

Significance
The movement of cells mechanically coupled by cell–cell adhesions, called collective cell migration (CCM), is critical in many biological processes. A current challenge is understanding how mechanical forces and biochemical regulation interact to control cell coupling. Using biosensors to probe the mechanical state of a key load-bearing protein, vinculin, we uncovered a molecular switch that toggles its ability to bear loads at cell–cell adhesions and controls speed and coupling of CCM. Molecular models of forces at cell–cell adhesions indicate that this switch controls the friction between cells, consistent with its effect on CCM. Together, this reveals a regulatory switch for controlling cell coupling and describes a paradigm for relating biochemical regulation, mechanical properties, and cell migration dynamics.
Keywords: collective cell migration, cell–cell adhesion, mechanical coupling, friction, molecular tension sensor
Abstract
The ability of cells to move in a mechanically coupled, coordinated manner, referred to as collective cell migration, is central to many developmental, physiological, and pathophysiological processes. Limited understanding of how mechanical forces and biochemical regulation interact to affect coupling has been a major obstacle to unravelling the underlying mechanisms. Focusing on the linker protein vinculin, we use a suite of Förster resonance energy transfer-based biosensors to probe its mechanical functions and biochemical regulation, revealing a switch that toggles vinculin between loadable and unloadable states. Perturbation of the switch causes covarying changes in cell speed and coordination, suggesting alteration of the friction within the system. Molecular scale modelling reveals that increasing levels of loadable vinculin increases friction, due to engagement of self-stabilizing catch bonds. Together, this work reveals a regulatory switch for controlling cell coupling and describes a paradigm for relating biochemical regulation, altered mechanical properties, and changes in cell behaviors.
The coordinated movements of groups of cells, termed collective cell migration (CCM), play important roles in many developmental, physiological and pathological processes, including tissue morphogenesis, wound healing, and the progression of cancer (1). CCM is distinguished from single cell migration by the presence of adhesive contacts between cells. The types of cell–cell adhesion, and the associated coupling across many cells, are often used to define the various modes of CCM, which range from weakly coupled neural crest cells undergoing streaming migration to the strongly coupled epithelial cells undergoing sheet migration (2–4). The differences between modes are thought to be due to expression of distinct sets of cell adhesion receptors, such as cadherin switching associated with full and partial epithelial–mesenchymal transitions (3, 5). In contrast, the molecular-scale processes enabling rapid tuning of coupling within a given migration mode are not as well understood. This tuning is particularly important in the case of epithelial sheet migration, where the rapid alteration in coupling enables both the long-scale organization of large groups of cells while also permitting local cellular rearrangements required for efficient migration and avoidance of obstacles (6–10).
Recent advances in the understanding of CCM have been driven by both screening-based approaches and mechanistic studies, which have identified key roles for many adhesive, scaffolding, and force-generating proteins, as well as physical models focusing on key cellular-scale mechanical properties, such as cell friction, polarity, and force-generation (11–15). However, how these key proteins give rise to larger-scale mechanical processes is unclear. Interestingly, the process of adhesion strengthening, where force application results in the reinforcement of adhesion structures through the stabilization of key linkages and/or the recruitment of more linkages, has been implicated in both modeling and screening efforts (6, 12, 13, 15).
We sought to determine whether a molecular-scale, physical understanding of adhesion strengthening could elucidate the connections between key molecular players, cell-scale mechanical properties, and the regulation of epithelial cell coupling during CCM. To do so, we focused on the mechanical linker protein vinculin, as it is shown to be involved in CCM-associated processes, such as embryogenesis and cancer invasion (16, 17). Furthermore, vinculin is also a key mediator of adhesion strengthening in two distinct ways. First, in response to force application, vinculin is recruited to the structures that link cells to the surrounding extracellular matrix (ECM), termed focal adhesions (FAs), as well as the structures that mediate linkages between neighboring cells, termed adherens junctions (AJs) (18–21). Second, vinculin’s bond to F-actin is among the strongest known catch-slip bonds, which exhibit increased binding lifetime in response to applied loads before eventually failing (22).
Consistent with a role as a mediator of coupling during CCM, we find that vinculin bears substantial load at AJs, FAs, and throughout the cytoplasm during epithelial sheet migration. Furthermore, we identify a key residue, S1033, whose mutation affects the ability of vinculin to transition between inactive, unloadable and active, loadable states within the AJs and cytoplasm, with smaller effects observed in FAs. Rescue of vinculin knockout (KO) cells with wild-type (WT), phosphomimetic (S1033D), or non-phosphorylatable (S1033A) vinculin results in covarying changes in cell speed and cell coupling, as measured by the length scale of correlated motion during CCM. Notably, these results are consistent with recent mechanical models of CCM, where variation in adhesion-based friction leads to covarying changes in cell speed and coordination (13). To assess the relationship between vinculin activation, vinculin load, and friction in adhesion structures, we created molecularly detailed friction clutch models that relate force-sensitive binding dynamics of key components of AJs and FAs to the friction at each structure. In these models, increases in vinculin activation and loading lead to increased friction, and these effects are stronger at AJs than FAs, as observed experimentally. Thus, this work reveals a regulatory switch that controls the mechanical functions of vinculin to alter cell adhesion-based friction and enable the rapid tuning of coupling during CCM.
Results
Vinculin Is Loaded and Conformationally Open at the Edge of Collectively Migrating Cells.
Key aspects of vinculin function are determined by the mechanical loads it experiences and its conformation (23). Previous work in single cells has shown that vinculin load bearing and conformational regulation are separable (19). Therefore, we sought to probe vinculin load and conformation during CCM. To do so, we developed a simple system of CCM where both of these characteristics of vinculin could be readily observed. As has been done previously, we created radially expanding cell sheets using a cell droplet–based assay with Madin–Darby canine kidney (MDCK) epithelial cells (SI Appendix, Fig. S1 A–C). As multiple MDCK strains have commonly been used in studies of epithelial dynamics (7, 9, 24, 25), we assessed both MDCK II and MDCK Parental cells. We verified that sheet expansion was primarily driven by migration, as reduction of cell proliferation with Actinomycin D caused no changes in dynamics (SI Appendix, Fig. S1 D and E). To assess vinculin loading and conformation, we expressed either a Förster resonance energy transfer (FRET)-based vinculin tension sensor (VinTS) or vinculin conformation sensor (VinCS) in each cell line (19, 26, 27). All constructs produced stable proteins with the expected molecular weights (SI Appendix, Fig. S1F) and localized as expected to FAs and AJs in both cell types (Fig. 1 and SI Appendix, Figs. S1I and S3). Overexpression of VinTS or VinCS did not alter migration dynamics or FA morphology in either cell line (SI Appendix, Fig. S1 G, H, and J–M). To interpret VinTS in this system, we verified that the cytosolic tension sensor module (TSMod) reported FRET efficiencies (~0.29) consistent with no mechanical loading (28) (SI Appendix, Fig. S2 A and B). Similarly, to interpret VinCS, we established a reference for the closed state by measuring the FRET efficiency of VinCS in single cells non-specifically adhered to poly-L-lysine surfaces (SI Appendix, Fig. S2 C and D), a condition in which vinculin is predominantly cytosolic and unloaded (19). Together, these data demonstrated that this system was sufficient for probing vinculin loading and conformation during CCM.
Fig. 1.
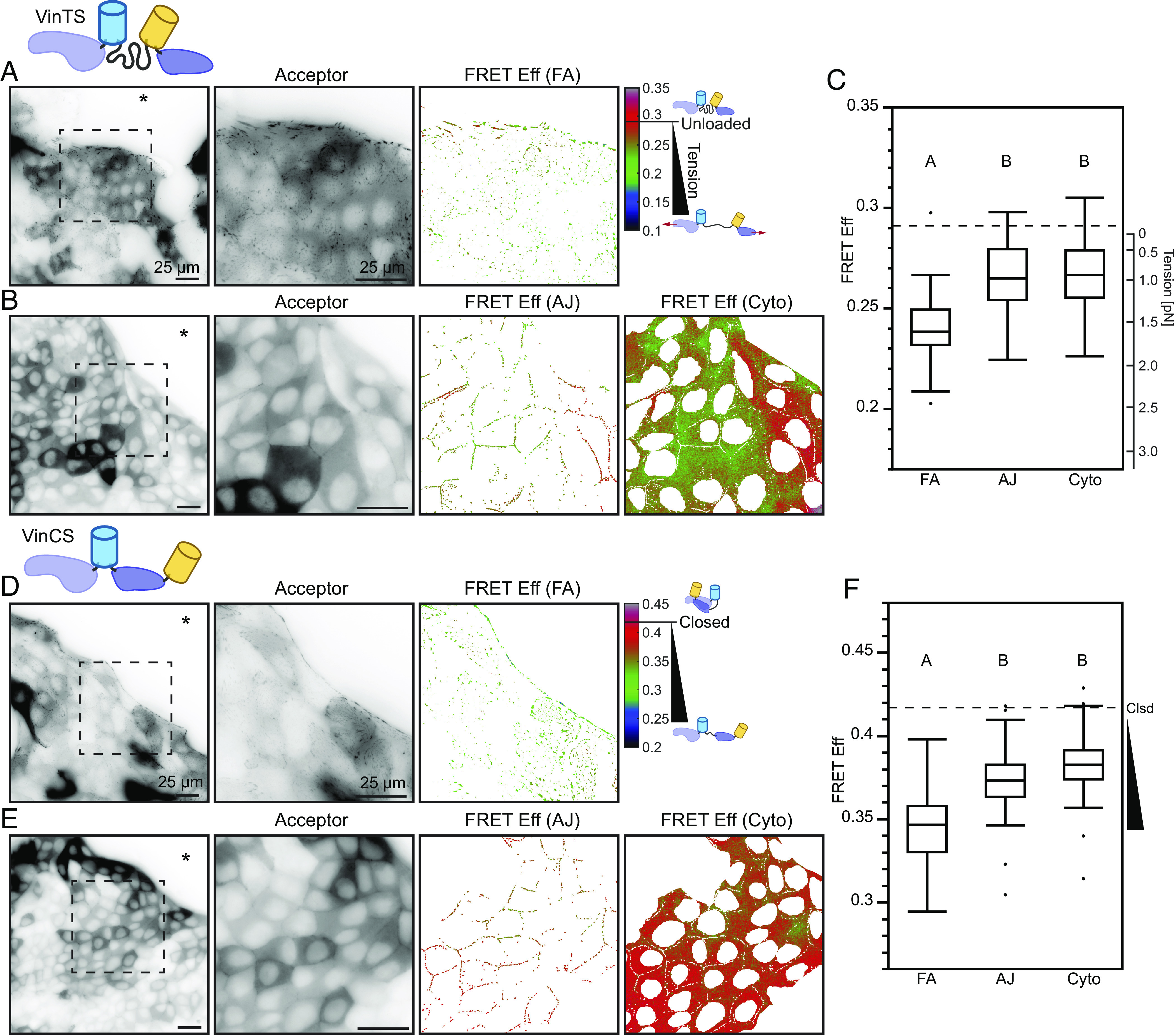
Vinculin is loaded and conformationally open at the edge of collectively migrating cells. Representative images of migrating MDCK II cell monolayers expressing VinTS taken in the basal (A) or apical (B) plane at the monolayer edge with acceptor channel indicating sensor localization followed by zoom-ins of the indicated region for acceptor channel and FRET efficiency in the FA mask (A) or AJ and cytoplasm masks (B). The asterisk indicates free space adjacent to monolayer edge. (C) Box plot of FRET efficiency for VinTS at FAs, AJs, and cytoplasm (n = 43, 34, and 34 images completed over at least 3 independent experiments) with unloaded reference level indicated (dotted line). Representative images of migrating MDCK II cell monolayers expressing VinCS taken in the basal (D) or apical (E) plane at the monolayer edge with acceptor channel indicating sensor localization followed by zoom-ins of the indicated region for acceptor channel and FRET efficiency in the FA mask (D) or AJ and cytoplasm masks (E). (F) Box plot of FRET efficiency for VinCS at FAs, AJs, and cytoplasm (n = 61, 51, and 52 images completed over at least 3 independent experiments) with closed reference level indicated (dotted line). Differences between groups were detected using the Steel–Dwass test. Levels not connected by the same letter are significantly different at P < 0.05. P-values for all comparisons can be found in SI Appendix, Supplementary Note 2 and Table S4 for VinTS and SI Appendix, Supplementary Note 2 and Table S5 for VinCS.
In confluent cells, vinculin exchanges between three sub-cellular compartments, the FAs, AJs, and cytoplasm, in a force-sensitive manner (29, 30). To probe the loads experienced by vinculin in these three compartments during CCM, we imaged VinTS-expressing MDCK cells at the leading edge of expanding epithelial sheets in the basal or apical plane. We employed standard image segmentation techniques to separate signals from the adhesion structures and cytoplasm in both focal planes. As the cytoplasmic signals in both planes were similar, we focused on the cytoplasm in the apical plane due to higher signal to noise in this compartment. Vinculin experiences the largest loads in the FAs, and lower but substantial loads in the AJs and cytoplasm of MDCK II cells (Fig. 1 A–C). Vinculin is also loaded in all three compartments in MDCK Parental cells (SI Appendix, Fig. S3 A–C). We note that vinculin loads at FAs, as well as AJs, do not vary with distance from the free edge (SI Appendix, Fig. S4), highlighting a difference between the tensions across a specific protein, vinculin, and the previously reported spatial trends in monolayer traction stresses (31). To determine whether vinculin loading was dependent on interactions with F-actin, we used VinTS-I997A (32–34). This point mutation strongly disrupts actin binding while maintaining the ability of vinculin to undergo conformational regulation, and VinTS-I997A has been shown to not bear detectable loads in the FAs of single cells. The I997A mutation resulted in the reduction of vinculin load in all compartments of both MDCK cell types (SI Appendix, Fig. S5), establishing that loads are transmitted through F-actin to vinculin in all compartments during CCM. Furthermore, as we observed actin-based loading of vinculin in the cytoplasm, we used STED imaging to confirm the existence of a cytoplasmic actin network in both MDCK cell types (SI Appendix, Fig. S6 A and B). We also observed comparable loading of VinTS at the AJs and in the cytoplasm using confocal microscopy (SI Appendix, Fig. S6 C–E), demonstrating that the VinTS signal at the AJs and in the cytoplasm was not due to out-of-plane optical effects from other sub-cellular structures.
As the exchange of vinculin between compartments is associated with conformation changes (23, 26), we also probed vinculin conformation in the FAs, AJs, and cytoplasm during CCM using MDCK cells stably expressing VinCS and the sheet expansion assay. Vinculin was the most open in FAs, and smaller but substantial portions of vinculin were open in both the AJs and in the cytoplasm (Fig. 1 D–F). FRET efficiency was highest in the cytoplasm but still significantly less than the closed reference. This demonstrates the existence of a cytoplasmic population of open vinculin and further supports a role for vinculin mediating mechanical connectivity in the cytoplasmic actin network. We found a similar trend in MDCK Parental cells (SI Appendix, Fig. S3 D–F).
Taken together, the VinTS and VinCS data demonstrate that in collectively migrating cells, vinculin is loaded and conformationally open in the FAs, AJs, and the cytoplasm, but to varying degrees in each compartment. This suggests that vinculin facilitates differential mechanical connectivity of key load-bearing sub-cellular structures within and between cells during CCM.
Vinculin S1033 Mediates a Regulatory Switch that Affects Vinculin Load and Conformation in Collectively Migrating Edges.
Previous work has shown that the phosphorylation state of vinculin affects its mechanical functions (23). Therefore, we sought to determine whether vinculin phosphorylation contributed to distinct loading and conformation observed in the various sub-cellular compartments. Trends for vinculin loading at FA, AJ, and cytoplasm were similar for the two MDCK variants, so we performed these experiments with one variant, MDCK Parental. First, we verified that paraformaldehyde fixation did not affect the FRET efficiency of VinTS (SI Appendix, Fig. S6F), consistent with previous reports (28). Fixation slightly decreased the FRET efficiency (by ~0.04) of VinCS, potentially due to a small perturbation from the fixation process. However, trends for VinCS across all subcellular compartments in monolayers were maintained, so a normalization approach was used for VinCS (SI Appendix, Fig. S6 G and H). Together, these data demonstrated that this system was sufficient for screening the effect of inhibitors and mutations on vinculin loading and conformation.
Phosphorylation sites implicated in the regulation of vinculin conformation, selective localization, or load-bearing include Y100, Y822, S1033, and Y1065 (23). Previously, Src and Abl have been shown to be the dominant regulators of phosphorylation at Y100/Y1065 and Y822, respectively (29, 35). To assess the role of these kinases and associated phosphorylation sites on the mechanical function of vinculin during CCM, we probed vinculin loading with VinTS. Inhibition of Src or Abl had little to no effect on VinTS FRET efficiency at FAs, AJs, or the cytoplasm (SI Appendix, Fig. S7 A–E). As phosphorylation at Y822 by Abl has been shown to affect vinculin mechanical function at the AJs of confluent epithelial cells, we also investigated the non-phosphorylatable point mutant (VinTS-Y822F) (29). Consistent with inhibitor studies, VinTS and VinTS-Y882F exhibited identical localization and loading in collectively migrating MDCK Parental cells (SI Appendix, Fig. S7 F–H). The primary regulators of vinculin S1033 phosphorylation are not well-established, so we employed non-phosphorylatable (S1033A) and phosphomimetic (S1033D) point mutations, as has been done previously (36). To assess the effects of these mutations on vinculin loading during CCM, we incorporated these mutations in VinTS, creating VinTS-S1033A and VinTS-S1033D, stably expressed these sensors in MDCK Parental cells, and performed sheet expansion assays. During CCM, both variants localized to FAs, AJs, and the cytoplasm. VinTS and the non-phosphorylatable VinTS-S1033A exhibited similar loading in all compartments (Fig. 2 A, B, and D and SI Appendix, Fig. S8 A, B, and D). In contrast, the phosphomimetic VinTS-S1033D exhibited drastically increased FRET efficiency at the AJs and in the cytoplasm compared to VinTS (Fig. 2 C and D), consistent with an apparent loss of loading. In FAs, VinTS-S1033D reported a partial loss of loading, suggesting a less-dominant regulatory role for S1033 phosphorylation in this compartment (SI Appendix, Fig. S8 C and D).
Fig. 2.
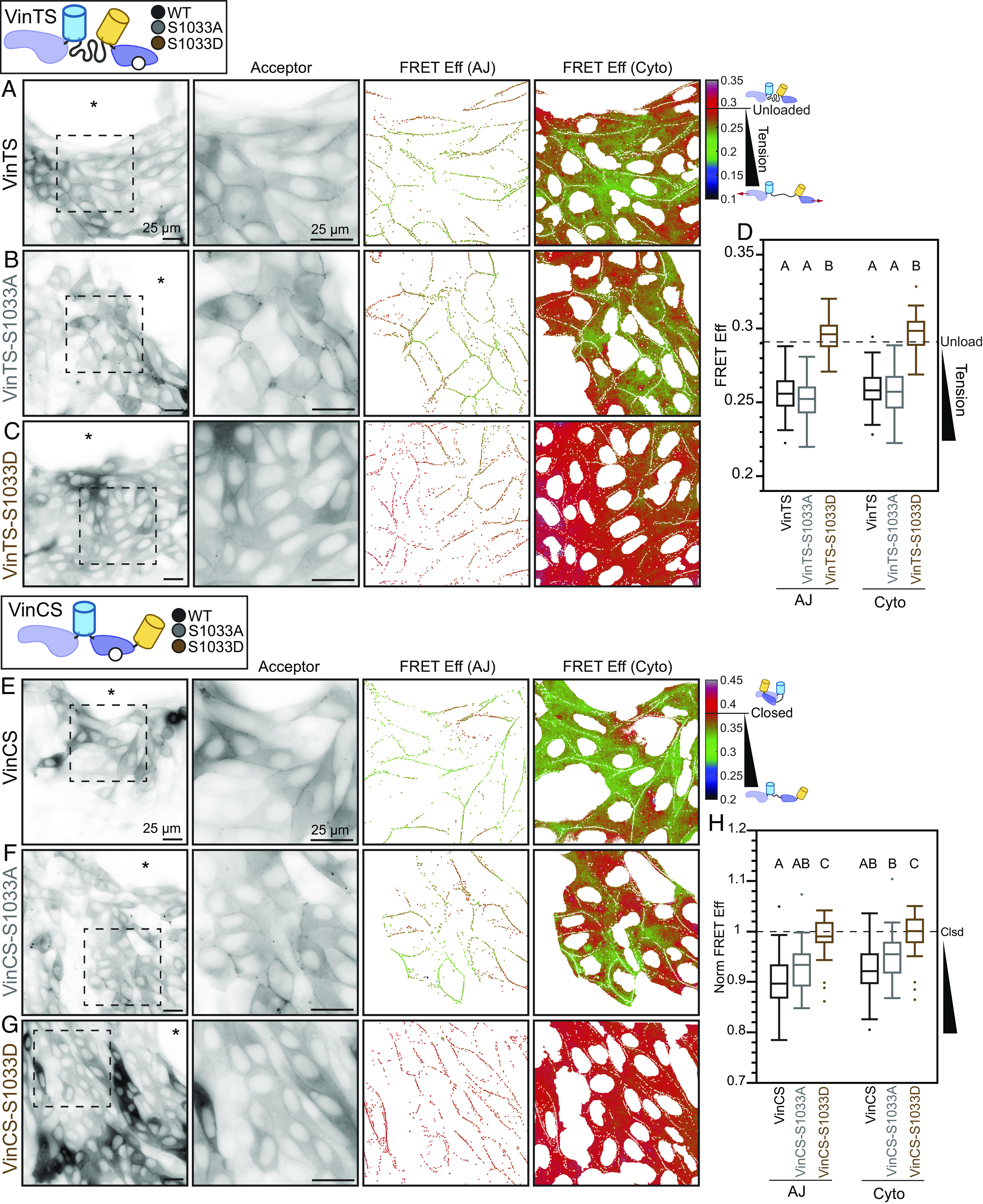
Vinculin S1033 mediates a regulatory switch that affects vinculin load and conformation at the edge of collectively migrating cells. Representative images of migrating MDCK Parental cell monolayers expressing VinTS (A), VinTS-S1033A (B), or VinTS-S1033D (C) taken in the apical plane at the monolayer edge with acceptor channel indicating sensor localization followed by zoom-ins of the indicated region for acceptor channel and FRET efficiency in AJ and cytoplasm masks. The asterisk indicates free space adjacent to monolayer edge. (D) Box plot of FRET efficiency for VinTS, VinTS-S1033A, and VinTS-S1033D at AJs and cytoplasm (n = 61/60, 55/55, and 48/48 AJ/cytoplasm images, respectively, over at least 3 independent experiments) with unloaded reference level indicated (dotted line). Representative images of migrating MDCK Parental cell monolayers expressing VinCS (E), VinCS-S1033A (F), or VinCS-S1033D (G) taken in the apical plane at the monolayer edge with acceptor channel indicating sensor localization followed by zoom-ins of the indicated region for acceptor channel and FRET efficiency in AJ and cytoplasm masks. The asterisk indicates free space adjacent to monolayer edge. (H) Box plots of normalized FRET efficiency for VinCS, VinCS-S1033A, and VinCS-S1033D at AJs and cytoplasm (n = 58/58, 26/26, and 37/37 AJ/cytoplasm images, respectively, over at least 3 independent experiments) with closed reference level indicated (dotted line). Differences between groups were detected using the Steel–Dwass test. Levels not connected by the same letter are significantly different at P < 0.05. P-values for all comparisons can be found in SI Appendix, Supplementary Note 2 and Tables S7 and S8.
In vinculin, the S1033 site is located at an interface between the tail and head domains that is critical for the regulation of conformation (37). To determine whether mutation of S1033 affects the conformation of vinculin during CCM, we created VinCS variants containing S1033A or S1033D, stably expressed them in MDCK Parental cells, and performed sheet expansion assays. VinCS and non-phosphorylatable VinCS-S1033A exhibited identical localization and conformation in all compartments (Fig. 2 E, F, and H and SI Appendix, Fig. S8 E, F, and H). In contrast, in AJs and the cytoplasm, the phosphomimetic VinCS-S1033D exhibited drastically higher FRET, consistent with complete closing of vinculin (Fig. 2 G and H). In FAs, VinCS-S1033D reported a partial reduction in the amount of open vinculin, again consistent with a less-dominant regulatory role for S1033 phosphorylation in this compartment (SI Appendix, Fig. S8 G and H).
Taken together, these data describe a regulatory switch for vinculin, where phosphorylation at S1033 biases vinculin toward a closed, unloaded state. Furthermore, this switch appears dominant at AJs and within the cytoplasm, but only partially reduces load and the amount of open vinculin in FAs. Thus, the switch mediates tuning of mechanical connectivity within and between cells, although with different strengths.
Vinculin Regulatory Switch Affects Speed and Correlation Length of CCM.
Next, we sought to determine the effects of this regulatory switch for vinculin on CCM. To do so, we first created a CRISPR-KO vinculin MDCK II cell line (SI Appendix, Fig. S9A) and rescued these cells with Vinculin-mVenus (VinV), VinV-S1033A, or VinV-S1033D. All constructs produced stable proteins with the expected molecular weights (SI Appendix, Fig. S9B). Furthermore, these variants localized to FAs, AJs, and the cytoplasm as expected (SI Appendix, Fig. S9 C–E) and exhibited broadly similar relationships between FA morphology and distance from the free edge compared to endogenous vinculin (SI Appendix, Figs. S1 J and L and S9 F and G). Expression of VinV, VinV-S1033A, or VinV-S1033D did not affect actin-based protrusions or actin belts at the leading edge of monolayers (SI Appendix, Fig. S10 A–F), and all four cell types exhibited similar trends in the morphology and intensity of paxillin-labeled FAs (SI Appendix, Fig. S11), indicating minimal effects on regulators of persistent motility in this system. Additionally, there were non-significant or small (<25%) differences in the abundance of E-cadherin (SI Appendix, Fig. S10 G–K), alpha-Catenin (SI Appendix, Fig. S10 L–P), or extended alpha-Catenin (SI Appendix, Fig. S10 Q–U) at the AJs across the four cell types. Together, these data demonstrated that this system lacks large defects in the assembly of sub-cellular structures and was sufficient for testing the effects of the regulatory switch for vinculin on CCM.
To characterize CCM dynamics, we observed the migration of monolayers in a previously described barrier assay (SI Appendix, Fig. S12 A and B) and measured velocity fields in the monolayer by optical flow constraint (25, 38) (Fig. 3 A–D and Movie S1–S4). To quantify effects on CCM, we analyzed large regions of the monolayer at the free edge (extending up to 500 µm or ~20 cells inward from the free edge, as indicated in SI Appendix, Fig. S12D) using two well-studied kinematic parameters for migrating monolayers: the speed (average velocity magnitude) and the correlation length of deviations in the lateral velocity component (direction indicated in SI Appendix, Fig. S12C), which is a previously described measure of mechanical coupling (7). Rescue of MDCK II vinculin CRISPR-KO cells with VinV reduced the average speed (Fig. 3E), consistent with previous findings that vinculin knockdown increased speed (12), and reduced the correlation length (Fig. 3F and SI Appendix, Fig. S12E). CCM of cells rescued with VinV-S1033A was comparable to those rescued with VinV, while rescue with VinV-S1033D was comparable to KO cells (Fig. 3 E and F and SI Appendix, Fig. S12F). Together, these data show that the regulatory switch controlling vinculin loading and conformation determines the speed and correlation length of collectively migrating MDCK cells.
Fig. 3.
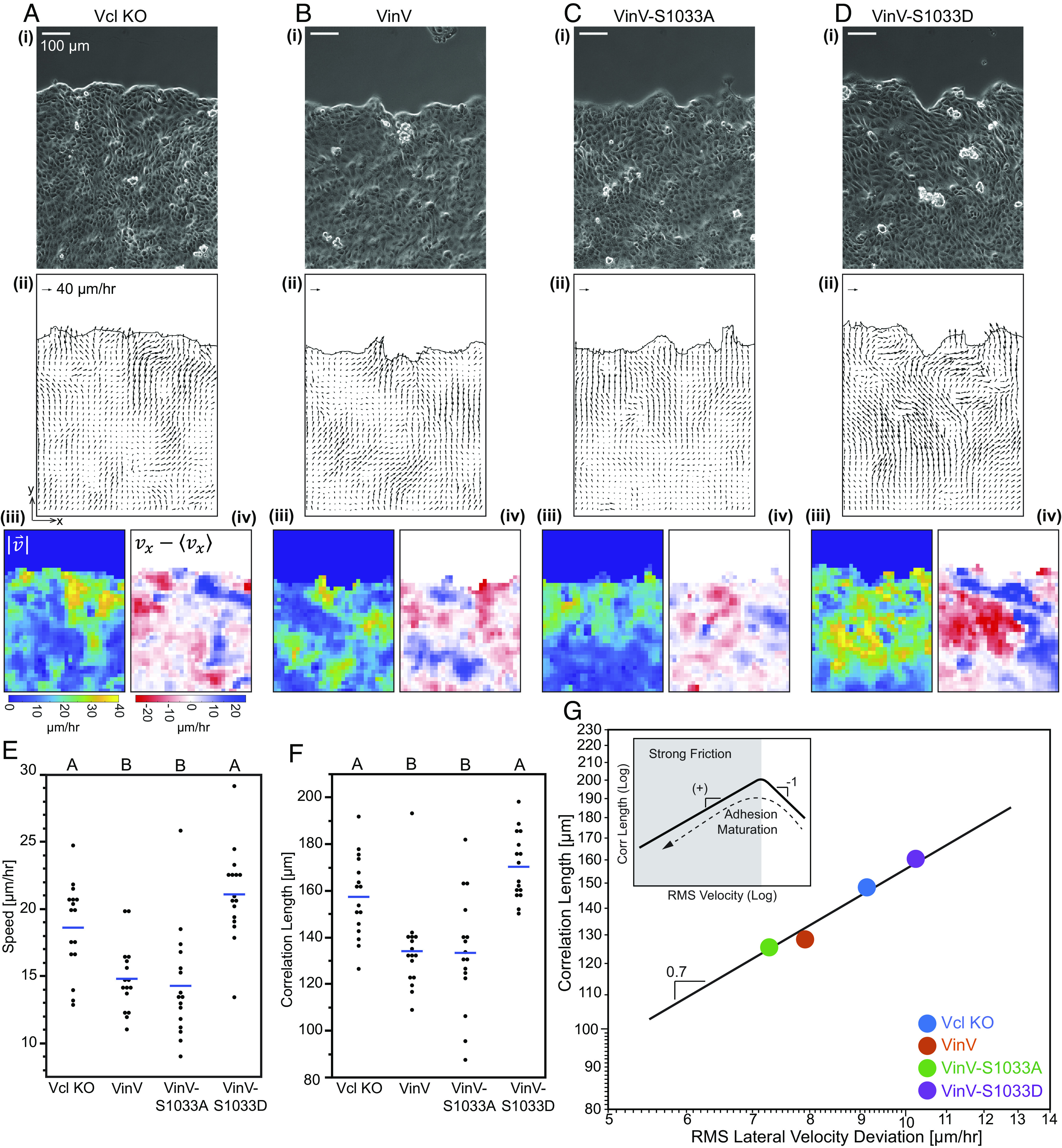
Vinculin regulatory switch affects speed and correlation length of CCM. Representative images of migrating MDCK II Vcl KO cells (A) or MDCK II Vcl KO cells rescued with VinV (B), VinV-S1033A (C), or VinV-S1033D (D) in the barrier migration assay. Shown for each are the following images: (i) phase contrast, (ii) velocity field, (iii) velocity magnitude, and (iv) lateral velocity deviation. Timelapses of phase contrast images with vector field overlay are available in Movie S1–S4. Plots showing mean (blue line) and all data points for velocity magnitude (E) and correlation length for lateral velocity deviations (F) (n = 16 monolayers for each cell line over 6 independent experiments). Differences between groups were detected using Tukey’s HSD test. Levels not connected by the same letter are significantly different at P < 0.05. P-values for all comparisons can be found in SI Appendix, Supplementary Note 2 and Tables S9 and S10. Log-log plot of correlation length for lateral velocity deviations vs. rms lateral velocity deviations (G), showing mean for each cell line and black line of slope 0.7 to guide the eye. See SI Appendix, Fig. S12 for separate plots by cell line. The Inset is a conceptual schematic.
Vinculin Regulatory Switch Controls Adhesion-Based Friction in Models of the FA and AJ.
We next leveraged recent modelling work to interpret the observed changes in CCM dynamics in terms of alterations in mechanical variables (11, 13). We favored a model that incorporated adhesive bond dynamics and had been applied in the context of the MDCK cells used here (13). This model has two regimes (Fig. 3 G, Inset). One regime is characterized by fast-moving cells and weak adhesion-based friction, with a constant effective friction coefficient. In the other regime, adhesion-based friction is dominant, with a velocity-dependent effective friction coefficient that arises from the force-sensitive bond dynamics of molecular linkers at cell adhesions. These two regimes, and the associated dominant mechanical variables, can be discriminated by the relationship between the speed and correlation length of CCM, being negative for the weak friction regime and positive for the strong friction regime. Their work showed that MDCK cells moving within a confluent monolayer (i.e., no free edge) exhibit a positive relationship, indicating adhesion-based friction plays a dominant role in resisting cell motion (13). We applied this framework to the lateral component of the velocity field (SI Appendix, Fig. S12C), which has previously been shown to be unbiased by the free edge (7). We found that the vinculin KO and all the rescued MDCK cells also exhibited a positive relationship between correlation length and speed, both within a given cell line (SI Appendix, Fig. S12G) and between cell lines (Fig. 3G). This is consistent with the strong adhesion-based friction regime and suggests a key role for velocity-dependent friction coefficients in our system (13). Furthermore, rescue of vinculin expression in MDCK KO cells promoted lower speeds and correlation lengths, and this required the ability of vinculin to become open and loaded. This suggests the vinculin switch can manipulate adhesion-based friction through changes to the friction coefficient.
To probe the relationship between force-activated binding dynamics and adhesion-based friction, we used friction clutch models, which predict the resistive force due to the sliding of two surfaces relative to each other at a particular speed as a function of the number and properties of adhesive linkages between these surfaces (39) (SI Appendix, Supplementary Note 1). To begin, we investigated previously developed models that contained linkages with a single simple bond type, such as an ideal bond that does not respond to force or a slip bond that weakens with force (39) (SI Appendix, Supplementary Note 1 and Fig. S15). To validate our implementation of a friction clutch, we simulated clutches comprised of linkages with ideal or slip bonds, and we observed agreement with previous predictions of the relationship between frictional force and speed (39). To gain intuition about the molecular determinants of friction, we also created a friction clutch based on catch-slip bonds, and for each scenario, we compared the mean engagement lifetime and the effective friction coefficient, which is a standard parameter describing the frictional resistance between interfaces (cell-ECM or cell–cell) in models of CCM (11). Notably, the qualitative shape of the friction coefficient–speed curve related to the individual linkage dynamics, being independent of speed for ideal bonds, monotonically decreasing for slip bonds, or biphasic for catch-slip bonds. Therefore, the underlying molecular-scale dynamics are indicative of the larger-scale mechanics of the friction clutch.
To probe the effects of complex connectivity and potential regulatability of load-bearing linkages within adhesion structures, we developed multi-component linkages for use in the friction clutch models. These multi-component linkages were based on integrin:talin:F-actin complexes in FAs or E-cadherin:beta-catenin:alpha-catenin:F-actin complexes in AJs, which could be reinforced through the incorporation of vinculin. The ability of these multi-component linkages to maintain connectivity under mechanical load was based on the force-dependent bond kinetics determined previously for key interfaces (22, 40–44) (SI Appendix, Supplementary Note 1 and Fig. S14). As these parameters were obtained from single molecule experiments characterizing the interfaces separately, we first assessed their suitability for use in combination to model multi-component linkages at the FA and AJ. The engagement lifetime of both linkages increased initially with loading rate and then decreased, indicating that the multi-component linkages possessed catch-slip characteristics, which were stronger for the integrin-based than cadherin-based linkage (SI Appendix, Supplementary Note 1 and Fig. S16). Furthermore, reinforcement of the F-actin interface with vinculin did not change the overall functional form, but instead increased the lifetime of both FA and AJ linkages across a wide range of loading rates, as expected for a mechanical stabilizer. We note that these behaviors of the multi-component linkages are not readily predictable from the force-sensitive dynamics of single components, as there is no single dominant interface (SI Appendix, Supplementary Note 1 and Fig. S14).
Next, we determined the qualitative relationships between force-activated bond dynamics and larger-scale mechanics in FAs and AJs using friction clutch models containing multi-component integrin- or cadherin-based linkages (Fig. 4 A and B and SI Appendix, Supplementary Note 1 and Figs. S17 and S18). We represented the action of the S1033-based vinculin regulatory switch by modeling vinculin in two states. As the mutation of S1033 affects vinculin load and conformation but not localization to FAs/AJs, vinculin is conceptualized as either 1) closed and unloadable in a localized pool at or near FAs/AJs (potentially bound to the membrane or another unloaded component of the FA/AJ), or 2) open and loadable within FAs/AJs (bound to an exposed vinculin binding site in talin/alpha-catenin and to F-actin) depending on the binding kinetics. In the absence of vinculin, the mean linkage engagement lifetime varied biphasically with velocity (Fig. 4 C and F), resembling the stronger/weaker catch-slip behaviors we found for individual integrin-/cadherin-linkages (SI Appendix, Supplementary Note 1 and Fig. S16). The friction coefficient–velocity relationships were also biphasic (Fig. 4 D and G). Thus, as was observed in the simpler friction clutch models, the multi-component linkage dynamics were predictive of the qualitative shape of the friction coefficient–speed relationships. Furthermore, over the range of speeds associated with epithelial sheet migration (1 to 30 µm/h), increasing the fraction of loadable vinculin substantially increased the magnitude of both the engagement lifetime and friction coefficient, without drastically altering their functional forms with respect to speed. The increase in friction due to the vinculin switch occurred over a wide range of other model parameters and was driven mainly by the force-dependent unbinding of vinculin from F-actin (SI Appendix, Supplementary Note 1 and Figs. S19 and S20), highlighting the force sensitivity of vinculin’s F-actin bond as a key model parameter. To assess the ability of the vinculin switch to tune friction, we assessed the effect of finer variations in the fraction of loadable vinculin at an intermediate speed in the range for CCM experimentally probed here (10 µm/h) (Fig. 4 E and H). In both FAs and AJs, the friction coefficient was tuned linearly by the amount of loadable vinculin, although vinculin’s effect was overall higher in AJs (~fourfold increase) than in FAs (~twofold increase). Similarly, the ensemble vinculin tension scaled linearly with the amount of loadable vinculin, and both the order of magnitude and prediction of higher vinculin tension at FAs vs. AJs were consistent with our experimental measurements of VinTS.
Fig. 4.
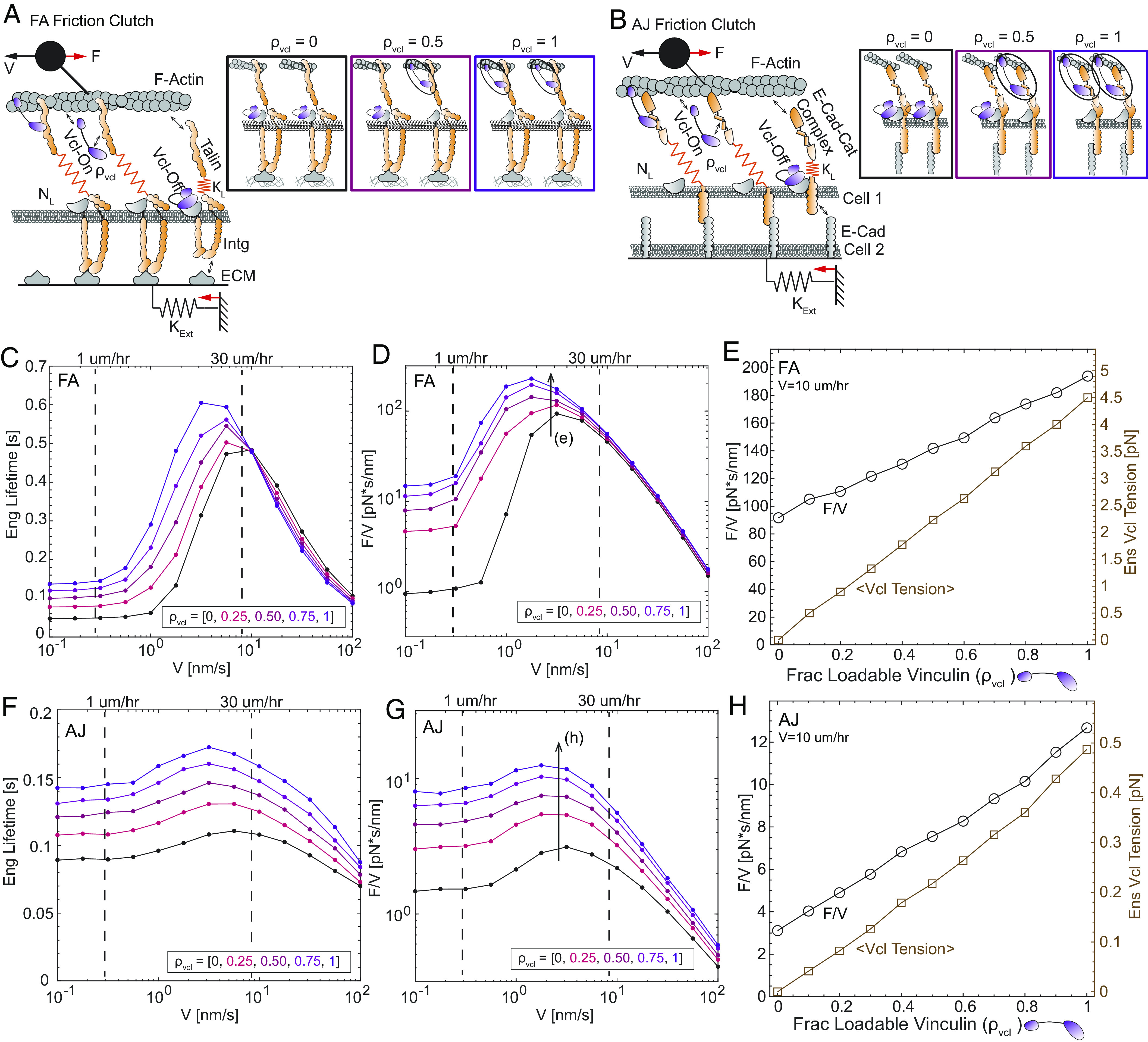
Vinculin regulatory switch controls adhesion-based friction in models of the FA and AJ. Schematics of FA (A) and AJ (B) friction clutch models. Linkage schematics depict different values for the fraction of linkages with loadable vinculin (ρVcl). Values for the external spring constant (KExt) were based on the stiffness of the substrate for the FA model or the stiffness of a cell monolayer for the AJ model (SI Appendix, Supplementary Note 1). For FA, plots of mean linkage engagement lifetime (C) and mean effective friction coefficient (F/V) (D) vs. speed (V) for 5 values of the fraction of loadable vinculin (ρVcl). For FA, plot of mean effective friction coefficient (Left Y axis) and mean ensemble vinculin molecular tension (Right Y axis) vs. fraction of loadable vinculin (E) for an intermediate speed (10 µm/h). (F–H) Analogous plots for the AJ friction clutch model. See SI Appendix, Supplementary Note 1 for the formulation, parameters, and additional analyses of the friction clutch models.
Together, these results suggest that regulation of vinculin mechanical reinforcement controls adhesion-based friction strongly at the cell–cell interface, and to a lesser extent at the cell-ECM interface, to affect the speed and coordination of collectively migrating epithelial sheets.
Discussion
This work reveals a regulatory, mechanochemical switch that determines the ability of vinculin to mediate mechanical connectivity within sub-cellular structures and cell coupling during CCM. This switch functions by toggling vinculin between closed, unloaded and open, loadable states at the AJs and in the cytoplasm. Manipulation of the switch through vinculin mutants mimicking the phosphorylation state of S1033 controlled the kinematics of CCM, with increased vinculin loading decreasing both the speed and length scale of mechanical coupling. This covariation indicates a regime of CCM dominated by adhesion-based friction (13), and suggests a role for the vinculin mechanochemical switch in determining this mechanical variable. Consistently, reinforceable friction clutch models based on force-sensitive binding dynamics of key components of AJs and FAs show that the presence of open, loadable vinculin increases friction in adhesion structures, with larger effects observed in AJs. These effects are driven by the stabilization of molecular linkages via the engagement of the Vcl:F-actin catch bond. Overall, this work elucidates how a regulatable, biological mechanism (vinculin mechanochemical switch) can tune a specific cell mechanical property (adhesion-based friction) to affect a larger-scale biological process (mechanical coupling in CCM).
As vinculin is a mechanical linker protein important in a variety of critical processes, several models have been put forth to explain the regulation of its conformation and ability to bear loads. The earliest models assumed that vinculin was closed in the cytoplasm and open in adhesion structures and focused on the co-localization of multiple binding partners to alleviate the autoinhibitory head-tail interaction of vinculin (26, 45). Other models focused on a key role for phosphorylation in regulating the activation of vinculin, the dominance of mechanical forces within adhesion structures “pulling” vinculin open, or combined mechanisms where vinculin phosphorylation primes mechanical-based activation (18, 37). Several recent publications have shown that vinculin conformation and loading are independently regulated, disfavoring purely mechanical models (19, 32, 46). The data in this manuscript are consistent with a model that builds upon these observations but is distinct from previous models. We propose that vinculin exists in closed, unloaded; open, unloaded; and open, loaded states within the adhesion structures as well as the cytoplasm. Furthermore, phosphorylation, mimicked by S1033D, can be inhibitory to both vinculin activation and loading but does not prevent vinculin localization to pools near or within adhesion structures. We propose the function of the localized pools of closed, unloaded vinculin is to mediate quick reinforcement of structures and the tuning of friction, particularly within AJs (SI Appendix, Fig. S13). This would facilitate responses that occur on faster time scales than regulatable by protein expression changes and that are insulated from signals associated with other load-bearing sub-cellular structures.
Several key points remain to be clarified about the regulation of vinculin as a mechanical linker. The first is the regulation of pools of closed vinculin within or near adhesion structures. Closed vinculin is known to localize to FAs, where it has been shown to be recruited via an interaction with phosphorylated paxillin (32, 47). In contrast, at the AJs, an interaction partner for closed vinculin has not been well characterized. Therefore, determining the molecular connectivity of closed vinculin at the AJs is an important future direction. The second is determining how adhesion-based friction and key regulators of vinculin localization, conformation, and loading vary in diverse contexts. We note that S1033 mutations had dominant effects in AJs and the cytoplasm, but not the FAs. In contrast to this work studying cells undergoing CCM, previous work in confluent epithelial cells has shown that Abl-mediated phosphorylation of vinculin at Y822 was required for its localization to AJs (29). This suggests context-dependent biochemical regulatory schemes that could enable differential regulation of vinculin-based mechanical connectivity, coupling, and friction in diverse adhesion structures and cell states.
The finding of a population of open, loaded vinculin in the cytoplasm was surprising. Current models for vinculin loading require a binding partner at the head in addition to F-actin at the tail (23). We propose that alpha-actinin could be a candidate, but the mediators of vinculin loading in the cytoplasm remain to be tested. While it has been noted that the mechanical loading of FAs and AJs are often co-regulated and integrated through the cytoplasm, the pertinent mechanisms and functional consequences of this co-regulation are not fully understood (48). One possible function is that this co-regulation facilitates the ability of sub-cellular structures to function in unison and prevent fragmentation, which is often referred to as mechanical coherence (49, 50). An interesting topic for the future will be determining whether loaded cytoplasmic vinculin plays a role in enabling the mechanical coherence of cells and tissues.
This work also has ramifications for the currently emerging molecular-scale understanding of adhesion strengthening as well as mechanical coupling during diverse forms of cell migration. First, adhesion strengthening can occur through the recruitment of new linkages or the reinforcement of existing linkages (51). Previous work has focused mainly on the recruitment of new linkages, especially integrins and cadherins (41, 52). On the other hand, the work here demonstrates the existence, tunability, and consequences of the reinforcement-based mechanism. Therefore, an important question is how these strengthening mechanisms interact and if they are regulated in a coordinated or independent fashion. Second, there are a plethora of primary mechanical linkages and mechanical linker proteins that localize to cellular adhesion structures in various biological contexts. These proteins are known to have differential interaction partners, binding dynamics, and load-bearing capabilities (6, 51, 53–56). Currently, the various types of primary linkages are thought to mediate specific interactions between various cell types (3, 5), but the biological relevance for the diversity of the mechanical linker proteins is currently unclear. Interestingly, we note that in our models of adhesion-based friction, control over properties of cell force transmission was decoupled in a similar manner at the AJs and FAs. The primary mechanical linkage (E-cadherin:beta-catenin:alpha-catenin:F-actin or integrin:talin:F-actin complexes) controlled the dependencies of linkage dynamics and friction coefficient on speed (e.g., the presence of a peak or broad optimum). The reinforcing protein (vinculin) modulated the magnitude of these two variables across a range of speeds associated with CCM, causing an approximately linear increase in their respective amplitudes as the fraction of loadable vinculin increased, with little effect on their functional forms. This suggests a control principle for adhesion-based forces where the functional form is encoded by the primary linkage and the magnitude is controlled via biochemical regulation of a reinforcing protein. If this paradigm is generalizable, it suggests that the various primary linkages, in addition to mediating specific interactions, could mediate distinct types of velocity dependences, such as effective slip bonding, while other reinforcing proteins could mediate differential strengths in the tuning of friction as a function of velocity, such as non-linear effects. Therefore, an attractive hypothesis for explaining the diversity of mechanical linker proteins could be to enable distinct and tunable relationships between cell force transmission and migration speed to mediate various biological processes. Lastly, the emerging complexity of the mechanical functions of adhesion structures during CCM indicates the need for more techniques capable of precisely measuring key mechanical variables associated with migration (e.g., friction, adhesion, motive force). This will be critical in informing physical models of CCM as well as further elucidating how molecular-scale processes, such as those mediated by mechanical linker proteins, regulate these mechanical variables.
Overall, this work demonstrates a biochemically regulatable switch for vinculin that enables control of friction and modulation of cell coupling during migration. Furthermore, the framework developed here integrates biosensors to probe the state and molecular loading of a linker protein, mathematical models to connect force-sensitive bond dynamics of the linker to adhesion level force transmission, and tests of the function of the linker in CCM. It provides a means for determining the relative importance of different mechanical linker proteins in specific contexts, which will likely impact a variety of future studies in mechanobiology and cell migration.
Materials and Methods
Cell Culture and Expression of DNA Constructs.
MDCK (NBL-2) Parental cells (ATCC® CCL-34™, obtained from Duke University Health System’s Cell Culture Facility) and MDCK II cells (generous gift from Dr. Adam Kwiatkowski, University of Pittsburgh) were maintained in a humidified 5% CO2 atmosphere at 37 °C in Dulbecco's Modified Eagle's Medium - low glucose (D6046; Sigma Aldrich) supplemented with 10% fetal bovine serum (HyClone), 1% antibiotic/antimycotic (Gibco), and 1 g/L sodium bicarbonate (Gibco). MDCK cell type was confirmed by probing expression of Claudin-2 (expressed by MDCK II cells but not MDCK Parental cells). DNA constructs were stably expressed in cell lines using viral transduction as previously described (46). See SI Appendix, Extended Methods for generation of DNA constructs and more information on expression of DNA constructs. CRISPR/Cas9-mediated knockout of vinculin in MDCK II cells was performed using the previously described guide RNA CACGAGGAAGGCGAGGTGGA (57). This sequence was cloned into All-in-One U6-gRNA/CMV-Cas9-tGFP (Sigma-Aldrich). MDCK II cells were subject to transient transfection of the plasmid using electroporation, followed by clonal selection to obtain a vinculin knockout line. Knockout of vinculin was confirmed by western blot analysis.
Migration Assays.
Droplet-based (31) and barrier-based (7) migration assays were conducted as previously described. Migration assays were conducted on fibronectin-coated glass. To quantify migration in the barrier-based assay, velocity fields were computed from timelapse images using a previously described implementation of the Optical Flow Constraint method (25). See SI Appendix, Extended Methods for complete information on the preparation, imaging, and analysis of the migration assays.
Western Blots and Immunofluorescent Labeling.
Western blot analysis and cell staining and immunofluorescence were performed using standard protocols as previously described (46). See SI Appendix, Extended Methods and Tables S11 and S12 for the identities and dilutions of the antibodies used.
Imaging of FRET-Based Sensors.
Imaging and analysis of FRET-based sensors was performed using previously published protocols (28). See SI Appendix, Extended Methods for more information.
Kinase Inhibitor Treatments.
Inhibitor treatments were performed on cells in the droplet assay 72 h post-seeding. To inhibit Src kinase, cells were treated with 10 μM PP2 (Abcam ab120308) for 1 h. To inhibit Abl kinase, cells were treated with 50 μM Imatinib (Sigma SML1027) for 1 h. After treatment, cells were washed, fixed, and imaged.
Statistics.
Statistical analyses were performed using JMP Pro (SAS, Cary, NC) software. Comparisons of data approximately normal and with equal variances, as determined by Levine’s test, were analyzed with ANOVA and, if necessary, Tukey’s Honest Significant Difference (HSD) test. Comparisons of data approximately normal and with unequal variances, as determined by Levine’s test, were analyzed with Welch’s ANOVA and, if necessary, Steel–Dwass non-parametric multiple comparisons test. Comparisons of data not approximately normal were analyzed with the Kruskal–Wallis test (one-way ANOVA on ranks) and, if necessary, Steel–Dwass non-parametric multiple comparisons test. A P-value of P < 0.05 was considered statistically significant. In figures, a single asterisk (*), double asterisk (**), triple asterisk (***), and quadruple asterisk (****) indicate P-values less than 0.05, 0.01, 0.001, and 0.0001 respectively, and ns indicates a P-value greater than or equal to 0.05. When letters were used instead to indicate statistical significance, levels not connected by the same letter are statistically different at P < 0.05. Standard box plots were created using JMP Pro, where the bottom and top of the box indicate the first and third quartiles, respectively, the middle line indicates the median, the whiskers extend to the outermost data points below the first quartile and above the third quartile that are within 1.5 times the interquartile range, and data outside the whiskers are indicated as points.
Computational Friction Clutch Models of FA and AJ.
The formulation, implementation, and parameter values for the FA and AJ friction clutch mathematical models are provided in SI Appendix, Supplementary Note I.
See SI Appendix, Extended Methods for the generation of DNA constructs; quantification of actin organization, vinculin FA morphology, and protein abundance at AJs; estimation of the closed FRET efficiency of VinCS; confocal imaging of FRET sensors; and STED imaging of actin.
Supplementary Material
Appendix 01 (PDF)
Representative phase contrast timelapse with vector field overlay for migrating MDCK II Vcl KO cells in the barrier migration assay. Length scale bar is 100 μm. Velocity scale arrow is 40 μm/hr. First timepoint is approximately 3 hours post-barrier lift and time between images is 10 min. Total elapsed time from first image is indicated at top right.
Representative phase contrast timelapse with vector field overlay for migrating MDCK II Vcl KO cells rescued with VinV in the barrier migration assay. Length scale bar is 100 μm. Velocity scale arrow is 40 μm/hr. First timepoint is approximately 3 hours post-barrier lift and time between images is 10 min. Total elapsed time from first image is indicated at top right.
Representative phase contrast timelapse with vector field overlay for migrating MDCK II Vcl KO cells rescued with VinV-S1033A in the barrier migration assay. Length scale bar is 100 μm. Velocity scale arrow is 40 μm/hr. First timepoint is approximately 3 hours post-barrier lift and time between images is 10 min. Total elapsed time from first image is indicated at top right.
Representative phase contrast timelapse with vector field overlay for migrating MDCK II Vcl KO cells rescued with VinV-S1033D in the barrier migration assay. Length scale bar is 100 μm. Velocity scale arrow is 40 μm/hr. First timepoint is approximately 3 hours post-barrier lift and time between images is 10 min. Total elapsed time from first image is indicated at top right.
Acknowledgments
We thank Dr. Adam Kwiatkowski (University of Pittsburgh) for providing MDCK II cells used in this study; Dr. Akira Nagafuchi (Nara Medical University) for providing the alpha-catenin conformation–sensitive antibody (α18); Vidya Venkatraman for production of stable cell lines and other technical support; and Dr. Trevor R. Ham for comments and discussion of the manuscript. T.C.S was supported by the NSF Graduate Research Fellowship under Grant No. (GRFP DGE 1644868). D.E.C. was supported by the National Institute of General Medical Sciences of the NIH under award number (R35GM119617). B.D.H. was supported by the National Institute of General Medical Sciences of the NIH under award number (R01GM121739).
Author contributions
B.D.H. designed research; T.C.S. and E.M.G. performed research; T.C.S., E.M.G., J.I.C., A.N.U., and D.E.C. contributed new reagents/analytic tools; T.C.S. and E.M.G. analyzed data; T.C.S. designed, implemented, and analyzed mathematical models; and T.C.S. and B.D.H. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All data supporting the findings of this study are available within the article or its supporting information. MATLAB codes used to perform image pre-processing (https://gitlab.oit.duke.edu/HoffmanLab-Public/image-preprocessing) (27), three channel sensitized emission FRET image analysis (https://gitlab.oit.duke.edu/HoffmanLab-Public/fret-analysis) (27), and simulation of the mathematical AJ and FA friction clutch models (https://gitlab.oit.duke.edu/HoffmanLab-Public/aj_fa_frictionclutchmodels) (58) are publicly available on GitLab.
Supporting Information
References
- 1.Friedl P., Gilmour D., Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 10, 445–457 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Friedl P., Locker J., Sahai E., Segall J. E., Classifying collective cancer cell invasion. Nat. Cell Biol. 14, 777–783 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Friedl P., Mayor R., Tuning collective cell migration by cell-cell junction regulation. Cold Spring Harb. Perspect. Biol. 9, a029199 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scarpa E., Mayor R., Collective cell migration in development. J. Cell Biol. 212, 143–155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell K., Casanova J., A common framework for EMT and collective cell migration. Development 143, 4291–4300 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Ladoux B., Mege R. M., Mechanobiology of collective cell behaviours. Nat. Rev. Mol. Cell Biol. 18, 743–757 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Petitjean L., et al. , Velocity fields in a collectively migrating epithelium. Biophys. J. 98, 1790–1800 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vedula S. R., et al. , Emerging modes of collective cell migration induced by geometrical constraints. Proc. Natl. Acad. Sci. U.S.A. 109, 12974–12979 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vishwakarma M., et al. , Mechanical interactions among followers determine the emergence of leaders in migrating epithelial cell collectives. Nat. Commun. 9, 3469 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaritsky A., et al. , Seeds of locally aligned motion and stress coordinate a collective cell migration. Biophys. J. 109, 2492–2500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alert R., Trepat X., Physical models of collective cell migration. Annu. Rev. Cond. Matt. Phys. 11, 77–101 (2020). [Google Scholar]
- 12.Bazellieres E., et al. , Control of cell-cell forces and collective cell dynamics by the intercellular adhesome. Nat. Cell Biol. 17, 409–420 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia S., et al. , Physics of active jamming during collective cellular motion in a monolayer. Proc. Natl. Acad. Sci. U.S.A. 112, 15314–15319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simpson K. J., et al. , Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat. Cell Biol. 10, 1027–1038 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Weber G. F., Bjerke M. A., DeSimone D. W., A mechanoresponsive cadherin-keratin complex directs polarized protrusive behavior and collective cell migration. Dev. Cell 22, 104–115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubashkin M. G., et al. , Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylinositol (3,4,5)-triphosphate. Cancer Res. 74, 4597–4611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu W., Baribault H., Adamson E. D., Vinculin knockout results in heart and brain defects during embryonic development. Development 125, 327–337 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Dumbauld D. W., et al. , How vinculin regulates force transmission. Proc. Natl. Acad. Sci. U.S.A. 110, 9788–9793 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grashoff C., et al. , Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature 466, 263–266 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.le Duc Q., et al. , Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J. Cell Biol. 189, 1107–1115 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leerberg J. M., et al. , Tension-sensitive actin assembly supports contractility at the epithelial zonula adherens. Curr. Biol. 24, 1689–1699 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang D. L., Bax N. A., Buckley C. D., Weis W. I., Dunn A. R., Vinculin forms a directionally asymmetric catch bond with F-actin. Science 357, 703–706 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bays J. L., DeMali K. A., Vinculin in cell-cell and cell-matrix adhesions. Cell Mol. Life Sci. 74, 2999–3009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dukes J. D., Whitley P., Chalmers A. D., The MDCK variety pack: Choosing the right strain. BMC Cell Biol. 12, 43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vig D. K., Hamby A. E., Wolgemuth C. W., On the quantification of cellular velocity fields. Biophys. J. 110, 1469–1475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen H., Cohen D. M., Choudhury D. M., Kioka N., Craig S. W., Spatial distribution and functional significance of activated vinculin in living cells. J. Cell Biol. 169, 459–470 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LaCroix A. S., Lynch A. D., Berginski M. E., Hoffman B. D., Tunable molecular tension sensors reveal extension-based control of vinculin loading. Elife 7, e33927 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gates E. M., LaCroix A. S., Rothenberg K. E., Hoffman B. D., Improving quality, reproducibility, and usability of FRET-based tension sensors. Cytometry A 95, 201–213 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bays J. L., et al. , Vinculin phosphorylation differentially regulates mechanotransduction at cell-cell and cell-matrix adhesions. J. Cell Biol. 205, 251–263 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sumida G. M., Tomita T. M., Shih W., Yamada S., Myosin II activity dependent and independent vinculin recruitment to the sites of E-cadherin-mediated cell-cell adhesion. BMC Cell Biol. 12, 48 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trepat X., et al. , Physical forces during collective cell migration. Nat. Phys. 5, 426–430 (2009). [Google Scholar]
- 32.Case L. B., et al. , Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions. Nat. Cell Biol. 17, 880–892 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rothenberg K. E., Scott D. W., Christoforou N., Hoffman B. D., Vinculin force-sensitive dynamics at focal adhesions enable effective directed cell migration. Biophys. J. 114, 1680–1694 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson P. M., et al. , Identification of an actin binding surface on vinculin that mediates mechanical cell and focal adhesion properties. Structure 22, 697–706 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z., et al. , The phosphorylation of vinculin on tyrosine residues 100 and 1065, mediated by SRC kinases, affects cell spreading. Mol. Biol. Cell 15, 4234–4247 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Auernheimer V., Goldmann W. H., Serine phosphorylation on position 1033 of vinculin impacts cellular mechanics. Biochem. Biophys. Res. Commun. 450, 1095–1098 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Golji J., Wendorff T., Mofrad M. R., Phosphorylation primes vinculin for activation. Biophys. J. 102, 2022–2030 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poujade M., et al. , Collective migration of an epithelial monolayer in response to a model wound. Proc. Natl. Acad. Sci. U.S.A. 104, 15988–15993 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sens P., Rigidity sensing by stochastic sliding friction. Europhys. Lett. 104, 38003 (2013). [Google Scholar]
- 40.Buckley C. D., et al. , Cell adhesion. The minimal cadherin-catenin complex binds to actin filaments under force. Science 346, 1254211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elosegui-Artola A., et al. , Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat. Cell Biol. 18, 540–548 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Le S., Yu M., Yan J., Direct single-molecule quantification reveals unexpectedly high mechanical stability of vinculin-talin/alpha-catenin linkages. Sci. Adv. 5, eaav2720 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Owen L. M., Bax N. A., Weis W. I., Dunn A. R., The C-terminal actin-binding domain of talin forms an asymmetric catch bond with F-actin. Proc. Natl. Acad. Sci. U.S.A. 119, e2109329119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rakshit S., Zhang Y., Manibog K., Shafraz O., Sivasankar S., Ideal, catch, and slip bonds in cadherin adhesion. Proc. Natl. Acad. Sci. U.S.A. 109, 18815–18820 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen H., Choudhury D. M., Craig S. W., Coincidence of actin filaments and talin is required to activate vinculin. J. Biol. Chem. 281, 40389–40398 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Tao A., et al. , Identifying constitutive and context-specific molecular-tension-sensitive protein recruitment within focal adhesions. Dev. Cell 58, 522–534 e527 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pasapera A. M., Schneider I. C., Rericha E., Schlaepfer D. D., Waterman C. M., Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 188, 877–890 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weber G. F., Bjerke M. A., DeSimone D. W., Integrins and cadherins join forces to form adhesive networks. J. Cell Sci. 124, 1183–1193 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai Y., Sheetz M. P., Force propagation across cells: Mechanical coherence of dynamic cytoskeletons. Curr. Opin. Cell Biol. 21, 47–50 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai Y., et al. , Cytoskeletal coherence requires myosin-IIA contractility. J. Cell Sci. 123, 413–423 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoffman B. D., Yap A. S., Towards a dynamic understanding of cadherin-based mechanobiology. Trends Cell Biol. 25, 803–814 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Malinverno C., et al. , Endocytic reawakening of motility in jammed epithelia. Nat. Mater 16, 587–596 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Case L. B., Waterman C. M., Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat. Cell Biol. 17, 955–963 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferrer J. M., et al. , Measuring molecular rupture forces between single actin filaments and actin-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 9221–9226 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoffman B. D., Grashoff C., Schwartz M. A., Dynamic molecular processes mediate cellular mechanotransduction. Nature 475, 316–323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leckband D. E., J. d. Rooij, Cadherin adhesion and mechanotransduction. Annu. Rev. Cell Dev. Biol. 30, 291–315 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Ozawa M., Nonmuscle myosin IIA is involved in recruitment of apical junction components through activation of alpha-catenin. Biol. Open 7, bio031369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shoyer T.C., et al. , HoffmanLab-Public/aj_fa_frictionclutchmodels. GitLab. https://gitlab.oit.duke.edu/HoffmanLab-Public/aj_fa_frictionclutchmodels. Deposited 2 November 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Representative phase contrast timelapse with vector field overlay for migrating MDCK II Vcl KO cells in the barrier migration assay. Length scale bar is 100 μm. Velocity scale arrow is 40 μm/hr. First timepoint is approximately 3 hours post-barrier lift and time between images is 10 min. Total elapsed time from first image is indicated at top right.
Representative phase contrast timelapse with vector field overlay for migrating MDCK II Vcl KO cells rescued with VinV in the barrier migration assay. Length scale bar is 100 μm. Velocity scale arrow is 40 μm/hr. First timepoint is approximately 3 hours post-barrier lift and time between images is 10 min. Total elapsed time from first image is indicated at top right.
Representative phase contrast timelapse with vector field overlay for migrating MDCK II Vcl KO cells rescued with VinV-S1033A in the barrier migration assay. Length scale bar is 100 μm. Velocity scale arrow is 40 μm/hr. First timepoint is approximately 3 hours post-barrier lift and time between images is 10 min. Total elapsed time from first image is indicated at top right.
Representative phase contrast timelapse with vector field overlay for migrating MDCK II Vcl KO cells rescued with VinV-S1033D in the barrier migration assay. Length scale bar is 100 μm. Velocity scale arrow is 40 μm/hr. First timepoint is approximately 3 hours post-barrier lift and time between images is 10 min. Total elapsed time from first image is indicated at top right.
Data Availability Statement
All data supporting the findings of this study are available within the article or its supporting information. MATLAB codes used to perform image pre-processing (https://gitlab.oit.duke.edu/HoffmanLab-Public/image-preprocessing) (27), three channel sensitized emission FRET image analysis (https://gitlab.oit.duke.edu/HoffmanLab-Public/fret-analysis) (27), and simulation of the mathematical AJ and FA friction clutch models (https://gitlab.oit.duke.edu/HoffmanLab-Public/aj_fa_frictionclutchmodels) (58) are publicly available on GitLab.