 Gag p24 DuoSet antibody (R&D Systems, Minneapolis, USA) with the LumiPhos-HRP substrate reagent was used for detection, following the manufacturer’s protocol. The luminescence was quantified on a GloMax® plate reader. For determination of CA-p24 antigen in cells, harvested cells were washed with PBS containing 1% FBS (Gibco™, Thermo Fisher Scientific, Waltham, Massachusetts, USA) and fixed with 4% PFA in PBS for 30 min at room temperature. Fixed cells were permeabilized with BD Perm/Wash buffer (BD Biosciences) and stained for 30 min at 4°C with the fluorochrome-labeled monoclonal antibody p24-APC (clone 28B7). Samples were analysed on a BD FACS Canto II flow cytometer equipped with FACSDiva 8.0 (BD Biosciences). Results were analyzed via FCS Express (DE Novo Software).
Flow cytometry to determine reporter gene transfer
Cells were washed twice in wash buffer (PBS, 2% FBS, 2 mM NaN3) prior to staining. Cells from the whole blood assay were stained with an antibody mix containing anti-CD8 (VioBlue, Mitlenyi Biotec, 130-110-683), anti-CD3 (PerCP, Mitlenyi Biotec, 130-113-131), anti-CD4 (PE-Cy7, BD Bioscience, 560644), anti-CD32 (APC, Biolegend, 303208) and viability dye eFluor780 (ebioscience, 65-0865-18).
For the binding assay of AAV particles to non-activated PBMC, bound cells were identified by staining of the AAV capsid with the A20 anti-AAV2 antibody (Progen, 61555) for 30 min at 37°C followed by incubation with the secondary antibody anti-mouse IgG (AlexaFluor 647, 0.5 μl per sample, Jackson immune research, 115-605-164) for 20 min at 4°C. After three washing steps cells were incubated with anti-CD4 (VioBlue, Mitlenyi Biotec, 130-113-219), anti-CD14 (VioGreen, Miltenyi Biotec, 130-113-153), anti-CD8 (FITC, Miltenyi Biotec, 130-113-157), anti-CD3 (PerCP, Miltenyi Biotec, 130-113-131), anti-CD19 (PE-Vio 770, Miltenyi Biotec, 130-113-170) and viability dye eFluor780 (ebioscience, 65-0865-18).
SupT1 cells used for vector titration were stained with anti-CD4 (VioBlue, Mitlenyi Biotec, 130-113-219), anti-CD32 (APC) and viability dye eFluor780.
Mouse-derived single cells were blocked with mouse Fc-block (Miltenyi Biotec, 130-092-575) and subsequently stained with anti-hCD45 (VioBlue, Mitlenyi Biotec, 130-113-122), anti-CD4 (Pe-Vio770, Mitlenyi Biotec, 130-113-216), anti-CD32a (APC) and viability dye eFluor780. After staining, cells were washed twice with wash buffer and fixed using 1% paraformaldehyde in PBS. Measurements were acquired on MACSQuant 10 (Miltenyi Biotec) and results were analyzed via FCS Express (De Novo Software) or FlowJo (BD Bioscience).
Western blotting
AAV vector stocks were lysed in 1x urea buffer containing 200 mM Tris/HCl [pH 8.0], 5% SDS, 8 M urea, 0.1 mM EDTA, 2.5% DTT, 0.03% bromophenol blue at 95°C for 10 min. Lysates were separated on a 4–15% gradient gel and afterwards blotted onto nitrocellulose membrane (Amersham Protran, 10600004). Subsequently, the membrane was blocked in 10% horse serum. Primary antibodies anti-AAV VP1/VP2/VP3 (1:50, clone B1, Progen, 690058) and anti-myc tag (1:100, clone: Myc.A7, Abcam, ab18185) were incubated with the membrane over night at 4°C followed by incubation with the secondary antibody rabbit anti-mouse-HRP (Agilent, P0260) for 2 hours at room temperature. All antibodies were diluted in TBS-T (50 mM Tris/HCl, 150 mM NaCl, 0.1% Tween-20 [pH 7.4]) containing 5% horse serum. Chemiluminescent signals were detected after incubation with HRP substrate (ECL western blotting substrate, Pierce, 32106) and visualized on the MicroChemi (DNR Bio-Imaging sysems) imaging system. Contrast of images was adjusted using Fiji retaining relative signal strength.
Transmission electron microscopy
20 μl of AAV stocks were placed on 200-mesh nickel grids and incubated for 10 min at RT. Grids were washed twice in PBS, once with ddH2O and negatively stained with 1% phosphotungstic acid for 20 s at RT. Samples were visualized using a JEM-1400Flash electron microscope (JEOL). Contrast of images was adjusted using ImageJ retaining relative signal strength.
Sample preparation for cryo-electron microscopy
Electron microscopy grids Cu 200 mesh, carbon R 2/2 (Quantifoil) were glow-discharged using Solarus 950 (Gatan). 5 μl of AAV stock were incubated on glow-discharged EM grids for 1–2 minutes on parafilm and blotted away using a Whatman No 1 paper. Subsequently the grid was placed into an automated plunge freezer FEI Vitrobot Mark IV and 3 μl of AAV preparation was applied onto the grid. The excess liquid was removed by 3 sec blotting using blot force 0 and the sample was vitrified by plunge-freezing into liquid ethane. 10 nm protein-A coated colloidal gold (Aurion) was added into the AAV preparations.
Cryo-electron tomography acquisition and tomogram reconstruction
Tilt series were collected on a Titan Krios Transmission Electron Microscope (FEI, now ThermoFisher Scientific) operated at 300 keV and equipped with a BioQuantum® LS energy filter with a slit width of 10 eV and K3 direct electron detector (Gatan). Tilt series were acquired at 81,000-fold magnification (1.106 Å pixel size) using a dose symmetric acquisition scheme [36] with an electron dose of approximately 3 e−/Å2 per projection with tilt ranges from 60° to −60° in 3° increments in SerialEM [37] with defocus −3 μm. Tomograms were reconstructed using the IMOD software package [38]. Tilt series were aligned using gold fiducials, corrected using a contrast transfer function (CTF), and dose-filtration implemented in IMOD. Reconstruction was performed by weighted back-projections with a simultaneous iterative reconstruction technique (SIRT)-like filter equivalent to 5 iterations. For visualization, 10 slices of the final tomogram were averaged.
Software
GraphPad Prism 9.5.0 was used to design graphs and calculate statistical differences. Flow cytometry data were analysed by FCS Express 6.06 (De Novo Software) or FlowJo 10.7.1 (BD Bioscience). AlphaFold run on the Colab-Fold server [39] were used to de novo fold VP3 conjugated to F11-55.2 DARPins and resulting structures were visualized with Chimera 1.16. Image histogram stretching was performed in Fiji (ImageJ 2.3.0) [40].
Results
Implementation and initial characterization of bispecific DARPin-displaying AAV vectors
As targeting ligands, we chose DARPins specific for CD4 (55.2) [41] and CD32a (F11) [27]. F11 reliably discriminates CD32a from the closely related CD32b receptor with an affinity of 6.1 nM. DARPin 55.2 has a higher affinity of 0.84 nM for its CD4 target. For display on the AAV2 capsid surface, the two DARPins were connected with each other in both orientations (55.2-F11 and F11-55.2) by a (G4S)3 linker. These bispecific DARPins were then inserted into the GH2/3 loop of VP1 on the genetic level as described previously for a mouse CD8 DARPin [16]. When combined with a plasmid encoding the VP2 and VP3 proteins whose HSPG-binding site was mutated, this resulted in particles displaying bispecific DARPins (Fig. 1A). To demonstrate proper DARPin insertion into VP1, AAV particles displaying either monospecific F11 or bispecific F11-55.2 were assessed by Western blot analysis. Both types of particles showed the expected signals for all VP proteins, including small shifts in electrophoretic mobility for VP2/VP3 due to the point mutations R585A and R588A as well as strong shifts for the DARPin-VP1 proteins as compared to unmodified AAV2 (Fig. 1B). Moreover, AAVs displaying the bispecific DARPins exhibited a supershifted signal according to the higher molecular weight. Detection of the DARPins via the conjugated myc-tag confirmed the identity of the bands (Fig. 1B). Cryo-electron tomography (cryo-ET) visualized the expected capsid morphology in 3D revealing that the DARPin modifications had not interfered with capsid integrity (Fig. 1C). While the 55.2-F11 DARPin was not clearly visible, most likely due to the highly flexible linker, the overall capsid morphology of 55.2-F11-AAV was indistinguishable from that of AAV2 (Fig. 1C). Using negative staining transmission electron microscopy, we could show homogeneity of the vector preparations and identified 98% particles containing a genome. Interestingly, 55.2-F11-AAV contained significantly more empty particles (92% full) (Fig. 1E). Genomic titers for all AAV types having the GFP reporter gene packaged of around 1×1010 genome copies (GC) per μl were in the range of what is usually obtained with unmodified AAV2 (Fig. 1F).
Figure 1:
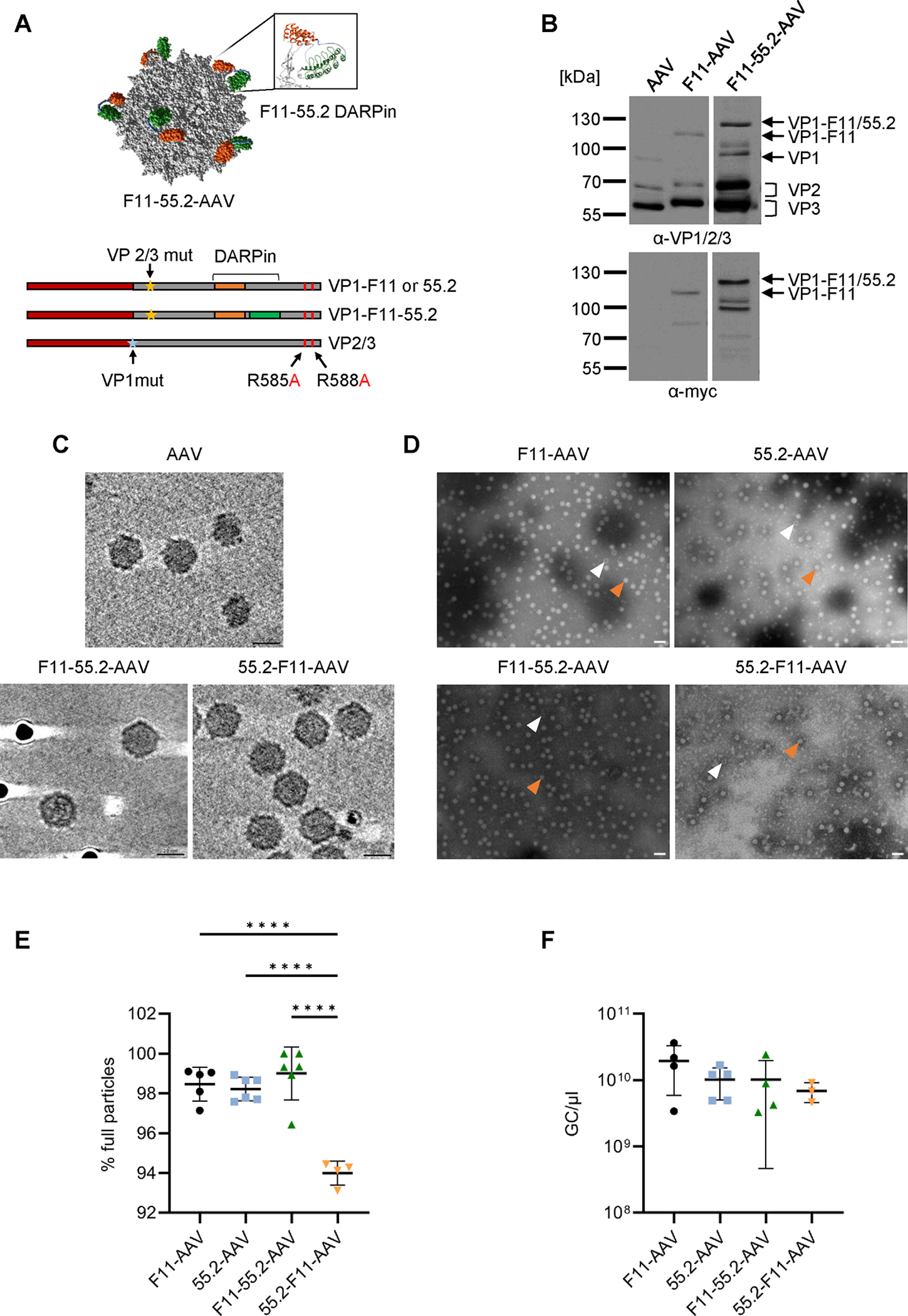
Biophysical characterization of bispecific AAV vectors.
A) Upper part: AlphaFold capsid structure prediction of bispecific CD32a-CD4-AAV based on the structure of AAV2 particles (PDB:1LP3). 55.2-F11 conjugated to VP3 was de novo folded and overlayed onto the particle crystal structure. DARPin F11 is shown in green, 55.2 in orange. The inlet shows the GH2/3 loop with the bispecific DARPin inserted. Lower part: Schematic drawings of reading frames in the plasmids encoding the capsid proteins. Yellow asterisks indicate disrupted splice acceptor sites preventing expression of VP2/3. The blue asterisk indicates the mutated start codon of VP1. DARPin reading frames are highlighted by orange and blue bars. The point mutations R585A and R588A ablating HSPG binding are labeled by arrows. B) Detection of capsid proteins in the indicated AAV stocks by Western blotting using the anti-VP1/VP2/VP3 B1 antibody (upper panel) and a myc-specific antibody (bottom panel). Contrast of images were adjusted retaining relative signal strength. C-E) Morphology and full/empty particle ratio of AAV stocks. C) Cryo-ET analysis of AAV2, F11-55.2-AAV and 55.2-F11-AAV. The dark spots are protein-A-colloidal gold particles added to the sample just before plunge-freezing. Scale bar indicates 20 nm. D) TEM analysis of vector stocks stained with phosphotungstic acid. Orange arrows point to full particles, white arrows to empty particles. Images were taken with a 50,000-fold magnification and the scale bar represents 50 nm. E) Numbers of full and empty particles of each vector type were determined by TEM. At least four images per vector stock were analyzed and plotted as percentage of full particles. F) Vector genome titers determined by ITR-specific qPCR. Each symbol represents one vector stock (F11-AAV n=4, 55.2-AAV n=5, F11-55.2-AAV n=4, 55.2-F11-AAV n=3). Statistical differences in E and F were calculated by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. Standard deviation is reported. P-value **** < 0.0001. Only significant differences are shown. The statistical comparison of all groups in each data set is available in the supplements.
The biological activities of the particles were assessed on a panel of SupT1 cells, a T cell line naturally expressing CD4 (below termed SupT1-CD4). SupT1-CD4 cells were modified such that they additionally expressed CD32a (SupT1-CD4/CD32a). By eliminating CD4 expression, SupT1-CD32a cells were generated (Fig. S1A). F11-AAV and 55.2-AAV were highly selective for SupT1 cells expressing their cognate receptors with negligible gene transfer activity detectable on target-receptor negative SupT1 cells (Fig. S1B, Table S1). In SupT1-CD32a cells, F11-AAV and F11-55.2-AAV showed similar titers of 11.5 and 13.9 transducing units (TU) per 1×105 GC, respectively. Transduction efficiency of 55.2-F11-AAV was significantly lower indicating that the orientation of the F11 and 55.2 DARPins on the bispecific AAV capsids influenced gene delivery (Fig. S1B left, Table S1). In SupT1-CD4 cells, the bispecific AAVs were more potent than 55.2-AAV. But by far the highest gene delivery rates were observed for the bispecific vectors on SupT1-CD4/CD32a cells (Fig. S1B, right diagram). This was not due to an overall enhanced susceptibility of this cell line for AAV-mediated gene delivery, since transfer rates for F11-AAV and 55.2-AAV were lower in these cells than in the respective SupT1 cells expressing CD4 or CD32a only. Additionally, bispecific AAVs exhibited an improved gene transfer activity in SupT1 CD4+CD32a+ cells compared to CD4+ or CD32a+ cells (Table S1). Overall, the results demonstrate that the DARPins were effectively presented on the surface of the AAV capsids and enabled efficient receptor-selective gene delivery.
Bispecific vectors are superior in transducing CD4+/CD32a+ cells
Based on the results from the titration in SupT1 cells, we determined whether the bispecific AAVs would preferably transduce CD4+/CD32a+ cells even in the presence of a surplus of cells positive for only one of the CD32a and CD4 target receptors. When exposing a mixture of all three cell types at an equal ratio to each of the vector particles, there was hardly any off-target activity detectable for the monospecific AAVs when gating all GFP-positive cells for CD4+ and CD32a+ cells (Fig. 2A). In each case, the fractions of off-target cells were below 0.3%. Interestingly, both bispecific AAVs were at least two-fold more efficient in transducing double-positive cells than the monospecific AAVs (Fig. 2A–B). This was confirmed when we serially reduced the number of SupT1-CD4/CD32a cells in the cell mix (Fig. S2). In this setting, F11-55.2- and 55.2-F11-AAV transduced up to 85% of SupT1-CD4/CD32a cells, while F11-AAV and 55.2-AAV reached only up to 45% of this cell population (Fig. 2B). Notably, single-target-receptor-positive cells were transduced much less efficiently by the bispecific vectors than double-target-receptor-positive cells. This was most pronounced in comparison to CD32a+ cells, where a preference of up to 30-fold was determined (Fig. 2C). Moreover, as compared to CD4+ cells, the preference for double-positive cells was significantly and up to 3.4-fold higher (Fig. 2D). Even in the presence of a large surplus of CD32a+ or CD4+ cells, with only 1.6% CD4+CD32a+ cells (ratio 1/48), the preference for double-positive cells was 4.5 (± 1.8)-fold for F11-55.2-AAV and 29.3 (± 11.9)-fold for 55.2-F11-AAV (Fig. 2C). These results show that bispecific AAVs have a preference for CD4+CD32a+ cells over CD32a+ or CD4+ cells, even if the double-receptor-displaying cells form only a small population in the presence of a large surplus of single-positive cells.
Figure 2:
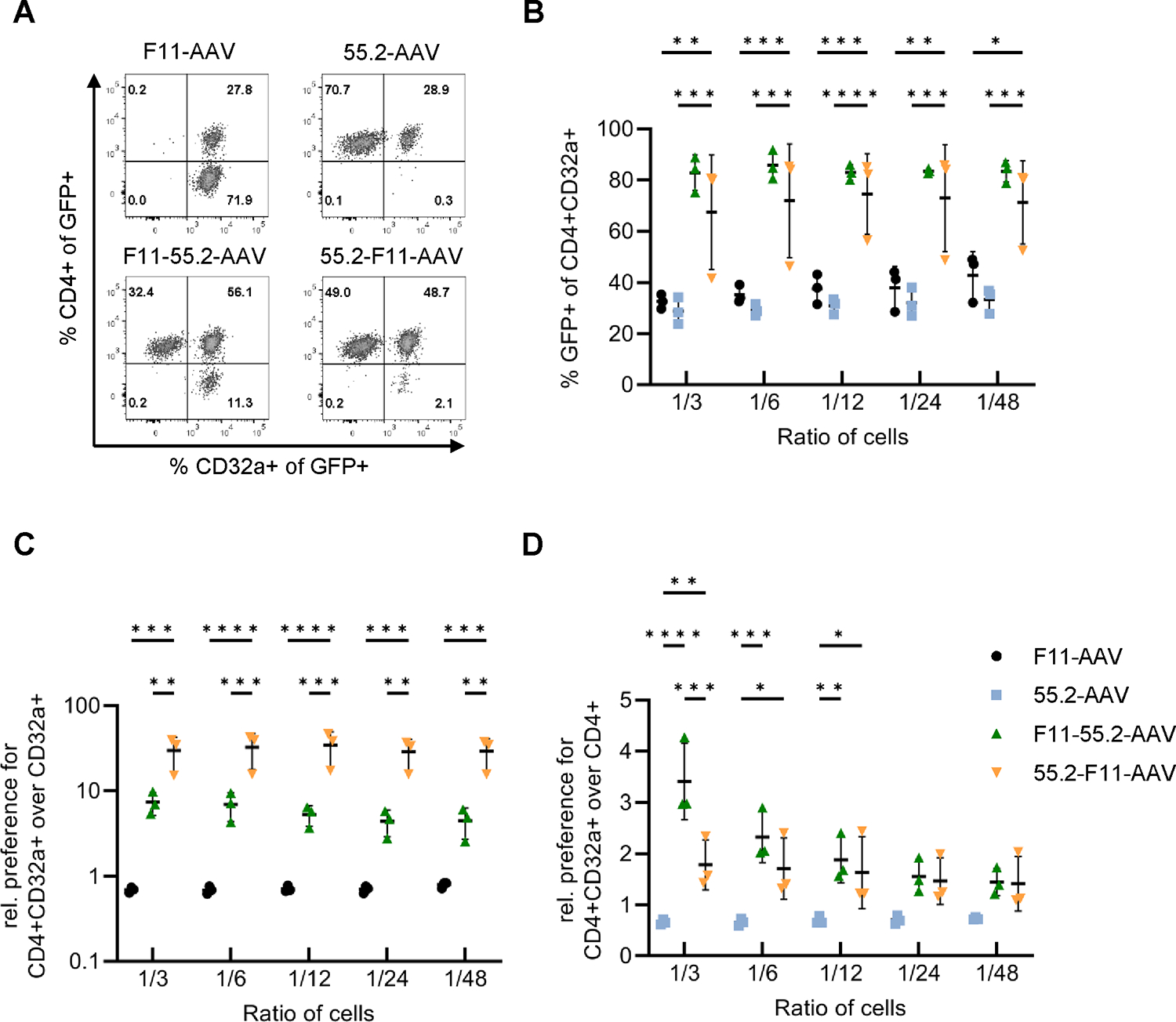
Bispecific AAVs preferentially transduce CD4+/CD32a+ cells in the presence of CD32a+ and CD4+ cells.
Gene transfer activities mediated by the indicated four AAV vectors (GC/cell of 2.67×104) into SupT1-CD4/CD32a cells were determined in the presence of SupT1-CD4 and SupT1-CD32a cells three days post-transduction. A) GFP expression was determined in a 1:1:1 mixture of SupT1-CD4, SupT1-CD32a and SupT1-CD4/CD32a cells. Flow cytometry plots provide the percentages of CD4+ and CD32a+ cells among all GFP+ cells. B) Percentage of GFP expression in SupT1-CD4/CD32a cells serially diluted into a 1:1 co-culture of SupT1-CD4 and SupT1-CD32a cells. C) Ratios of CD4+CD32a+GFP+ cells over CD32a+GFP+ cells. D) Ratios of CD4+CD32a+GFP+ cells over CD4+GFP+ cells. B-D) Experiments were performed in three independent replicates, and statistical differences were calculated using two-way ANOVA followed by Tukey’s multiple comparisons test. The standard deviation is reported. P-value **** < 0.0001, p-value *** = 0.0001–0.001, p-value ** = 0.001–0.01, p-value * = 0.01–0.05. Only significant and relevant statistical comparisons are shown. The statistical comparison of all groups in each data set is available in the supplements.
To mediate gene delivery in vivo, vector particles have to be active at relatively low GC per cell (GC/cell). Therefore, we next exposed the cell mixture with 2% double-positive cells to lower vector doses (Fig. 3A). For comparison, a 1:1 mixture of F11-AAV and 55.2-AAV was applied. Remarkably, transduction efficiencies of the bispecific AAVs on SupT1-CD4/CD32a cells were significantly higher than those mediated by the mix of 55.2-AAV and F11-AAV throughout all experimental conditions (Fig. 3B). Even at the low GC/cell of 107, both bispecific AAVs showed a preferential transduction of CD4+CD32a+ over CD32a+ cells (27.3 (± 3.3)-fold and 66.5 (± 35.3)-fold increased efficiency, respectively) (Fig. 3C). The preference over CD4+ cells was less pronounced, but still high reaching 9.4 (± 0.8)-fold for F11-55.2-AAV and 3.6 (± 0.4)-fold for 55.2-F11-AAV (Fig. 3D). These findings further highlight the specificity of bispecific AAVs for CD4+CD32a+ cells and suggest that they are more effective vectors for transducing these cells than monospecific vectors.
Figure 3:
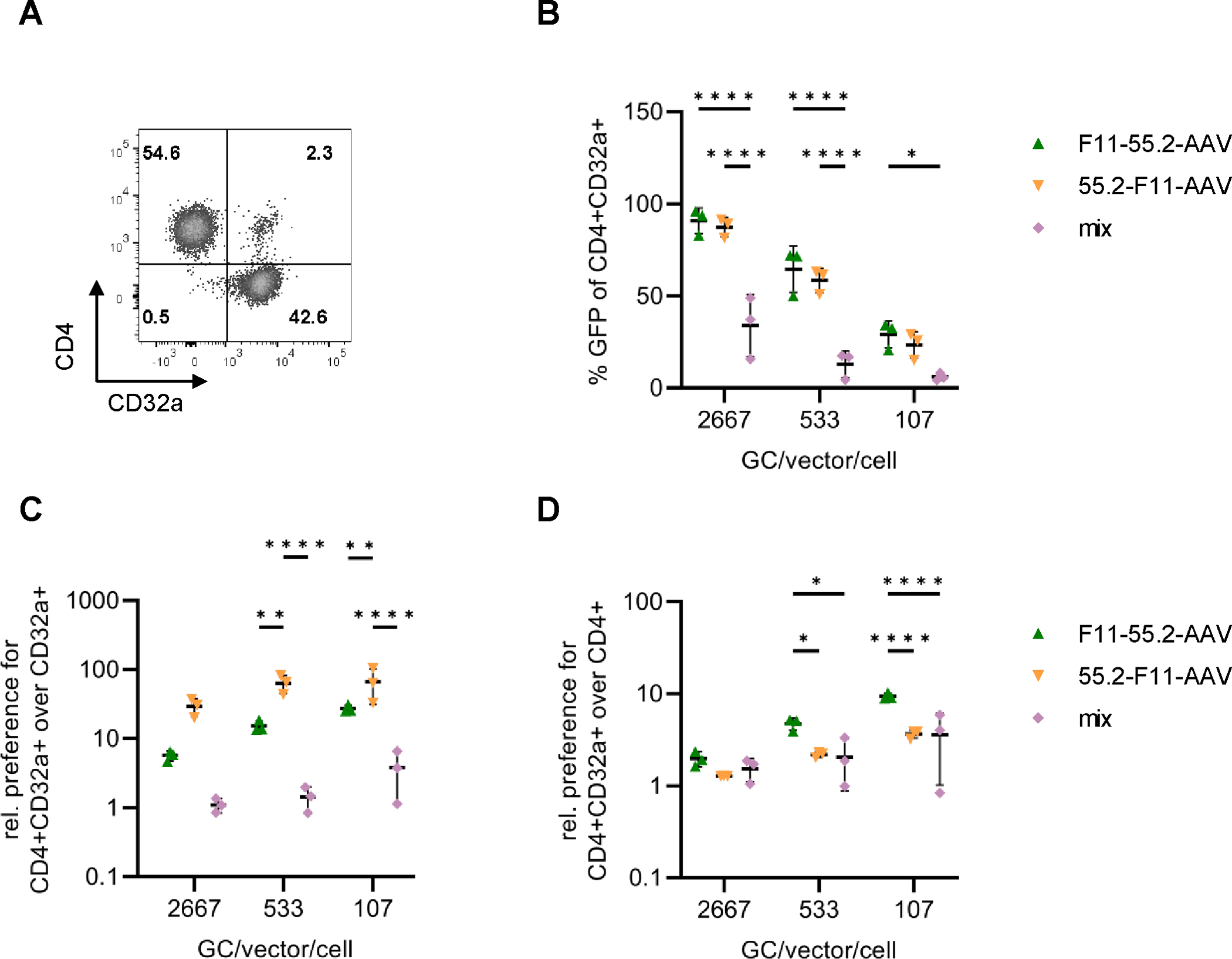
Transduction of underrepresented CD4+CD32a+ at low GC/cell.
A) Composition of the cell mixture used to assess transduction efficiencies. The percentages of SupT1-CD4, SupT1-CD32a and SupT1-CD4/CD32a cells are indicated in the respective quadrants. B-D) The cell mixture was exposed to 55.2-F11-AAV, F11-55.2-AAV or a 1:1 mixture of F11-AAV and 55.2-AAV (mix) at the indicated GC/cell, respectively. For the mixture, the dose for each vector type is provided. Percentages of GFP+ cells among CD4+/CD32a+ cells were determined and ratios over CD32a+/GFP+ cells (C) or over CD4+/GFP+ cells calculated (D). Statistical differences of three independent experiments were calculated using two-way ANOVA followed by Tukey’s multiple comparisons test. The standard deviation is reported. P-value **** < 0.0001, p-value ** = 0.001–0.01, p-value * = 0.01–0.05. Only significant and relevant statistical comparisons are shown. The statistical comparison of all groups in each data set is available in the supplements.
Vector distribution in cells and blood
Having determined a clear preference in reporter gene expression for bispecific AAVs for cells expressing both target receptors, we next assessed whether this could be confirmed on the vector genome level. To this end, the various vector types were mixed with SupT1-CD4/CD32a cells at 4°C to allow vector binding but prevent endocytosis. After removing unbound particles, cells were incubated at 37°C, before vector genomes present in cytosol, membrane and nucleus were quantified. Notably, F11-55.2-AAV exhibited the by far highest abundance of GC in each of the cellular compartments, with 2.3% (± 0.5) of the input dose having reached the cell nucleus (Fig. 4A). While less active than F11-55.2-AAV, also higher amounts of 55.2-F11-AAV vector genomes had reached the cell nucleus as compared to 55.2-AAV or F11-AAV (Fig. 4A). This suggests that more efficient uptake of the bispecific AAVs contributed to the preferred transduction of CD4+/CD32a+ cells.
Figure 4:

Quantification of vector uptake and binding to primary cells.
A) Uptake of the indicated AAVs into subcellular fractions of SupT1-CD4/CD32a cells. After internalization, vector genomes present in DNA isolated from cytoplasm, membrane and nucleus were quantified by GFP-specific qPCR. Shown is the percentage of input GC (3×1010). Statistical differences were calculated by two-way ANOVA followed by Tukey’s multiple comparisons test. The standard deviation is reported. ****p < 0.0001. B) AAV binding to primary PBMC. Human donor PBMC were incubated with the indicated vector particles at a GC/cell of 5×105 or medium as control (mock) for 2 h at 37°C. Vector-bound cells were quantified by A20 antibody staining. Monocytes were identified by staining for CD14 and T cells by staining for CD3 and CD4. Mean fluorescence intensities (MFIs) of single, viable cells are reported. The gating strategy is represented in Fig. S5. C-D) MFIs of vector-bound CD4+ T cells (C) and monocytes (D). Data for three different donors are shown. Statistical differences were calculated by one-way ANOVA followed by Tukey’s multiple comparisons test. The standard deviation is reported. ****p < 0.0001, *p = 0.01–0.05. Only significant and relevant statistical comparisons are shown. The statistical comparison of all groups in each data set is available in the supplements.
We next assessed binding of the vectors to primary human blood mononuclear cells (PBMC). Here, most frequent double-positive cells are monocytes (CD32ahigh/CD4dim), while CD32a-expressing T lymphocytes are rare in whole blood [27]. Both bispecific AAVs bound monocytes with much higher efficiencies than the monospecific vectors (Fig. 4B/D). Binding to T cells differed between the two bispecific vectors. While 55.2-F11-AAV was most efficient in binding T cells, the activity of F11-55.2-AAV was similar to that of the monospecific 55.2-AAV (Fig. 4B–C).
To assess whether monocytes would negatively influence the transduction of double-positive T cells in human whole blood, we spiked donor blood with a small amount of SupT1-CD4/CD32a cells. After incubation with the vectors, mononuclear cells including SupT1 cells were isolated and activated to follow GFP expression (Fig. 5A). In agreement with the binding data, F11-55.2-AAV exhibited the highest gene delivery activity. Even under this in vivo-like condition, it reached 45% of SupT1 cells (Fig. 5B). Both monospecific vectors were substantially less active. This was especially pronounced for F11-AAV which reached about ten-fold less SupT1-CD4/CD32a cells than F11-55.2-AAV. In summary, our results show that bispecific vectors reach their target cells also in human whole blood with high efficiency and specificity.
Figure 5:
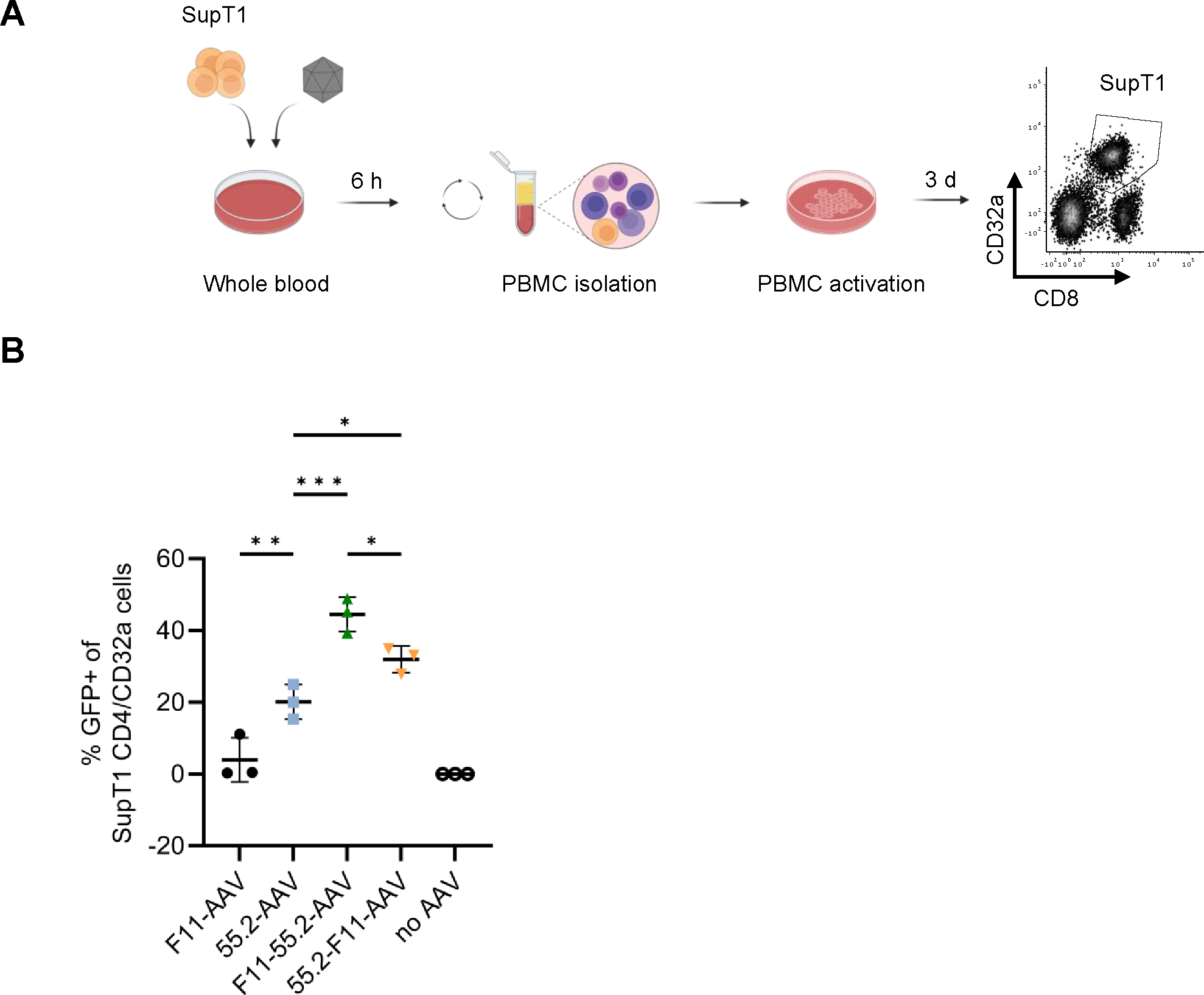
Transduction of CD4+/CD32a+ cells in human blood.
Four hundred μl whole blood spiked with 1×105 SupT1-CD4/CD32a cells was mixed with 8×108 GC of the indicated AAVs for 6 h at 37°C while shaking. Subsequently, SupT1 cells were isolated by density centrifugation, activated with anti-CD3 and anti-CD28 antibodies and cultured for three days in Nutri-T media supplemented with IL7 + IL15. At the time of analysis, SupT1 cells (CD8dim/CD32a+) were identified by staining with anti-CD32a and anti-CD8 using flow cytometry. The workflow is shown in (A). The percentages of GFP-positive SupT1-CD4/CD32a cells are shown in (B). The gating strategy is represented in Fig. S6. Three blood donors were used for this experiments and statistical differences were calculated using one-way ANOVA followed by Tukey’s multiple comparisons test. The standard deviation is reported. ***p = 0.0001–0.001, **p = 0.001–0.01, *p = 0.01–0.05. Only significant and relevant statistical comparisons are shown. The statistical comparison of all groups in each data set is available in the supplements.
In vivo gene delivery
To characterize the novel AAV vectors in vivo, we started out with F11-AAV to obtain an initial idea about the possibility to target CD32a-positive cells in vivo. Immunodeficient mice transplanted with a mixture of SupT1-CD4/CD32a and CD32a-negative HUT78 cells were intravenously injected with GFP-encoding F11-AAV. We analyzed GFP expression in cells harvested from the bone marrow on day three and seven after vector injection (Fig. 6A). Flow cytometry revealed a very high gene transfer rate into SupT1-CD4/CD32a cells at day three after vector injection, evidenced by 55.5% (± 3.4) GFP-positive cells. This signal declined but remained at a substantial high level of 9.6% (± 2.3) at day seven, most likely due to the non-integrating gene delivery mediated by AAVs (Fig. 6B–C; Fig. S3A). Off-target transduction into CD32a-negative cells was close to background demonstrating highly selective gene delivery in vivo. Furthermore, we observed a robust on-target efficiency of 30-fold above CD32a-negative cells on day 3 and 16-fold on day 7 (see Fig. 6D). Importantly, we did not observe GFP-positive cells in liver, implying that receptor targeting had effectively abolished liver tropism (Fig. S3B).
Figure 6:
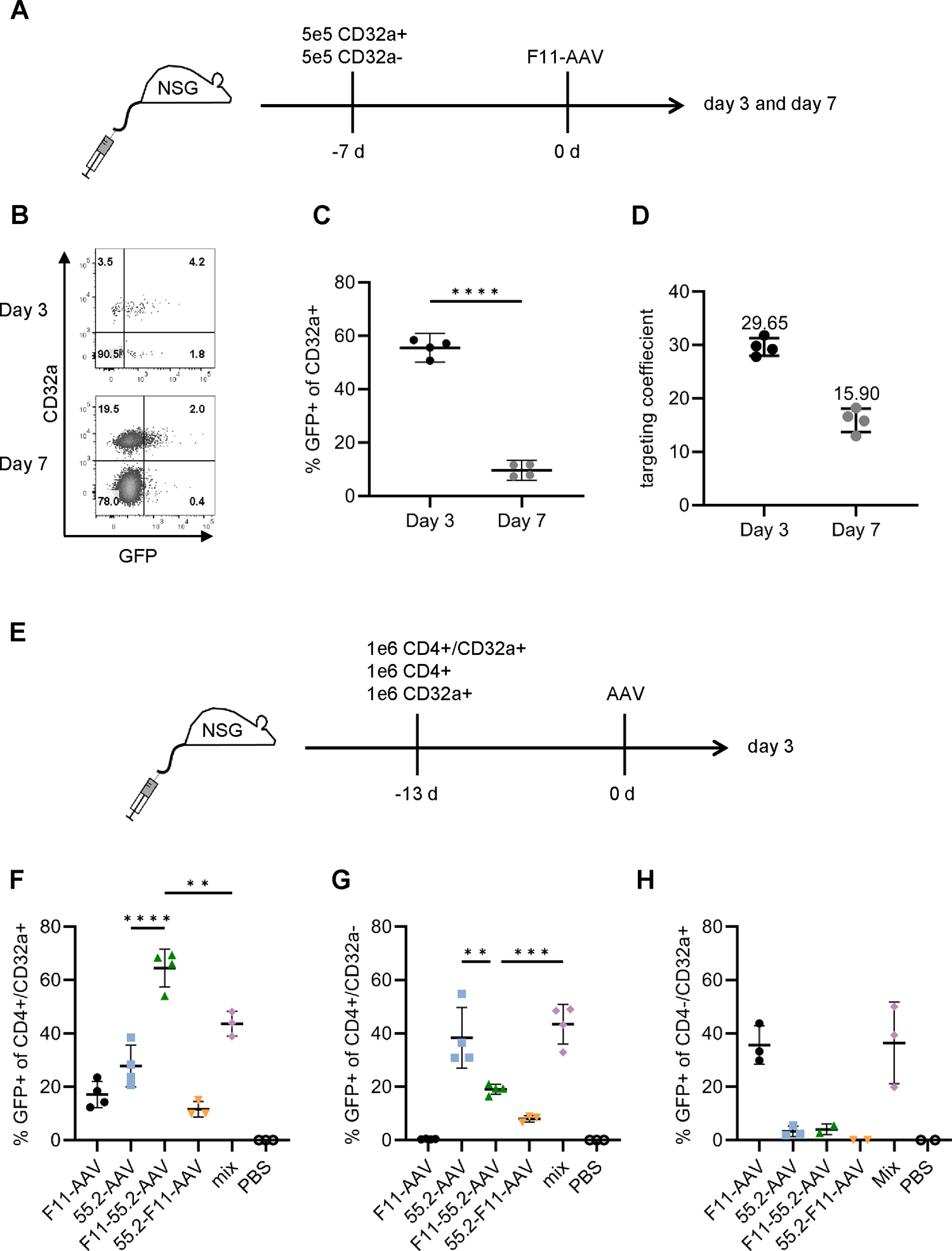
In vivo gene delivery with mono- and bispecific vectors.
A-C) NSG mice were intravenously injected with 5×105 SupT1-CD4+/CD32a+ and 5×105 CD32-negative HUT78 cells followed by administration of F11-AAV (2×1011 GCs) seven days later. Isolated cells from bone marrow or liver were analyzed for GFP expression three and seven days after vector application. B) Flow cytometry plots showing the fraction of GFP+ cells among CD45+/CD32a+ cells isolated from bone marrow at day 3 (upper) and 7 (lower) of one representative mouse, respectively. For the gating strategy and plots of other individuals, see Fig. S3A. C) Percentages of GFP-positive CD32a+ cells in bone marrow on day 3 and day 7 of vector-injected mice. n=4. Statistical differences were calculated using unpaired t-test. The standard deviation is reported. ****p < 0.0001. D) The targeting coefficient of F11-AAV three days and seven days after vector injection was calculated as ratio of GFP+ cells within CD32a-positive and within CD32a-negative cells. n=4. E-H) In vivo gene delivery of bispecific AAVs. NSG mice were intravenously (i.v.) injected with a mixture of 1×106 SupT1-CD32a, SupT1-CD4 and SupT1-CD4/CD32a cells, respectively. After 13 days, 2×1011 GCs of F11-AAV, 55.2-AAV, F11-55.2-AAV, 55.2-F11-AAV, or a 1:1 mix of 2×1011 GCs of each, F11-AAV and 55.2-AAV (mix), respectively, were intravenously injected. PBS-injected mice served as control. Isolated cells from bone marrow were analyzed for GFP expression three days after vector injection. Percentages of GFP+ cells among CD4+/CD32a+ cells (F), CD4+/CD32a− cells (G), and CD4−/CD32a+ cells (H) are shown. The gating strategy is represented in Fig. S7. N=4 (F11-AAV, 55.2-AAV, F11-55.2-AAV, mix) and n=3 (55.2-F11-AAV, PBS) in F. Data points were excluded from statistical analysis if a cell population had less than ten events (Fig. S4A). N=4 (F11-AAV, 55.2-AAV, F11-55.2-AAV, mix) and n=3 (55.2-F11-AAV, PBS) in G, and n=3 (F11-AAV, 55.2-AAV, mix) and n=2 (F11-55.2-AAV, 55.2-F11-AAV, PBS) in H. Statistical differences were calculated using one-way ANOVA followed by Tukey’s multiple comparisons test. The standard deviation is reported. ****p < 0.0001, ***p = 0.0001–0.001, **p = 0.001–0.01, *p = 0.01–0.05. Only significant and relevant statistical comparisons are shown and complementary statistics are available in the supplements.
Following the demonstration of in vivo gene delivery using F11-AAV, we assessed the biodistribution of the bispecific AAVs. For this purpose, we engrafted NSG mice with a mixture of SupT1-CD4, SupT1-CD32a and SupT1-CD4/CD32a cells. Afterwards, mice were injected with each of the four vector types or a mixture of F11-AAV and 55.2-AAV (mix). Three days after vector injection, we analyzed GFP expression in cells isolated from the bone marrow (Fig. 6E). SupT1-CD4 cells were most abundant (2% ± 1.4 of viable cells) followed by SupT1-CD4/CD32a cells (0.21% ± 0.22 of viable cells), while SupT1-CD32a cell engraftment was substantially lower (on average 0.04% ± 0.06 of viable cells) (Fig. S4A). The highest in vivo gene delivery rate was achieved by F11-55.2-AAV transducing on average more than 60% of the SupT1-CD4/CD32a cells present in bone marrow. In contrast, 55.2-F11-AAV was less efficient and remained below the gene transfer activities of the monospecific vectors (Fig. 6F). Notably, none of the monospecific vector types including their combination reached the transduction level achieved by F11-55.2-AAV. Accordingly, F11-55.2-AAV exhibited a high preference for double-positive SupT1 cells, while mix and monospecific vectors had no preference or preferred single-receptor positive cells (Fig. S3F–H). These data demonstrate that F11-55.2-AAV preferentially transduced CD4+CD32a+ cells in vivo and was detargeted from cells positive only for CD4 or CD32a, respectively.
Cas9 delivered by targeted AAVs inhibits HIV replication
Lastly, we assayed the vectors for the delivery of HIV-directed CRISPR/Cas9 cassettes as therapeutically relevant application. Due to the size constraints for AAV packaging, we initially kept spCas9 and gRNAs separated in two capsids. The gRNA cassette consisted of one gRNA driven by the U6 and 7SK promoters, which targeted the HIV-1 LTR regions, while a second gRNA, under the control of the H1 promoter, was directed against the HIV-1 gag gene. The Cas9 and gRNA constructs were packaged in CD32a-targeted AAVs resulting in F11-AAV-Cas9 and F11-AAV-gRNA. SupT1 cells were incubated for 2 hours with both AAV vectors and then infected with HIV at a MOI of 0.02. To assess the impact on HIV replication, we determined the CA-p24 protein levels up to 12 days after infection, both intracellularly and in the supernatant (Fig. 7A). We found a highly significant reduction of the CA-p24 protein level after transduction with both AAV vectors. As expected, delivering only spCas9 and no gRNAs did not impair HIV replication (Fig. 7B).
Figure 7:
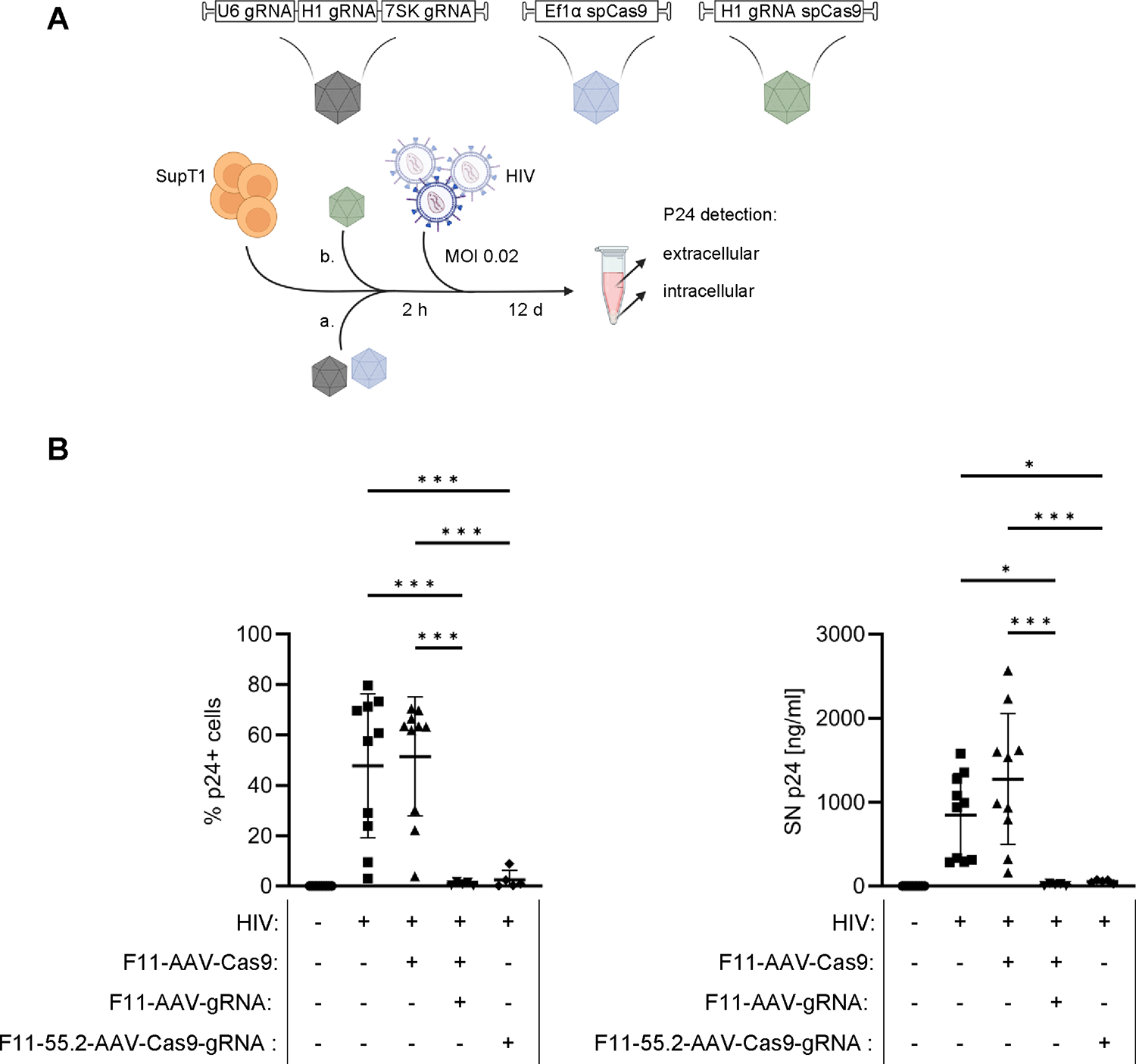
HIV-targeted endonuclease delivery by F11-AAV and F11-55.2-AAV.
A) Workflow for anti-HIV assay. SupT1-CD4/CD32a cells were incubated with F11-AAVs separately encoding Cas9 and gRNAs directed against two different sequences of the HIV-1 genome (a) or an all-in-one vector packaged in F11-55.2-AAV (green) (b). Two hours after transduction, cells were infected with HIV at an MOI of 0.02. Subsequently, CA-p24 protein concentration was analyzed in supernatant via ELISA and by intracellular staining at 12 days post infection. B) Percentages of CA-p24-positive cells (left) and CA-p24 levels in cell supernatants (right). Uninfected n=10, HIV infected n=10, HIV infected + F11-AAV-Cas9 n=10, HIV infected + F11-AAV-Cas9 + F11-AAV-gRNA n=5, HIV infected+ F11-55.2-AAV-Cas9-gRNA n=5. Statistical differences of technical replicates were calculated by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. SD = standard deviation. ***p = 0.0001–0.001, *p = 0.01–0.05. Only significant and relevant statistical comparisons are shown. The statistical comparison of all groups in each data set is available in the supplements.
Having seen efficient HIV inhibition with F11-AAV we next proceeded with F11-55.2-AAV. For this purpose, we designed an all-in-one cassette delivering both Cas9 and a single gRNA packaged into the same particle. In this setting, the gRNA was directed against the overlapping reading frames of the tat and rev genes [32]. We took advantage of the recently described miniature H1 promoter [31], which allowed expression of Cas9 and transcription of the gRNA due to its combined pol II and pol III activities. Utilizing only the small H1 promoter reduced the size of the transfer gene, making it comparable in size to the transfer cassette used in the previous F11-AAV-Cas9 construct and consequently allowed for packaging into a single AAV capsid. The anti-HIV activity of F11-55.2-AAV-Cas9-gRNA was assessed as described above (Fig. 7A). Here, we observed highly efficient inhibition of HIV replication as demonstrated by the reduction of CA-p24 signals to levels indistinguishable from the background (Fig. 7B). Thus, monospecific CD32a-targeted as well as bispecific AAVs successfully deliver therapeutic genes in the context of HIV gene therapy.
Discussion
Gene delivery precisely to the therapy-relevant cell type in the human organism following systemic vector delivery is a prime goal in current gene therapy research activities. Enormous progress has been made, be it by directed evolution of libraries of AAV capsid variants or by rational design through display of high-affinity ligands for the receptor of choice [9,42]. Through the latter approach, vectors highly selective for subtypes of T lymphocytes have been generated [13,15,16]. However, only a few cell types can be identified through a single surface marker, while many more are characterized by a defined combination of two or more markers. Vectors distinguishing such cells from cells expressing just one or none of the two markers have not yet been described, but would greatly expand the cell types that can be targeted by gene therapy in a highly selective way.
Here, we describe bispecific AAV2 vectors displaying two DARPins recognizing CD4 and CD32a, and compared them to their monospecific counterparts. Among the latter, F11-AAV is the first description of a vector targeted to CD32a making use of the recently selected DARPin F11 that binds CD32a, but not CD32b, with low nanomolar affinity [27]. Display of mono- and bispecific DARPins was made possible through the use of the GH2/3 loop on the VP1 capsid protein, an insertion site that has so far only rarely been used for insertion of targeting ligands [14–17]. Accordingly, we assessed 55.2-F11-AAV particles by cryo-electron tomography, which revealed no capsid alterations induced by DARPin display. While biochemical data demonstrated a high level of DARPin incorporation into particles, the absence of any large protrusions in cryo-electron tomography was not unexpected, since there are only five DARPins per capsid on average. Moreover, their random positioning on the capsid surface and anchoring via flexible linkers make their microscopic detection difficult. Yet, their gene transfer activities together with the biochemical characterization demonstrate that the GH2/3 loop appears ideal for DARPin insertions excluding any interference with the AAV capsid structure. Notably, the insertion of the 55.2-F11 DARPins represents the largest protein construct (342 amino acids) successfully inserted into the GH2/3 loop thus far, even when compared to the initial screening experiment using random insertions of mCherry (236 amino acids) [43].
The most important finding of our study was that bispecific AAV particles displaying both DARPins exhibited a clear preference for transducing CD4/CD32a double-positive cells over cells expressing only one of the target receptors. This behavior is reminiscent of biological medicinal products designed to follow AND-gated Boolean logic. In terms of molecular binder development, AND-gated concepts are used to enhance the selectivity to a target cell. This strategy involves at least two binders recognizing different receptors in such a way that they require the simultaneous presence of both target receptors on the cell surface for efficient activity. AND-gated engineering strategies for the elimination of cancer cells are pursued for chimeric antigen receptor (CAR) T cells as well as various antibody formats to avoid toxicity in healthy cells expressing only one of the markers [44,45]. For CAR T cells, a wide variety of strategies have enabled selective cytotoxic activity against two-marker-positive cells [46,47]. These strategies utilize the signaling machinery of the CAR T cell. Only when both antigens are present on the target cell, the two different CARs and their signaling domains are brought in sufficiently close proximity to trigger signaling and cytotoxic activities of the T cell. While AND gating strategies pursued for CAR T cells are rather not transferable to gene transfer vectors, antibodies and related molecules, for which AND gated bivalent or trivalent formats have been described, are more comparable. Since there are only two binding sites per antibody, engineering can be designed such that simultaneous binding to both surface antigens provides a strong preference over binding to only one of the two [48,49]. This preference can be optimized by combining binders with low affinities for their target cell surface antigens. Yet, binding to single target-receptor positive cells can usually not be completely eliminated [48]. Since vector particles carry a multitude of receptor-binding proteins on their surface, display of bispecific binders recognizing two different cell surface proteins is expected to expand rather than restrict the tropism to cells carrying either of the two markers. Among the few examples which tested this experimentally, oncolytic measles viruses displaying bispecific DARPins directed against Her2/neu and EpCAM behaved, as expected, in an OR-gated manner. While this result was beneficial for the oncolytic activity by reducing the risk for therapy-resistance through loss of one of the two surface antigens [50,51], it suggested that AND gating may be more challenging to achieve with viral vectors.
Therefore, the preference for cells expressing both target receptors exhibited by F11-55.2-AAV and 55.2-F11-AAV came with some surprise. This preference reached up to 66-fold when compared to cells expressing CD32a only and when a low vector dose was applied, while the preference over CD4-only expressing cells was less pronounced. Remarkably, also after systemic in vivo administration, F11-55.2-AAV exhibited a more than three-fold preference for double-positive cells over CD4- and an even ten-fold preference over CD32a-positive cells. Notably, a 1:1 mixture of the two monospecific AAVs was non-selective for double-positive cells and also less efficient in transducing them in vivo. In this context it is important to emphasize that the double-positive cells were not intrinsically more susceptible for AAV particles, since in all experimental settings the monospecific vectors were less active in double-positive than in single-positive cells. While the binding of the bispecific vectors was not absolutely binary and thus did not completely fulfil AND-gating, it achieved enhanced selectivity through an increased preference for double-positive cells and less frequent attachment to cells expressing only one of the receptors. Thus, this targeting approach is one step closer towards the desired AND-gated behavior.
What caused the AAV vectors in this work to exhibit a preference for double-positive cells and behave differently from oncolytic measles viruses, which rather follow OR-gating? AAV particles are non-enveloped and much smaller in diameter with only 60 capsid subunits per particle, while measles virus carries several hundred envelope glycoproteins [52]. While recent mass spectrometry data revealed some variability in the polypeptide composition of AAV particles, VP1, which we used for inserting the bispecific F11-55.2 DARPin, makes up on average five molecules per AAV2 particle [53]. Thus, F11-55.2-AAV contains at most ten binding sites for CD4 and CD32a randomly distributed over the entire capsid surface. Sterically, it will not be possible that at the given random distribution of the five VP1 proteins over the surface of the spherical particle all ten binding sites contact the cell surface simultaneously. Second, the two DARPins are connected by a (G4S)3-linker, which covers a maximal distance of 50 Å when stretched out [54]. It is rather unlikely that CD32a and CD4 are in such close proximity on the cell surface that both can be bound by a single VP1 protein containing the two DARPins. Thus, an educated guess ends up with a tetra- or trivalent interaction of a single F11-55.2-AAV particle with the cell surface. This makes AAVs displaying DARPins on VP1 clearly more comparable to antibody-like molecules than to CAR T cells. The preferred gene delivery into double-positive cells mediated by bispecific AAV particles could thus simply be the consequence of a higher avidity and thus more stable cell association mediated by the bispecific DARPin, or, alternatively, of a more rapid cell entry mediated by a concerted action of CD4 and CD32a. When systemically injected into mice with target cells in bone marrow, as in the mouse models used here, these vectors are transported through the whole body by the circulation. Due to the detargeting from liver by ablating the binding site for HSPG this likely results in an increased persistence and thus enhanced likelihood to finally reach the target cells. Clearly, more research will be required to elucidate the differences in biodistribution and cell entry mechanisms of mono- and bispecific AAVs versus untargeted AAVs.
This implies that optimizing affinities of binder pairs will make the discrimination between double- and single-positive cells even more pronounced than what was achieved in this proof-of-concept study. Indeed, the preference of the bispecific vectors for CD4-positive over CD32a-positive cells was most likely due to the higher affinity of 55.2 for CD4 (0.84 nM) [41] than that of F11 for CD32a (6.1 nM) [27]. In line with this finding, the preference for double-positive cells was clearly influenced by the orientation of the two DARPins to each other. This was especially pronounced in the in vivo setting, suggesting that the N-terminal position of F11 at least partly compensated for its lower affinity. In future, selection of a CD4-specific DARPin with a substantially lower affinity will likely further detarget the vectors from CD4 single-positive cells and thus improve selectivity for double-positive cells. DARPins are probably most suited for this task due to their rigid structure and high stability, which makes them less prone to misfolding, in turn enabling combinations of multiple DARPins with various specificities. Recently, this has been shown for Ensovibep, which comprises five connected DARPins targeting the SARS-CoV-2 spike protein receptor-binding domain and serum albumin to increase its half-life [55]. Accordingly, the generation of tri- or even tetra-specific AAVs is possible.
Applications for multi-specific AAVs will be plentiful, ranging from basic gene-function studies to novel therapeutic strategies. For the latter, careful safety testing in preclinical models will be necessary for this novel type of targeted AAVs. While there is in depth experience with clinical applications for in vivo gene delivery with AAVs, surface engineered particles binding to several surface receptors were not yet among them. Although such particles are more selective for their target cells, thus allowing highly precise gene delivery and potentially reduced vector doses, certain combinations of DARPins displayed on AAV might lead to receptor crosslinking followed by over-activation of target cells. In case of T cells, this could lead to unwanted immune responses and/or exhaustion. This scenario can be tested by a combination of in vitro and in vivo assays. Depending on the animal chosen and the cross-reactivity of the DARPins, it may require the generation of species adapted vectors.
Among the therapeutic applications, the targeted transduction of latently HIV-infected reservoir cells to inactivate the proviral genome is particularly promising. We demonstrate here that the bispecific F11-55.2-AAV can package and deliver an all-in-one vector encoding the endonuclease Cas9 plus gRNAs targeted to the integrated HIV DNA genome. While the split-AAV system showed high activity in vitro, the all-in-one is expected to be advantageous in vivo, since it does not require simultaneous transduction by at least two particles. The molecule CD32a was proposed as a marker of the HIV reservoir as these cells showed a very high enrichment for HIV DNA [19]. While subsequent work challenged this finding [20,56] other could confirm its relevance by using independent methods [21,22,57]. Anyhow, it remains undisputed that HIV reservoir cells are highly heterogenous in cell surface markers not only between patients but also within single individuals [58–60]. F11-55.2-AAV represents the very first example for a vector targeted to two surface markers of HIV reservoir cells. While CD4- and CD32a-targeted vector particles may also be bound by monocytes, functional transduction of these cells is unlikely but remains to be investigated. For monocyte-derived dendritic cells it was recently reported that intracellular restriction factors effectively prevent transduction [61]. Subsequent studies analyzing its distribution in patient blood will reveal its true therapeutic relevance with respect to off-target and on-target delivery. Regarding the latter, reduced expression levels of CD4 (and CD32a), which occur during acute infection are not expected to be an issue in reservoir cells [59]. Until then, the flexible adaptation of the system to other target receptors and the proof-of-concept data provided here pave the way for the exploration of further marker combinations in the context of HIV gene therapy and beyond.
Supplementary Material
Research Highlights.
Preferred transduction of cells expressing two surface markers by bispecific AAVs
Incorporation of two DARPins without detectable structural changes in the capsid
Increased uptake rates of bispecific versus monospecific AAVs
New options for precise in vivo gene delivery to therapy-relevant cells
Targeting of the HIV reservoir as potential application
Acknowledgments
The authors acknowledge Gundula Braun and Manuela Gallet (Paul-Ehrlich-Institut) for their excellent technical assistance in AAV vector generation and quantification of VCNs, as well as Regina Eberle (Paul-Ehrlich-Institut) for technical assistance at the electron microscope. The authors gratefully access to the infrastructure and support provided by the Cryo-EM Network at the Heidelberg University (HD-cryoNet) and the data storage service SDS@hd supported by the Ministry of Science, Research and the Arts Baden-Württemberg (MWK) and the German Research Foundation (DFG) through grant INST 35/1314-1 FUGG and INST 35/1503-1 FUGG. Figures in the manuscript were partially created with BioRender.com.
Funding
This work was supported by grant 1R01AI145045-01 from the National Institutes of Health (NIH) to B.B., E.H., and C.J.B. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. F.B.T. and C.J.B. received funding by the State of Hessen within the LOEWE program. D.G. and C.J.B. are funded by the BMBF project COMMUTE (16GW0339). P.C. received funding from the Chica and Heinz Schaller Foundation and acknowledges support from the German Research Foundation (DFG) project number 437060729. P.C. and D.G. acknowledge support by the DFG SFB1129 (project number 240245660). K.B. and D.G. are grateful for support from the German Center for Infection Research (DZIF, BMBF; TTU-HIV 04.819).
Footnotes
Declaration of interest
V. R. and C. J. B. are listed as inventors on a filed patent describing FcγRIIA-specific DARPins. All other authors declare no conflicts of interest.
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Christian J Buchholz reports financial support was provided by National Institutes of Health. Christian J Buchholz, Vanessa Riechert has patent pending to Assignee.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.
References
- [1].Pupo A, Fernández A, Low SH, François A, Suárez-Amarán L, Samulski RJ, AAV vectors: The Rubik’s cube of human gene therapy, Mol. Ther. 30 (2022) 3515–3541. 10.1016/j.ymthe.2022.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nathwani AC, Gene therapy for hemophilia, Hematology Am. Soc. Hematol. Educ. Program 2022 (2022) 569–578. 10.1182/hematology.2022000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, Goodspeed K, Gray SJ, Kay CN, Boye SL, Boye SE, George LA, Salabarria S, Corti M, Byrne BJ, Tremblay JP, Current Clinical Applications of In Vivo Gene Therapy with AAVs, Mol. Ther. 29 (2021) 464–488. 10.1016/j.ymthe.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Maguire AM, Bennett J, Aleman EM, Leroy BP, Aleman TS, Clinical Perspective: Treating RPE65-Associated Retinal Dystrophy, Mol. Ther. 29 (2021) 442–463. 10.1016/j.ymthe.2020.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li C, Samulski RJ, Engineering adeno-associated virus vectors for gene therapy, Nat Rev Genet 21 (2020) 255–272. 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- [6].Kishimoto TK, Samulski RJ, Addressing high dose AAV toxicity - ‘one and done’ or ‘slower and lower’?, Expert Opin Biol Ther 22 (2022) 1067–1071. 10.1080/14712598.2022.2060737. [DOI] [PubMed] [Google Scholar]
- [7].Watkins PB, Liver Injury Due to Drugs and Viruses: Mechanistic Similarities and Implications for AAV Gene Therapy, Clin. Pharmacol. Ther 112 (2022) 751–753. 10.1002/cpt.2500. [DOI] [PubMed] [Google Scholar]
- [8].High-dose AAV gene therapy deaths, Nat. Biotechnol. 38 (2020) 910. 10.1038/s41587-020-0642-9. [DOI] [PubMed] [Google Scholar]
- [9].Becker J, Fakhiri J, Grimm D, Fantastic AAV Gene Therapy Vectors and How to Find Them-Random Diversification, Rational Design and Machine Learning, Pathogens 11 (2022). 10.3390/pathogens11070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Buchholz CJ, Friedel T, Büning H, Surface-Engineered Viral Vectors for Selective and Cell Type-Specific Gene Delivery, Trends Biotechnol 33 (2015) 777–790. 10.1016/j.tibtech.2015.09.008. [DOI] [PubMed] [Google Scholar]
- [11].Opie SR, Warrington KH, Agbandje-McKenna M, Zolotukhin S, Muzyczka N, Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding, J. Virol. 77 (2003) 6995–7006. 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Münch RC, Janicki H, Völker I, Rasbach A, Hallek M, Büning H, Buchholz CJ, Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer, Mol Ther 21 (2013) 109–118. 10.1038/mt.2012.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Münch RC, Muth A, Muik A, Friedel T, Schmatz J, Dreier B, Trkola A, Plückthun A, Büning H, Buchholz CJ, Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors, Nat Commun 6 (2015) 6246. 10.1038/ncomms7246. [DOI] [PubMed] [Google Scholar]
- [14].Eichhoff AM, Börner K, Albrecht B, Schäfer W, Baum N, Haag F, Körbelin J, Trepel M, Braren I, Grimm D, Adriouch S, Koch-Nolte F, Nanobody-Enhanced Targeting of AAV Gene Therapy Vectors, Mol Ther Methods Clin Dev 15 (2019) 211–220. 10.1016/j.omtm.2019.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hamann MV, Beschorner N, Vu X-K, Hauber I, Lange UC, Traenkle B, Kaiser PD, Foth D, Schneider C, Büning H, Rothbauer U, Hauber J, Improved targeting of human CD4+ T cells by nanobody-modified AAV2 gene therapy vectors, PLoS ONE 16 (2021) e0261269. 10.1371/journal.pone.0261269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Michels A, Frank AM, Günther DM, Mataei M, Börner K, Grimm D, Hartmann J, Buchholz CJ, Lentiviral and adeno-associated vectors efficiently transduce mouse T lymphocytes when targeted to murine CD8, Mol Ther Methods Clin Dev 23 (2021) 334–347. 10.1016/j.omtm.2021.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Günther DM, Kovacs R, Wildner F, Salivara A, Thalheimer FB, Fries P, Geiger JRP, Buchholz CJ, Substantially improved gene transfer to interneurons with second-generation glutamate-receptor targeted DART-AAV vectors, J Neurosci Methods (2023) 109981. 10.1016/j.jneumeth.2023.109981. [DOI] [PubMed] [Google Scholar]
- [18].Fromentin R, Chomont N, HIV persistence in subsets of CD4+ T cells: 50 shades of reservoirs, Semin. Immunol. 51 (2021) 101438. 10.1016/j.smim.2020.101438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Descours B, Petitjean G, López-Zaragoza J-L, Bruel T, Raffel R, Psomas C, Reynes J, Lacabaratz C, Levy Y, Schwartz O, Lelievre JD, Benkirane M, CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses, Nature 543 (2017) 564–567. 10.1038/nature21710. [DOI] [PubMed] [Google Scholar]
- [20].Badia R, Ballana E, Castellví M, García-Vidal E, Pujantell M, Clotet B, Prado JG, Puig J, Martínez MA, Riveira-Muñoz E, Esté JA, CD32 expression is associated to T-cell activation and is not a marker of the HIV-1 reservoir, Nat. Commun. 9 (2018) 2739. 10.1038/s41467-018-05157-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Darcis G, Kootstra NA, Hooibrink B, van Montfort T, Maurer I, Groen K, Jurriaans S, Bakker M, van Lint C, Berkhout B, Pasternak AO, CD32+CD4+ T Cells Are Highly Enriched for HIV DNA and Can Support Transcriptional Latency, Cell Rep. 30 (2020) 2284–2296.e3. 10.1016/j.celrep.2020.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Adams P, Fievez V, Schober R, Amand M, Iserentant G, Rutsaert S, Dessilly G, Vanham G, Hedin F, Cosma A, Moutschen M, Vandekerckhove L, Seguin-Devaux C, CD32+CD4+ memory T cells are enriched for total HIV-1 DNA in tissues from humanized mice, iScience 24 (2021) 101881. 10.1016/j.isci.2020.101881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Abdel-Mohsen M, Kuri-Cervantes L, Grau-Exposito J, Spivak AM, Nell RA, Tomescu C, Vadrevu SK, Giron LB, Serra-Peinado C, Genescà M, Castellví J, Wu G, Del Rio Estrada PM, González-Navarro M, Lynn K, King CT, Vemula S, Cox K, Wan Y, Li Q, Mounzer K, Kostman J, Frank I, Paiardini M, Hazuda D, Reyes-Terán G, Richman D, Howell B, Tebas P, Martinez-Picado J, Planelles V, Buzon MJ, Betts MR, Montaner LJ, CD32 is expressed on cells with transcriptionally active HIV but does not enrich for HIV DNA in resting T cells, Sci Transl Med 10 (2018). 10.1126/scitranslmed.aar6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vásquez JJ, Aguilar-Rodriguez BL, Rodriguez L, Hogan LE, Somsouk M, McCune JM, Deeks SG, Laszik ZG, Hunt PW, Henrich TJ, CD32-RNA Co-localizes with HIV-RNA in CD3+ Cells Found within Gut Tissues from Viremic and ART-Suppressed Individuals, Pathog. Immun 4 (2019) 147–160. 10.20411/pai.v4i1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cantero-Pérez J, Grau-Expósito J, Serra-Peinado C, Rosero DA, Luque-Ballesteros L, Astorga-Gamaza A, Castellví J, Sanhueza T, Tapia G, Lloveras B, Fernández MA, Prado JG, Solé-Sedeno JM, Tarrats A, Lecumberri C, Mañalich-Barrachina L, Centeno-Mediavilla C, Falcó V, Buzon MJ, Genescà M, Resident memory T cells are a cellular reservoir for HIV in the cervical mucosa, Nat Commun 10 (2019) 4739. 10.1038/s41467-019-12732-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Astorga-Gamaza A, Grau-Expósito J, Burgos J, Navarro J, Curran A, Planas B, Suanzes P, Falcó V, Genescà M, Buzon MJ, Identification of HIV-reservoir cells with reduced susceptibility to antibody-dependent immune response, eLife 11 (2022). 10.7554/eLife.78294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Riechert V, Hein S, Visser M, Zimmermann M, Wesche J, Adams PA, Theuerkauf SA, Jamali A, Wangorsch A, Reuter A, Pasternak AO, Hartmann J, Greinacher A, Herrera-Carrillo E, Berkhout B, Cichutek K, Buchholz CJ, FcγRIIA-specific DARPins as novel tools in blood cell analysis and platelet aggregation, J Biol Chem 299 (2023) 104743. 10.1016/j.jbc.2023.104743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Patel P, Michael JV, Naik UP, McKenzie SE, Platelet FcγRIIA in immunity and thrombosis: Adaptive immunothrombosis, J. Thromb. Haemost. 19 (2021) 1149–1160. 10.1111/jth.15265. [DOI] [PubMed] [Google Scholar]
- [29].Balakrishnan A, Rajan A, Salter AI, Kosasih PL, Wu Q, Voutsinas J, Jensen MC, Plückthun A, Riddell SR, Multispecific Targeting with Synthetic Ankyrin Repeat Motif Chimeric Antigen Receptors, Clin. Cancer Res. (2019). 10.1158/1078-0432.CCR-19-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Plückthun A, Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy, Annu Rev Pharmacol Toxicol 55 (2015) 489–511. 10.1146/annurev-pharmtox-010611-134654. [DOI] [PubMed] [Google Scholar]
- [31].Gao Z, van der Velden YU, Fan M, van der Linden CA, Vink M, Herrera-Carrillo E, Berkhout B, Engineered miniature H1 promoters with dedicated RNA polymerase II or III activity, J Biol Chem 296 (2021) 100026. 10.1074/jbc.RA120.015386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang G, Zhao N, Berkhout B, Das AT, CRISPR-Cas9 Can Inhibit HIV-1 Replication but NHEJ Repair Facilitates Virus Escape, Mol. Ther. 24 (2016) 522–526. 10.1038/mt.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Reul J, Muik A, Buchholz CJ, Ligand Coupling to the AAV Capsid for Cell-Specific Gene Transfer, Methods Mol Biol 1950 (2019) 35–50. 10.1007/978-1-4939-9139-6_3. [DOI] [PubMed] [Google Scholar]
- [34].Weidner T, Agarwal S, Perian S, Fusil F, Braun G, Hartmann J, Verhoeyen E, Buchholz CJ, Genetic in vivo engineering of human T lymphocytes in mouse models, Nat Protoc 16 (2021) 3210–3240. 10.1038/s41596-021-00510-8. [DOI] [PubMed] [Google Scholar]
- [35].Herrera-Carrillo E, Paxton WA, Berkhout B, The search for a T cell line for testing novel antiviral strategies against HIV-1 isolates of diverse receptor tropism and subtype origin, J. Virol. Methods 203 (2014) 88–96. 10.1016/j.jviromet.2014.03.021. [DOI] [PubMed] [Google Scholar]
- [36].Hagen WJH, Wan W, Briggs, John AG, Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging, J. Struct. Biol. 197 (2017) 191–198. 10.1016/j.jsb.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mastronarde DN, Automated electron microscope tomography using robust prediction of specimen movements, J. Struct. Biol. 152 (2005) 36–51. 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- [38].Mastronarde DN, Held SR, Automated tilt series alignment and tomographic reconstruction in IMOD, J. Struct. Biol. 197 (2017) 102–113. 10.1016/j.jsb.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M, ColabFold: making protein folding accessible to all, Nat. Methods 19 (2022) 679–682. 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Fiji: an open-source platform for biological-image analysis, Nat. Methods 9 (2012) 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Schweizer A, Rusert P, Berlinger L, Ruprecht CR, Mann A, Corthésy S, Turville SG, Aravantinou M, Fischer M, Robbiani M, Amstutz P, Trkola A, CD4-specific designed ankyrin repeat proteins are novel potent HIV entry inhibitors with unique characteristics, PLoS Pathog. 4 (2008) e1000109. 10.1371/journal.ppat.1000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Büning H, Srivastava A, Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors, Mol Ther Methods Clin Dev 12 (2019) 248–265. 10.1016/j.omtm.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Judd J, Wei F, Nguyen PQ, Tartaglia LJ, Agbandje-McKenna M, Silberg JJ, Suh J, Random Insertion of mCherry Into VP3 Domain of Adeno-associated Virus Yields Fluorescent Capsids With no Loss of Infectivity, Mol Ther Nucleic Acids 1 (2012) e54. 10.1038/mtna.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Saleh HA, Mitwasi N, Ullrich M, Kubeil M, Toussaint M, Deuther-Conrad W, Neuber C, Arndt C, R Loureiro L, Kegler A, González Soto KE, Belter B, Rössig C, Pietzsch J, Frenz M, Bachmann M, Feldmann A, Specific and safe targeting of glioblastoma using switchable and logic-gated RevCAR T cells, Front Immunol 14 (2023) 1166169. 10.3389/fimmu.2023.1166169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tousley AM, Rotiroti MC, Labanieh L, Rysavy LW, Kim W-J, Lareau C, Sotillo E, Weber EW, Rietberg SP, Dalton GN, Yin Y, Klysz D, Xu P, de La Serna EL, Dunn AR, Satpathy AT, Mackall CL, Majzner RG, Co-opting signalling molecules enables logic-gated control of CAR T cells, Nature 615 (2023) 507–516. 10.1038/s41586-023-05778-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhao Z, Sadelain M, CAR T cell design: approaching the elusive AND-gate, Cell Res. (2023). 10.1038/s41422-023-00828-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hamieh M, Mansilla-Soto J, Rivière I, Sadelain M, Programming CAR T Cell Tumor Recognition: Tuned Antigen Sensing and Logic Gating, Cancer Discov 13 (2023) 829–843. 10.1158/2159-8290.CD-23-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mazor Y, Hansen A, Yang C, Chowdhury PS, Wang J, Stephens G, Wu H, Dall’Acqua WF, Insights into the molecular basis of a bispecific antibody’s target selectivity, MAbs 7 (2015) 461–469. 10.1080/19420862.2015.1022695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mazor Y, Sachsenmeier KF, Yang C, Hansen A, Filderman J, Mulgrew K, Wu H, Dall’Acqua WF, Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence, Sci Rep 7 (2017) 40098. 10.1038/srep40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hanauer JD, Rengstl B, Kleinlützum D, Reul J, Pfeiffer A, Friedel T, Schneider IC, Newrzela S, Hansmann M-L, Buchholz CJ, Muik A, CD30-targeted oncolytic viruses as novel therapeutic approach against classical Hodgkin lymphoma, Oncotarget (2018). 10.18632/oncotarget.24191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Friedrich K, Hanauer JR, Prüfer S, Münch RC, Völker I, Filippis C, Jost C, Hanschmann K-M, Cattaneo R, Peng K-W, Plückthun A, Buchholz CJ, Cichutek K, Mühlebach MD, DARPin-targeting of measles virus: unique bispecificity, effective oncolysis, and enhanced safety, Mol. Ther. 21 (2013) 849–859. 10.1038/mt.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cox RM, Plemper RK, Structure and organization of paramyxovirus particles, Curr Opin Virol 24 (2017) 105–114. 10.1016/j.coviro.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wörner TP, Bennett A, Habka S, Snijder J, Friese O, Powers T, Agbandje-McKenna M, Heck AJR, Adeno-associated virus capsid assembly is divergent and stochastic, Nat Commun 12 (2021) 1642. 10.1038/s41467-021-21935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jost C, Schilling J, Tamaskovic R, Schwill M, Honegger A, Plückthun A, Structural basis for eliciting a cytotoxic effect in HER2-overexpressing cancer cells via binding to the extracellular domain of HER2, Structure 21 (2013) 1979–1991. 10.1016/j.str.2013.08.020. [DOI] [PubMed] [Google Scholar]
- [55].Rothenberger S, Hurdiss DL, Walser M, Malvezzi F, Mayor J, Ryter S, Moreno H, Liechti N, Bosshart A, Iss C, Calabro V, Cornelius A, Hospodarsch T, Neculcea A, Looser T, Schlegel A, Fontaine S, Villemagne D, Paladino M, Schiegg D, Mangold S, Reichen C, Radom F, Kaufmann Y, Schaible D, Schlegel I, Zitt C, Sigrist G, Straumann M, Wolter J, Comby M, Sacarcelik F, Drulyte I, Lyoo H, Wang C, Li W, Du W, Binz HK, Herrup R, Lusvarghi S, Neerukonda SN, Vassell R, WANG W, Adler JM, Eschke K, Nascimento M, Abdelgawad A, Gruber AD, Bushe J, Kershaw O, Knutson CG, Balavenkatraman KK, Ramanathan K, Wyler E, Teixeira Alves LG, Lewis S, Watson R, Haeuptle MA, Zürcher A, Dawson KM, Steiner D, Weiss CD, Amstutz P, van Kuppeveld FJM, Stumpp MT, Bosch B-J, Engler O, Trimpert J, The trispecific DARPin ensovibep inhibits diverse SARS-CoV-2 variants, Nat. Biotechnol. 40 (2022) 1845–1854. 10.1038/s41587-022-01382-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Osuna CE, Lim S-Y, Kublin JL, Apps R, Chen E, Mota TM, Huang S-H, Ren Y, Bachtel ND, Tsibris AM, Ackerman ME, Jones RB, Nixon DF, Whitney JB, Evidence that CD32a does not mark the HIV-1 latent reservoir, Nature 561 (2018) E20–E28. 10.1038/s41586-018-0495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Thornhill JP, Pace M, Martin GE, Hoare J, Peake S, Herrera C, Phetsouphanh C, Meyerowitz J, Hopkins E, Brown H, Dunn P, Olejniczak N, Willberg C, Klenerman P, Goldin R, Fox J, Fidler S, Frater J, CD32 expressing doublets in HIV-infected gut-associated lymphoid tissue are associated with a T follicular helper cell phenotype, Mucosal Immunol. 12 (2019) 1212–1219. 10.1038/s41385-019-0180-2. [DOI] [PubMed] [Google Scholar]
- [58].Dufour C, Richard C, Pardons M, Massanella M, Ackaoui A, Murrell B, Routy B, Thomas R, Routy J-P, Fromentin R, Chomont N, Phenotypic characterization of single CD4+ T cells harboring genetically intact and inducible HIV genomes, Nat. Commun. 14 (2023) 1115. 10.1038/s41467-023-36772-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wu VH, Nordin JML, Nguyen S, Joy J, Mampe F, Del Rio Estrada PM, Torres-Ruiz F, González-Navarro M, Luna-Villalobos YA, Ávila-Ríos S, Reyes-Terán G, Tebas P, Montaner LJ, Bar KJ, Vella LA, Betts MR, Profound phenotypic and epigenetic heterogeneity of the HIV-1-infected CD4+ T cell reservoir, Nat. Immunol. 24 (2023) 359–370. 10.1038/s41590-022-01371-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sun W, Gao C, Hartana CA, Osborn MR, Einkauf KB, Lian X, Bone B, Bonheur N, Chun T-W, Rosenberg ES, Walker BD, Yu XG, Lichterfeld M, Phenotypic signatures of immune selection in HIV-1 reservoir cells, Nature 614 (2023) 309–317. 10.1038/s41586-022-05538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rossi A, Dupaty L, Aillot L, Zhang L, Gallien C, Hallek M, Odenthal M, Adriouch S, Salvetti A, Büning H, Vector uncoating limits adeno-associated viral vector-mediated transduction of human dendritic cells and vector immunogenicity, Sci Rep 9 (2019) 3631. 10.1038/s41598-019-40071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.