

Abstract
Emergent resistance to all clinical antibiotics calls for the next generation of therapeutics. Here we report an effective antimicrobial strategy targeting the bacterial H2S-mediated defense system. We identified cystathionine-γ-lyase (CSE) as the primary generator of H2S in two major human pathogens, Staphylococcus aureus and Pseudomonas aeruginosa, and discovered small molecules inhibiting bacterial CSE by a novel mechanism. These inhibitors potentiate bactericidal antibiotics against both pathogens in vitro, and in mouse models of infection. Remarkably, the CSE inhibitors also suppress bacterial tolerance, disrupting biofilm formation and drastically reducing the number of “persister” bacteria surviving antibiotic treatment. Our results establish bacterial H2S as a multifunctional defense factor, and CSE as a drug target for versatile antibiotic enhancers.
One Sentence Summary:
Versatile antibiotic potentiators targeting H2S biosynthesis in bacteria
Introduction
The twin trends of increasingly prevalent pathogens resistant to multiple antibiotics, and a dwindling number of new antimicrobials reaching the clinic (1–3), have been projected to result in antibiotic-resistant pathogens killing 10 million people annually by the year 2050 (4). Antibiotic resistance is a stable, heritable ability of a microorganism to proliferate in the presence of a high level of antibiotic (3, 5).
Yet, bacteria also have an innate ability to survive normally lethal antibiotic challenge without genetic resistance, by transiently stopping growth and slowing down their metabolism. This is known as antibiotic tolerance, or persistence, if only a fraction of the bacterial population acquires the phenotype; the surviving bacteria are called persisters (6). Persistence can be triggered by a range of stressors besides the antibiotic insult; moreover, bacterial populations can spontaneously generate low levels of persisters as an apparent hedging strategy (7).
Antibiotic tolerance received little attention until the early 2000s (8–11), when the ability of bacterial biofilms to withstand high levels of antibiotics was explained by the presence of persisters. Biofilms are prevalent in human disease, particularly in the hospital setting and associated with implantable medical devices (8, 12). Persisters are also implicated in other chronic disease (7, 13–16); and in acute infections (17), antibiotic tolerance may explain the paradoxical therapy failures of antibiotic-sensitive infections.
In addition to their direct role in disease, the potentially critical role of persister bacteria in the emergence and spreading of antibiotic resistance is becoming increasingly clear (18–20). Yet to date, no persister-targeting therapeutic has received FDA approval. Using potentiator drugs to sensitize persister bacteria to the available clinical antibiotics is an attractive strategy for addressing this need.
A conceptually compelling but sparsely investigated approach to the identification of useful potentiators involves disrupting the general defense systems that protect pathogens from diverse antibiotics. One such system produces hydrogen sulfide (H2S), which protects bacteria against oxidative stress (21). Virtually all bacteria generate H2S via enzymes orthologous to the mammalian cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), or 3-mercaptopyruvate sulfurtransferase (3MST) (21–24). Genetic disruption of H2S biogenesis sensitizes a wide range of pathogens, including S. aureus and P. aeruginosa, to different classes of bactericidal drugs, and to the host immune response (23, 25–27).
We set out to block the source of bacterial H2S pharmacologically, aiming to potentiate bactericidal antibiotics. To this goal, we first established the primary enzymatic source of H2S in two common human pathogens, Gram-positive S. aureus and Gram-negative P. aeruginosa that are among the leading causes of hospital-acquired infections and are characterized by pervasive multi-drug resistance. We then combined computational, structural, biochemical, and in vivo approaches to screen, select, and validate small molecule compounds targeting this source. The lead inhibitors synergized with diverse clinical antibiotics in vitro and in vivo. Furthermore, combined with antibiotics, they markedly diminished persisters and biofilm formation in the two model pathogens. These observations establish H2S biogenesis as an important contributor to bacterial tolerance and the target of a new class of versatile antibiotic potentiators.
CSE is the primary source of H2S in S. aureus and P. aeruginosa
Previously we have shown that many bacterial species, including S. aureus and P. aeruginosa, possess cse and cbs genes organized in a single operon responsible for bulk H2S production under normal growth in rich media or under antibiotic stress (23). To determine the relative contribution of bacterial CSE (bCSE) to H2S production, the corresponding genes in both species were inactivated via Tn insertions (Fig. 1A; Table S1) or Tn-based gene replacements/deletions (Figs. S1, S2; Table S1). Using the classic lead acetate reactivity test for H2S detection, as well as H2S-specific twisted internal charge transfer (TICT)-based (28) and monobromobimane (MBB)-based (29) fluorescent probes, we showed that lack of bCSE results in major deficiency of H2S production in S. aureus and P. aeruginosa strains of clinical origin (Figs. 1B and S1). The contribution of bacterial CBS (bCBS), under the same standard conditions, is relatively insignificant (Figs. S1 and S2). While growth of the mutant strains in rich media was unaffected without antibiotics, the inactivation of cse alone was sufficient to sensitize S. aureus and P. aeruginosa to low doses of representative bactericidal antibiotics from different classes, including gentamycin (Gm), norfloxacin (Nor), and ampicillin (Amp) (Fig. 1C, Figs. S1 and S2, Tables S2 and S3).
Fig. 1. bCSE is a predominant source of H2S in S. aureus and P. aeruginosa.
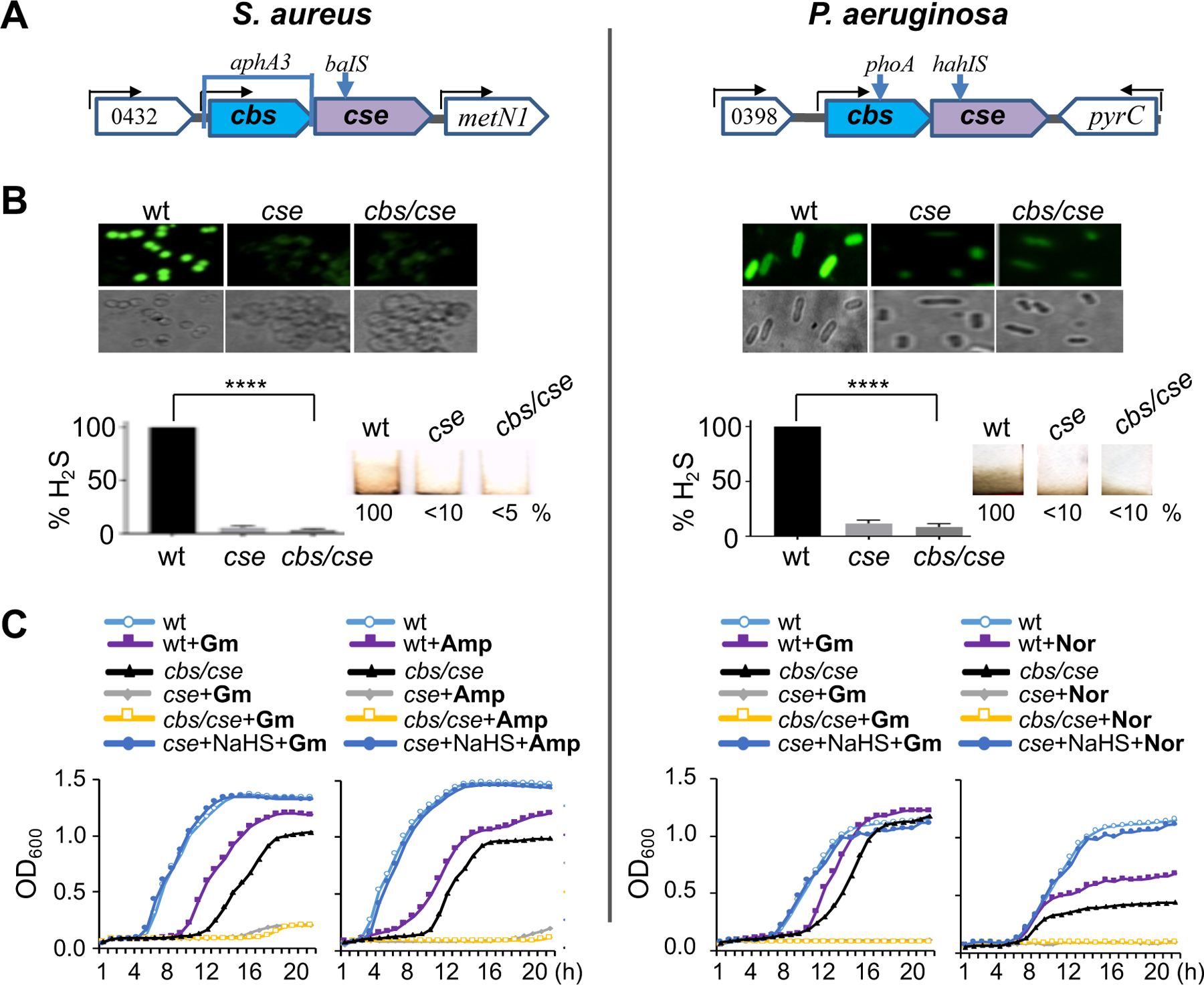
(A) Genomic organization of the cbs/cse operon in S. aureus (USA300) and P. aeruginosa (PAO1) and genetic tools used to selectively disrupt cbs and cse genes. A bracket shows gene replacement/deletion and arrows indicate transposon insertions (gene disruption) (see also Figs. S1, S2, and Table S1). (B) Quantitation of H2S produced by wild type (wt) and cbs/cse(−) S. aureus USA300 (MRSA isolate JE2) and P. aeruginosa (PAO1). Top: Representative fluorescence images demonstrate H2S production by live wt and mutant S. aureus (USA300) and P. aeruginosa (PAO1) cells treated with the TICT-based fluorescent H2S probe. Bottom left: Quantification of images. Values are means ± SD (n = 3), ****P < 0.0001 (Student’s t-test; equal variance). Bottom right: Representative Pb-acetate-soaked paper strips show a brown stain of PbS as a result of the reaction with gaseous H2S exiting liquid bacterial cultures. Numbers show H2S production relative to wt cells. (C) Representative growth curves of wt and cbs/cse(–) S. aureus (USA300) and P. aeruginosa (PAO1) in the presence of gentamycin (Gm, 1 and 2 µg/ml, respectively), ampicillin (Amp, 0.1 µg/ml), or norfloxacin (Nor, 1 µg/ml), without or with the H2S donor NaHS (0.2 and 0.4 mM, respectively). The data points are averages of optical density (OD) at 600 nm wavelength with a margin of error of less than 5%. See also Figs. S1 and S2.
Inhibition of bCSE holoenzyme requires novel types of drugs
The above results provided the rationale for designing specific small molecule inhibitors of bCSE. Given the high sequence identity and structural similarity of the catalytic site between human CSE (hCSE) and bCSEs (Fig. S3A- B, Table S4) (30, 31), we first examined whether S. aureus CSE (SaCSE) and P. aeruginosa CSE (PaCSE) activity could be suppressed by the established and clinically relevant inhibitors of hCSE (32–34). DL-propargylglycine (PAG), β-cyano-L-alanine (BCA), hydroxylamine (HA), aminooxyacetic acid (AOAA), and L-aminoethoxyvinylglycine (AVG) inhibited the production of H2S from L-cysteine by hCSE with the IC50 of ~1–18 μM (Fig. S3C). However, only AOAA, AVG and HA at considerably higher concentrations were capable of inhibiting bCSEs (Figs. S3C and S4).
Most hCSE inhibitors act via targeting the coenzyme pyridoxal phosphate (PLP) and therefore can interfere with the activity of other PLP enzymes (32, 33, 35). In addition, PAG can covalently modify a residue in the active site of hCSE (30). To determine whether AOAA, the most efficient bCSE inhibitor, has a special mechanism of bCSE suppression, we determined the crystal structure of the SaCSE-AOAA complex. AOAA forms an external aldimine with PLP that prevents catalytically essential formation of an internal aldimine between PLP and K196 (Fig. S3D- H). As this mode of inhibition was proposed for various PLP-dependent enzymes (36–38) (Fig. S3I), which are abundant in humans, the development of PLP-targeting drugs, either de novo or based on the available hCSE inhibitors, would be unlikely to yield bCSE-specific low toxicity inhibitors.
Structure-based virtual screening for allosteric bCSE inhibitors
To identify other potential drug-binding sites, we examined the available structures of CSE and “CSE-like enzymes” (31, 39, 40) and determined X-ray crystallographic structures of SaCSE in different states (Table S4). SaCSE holoenzyme has a large substrate entry tunnel and a crevice oriented approximately perpendicularly to the tunnel, with a total volume ~1,500–1,600 Å3, depending on the location of the flexible side chains (Fig. S3J). Apart from reaction substrates, the tunnel can accommodate other small molecules, such as HEPES or cacodylate, albeit at low occupancy even in the absence of a substrate (Fig. S3J). A characteristic feature of the crevice is parallelly oriented Y103 and H339 (Fig. S3K). These amino acids sandwich Y99 in the apo SaCSE (30, 31) and PLP-bound SaCSE, prior to the formation of the internal aldimine that requires movement of Y99 into the catalytic site to assist in proper positioning of PLP for catalysis (Movie S1).
Accordingly, our in silico predictions highlighted two potential drug-binding areas (I and II) in the SaCSE holoenzyme (Fig. S3L). Area I overlaps with the substrate entry tunnel and Area II mostly coincides with the crevice. We reasoned that PLP and reaction substrates bind to the catalytic site with high affinity. Therefore, it would be difficult to identify competitive inhibitors targeting Area I in the initial screens. In contrast, Area II appears amenable for small aromatic or planar molecules binding Y103 through π-stacking when Y99 moves inwards for catalysis (Fig. S3K, Movie S2). Notably, an aromatic amino acid at position 103 in bCSEs (tyrosine in S. aureus and phenylalanine in P. aeruginosa) is substituted by a non-aromatic asparagine in mammalian CSEs. Given distinct binding properties of asparagine and aromatic amino acids, this observation suggests that small molecules preferentially targeting bCSEs may exist.
To identify such compounds, we conducted structure-based virtual screening (SBVS) of ~3.2 million commercially available small molecules by docking the representative “cluster-centroid” compounds using the AutoDock Vina software package (41) (Fig. S5A). Close analogs to the top-rated hits were also selected and run through the docking pipeline in two rounds (42). SBVS resulted in the selection of over 40 hits, most of which contained planar aromatic functionalities, amide groups, and carboxylate moieties.
In vitro and in vivo validation of selected bCSE inhibitors
SBVS hits and over 100 related compounds were purchased and/or chemically synthesized and screened in a variety of functional assays (Fig. S5B). The three most promising compounds, designated NL1, NL2, and NL3, belong to the same chemotype (Fig. 2A). They contain a central indole moiety with acetyl glycine (NL1), furan carboxylate (NL2) or pyrazole carboxylate (NL3) at the N1 position and bromine (NL1 and NL2) or chlorobenzothiophene (NL3) at the C6 position.
Fig. 2. Target validation of selected bCSE inhibitors.
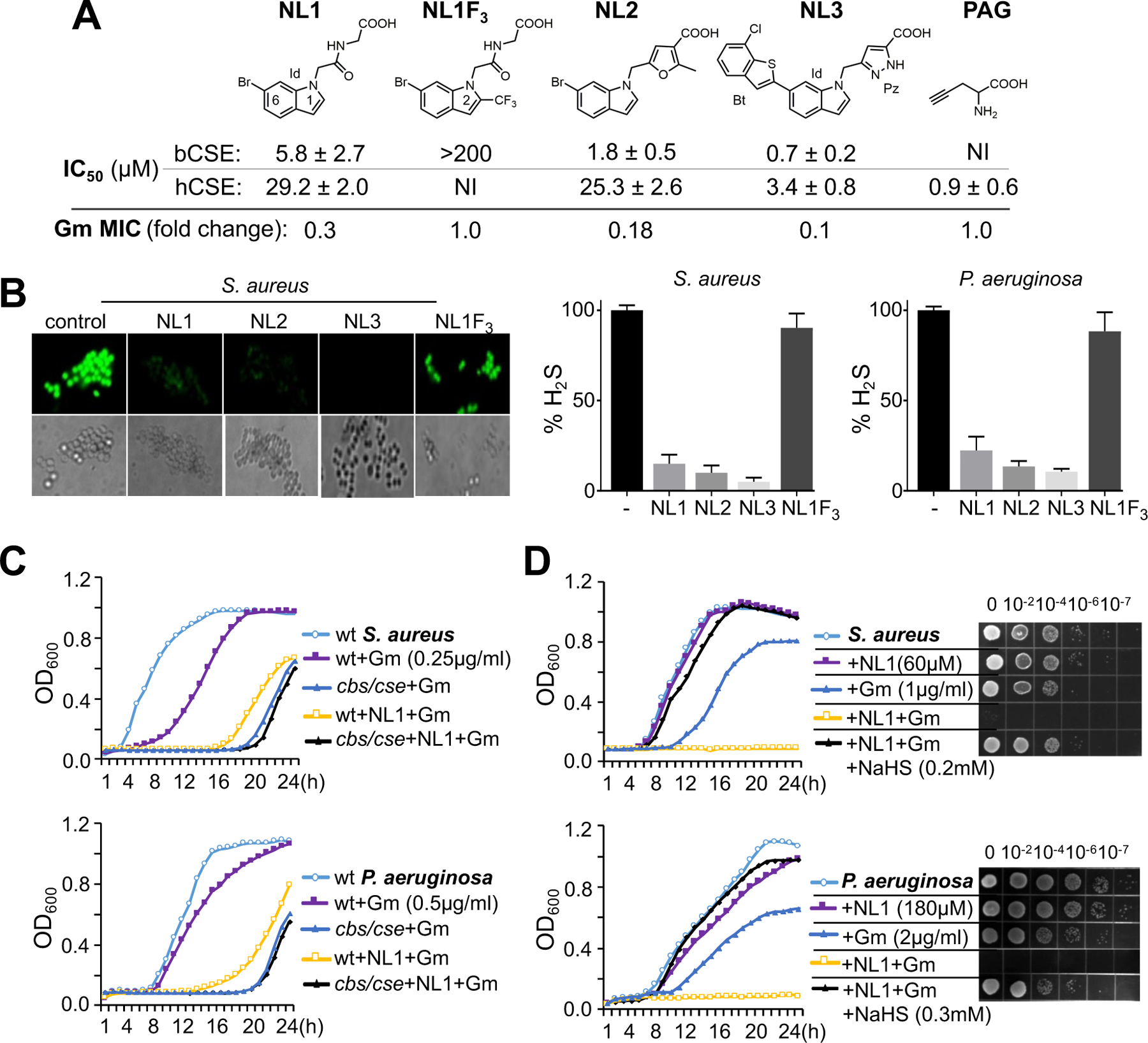
(A) Chemical structures of selected bCSE inhibitors, hCSE inhibitor (PAG), and a negative control (NL1F3). Id, indole; Bt, benzothiophene; Pz, pyrazole. IC50 measurements were conducted with 1,000× and 100× excess of substrate Cys and coenzyme PLP, respectively, over SaCSE (mean ± SE, n=2–4). NI, no inhibition. S. aureus RN4220 was used for representative gentamycin (Gm) MIC potentiation by 10 µM of each inhibitor. (B) Inhibition of cellular H2S production by the NL1–3 compounds. Representative fluorescence images and the bar plots demonstrate H2S production by NL1-treated and untreated S. aureus (RN4220) and P. aeruginosa (PA14) cells using TICT-based fluorescent H2S probe. Values are means ± SD (n=3). (C) Representative growth curves of wt and cbs/cse S. aureus RN4220 and P. aeruginosa PA14 in the presence of Gm and NL1. Cells were grown in triplicate at 37 °C with aeration using a Bioscreen C automated growth analysis system. The curves represent averaged values with a margin of error of less than 5%. (D) Representative growth curves and efficiencies of colony formation of S. aureus RN4220 and P. aeruginosa PA14 in the presence of Gm, NL1, and NaHS.
In the H2S production enzymatic assay, these leads inhibited 50% of SaCSE and PaCSE activity at low micromolar concentrations in the presence of the natural substrate L-cysteine at 100 μM, the intracellular concentration reported for bacterial cells (43). hCSE was inhibited less efficiently (Fig. 2A and Fig. S6). NL3 was the most potent inhibitor with IC50 values at 0.7 and 1.2 μM for SaCSE and PaCSE, respectively. A much higher, protein-saturating, substrate concentration (2.2 mM) did not affect the IC50 value, which was 1.2±0.7 μM (mean±SE, n=2) for the NL3 and SaCSE combination. An apparent lack of competition between a substrate and the inhibitor suggests an allosteric mode of inhibition. This data generally agrees with our Microscale Thermophoresis (MST) binding affinity measurements: for SaCSE, the apparent KD value for NL1 was ~0.4 µM (Fig. S7).
We also synthesized an analog of NL1, designated NL1F3, which adds a −CF3 moiety at position 2 of the indole ring (Fig. 2A). According to the structural model of the binding site for NL1 in SaCSE presented below, this moiety sterically clashes with the protein, precluding NL1F3 from fitting in the NL1 binding pocket. Indeed, we failed to detect any SaCSE inhibition by NL1F3 at the concentration up to at least 200 µM (Fig. 2A), or to determine NL1F3 KD in the µM range using MST (Fig. S7).
We next examined the ability of NL1–3 and NL1F3 to inhibit H2S synthesis in live S. aureus RN4220 and P. aeruginosa PA14. NL1–3, but not NL1F3, eliminated most H2S production as determined by an H2S-specific fluorescent probe (Fig. 2B); the effect was similar to that of cse genetic inactivation (Fig. 1). To confirm that the inhibition of H2S production was due to targeting of bCSE, we compared the ability of NL1 to sensitize wild type (wt) and cse RN4220 and PA14 to Gm. NL1 enhanced the killing effect of Gm on S. aureus and P. aeruginosa, respectively, to approximately the same extent as did genetic inactivation of bCSE in both species (Fig. 2C, Tables S2 and S3). Notably, NL1 failed to sensitize bCSE-deficient bacteria to Gm any further (Fig. 2C; Tables S2 and S3), arguing that bCSE is indeed the endogenous target for NL1. Furthermore, as in the case of bCSE-deficient cells (Fig. 1C), exogenous sulfide (NaHS) largely suppressed Gm potentiation (Fig. 2D), indicating that NL1 acts by suppressing the H2S-medated cellular defense against the antibiotic.
Allosteric mechanism and specificity of selected bCSE inhibitors
To understand the molecular basis of bCSE inhibition, we determined high-resolution X-ray structures of SaCSE with bound leads (Table S5). Strong anomalous signal generated by the bromine atom present in NL1 and NL2 (Fig. 2A) and excellent electron density maps allowed for unambiguous localization of these ligands at a single site (Site 1) (Fig. S8A-D). Large (20×) molar excess of NL1 during crystallization and high sensitivity analysis also revealed several minor binding sites with much weaker anomalous signals (Fig. S8A) and insufficient density for NL1 modelling ( Fig. S8E-G).
Site 1 is located in the Area II crevice and organized predominantly by hydrophobic and polar residues and aliphatic moieties of charged residues of a single protomer (Fig. 3A,B and Fig. S8H,I). The indole binds in a semi-open cavity so that the short edges of the ring system are braced by the “ceiling” and “floor” of the pocket. The ceiling holds Br atom in a cavity formed by I342, H339, I346, and E350. The opposite edge forms CH–π interactions with Y103 of the floor (Fig. 3A and Fig. S8I). Substitution of −H by a bulkier −CF3 group prevented binding of NL1F3 to Site 1 because of clashing with Y103 (Fig. S7J). The long (C3-C4) edge of NL1 is oriented to the outside, towards solution (Fig. 3A and Fig. S8H). The acetyl glycine moiety, the “tail” of NL1, is sandwiched near the substrate entry tunnel by Y103 and G100 from the bottom, and H339 and T338 from the top. Most notably, NL1 binds only 4.7 Å away from the catalytic Y99 and at a hydrogen bond distance from the neighboring G100.
Fig. 3. Co-crystal structures of SaCSE bound to bCSE inhibitors.
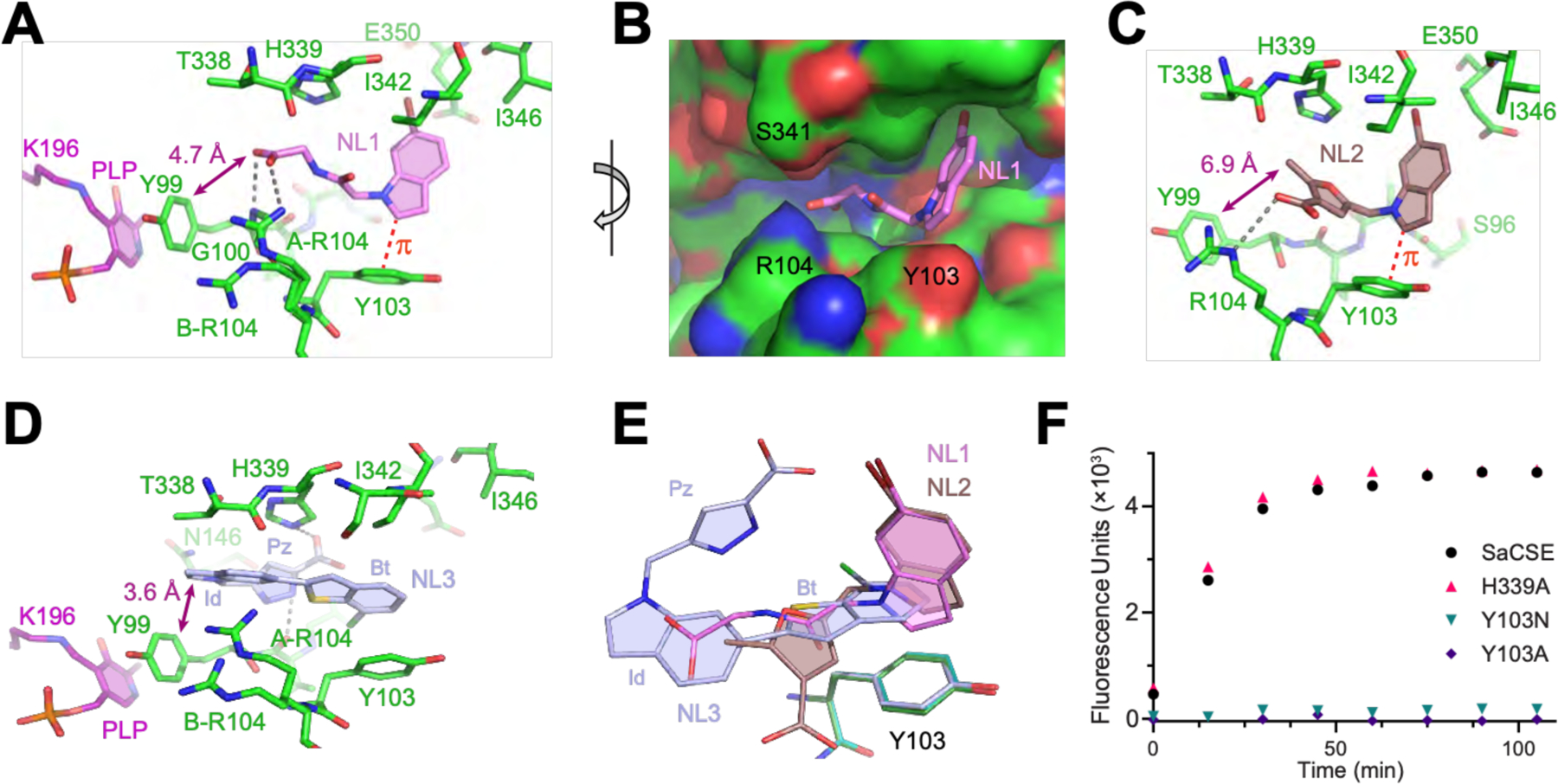
(A) Interactions of NL1 (violet) with the SaCSE monomer. Carbon, nitrogen and oxygen atoms are in green, blue and red, respectively. Gray dashed lines depict putative hydrogen bonds. Red dashed line indicates CH-π interactions. (B) View of NL1 (sticks) in the CSE binding pocket (surface representation). (C) Details of NL2 (brown) binding. (D) Details of NL3 (light blue) binding. Benzothiophene (Bt), indole (Id) and pyrazole (Pz) moieties of NL3 are indicated. (E) Bound leads (same colors) after all-atom superposition of the lead-bound structures shown with Y103 from the NL1-, NL2-, and NL3-bound structures (green, cyan, and light blue, respectively). (F) Representative data for the H2S generating activity of SaCSE mutants evaluated by the fluorescence assay.
NL2 contains two methylene-bridged ring systems that adopt a rather rigid triangle-shaped conformation. As a result, NL2 binds SaCSE only in Site 1 (Fig. S8K) and in a conformation similar to the bound NL1 (Fig. 3C and Fig. S8L).
The bulkier NL3 occupies Site 1 in a different manner; its benzothiophene moiety stacks on Y103 (Fig. 3D and Fig. S8M,N). The indole shifts towards the catalytic site and occupies the position of the tail of NL1. The pyrazole bends towards the interior, where it has a potential for hydrogen bonding. This moiety is positioned almost parallel to benzothiophene and at ~50° angle to its plane, thereby completing a compact semi-circular conformation of the lead.
Notwithstanding the chemical and conformational differences, NL1–3 bind SaCSE in the same site (Fig. 3E), form large intermolecular interfaces (342, 378, and 442 Å2, respectively), have large buried areas (72%, 75%, 79%, respectively), and likely inhibit bCSE via a common mechanism. At merely 3.6–6.9 Å from the side chain of the PLP-coordinating Y99, they can interfere with the dynamics of the catalytic site. In addition, the leads bound between Y103 and H339 can block the conformational rearrangement of Y99, a residue essential for catalysis (44, 45) ( Movie S3). To evaluate the role of the lead-binding amino acids, we mutated Y103 and H339 in SaCSE (Fig. 3F). The H339A mutant exhibited the same H2S producing activity, indicating that H339 is not critical for catalysis. The mutant also responded to NL3 similarly to the wt (IC50= 1.7 ± 0.1 μM, mean ± SE, n=2), although inhibition by smaller in size NL2 was reduced by ~70-fold (IC50= 154.0 ± 94.8 μM). In contrast, the Y103A mutation, or even the “humanizing” variant Y103N, abolished SaCSE activity, demonstrating the critical role of an aromatic residue at position 103 for bCSE, but not hCSE, functioning. Tetramerization (Fig. S9A) and overall SaCSE structure were unaffected by Y103N and Y103A mutations (Fig. S9B), arguing for a specific contribution of Y103 to bCSEs catalysis. Thus, despite being bound outside of the active site, the leads directly engage an amino acid that is essential for bCSE catalysis. Conformational differences around Y103 could explain the essentiality and dispensability of this residue in bCSE and hCSE, respectively (Fig. S9C).
To examine the importance of Y103 for lead binding, we determined co-crystal structures of the Y103N and Y103A mutants with NL1 and NL2 (Table S6), which have higher aqueous solubility than NL3. Co-crystallization with large excess of the leads revealed weak or no binding in Site 1 of the mutants (Fig. S9D-K), providing possible explanation for less efficient inhibition of hCSE by the leads (Fig. 2A).
bCSE inhibitors potentiate diverse types of bactericidal antibiotics
Using methicillin-sensitive S. aureus RN4220 (MSSA) and methicillin-resistant USA300 (MRSA), as well as P. aeruginosa PA14 and PAO1, we evaluated the potentiation of selected members of different classes of bactericidal and bacteriostatic antibiotics by NL1–3 bCSE inhibitors in a standard MIC and MBC assays (Tables S2 and S3). We first established the minimal concentrations of NL1, NL2, and NL3, which cause maximum potentiation of Gm at 0.1×MIC for each strain (Table S7). At these concentrations, the bCSE inhibitors did not affect bacterial growth on their own, but enhanced Gm toxicity during exponential growth (Fig. S10). The extent of Gm potentiation by NL1, NL2, and NL3 parallels the inhibitors’ IC50 values for bCSE (Fig. 2A), further supporting the target specificity.
Next, we examined the effect of these CSE inhibitors on bacterial growth in the presence of different antibiotics (Table S2). As positive control, we used S. aureus and P. aeruginosa strains lacking CSE/CBS. All three selected CSE inhibitors potentiated the members of major bactericidal classes, including fluoroquinolones (ciprofloxacin, norfloxacin), beta-lactams (ampicillin), and aminoglycosides (gentamycin and kanamycin). The potentiation varied from approximately 2 to 5-fold, reaching over 15-fold, depending on the specific combination of antibiotic, bacterial strain, and bCSE inhibitor (Fig. 2A, Tables S2 and S3). Similar levels of MIC change were observed as a result of genetic inactivation of CSE, supporting the target specificity of CSE inhibitors (Table S2).
Notably, no potentiation by bCSE inhibitors was observed with the bacteriostatics tetracycline and chloramphenicol (Table S2). Likewise, S. aureus and P. aeruginosa lacking CSE were no more sensitive to these bacteriostatics than their wild type counterparts (Table S2). These observations support the role of H2S in partially neutralizing the critical element of toxicity that is common between different classes of bactericidals. Such toxicity has been proposed to depend, at least in part, on Fenton chemistry (23, 25, 46, 47). Indeed, we observed little, if any, potentiation by the bCSE inhibitors in the presence of the iron chelator dipyridyl (Fig. S11).
bCSE inhibitors promote antibiotic potency in murine models of infection
The synergy between NL1–3 and antibiotics observed in vitro suggests that bCSE inhibitors may enhance the effectiveness of antibiotic treatment in vivo. NL1 is non-toxic to different types of human cells in various assays to at least 25× of its minimal potentiator concentration (Fig. S12A-G). No adverse effects of chronic exposure to NL1 were detected in a stringent animal model (Fig. S12H,I). To evaluate the efficacy of NL1 in vivo, we adopted two murine models of infection: a S. aureus sepsis model, and a P. aeruginosa lung infection model (Fig. 4).
Fig. 4. bCSE inhibition synergizes gentamycin in murine models of infection.
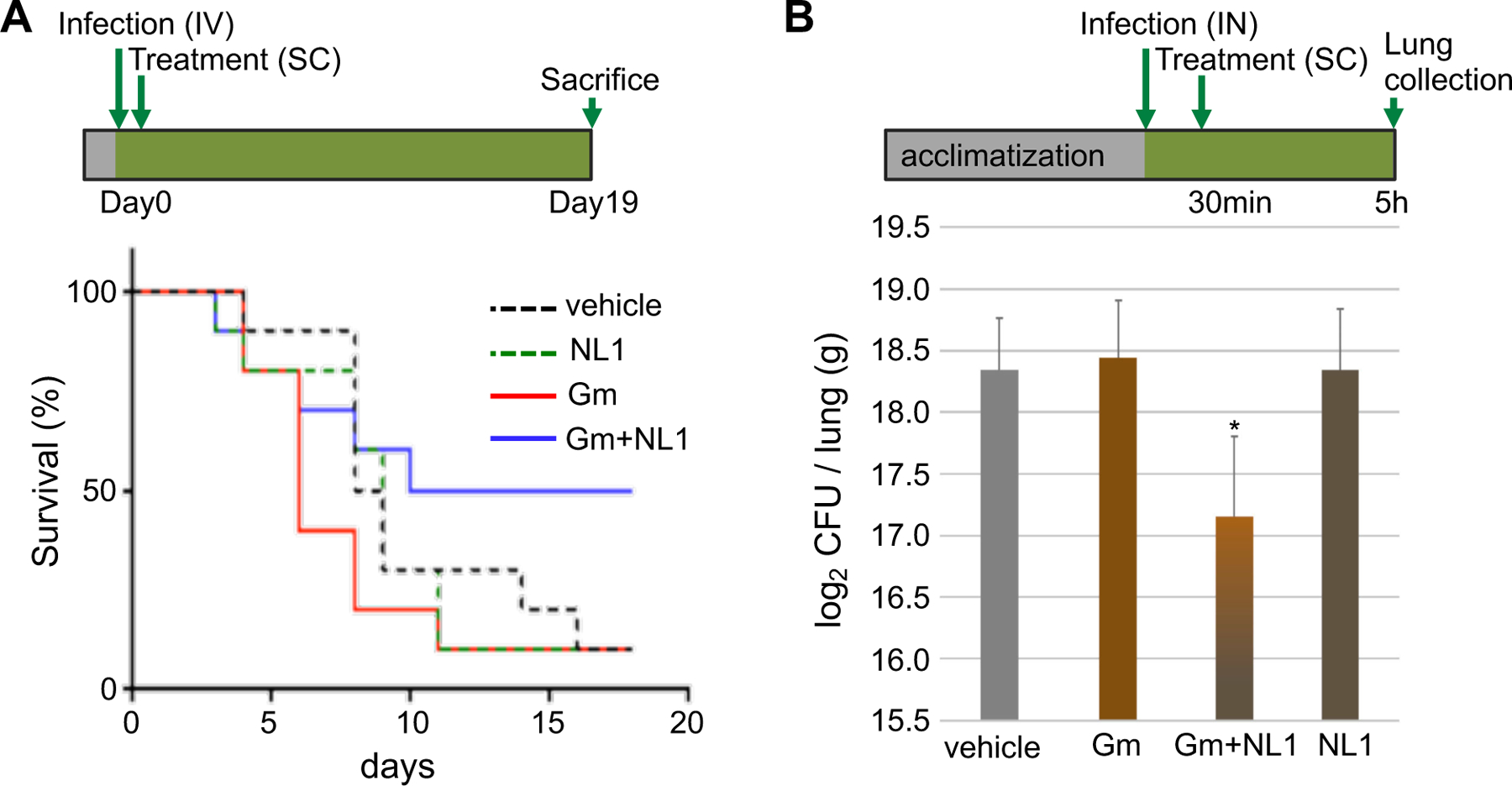
(A) Acute S. aureus-induced bacteremia model. Treatment with Gm or NL1 alone did not increase survival of mice after lethal infection with S. aureus. Treatment with NL1+Gm improved survival to 50%. Study design is shown on top. (B) P. aeruginosa intranasal lung infection model. Treatment with the combination of Gm and NL1 significantly decreased bacteria burden in lungs. Graph represents the mean ± SE, *P<0.05.
For the sepsis model, Swiss Webster mice were challenged with S. aureus Newman via intravenous injection at approximately 2.5 × 107 colony forming units (CFU)/mouse. NL1 was administered at 60 mg/kg together with a subactive dose of Gm (2 mg/kg) via a single 100 µL subcutaneous injection on Day 0 (30 min after the challenge) and animals were monitored for 19 days following the challenge. The single bolus treatment of NL1 and Gm resulted in 50% survival, while 90% of the control animals died, and neither NL1 nor Gm alone increased survival (Fig. 4A).
For the lung model, Swiss Webster mice were infected with P. aeruginosa PA14 (1.3 × 106 CFU/mouse) by intranasal route. At 30 min after infection, mice were treated with a single dose of NL1 at 60 mg/kg together with a subactive dose of Gm (2 mg/kg) by subcutaneous route. Mice were sacrificed to collect lungs 5h after infection. The lung bacterial burden did not change significantly in response to NL1 or Gm administered separately; however, the combination of Gm with NL1 markedly decreased the lung bacterial burden (Fig. 4B).
Even though Gm potentiation in vitro was substantial based on standard MIC and MBC tests (Fig. 2 and Table S2 and S3), we were surprised by the extent of synergy displayed by NL1 and Gm in vivo after only a single bolus administration. Thus, we hypothesized that there could be additional benefits to NL1 in combination with antibiotics not reflected by the MIC and MBC assays. We therefore examined the effects of CSE inhibitors on bacterial tolerance, which, by definition, is uncoupled from MIC/MBC.
bCSE inactivation reduces persisters and disrupts biofilm formation
Ciprofloxacin (10 µg/ml) and Gm (40 µg/ml) rapidly kill the vast majority of S. aureus and P. aeruginosa cells in exponentially growing culture. However, after approximately 30 min of treatment, the killing rate declines profoundly in the wt, but not in cells deficient in bCSE (Fig. 5A and Fig. S13). The tails of the killing curves represent the clonal population of rare antibiotic-tolerant cells – i.e., persisters - capable of surviving majority-lethal doses of the antibiotic (7,10). The population of persisters was approximately 2 orders of magnitude smaller in mutants lacking bCSE (Fig. 5A), arguing that endogenous H2S production is critical for establishing the persister population. Congruently, treating wt cells with NL1 eliminated approximately the same amount of persisters as did genetic inactivation of cse/cbs (Fig. 5A). The slow releasing H2S donor diallyl trisulfide (DATS; Fig. S13A) prevented the anti-persister effect of NL1 (Fig. S13B), supporting the role of bCSE-derived H2S in establishing and/or maintaining the persister state.
Fig. 5. bCSE inhibition interferes with persistence and biofilm formation.
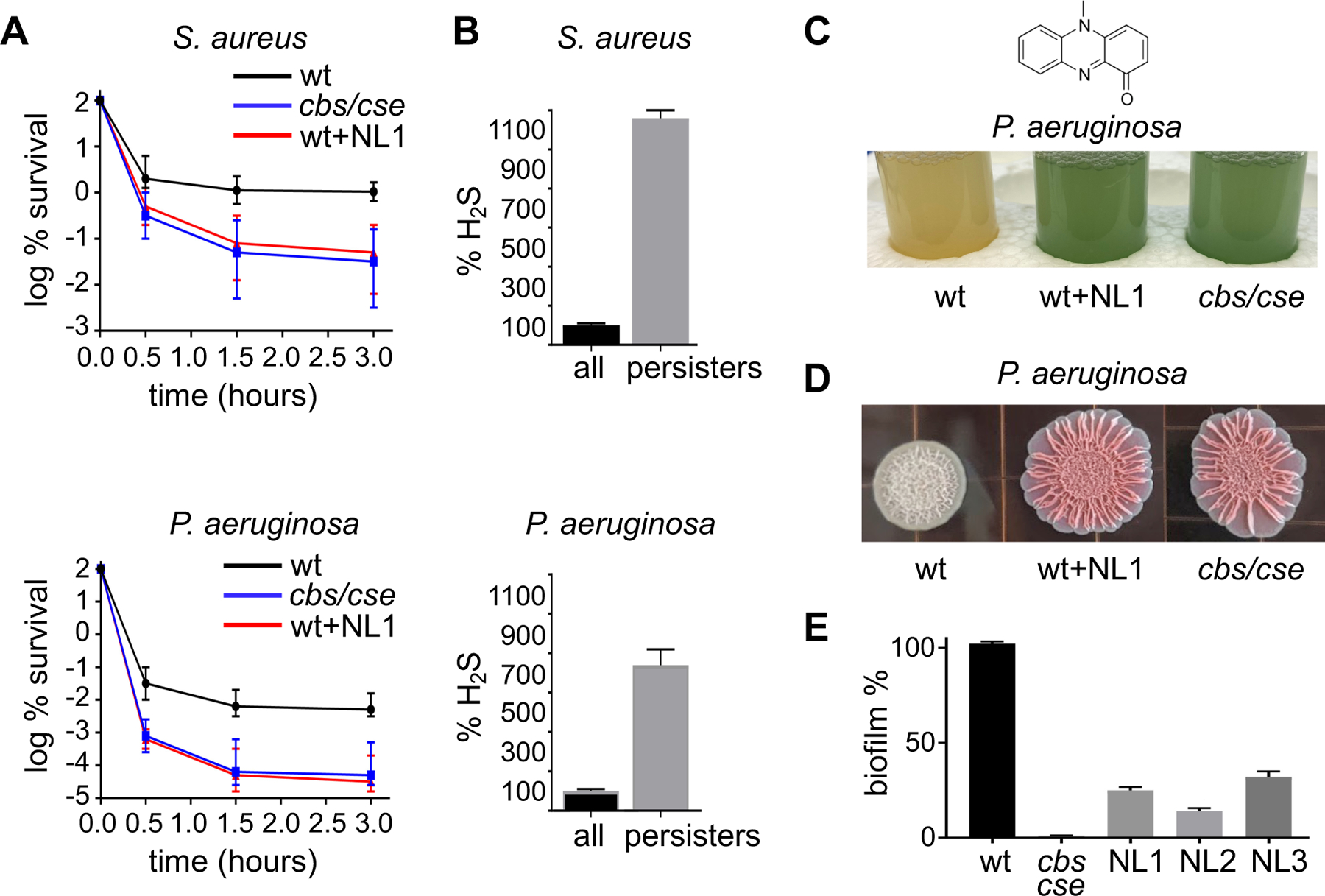
(A) Dynamics of persister viability in wt, bCSE-deficient, or NL1-treated S. aureus USA300 and P. aeruginosa PA14. The graphs show changes in the viable cell fraction of exponentially growing cultures challenged with 10 µg/ml (10× MIC) ciprofloxacin. Data points are mean ± SE (n=3). (B) The bar plots demonstrate H2S production by S. aureus USA300 and P. aeruginosa PA14 cells before (black) and after (gray) 3 h incubation with 10 µg/ml ciprofloxacin. Cells were treated with the TICT-based fluorescent H2S probe and the fluorescence signal was normalized to CFU. Data are mean ± SE (P<0.01) (n=3). (C) Inactivation of bCSE depletes P. aeruginosa of the reduced pyocyanin toxin. Pyocyanin (1-hydroxy-5-methyl-phenazine) chemical structure is shown on top. Oxidized pyocyanin gives the P. aeruginosa culture blue/green color. The tubes show stationary phase P. aeruginosa PA14 cultures (OD600~1.5): untreated wt, untreated cbs/cse(−) mutant, and wt treated with NL1 (180 µM) for 18 h. (D) bCSE modulates colony morphology of P. aeruginosa PA14. wt and cse/cbs(−) cultures were spotted onto agar plates containing Congo Red and Coomassie Blue, and incubated with or without NL1 for 3 days prior to taking pictures. (E) Genetic or chemical inactivation of bCSE compromises P. aeruginosa PA14 biofilm formation, as determined by the crystal violet assay. Data are mean ± SE (n=3).
Persisters are a stress-triggered or stochastically arising fraction of metabolically less active cells. One of the common features proposed to explain the induction of persistence is the inhibition of ATP production (16, 48–50). Indeed, the proportion of the antibiotic-tolerant cells greatly increases following the transition to the stationary phase of growth, where the metabolic rate is progressively decreased. Notably, the amount of H2S also increased substantially in the stationary phase (Fig. S14A), which correlated with the increased intracellular level of bCSE and bCBS (Fig. S14B) likely because of posttranscriptional regulation (Fig. 14C). H2S is a well-known inhibitor of ATP synthesis. At low µM concentrations, H2S readily inactivates heme-copper terminal oxidases, but not the cytochrome bd oxidases (51). This allows the bacteria to sustain growth and respiration in the presence of high H2S production, albeit at a much lower rate of ATP synthesis. We, therefore, hypothesized that persister cells may display higher levels of H2S than the rest of the population, resulting in “self-poisoning” and, hence, slow metabolism and high tolerance. Indeed, persisters (cells that survived ciprofloxacin challenge for 3 h) generated substantially more H2S than non-persisters (Fig. 5B).
Stationary-phase P. aeruginosa produce and secrete the secondary metabolite pyocyanin (Fig. 5C, top), which functions as a signaling molecule (52) and virulence factor (53). Pyocyanin cycles between redox states and, therefore, can generate reactive oxygen species, which contribute to killing mammalian cells (reviewed in [(54)]). Stationary-phase P. aeruginosa PA14 cells generate a large amount of pyocyanin (55), and the amount of pyocyanin is the same in wt and bCSE/CBS-deficient cells (Fig. S15). The wt PA14 culture, regardless of the level of aeration, is light yellow in color due to the cells containing reduced pyocyanin. In contrast, well-aerated PA14 cells deficient in bCSE/CBS, or treated with NL1, display green-blue coloration characteristic of the oxidized pyocyanin (Fig. 5C). Remarkably, even a brief aeration by shaking of a non-aerated mutant culture results in a rapid emergence of the green-blue coloration (Movie S4) indicative of pyocyanin oxidation. Thus, endogenous H2S helps to continuously maintain the reduced form of pyocyanin in P. aeruginosa. Since wt P. aeruginosa can increase the fraction of persister cells up to 90-fold in response to pyocyanin (56), the lack of the redox activity of pyocyanin may explain, at least in part, the anti-persister effect of bCSE inhibition in P. aeruginosa.
Pyocyanin has been also associated with P. aeruginosa biofilm formation (57, 58). We therefore examined the effect of endogenous H2S on PA14 biofilm development. Strikingly, both genetic and chemical inactivation of bCSE resulted in the same dramatic change in colony morphology on agar plates (Fig. 5D) and the drastic reduction in the overall static biofilm formation (Fig. 5E). Genetic or chemical inactivation of bCSE also inhibited biofilm formation in S. aureus strains (Fig. S16). Comparative transcriptomic analysis of the wt and cse/cbs P. aeruginosa PA14 in the stationary phase revealed that genes involved in biofilm formation, including alginate and other exopolysaccharide biosynthesis genes, are among the most downregulated categories in H2S-deficent cells (Table S8).
This data warrants future studies to elucidate the H2S-mediated signaling pathways contributing to bacterial tolerance, biofilms, and virulence, some of which may rely on persulfidation of particular transcriptional regulators (59).
Discussion
Antibiotic tolerance is implicated in difficult-to-treat infections, as well as in the development and spread of antibiotic resistance. Defeating bacterial tolerance to antibiotics using an adjuvant drug could make clinical antibiotics more efficacious, while protecting them from antibiotic resistance. Frustratingly, no “tolerance pathway” has emerged as a viable target (60).
Here we explored the strategy of potentiating existing antibiotics by attacking a general defense mechanism of pathogenic bacteria (21,23): the H2S biogenesis system. We demonstrate the key role of bCSE in H2S biogenesis of S. aureus and P. aeruginosa, and describe drug-like inhibitors of bCSE that potentiate diverse bactericidal antibiotics. Furthermore, we discovered the role of H2S in bacterial antibiotic tolerance, including persister and biofilm formation, and in associated aspects of bacterial virulence.
Our data indicate that persisters generate more H2S than non-persisters, suggesting that endogenous H2S helps to trigger and/or maintain the persistent state. Possibly, being a potent inhibitor of heme-copper cytochrome oxidases (48), H2S helps establishing the characteristic low metabolic state of persisters, particularly in the stationary phase when the level of H2S markedly increases.
We also report a role for the H2S biogenesis system in P. aeruginosa virulence. In stationary culture of P. aeruginosa, bCSE inactivation diminished the level of reduced pyocyanin, a toxin associated with P. aeruginosa virulence. This may be a result of the altered intracellular redox state, e.g. the NADH/NAD+ ratio (61), which controls persistence (62). As pyocyanin helps P. aeruginosa to persist during cystic fibrosis and is positively correlated with infection severity (54), our data also suggest that inhibiting H2S biogenesis may improve treatment of chronic infection.
Another striking outcome of bCSE inactivation that we observed is biofilm deficiency. A hallmark of lingering lung infection in cystic fibrosis patients is the ability of P. aeruginosa to form robust biofilm that facilitates bacterial adhesion and imparts recalcitrance to both host immunity and antibiotic treatment (63–65). Our transcriptomic analysis of P. aeruginosa PA14 revealed multiple genes involved in biofilm formation to be profoundly affected by suppressing H2S production.
In the aggregate, our proof-of-concept results suggest several directions for exploring the combinations of bCSE inhibitors and approved or new antibiotics. Because bCSE inhibitors can potentiate bactericidal antibiotics (at least in P. aeruginosa and S. aureus), the combinations may prove useful against intermediate-level resistant strains; or the antibiotic dose might be reduced while keeping the same effectiveness with lower toxicity. Furthermore, by exploiting the ability of bCSE inhibitors to interfere with bacterial tolerance, it may be possible to use antibiotic – bCSE inhibitor combinations to reduce treatment failures in acute infections; reduce colonization, transition to chronicity, and recurrence; and shorten the course of treatment. bCSE inhibitors may also have a role in treating established chronic diseases, including those involving biofilms. For antibiotics used in combination with bCSE inhibitors, the chance of emergence or spreading of antibiotic resistance may be reduced. These are tantalizing possibilities that await exploration.
Supplementary Material
Acknowledgements:
We thank Richard Novick, Victor Torres, Bo Shopsin, Olga Zaborina, and Yongzhen Xia for bacterial strains and discussions, Ming Xian for the H2S fluorescence probe, and Nadim Shohdy for his support. This work used NE-CAT beamlines (GM124165), a Pilatus detector (RR029205), an Eiger detector (OD021527) at the Advanced Photon Source (DE-AC02-06CH11357) of the Argonne National Laboratory; and FMX (17ID-2) and AMX (17ID-1) beamlines at NSLS-II (Brookhaven National Laboratory), supported by the NIH NIGMS (1P30GM133893) and BER- BO 070. NSLS-II is supported by DOE, BES-FWP-PS001. Coordinates and structure factors of the SaCSE structures were deposited in the Protein Data Bank (PDB) under the following accession numbers: 7MCL, PLP-bound protein; 7MCB holoenzyme; 7MCN, holoenzyme at high HEPES; 7MCP, holoenzyme dimer; 7MCQ, AOAA-bound dimer; 7MCT, bound to NL1; 7MCU, bound to NL2; 7MCY, bound to NL3; 7MD0, crystallized in the presence of NL1F3; 7MD1, Y103N mutant; 7MD6, Y103N mutant in the presence of NL1; 7MD8, Y103N mutant in the presence of NL2; 7MD9, Y103A mutant; 7MDA, Y103A mutant in the presence of NL1; and 7MDB, Y103A mutant in the presence of NL2.
Funding:
This work was supported by Gero LLC (D.S. and P.F.), the Russian Science Foundation grant 17-74-30030 (A.M.), Grant 075-15-2019-1660 from the Ministry of Science and Higher Education of the Russian Federation (A.M.), NYU Therapeutic Alliances (K.S.), DoD grants PR171734 (E.N.) and PR171734P1 (A.S.), Blavatnik Family Foundation (E.N.), and by the Howard Hughes Medical Institute (E.N.).
Footnotes
Competing interests: NYUSoM has filed a related patent application with EN, KS, DS, PF as co-inventors.
Data and material availability: Coordinates and structure factors of 15 structures determined in the study are deposited in the Protein Data Bank (PDB) under accession numbers listed in Tables S4-S6. RNA sequencing raw data was deposited to the NIH Sequence Read Archive (accession number PRJNA637626).
References
- 1.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL, Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6, 29–40 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Ribeiro da Cunha B, Fonseca LP, Calado CRC, Antibiotic discovery: where have we come from, where do we go? Antibiotics (Basel) 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schrader SM, Vaubourgeix J, Nathan C, Biology of antimicrobial resistance and approaches to combat it. Sci Transl Med 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Neill J, “Tackling drug-resistant infections globally: Final report and recommendations “ (2016). [Google Scholar]
- 5.Brauner A, Fridman O, Gefen O, Balaban NQ, Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol 14, 320–330 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Balaban NQ et al. , Definitions and guidelines for research on antibiotic persistence. Nat Rev Microbiol 17, 441–448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michiels JE, Van den Bergh B, Verstraeten N, Michiels J, Molecular mechanisms and clinical implications of bacterial persistence. Drug Resist Updat 29, 76–89 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Costerton JW, Stewart PS, Greenberg EP, Bacterial biofilms: a common cause of persistent infections. Science 284, 1318–1322 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Kirby AE, Garner K, Levin BR, The relative contributions of physical structure and cell density to the antibiotic susceptibility of bacteria in biofilms. Antimicrob Agents Chemother 56, 2967–2975 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meredith HR, Srimani JK, Lee AJ, Lopatkin AJ, You L, Collective antibiotic tolerance: mechanisms, dynamics and intervention. Nat Chem Biol 11, 182–188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dewachter L, Fauvart M, Michiels J, Bacterial heterogeneity and antibiotic survival: understanding and combatting persistence and heteroresistance. Mol Cell 76, 255–267 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Schulze A, Mitterer F, Pombo JP, Schild S, Biofilms by bacterial human pathogens: Clinical relevance - development, composition and regulation - therapeutical strategies. Microb Cell 8, 28–56 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conlon BP, Staphylococcus aureus chronic and relapsing infections: evidence of a role for persister cells: an investigation of persister cells, their formation and their role in S. aureus disease. Bioessays 36, 991–996 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Donlan RM, Costerton JW, Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev 15, 167–193 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mulcahy LR, Burns JL, Lory S, Lewis K, Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J Bacteriol 192, 6191–6199 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Percival SL, Hill KE, Malic S, Thomas DW, Williams DW, Antimicrobial tolerance and the significance of persister cells in recalcitrant chronic wound biofilms. Wound Repair Regen 19, 1–9 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Huemer M et al. , Molecular reprogramming and phenotype switching in Staphylococcus aureus lead to high antibiotic persistence and affect therapy success. Proc Natl Acad Sci U S A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies J, Davies D, Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev 74, 417–433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levin BR, Rozen DE, Non-inherited antibiotic resistance. Nat Rev Microbiol 4, 556–562 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Sebastian J et al. , De Novo emergence of genetically resistant mutants of Mycobacterium tuberculosis from the persistence phase cells formed against antituberculosis drugs In vitro. Antimicrob Agents Chemother 61, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luhachack L, Nudler E, Bacterial gasotransmitters: an innate defense against antibiotics. Curr Opin Microbiol 21, 13–17 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Kimura H, Signaling of hydrogen sulfide and polysulfides. Antioxid Redox Signal 22, 347–349 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shatalin K, Shatalina E, Mironov A, Nudler E, H2S: a universal defense against antibiotics in bacteria. Science 334, 986–990 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Szabo C, A timeline of hydrogen sulfide (H2S) research: From environmental toxin to biological mediator. Biochem Pharmacol 149, 5–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mironov A et al. , Mechanism of H2S-mediated protection against oxidative stress in Escherichia coli. Proc Natl Acad Sci U S A 114, 6022–6027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nzungize L et al. , Mycobacterium tuberculosis metC (Rv3340) derived hydrogen sulphide conferring bacteria stress survival. J Drug Target 27, 1004–1016 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Toliver-Kinsky T et al. , H2S, a bacterial defense mechanism against the host immune response. Infect Immun 87, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ren M et al. , A TICT-based fluorescent probe for rapid and specific detection of hydrogen sulfide and its bio-imaging applications. Chem Commun (Camb) 52, 6415–6418 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Shen X, Kolluru GK, Yuan S, Kevil CG, Measurement of H2S in vivo and in vitro by the monobromobimane method. Methods Enzymol 554, 31–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Q et al. , Structural basis for the inhibition mechanism of human cystathionine gamma-lyase, an enzyme responsible for the production of H2S. J Biol Chem 284, 3076–3085 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Lee D, Jeong S, Ahn J, Ha N-C, Kwon A-R, Crystal structure of bacterial cystathionine gamma γ-lyase in the cysteine biosynthesis pathway of Staphylococcus aureus. Crystals 9, 656 (2019). [Google Scholar]
- 32.Asimakopoulou A et al. , Selectivity of commonly used pharmacological inhibitors for cystathionine beta synthase (CBS) and cystathionine gamma lyase (CSE). Br J Pharmacol 169, 922–932 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang R, Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92, 791–896 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Whiteman M, Le Trionnaire S, Chopra M, Fox B, Whatmore J, Emerging role of hydrogen sulfide in health and disease: critical appraisal of biomarkers and pharmacological tools. Clin Sci (Lond) 121, 459–488 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Yadav PK et al. , S-3-Carboxypropyl-l-cysteine specifically inhibits cystathionine gamma-lyase-dependent hydrogen sulfide synthesis. J Biol Chem 294, 11011–11022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clausen T, Huber R, Messerschmidt A, Pohlenz HD, Laber B, Slow-binding inhibition of Escherichia coli cystathionine beta-lyase by L-aminoethoxyvinylglycine: a kinetic and X-ray study. Biochemistry 36, 12633–12643 (1997). [DOI] [PubMed] [Google Scholar]
- 37.Lowther J et al. , Inhibition of the PLP-dependent enzyme serine palmitoyltransferase by cycloserine: evidence for a novel decarboxylative mechanism of inactivation. Mol Biosyst 6, 1682–1693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mihara H et al. , Structure of external aldimine of Escherichia coli CsdB, an IscS/NifS homolog: implications for its specificity toward selenocysteine. J Biochem 131, 679–685 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Ngo HP et al. , PLP undergoes conformational changes during the course of an enzymatic reaction. Acta Crystallogr D Biol Crystallogr 70, 596–606 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Sagong HY, Kim KJ, Structural Insights into substrate specificity of cystathionine γ-synthase from Corynebacterium glutamicum. J Agric Food Chem 65, 6002–6008 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Trott O, Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31, 455–461 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joce C et al. , Application of a novel in silico high-throughput screen to identify selective inhibitors for protein-protein interactions. Bioorg Med Chem Lett 20, 5411–5413 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smirnova GV et al. , Cysteine homeostasis under inhibition of protein synthesis in Escherichia coli cells. Amino Acids 51, 1577–1592 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Fesko K, Suplatov D, Svedas V, Bioinformatic analysis of the fold type I PLP-dependent enzymes reveals determinants of reaction specificity in l-threonine aldolase from Aeromonas jandaei. FEBS Open Bio 8, 1013–1028 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang S et al. , Site-directed mutagenesis on human cystathionine-gamma-lyase reveals insights into the modulation of H2S production. J Mol Biol 396, 708–718 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Dwyer DJ, Collins JJ, Walker GC, Unraveling the physiological complexities of antibiotic lethality. Annu Rev Pharmacol Toxicol 55, 313–332 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Zhao X, Hong Y, Drlica K, Moving forward with reactive oxygen species involvement in antimicrobial lethality. J Antimicrob Chemother 70, 639–642 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwan BW, Valenta JA, Benedik MJ, Wood TK, Arrested protein synthesis increases persister-like cell formation. Antimicrob Agents Chemother 57, 1468–1473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shan Y et al. , ATP-dependent persister formation in Escherichia coli. mBio 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilmaerts D, Windels EM, Verstraeten N, Michiels J, General mechanisms leading to persister formation and awakening. Trends Genet 35, 401–411 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Forte E et al. , The terminal oxidase cytochrome bd promotes sulfide-resistant bacterial respiration and growth. Sci Rep 6, 23788 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dietrich LE, Price-Whelan A, Petersen A, Whiteley M, Newman DK, The phenazine pyocyanin is a terminal signalling factor in the quorum sensing network of Pseudomonas aeruginosa. Mol Microbiol 61, 1308–1321 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Lau GW, Hassett DJ, Ran H, Kong F, The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol Med 10, 599–606 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Rada B, Leto TL, Pyocyanin effects on respiratory epithelium: relevance in Pseudomonas aeruginosa airway infections. Trends Microbiol 21, 73–81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rahme LG et al. , Common virulence factors for bacterial pathogenicity in plants and animals. Science 268, 1899–1902 (1995). [DOI] [PubMed] [Google Scholar]
- 56.Moker N, Dean CR, Tao J, Pseudomonas aeruginosa increases formation of multidrug-tolerant persister cells in response to quorum-sensing signaling molecules. J Bacteriol 192, 1946–1955 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Das T et al. , Phenazine virulence factor binding to extracellular DNA is important for Pseudomonas aeruginosa biofilm formation. Sci Rep 5, 8398 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dietrich LE, Teal TK, Price-Whelan A, Newman DK, Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science 321, 1203–1206 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walsh BJC et al. , The response of Acinetobacter baumannii to hydrogen sulfide reveals two independent persulfide-sensing systems and a connection to biofilm regulation. mBio 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaldalu N et al. , In Vitro Studies of Persister Cells. Microbiol Mol Biol Rev 84, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Price-Whelan A, Dietrich LE, Newman DK, Pyocyanin alters redox homeostasis and carbon flux through central metabolic pathways in Pseudomonas aeruginosa PA14. J Bacteriol 189, 6372–6381 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Allison KR, Brynildsen MP, Collins JJ, Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 473, 216–220 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jensen PO, Givskov M, Bjarnsholt T, Moser C, The immune system vs. Pseudomonas aeruginosa biofilms. FEMS Immunol Med Microbiol 59, 292–305 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Mah TF, O’Toole GA, Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9, 34–39 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Stewart PS, Costerton JW, Antibiotic resistance of bacteria in biofilms. Lancet 358, 135–138 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.