

The surface area of human skin is substantial, making up approximately 30 square meters and representing one of the largest epithelial surfaces in the human body for which microbes can interact (1). For microbial colonizers, the skin is a laterally and vertically heterogeneous environment, where these organisms are constantly exposed to environmental insults such as changes in temperature, pH, nutrient availability, and light exposure. In addition, microbes that colonize the skin encounter diverse competitors and antagonists in the form of bacteria, viruses, fungi, and archaea. Despite this relatively hostile and dynamic environment, human skin is home to a resident microbiome that interacts with epithelial cells, impacting health and disease. Research into the skin microbiome has historically focused on pathogens of the skin, but recent work has led to an appreciation that the skin microbiota plays a pivotal role in skin health. The specificity of these effects varies by species and strain, hinting at the potential to identify microbe–microbe and microbe–host interactions that can foster beneficial physiological processes in the skin, while also enhancing skin defense mechanisms. Some of the most abundant members of the skin microbiome are members of the genus Staphylococci. These organisms interact with microbial and host cells in a strain-specific manner to influence the host immune response and tissue repair and promote colonization resistance against pathogenic microbes. In this issue of PNAS, Joglekar et al. define the most common members of the Staphylococcal skin microbiome and provide valuable information regarding the physiological role of the Staphylococci in skin health in their manuscript titled “Integrated genomic and functional analyses of human skin associated Staphylococcus reveals extensive inter- and intra-species diversity” (2).
Although Staphylococcal skin interactions have been most widely studied in the context of the prolific skin pathogen Staphylococcus aureus, emerging research has revealed important roles for other Staphylococci in skin health and homeostasis. For instance, commensal Staphylococci represent a potent defense against colonization by the pathogenic S. aureus species (3). Some strains of Staphylococci are directly antimicrobial through the production of antimicrobial peptides to target invading and competing microorganisms or the activities of biofilm degrading proteases (4, 5). For example, the nasal colonizer Staphylococcus lugdunensis produces lugdunin, a thiazolidine-containing cyclic peptide antibiotic that inhibits colonization by S. aureus (6). In another example, Staphylococcus epidermidis directly affects skin lipid levels to promote a strong barrier for osmoregulation (7) and immune function (8). Despite this increased appreciation for the role of Staphylococcal species in colonizing human skin and affecting skin physiology, a comprehensive analysis of the Staphylococcal species that inhabit human skin, which anatomical sites they colonize, and what metabolic processes are required for site-specific colonization has not been performed.
In PNAS, Joglekar et al. define the most common members of the Staphylococcal skin microbiome and provide valuable information regarding the physiological role of the Staphylococci in skin health.
Joglekar et al. set out to define the diversity of the Staphylococcus species on human skin, reveal the core genome of the most abundant members of the genera that colonize skin, and uncover the gene expression programs driven by these organisms in response to skin exposure. This work built upon the strong foundation generated by the group where they established V1-V3-based 16S rRNA analysis that allows for species-level discrimination of Staphylococci (9). The group employed this powerful assay to analyze 16S rRNA amplicon datasets representing a variety of human body sites from 22 male and female healthy volunteers to identify the abundant species, define the pan-genome, and analyze in vitro gene expression in skin-like medium.
The authors concluded that 17 Staphylococcal species were resident or indigenous to these donors spanning body sites from head to toe. The most abundant species were S. epidermidis, Staphylococcus capitis, and Staphylococcus hominis which were present across most individuals and only S. epidermidis, S. capitis, S. hominis, and Staphylococcus warneri were present on all volunteers. The regional specificity of some organisms was made apparent by the authors’ analyses. For example, Staphylococcus auricularis was specifically present in the auditory canal raising interesting questions regarding what is specific about S. auricularis that results in this anatomic site tropism. The group paid special attention to S. aureus, as this organism is one of the leading causes of bacterial infection while also being a common member of the skin microbiome. They found that S. aureus was most abundant in the nares, and this organism was only detected in 22% of individuals, consistent with prior literature (10).
The authors then asked what core genomic elements are conserved across the organisms that live on human skin and they focused this effort on S. epidermidis, S. capitis, and S. hominis as the three most commonly detected organisms in their analyses. For this effort, the authors took advantage of a previously collected isolate libraries of these organisms. One hundred and twenty-six isolates taken from diverse volunteers and body sites were genome sequenced and analyzed, leading to the designation of a genus core genome of 1,660 genes. This analysis not only allowed the authors to define core metabolic processes that are conserved across the species but conversely enabled the definition of species-specific metabolic processes that are restricted to one species. Forty-six metabolic modules were complete in all genomes as well as several pathways that are important for skin colonization. Through comparative pan-genome analysis, the group revealed that most genes were not uniformly shared across species. Core metabolic modules that were restricted in individual species include histidine biosynthesis in S. epidermidis, histidine degradation in S. capitis and thiamine biosynthesis in S. hominis (Fig. 1). These findings highlight the diversity of the skin microenvironment and support the idea that individual Staphylococcal species have evolved to colonize distinct skin sites.
Fig. 1.
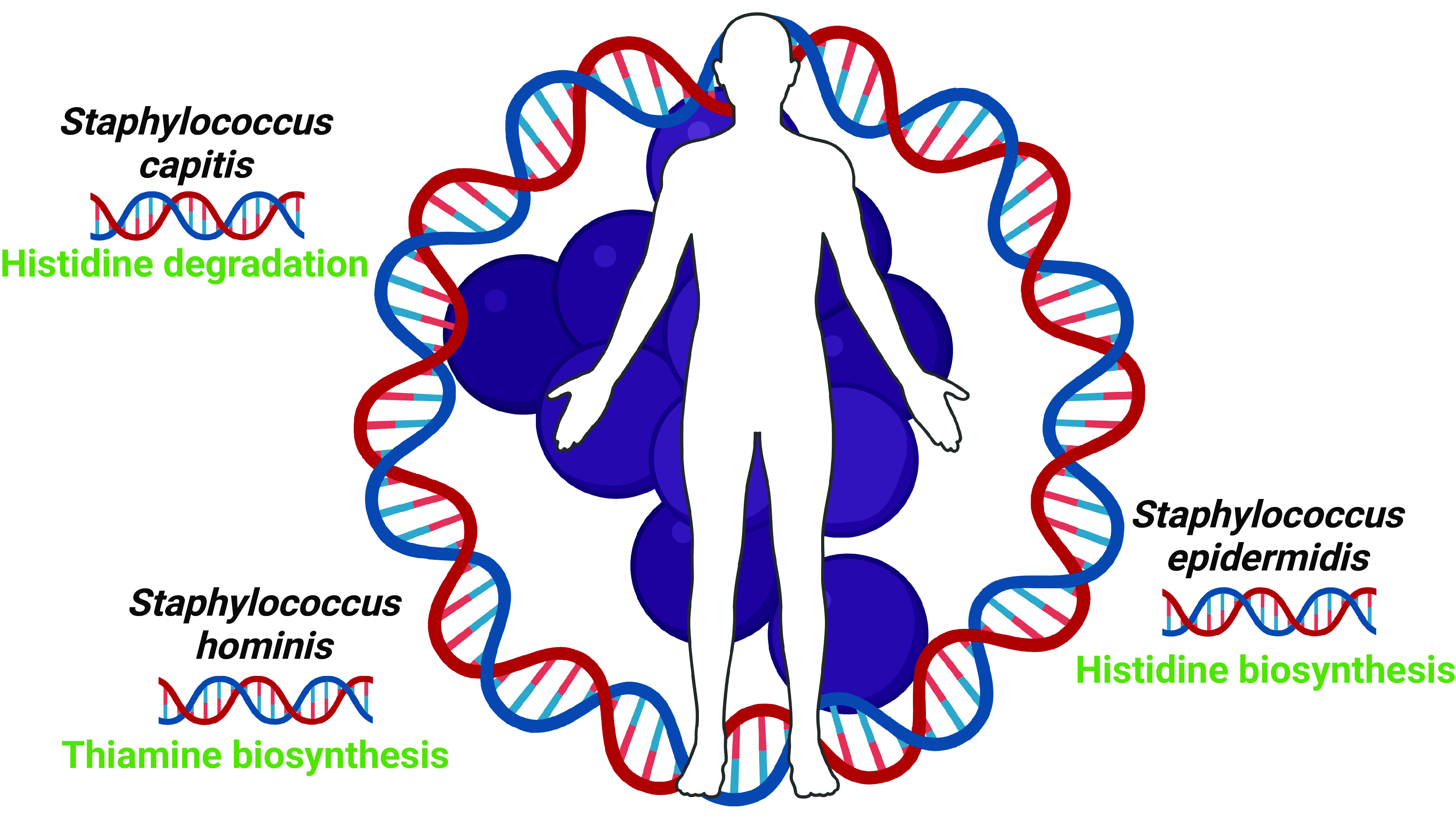
The most abundant members of the genus Staphylococcus present on human skin share a core genome while also encoding specific biochemical functions. Figure created in Biorender.
To begin to define the transcriptional programs of these important Staphylococcal skin species, the group performed in vitro growth analysis on representative isolates of each species using a synthetic skin-like medium. Organisms grown in this media were subjected to RNA sequencing analysis to define the transcriptional changes that occur during growth in skin-like media, as compared to control growth media. The group focused their analysis on the 1,647 core genes that had at least one read in all samples. Similar to the analysis of genomes, this transcriptional analysis positioned the group to define transcriptional changes that were conserved across species as well as those that were species-specific. Conserved expression patterns were observed in genes involved in gluconeogenesis, TCA cycle, amino acid and nucleotide metabolism, glycolysis, and zinc transport, among others. Notably, the most affected genes in this analysis were disproportionately found within the species-restricted core or accessory genes, suggesting elegant evolution of each species to their specific niches.
A theme that emerged from this work was the importance of metal metabolism to skin colonization by Staphylococci. Genes involved in iron acquisition were part of the genus-specific core genome, implying that the skin is an iron poor environment for colonizing microbes. In addition, the metallophore staphylopine was present in all strains of S. epidermidis, and a subset of S. capitis genomes (11). One of the metals imported by staphylopine is nickel, which is a critical cofactor for the genus core genes that encode urease, an enzyme that enables Staphylococci to survive on the acidic skin environment (12). Together, these findings lead to the possibility that Staphylococci either use staphylopine to obtain iron from the skin or alternatively using this metallophore to acquire other metals that are important cofactors for enzyme function. Conversely, adcA was one of the genes whose transcription was down-regulated in skin-like medium suggesting that the Staphylococci are zinc replete when exposed to skin, supporting the fact that the skin is one of the most zinc abundant tissues in the body (13). Together, these observations reveal the impact of metal to the evolution of the Staphylococcal genus core genome in these skin commensals and highlight the importance of metal for microbe–host interactions on the skin (14).
Through this comprehensive analysis, the authors have created a genomic atlas of one of the most common genera inhabiting the human skin. Expansion of this study could include additional research to determine how well the species focused on in this work represent the entirety of the Staphylococcal genus colonizing the skin. In addition, it will be interesting to determine how well the gene expression changes observed in synthetic skin media represent the transcriptional changes that occur during microbial colonization of human skin. Environmental factors including the specific body site, acidity, moisture, light, and antimicrobial peptides shape the microbiome of the skin making it difficult to define the species that are often present and of considerable importance to human health. Understanding the members of the skin microbiota and their gene expression profiles is a step toward engineering skin microbial communities to promote health. This is particularly true in the case of Staphylococci, which are largely genetically tractable and have the potential to be overtly antimicrobial. Information obtained in the study by Joglekar et al. is a significant step toward the rationale design of beneficial Staphylococci that could be used as microbial therapeutics.
Acknowledgments
E.P.S. is supported by R01AI150701, R01AI138581, R01AI17829, R01AI073843, U19AI174999, R01AI164587, and R01AI145992.
Author contributions
E.P.S. wrote the paper.
Competing interests
The author declares no competing interest.
Footnotes
See companion article, “Integrated genomic and functional analyses of human skin–associated Staphylococcus reveal extensive inter- and intra-species diversity,” 10.1073/pnas.2310585120.
References
- 1.Gallo R. L., Human skin is the largest epithelial surface for interaction with microbes. J. Invest. Dermatol. 137, 1213–1214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joglekar P., et al. , Integrated genomic and functional analyses of human skin–associated Staphylococcus reveal extensive inter- and intra-species diversity. Proc. Natl. Acad. Sci. U.S.A. 120, e2310585120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parlet C. P., Brown M. M., Horswill A. R., Commensal Staphylococci influence Staphylococcus aureus skin colonization and disease. Trends Microbiol. 27, 497–507 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwase T., et al. , Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 465, 346–349 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Severn M. M., et al. , The commensal Staphylococcus warneri makes peptide inhibitors of MRSA quorum sensing that protect skin from atopic or necrotic damage. J. Invest. Dermatol. 142, 3349–3352.e3345 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zipperer A., et al. , Human commensals producing a novel antibiotic impair pathogen colonization. Nature 535, 511–516 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Zheng Y., et al. , Commensal Staphylococcus epidermidis contributes to skin barrier homeostasis by generating protective ceramides. Cell Host Microbe 30, 301–313 e309 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naik S., et al. , Compartmentalized control of skin immunity by resident commensals. Science 337, 1115–1119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conlan S., Kong H. H., Segre J. A., Species-level analysis of DNA sequence data from the NIH Human Microbiome Project. PLoS ONE 7, e47075 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grundmann H., Aires-de-Sousa M., Boyce J., Tiemersma E., Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet 368, 874–885 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Ghssein G., et al. , Biosynthesis of a broad-spectrum nicotianamine-like metallophore in Staphylococcus aureus. Science 352, 1105–1109 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Zhou C., et al. , Urease is an essential component of the acid response network of Staphylococcus aureus and is required for a persistent murine kidney infection. PLoS Pathog. 15, e1007538 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogawa Y., Kinoshita M., Shimada S., Kawamura T., Zinc and skin disorders. Nutrients 10, 199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murdoch C. C., Skaar E. P., Nutritional immunity: The battle for nutrient metals at the host–pathogen interface. Nat. Rev. Microbiol. 20, 657–670 (2022), 10.1038/s41579-022-00745-6. [DOI] [PMC free article] [PubMed] [Google Scholar]