

Abstract
Fms-like tyrosine kinase 3 (Flt3) tyrosine kinase inhibitors (Flt3-TKI) have improved outcomes for patients with Flt3-mutated acute myeloid leukemia (AML) but are limited by resistance and relapse, indicating persistence of leukemia stem cells (LSC). Here utilizing a Flt3-internal tandem duplication (Flt3-ITD) and Tet2-deleted AML genetic mouse model we determined that FLT3-ITD AML LSC were enriched within the primitive ST-HSC population. FLT3-ITD LSC showed increased expression of the CXCL12 receptor CXCR4. CXCL12-abundant reticular (CAR) cells were increased in Flt3-ITD AML marrow. CXCL12 deletion from the microenvironment enhanced targeting of AML cells by Flt3-TKI plus chemotherapy treatment, including enhanced LSC targeting. Both treatment and CXCL12 deletion partially reduced p38 mitogen-activated protein kinase (p38) signaling in AML cells and further reduction was seen after treatment in CXCL12 deleted mice. p38 inhibition reduced CXCL12-dependent and -independent maintenance of both murine and human Flt3-ITD AML LSC by MSC and enhanced their sensitivity to treatment. p38 inhibition in combination with chemotherapy plus TKI treatment leads to greater depletion of Flt3-ITD AML LSC compared with CXCL12 deletion. Our studies support roles for CXCL12 and p38 signaling in microenvironmental protection of AML LSC and provide a rationale for inhibiting p38 signaling to enhance Flt3-ITD AML targeting.
Introduction
Acute myeloid leukemia (AML) is an aggressive hematopoietic malignancy with poor survival and high rates of relapse and treatment resistance. For years, the standard-of-care for AML was a combination of cytarabine and daunorubicin. With further understanding of recurrent AML mutations, more sophisticated prognostic schema emerged and molecularly-targeted therapies have been developed and approved for clinical use.
Internal tandem duplications in Fms-like tyrosine kinase 3 receptor (Flt3-ITD) are common mutations in adult AML(1), and are also seen in pediatric AML(2). Flt3-ITD correlates with poor prognosis due to increased relapse risk. Flt3-specific tyrosine kinase inhibitors (TKI) were among the first molecularly-targeted therapeutics developed and FDA-approved in AML. The Flt3-TKI, midostaurin, was approved after a phase 3 trial showed that addition to first-line chemotherapy led to a modest increase in overall survival (OS) (3). The more potent and selective Flt3 TKI, Gilteritinib, was FDA approved for relapsed/refractory FLT3-mutated AML based on a phase 3 trial that showed increased median OS vs. chemotherapy and complete response rate (4). These results, while promising, indicate a need for better understanding of resistance and relapse with targeted therapies.
AML arises from leukemia stem cells (LSC)(5) which result from accumulation of mutations in hematopoietic stem and progenitor cells (HSPC)(6) in bone marrow (BM), and are a source of drug resistance and relapse(7). LSC are supported by BM microenvironment (BMM) niches which are remodeled to selectively support LSC over normal HSC and contribute to drug resistance(8). The C-X-C motif chemokine ligand 12 (CXCL12) is important for maintaining the localization and function of HSPC in specific BMM niches(9, 10). Others and we have also identified a role for CXCL12-expressing niches in supporting LSC(11, 12).
Animal studies suggest that Flt3-ITD is insufficient to give rise to acute leukemia(13). Flt3-ITD mutations often follow mutations in enzymes that alter DNA methylation (DNMT3A, IDH1, IDH2, TET2) during AML development. Mouse models have been created with several of these cooperating mutations in combination with Flt3-ITD(14–17). Here we characterized a genetically-engineered mouse model (GEMM) of Flt3-ITD AML containing a hemizygous Flt3-ITD knock-in(13), homozygous flox/flox TET2(18), and Mx-1 cre(19) and utilized UBC-ERT2-cre(20) CXCL12(9) knock-out mice to study microenvironmental mechanisms of resistance to Flt3-TKI, and validated our results using primary human Flt3-ITD AML cells.
Methods
Patient samples
BM samples were collected from treatment-naive AML patients at the University of Alabama at Birmingham (UAB) after obtaining informed consent. Sample acquisition was approved by the Institutional Review Board at UAB in accordance with assurances filed with the Department of Health and Human Services and met all requirements of the Declaration of Helsinki. Patient characteristics are listed in Supplementary Table 1.
Mice
B6.129-Flt3tm1Dgg/J mice (Flt3-ITD) (The Jackson Laboratory (JAX), Stock no. 011112) and B6.129S-Tet2tm1.1Iaai/J mice (TET2flox/flox) (JAX, Stock no. 017573) were crossbred to obtain double heterozygous mice, back-crossed into C57BL/6j mice (JAX, Stock no. 000664) for 5 generations, and crossbred with B6.Cg-Tg(Mx1-cre)1Cgn/J mice (JAX, Stock no. 003556) to obtain Flt3-ITDwt/ki TET2flox/flox Mx1-cre mice. Homozygous B6(FVB)-Cxcl12tm1.1Link/J mice (JAX, Stock no. 021773), with exon 2 of the mouse Cxcl12 gene flanked by loxP sites, were crossed with UBC-ERT2-cre mice (JAX stock no. 008085) to obtain UBC-ERT2-cre Cxcl12tm1.1Link mice. CXCL12-GFP knock-in mice (exon 2 of CXCL12 gene replaced by GFP expression cassette; CD45.2+) were from Dr. Takashi Nagasawa (Kyoto University, Japan) and maintained in heterozygous state(21). NSG mice were from JAX (Stock no. 005557). Wild-type (WT) CD45.2 and CD45.1 mice were from Charles River. Sample size was based on previous studies that established numbers of mice per group required to detect significant differences. Mice were randomized between treatment groups based on sex and engraftment levels. Blinding was not used. Mouse care and procedures were in accordance with federal guidelines and protocols approved by the Institutional Animal Care and Use Committee (IACUC) at UAB. See Supplemental Methods for details of hematopoietic cell analysis and for all antibodies used (Supplementary Table 2).
Murine BM transplantation
For syngeneic transplants, recipients received two doses of 4Gy total body radiation, 4 hours apart, and were transplanted by tail vein injection 2-4 hours after second radiation dose. For BM mononuclear cell (MNC) transplants, 106 - 107 donor cells were transplanted into each recipient. Purified HSPC populations were sorted using a BD FACS ARIA cell sorter and mice were transplanted with 2 x 105 CD45.1 support BM cells plus 5x102, 1.5x103, or 4.5x103 ST-HSC; 5x103, 1.5x104, or 4.5x104 MPP; or 2x104, 6x104, 1.8x105 GMP (details provided in Supplemental Methods).
Patient derived Xenografts
NSG mice were irradiated with 1.5Gy total body radiation 24 hours before transplant. BM MNC from AML patient samples were mixed with OKT3 anti-CD3 antibody (1 μg/million cells, BioXCell) prior to injection (5-10 million cells/mouse). For xenografts of purified HSPC, CMP, GMP and MPP populations were sorted from patient BM MNC using a BD FACS ARIA cell sorter (details provided in Supplemental Methods). Each mouse received CMP, GMP, or MPP sorted from 107 BM MNC.
Drug Treatments
Mx1-cre mice were injected intraperitoneally with 5 mg/kg polyinosinic-polycytidylic acid (pIpC, Invivogen) every 48 hours for 7 doses to induce Cre-recombinase activity. UBC-Cre mice were injected intraperitoneally with 1 mg tamoxifen (10 mg/mL in sterile corn oil) daily for 5 days to induce cre-recombinase activity. AC220 (Quizartinib, AdooQ) was administered by gavage at 5-10 mg/kg/day for 21-28 days. Cytarabine (Cayman Chemical) and Doxorubicin (Cayman Chemical) were injected intraperitoneally with cytarabine, 50 mg/kg/day for 7 days and doxorubicin, 1.5 mg/kg/day for 3 days (details of drug preparation provided in Supplemental Methods).
Stromal cell analysis
Bone fragments were incubated with collagenase for 1 hour in a shaking incubator at 37°C, then washed 3 times with sterile PBS. Suspended cells were filtered through a 70 micron filter, added to total BM MNC, and stained with antibodies to analyze BM stromal populations: CD45-AF700, Ter119-AF700, Tie2-APC, VE-Cadherin-PerCp-eFluor710, CD31-eFluor450, CD51-biotin, Sca-1-APC-Cy7, CD140a-PE-Cy7, and streptavidin-BV605.
RNASeq analysis
RNA was extracted using RNeasy Plus Micro Kit (Qiagen). Libraries were prepared using SMARTer Ultra Low Input RNA Kits for Sequencing (v4, TaKaRa Clontech) and Nextera XT DNA Library Preparation Kit (Illumina). Paired-end sequencing with 150 bp reads was performed using the Novaseq 6000 Sequencing System, S4 flow cell. RNA-Seq FASTQ reads were aligned to the mouse reference genome (Gencode GRCm39 Release M26) using STAR (version 2.7.9a) and reads mapping to each gene enumerated using HTSeq-count (version 0.6.7). Normalization and differential expression were calculated using DESeq2. Pathway analysis was performed using gene set enrichment analysis (GSEA). Partek software was used to generate Venn diagrams.
Intracellular flow cytometry
BM MNC were stained for surface markers as above, stimulated with Phorbol 12-myristate-13-acetate (PMA, MedChemExpress) for 15 min, fixed with 16% paraformaldehyde, permeabilized with Cytofix/Cytoperm (BD), and stained with phospho-p38 MAPK-PE (1:30 dilution, Santa Cruz Biotechnology) or phospho-ERK-PE (1:10 dilution, Cell Signaling Technology).
Stromal co-culture
Murine BM stromal layers were generated from BM cells from CXCL12-KO mice and CXCL12-WT littermates. Expanded stromal cells were plated on 96-well plates in a hypoxic incubator. After 3 days, FACS-sorted LSK cells were added and exposed to drugs for 3 days followed by flow cytometry analysis. Human AML BM stromal layers were generated from BM MNC and frozen in aliquots. For co-culture, stromal cells were thawed and plated in 96-well plates in a hypoxic incubator. After 4 days, CD34+ cells from the same patient were added and exposed to drugs for 3 days, followed by flow cytometry analysis. Details are provided in Supplemental Methods.
Statistics
Results are shown as mean plus/minus standard error of the mean (SEM). Significance values were calculated using Prism version 9.0 software (GraphPad Prism, La Jolla, CA) using parametric and nonparametric t tests (Mann-Whitney test), or analysis of variance as appropriate based on normality of distribution. Survival was analyzed using Kaplan-Meier analysis.
Data availability statement
The datasets generated during during the current study are available from the corresponding author on reasonable request.
Data Sharing Statement
RNAseq data is deposited in NCBI’s Gene Expression Omnibus (GEO) and is accessible through GEO Series accession number: GSE212428
Results
AML development in Flt3-ITD TET2 flox/flox Mx-1 cre mice
We developed a Flt3ITD/WT TET2flox/flox Mx-1 cre mouse model of AML that allowed inducible Tet2 deletion from HSC (see methods). A similar model, using Vav-cre to constitutively target TET2 excision in HSC, was previously described(14). Partial excision of Tet2 was seen without pIpC induction, but complete excision after pIpC treatment (Figure S1A). Flt3-ITD TET2-KO mice developed a 100% penetrant, lethal (Figure 1A) hematological disorder consistent with AML with leukocytosis (Figure S1B), anemia (Figure S1C), thrombocytopenia (Figure S1D), and increased immature Gr1-intermediate (Figure 1B, left), c-Kit+ (Figure 1B, center), and Lin−Sca1+cKit+ (LSK) cells in PB (Figure 1B, right). Mice exhibited increased BM cellularity (Figure 1C, left) and splenomegaly (Figure 1C, center and right). These features were not observed in FLT3-ITD mice without Tet2 deletion.
Figure 1. AML development in Flt3-ITD TET2 flox/flox Mx-1 cre mice.
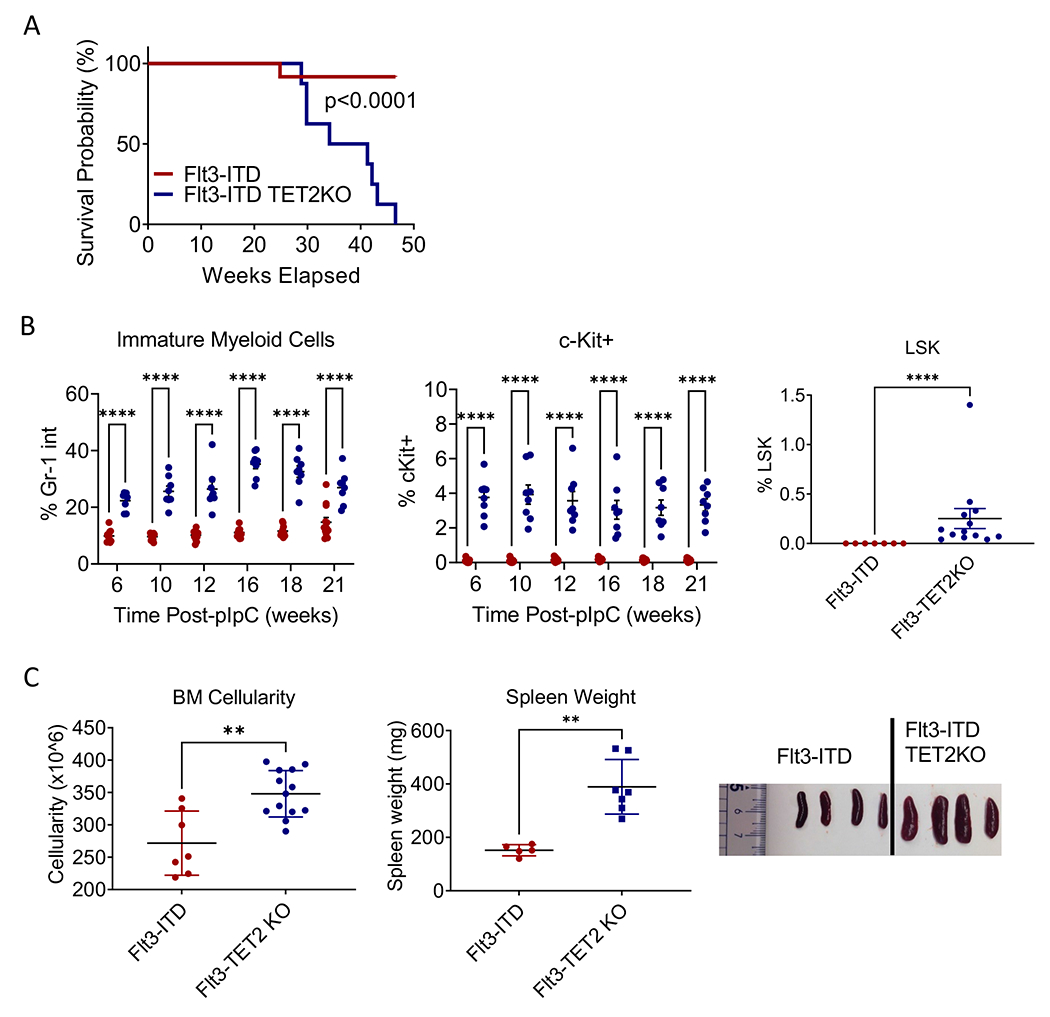
(A) Kaplan-Meier survival analysis for Flt3-ITD and Flt3-ITD TET2-KO mice. (B) Percentage of Gr-1 intermediate immature myeloid cells (left) and percentage of c-Kit positive immature cells (center) in peripheral blood of Flt3-ITD and Flt3-ITD TET2-KO mice over time post administration of pIpC, and frequency of LSK cells in peripheral blood 10 weeks post-pIpC (right). (C) Bone marrow cellularity (left, spleen weights (center), and representative spleen images (right) of Flt3-ITD and Flt3-ITD TET2-KO mice. Significance values: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. Results represent mean ± SEM of multiple replicates.
Flt3-ITD TET2-KO LSC reside in primitive phenotypic HSPC compartments
Evaluation of BM HSPC populations revealed that Flt3-ITD TET2-KO mice had increased GMP and MPP3 but not ST-HSC, and significant reduced LT-HSC compared to FLT3-ITD TET2-WT littermates or WT mice (Figure 2A). Transplantation of BM MNC from Flt3-ITD TET2-KO mice into WT recipients consistently resulted in leukemia development (Figure S2A–F). To identify LSC-containing subpopulation(s), we transplanted FACS-sorted GMP, MPP, and ST-HSC from Flt3-ITD TET2-KO AML mice into WT recipients (Figure 2B, left). Phenotypic LT-HSC (CD48-CD150+LSK) were almost absent in Flt3-ITD TET2-KO mice, consistent with previous reports, and were not transplanted(14, 22). We performed limiting dilution transplantation guided by the average frequency of each population in 1, 3, or 9x106 BM MNC (Figure 2B, left). GMP did not engraft at any cell dose tested (Figure S2G), MPP exhibited low levels of engraftment at the highest cell dose tested (Figure S2H), and ST-HSC showed dose-dependent engraftment at all cell doses tested (Figure S2I). The stem cell frequency within MPP was 1:63,635 and in ST-HSC was 1:2,730 (Figure 2B, right).
Figure 2. Flt3-ITD TET2-KO LSC reside in primitive phenotypic HSPC compartments.
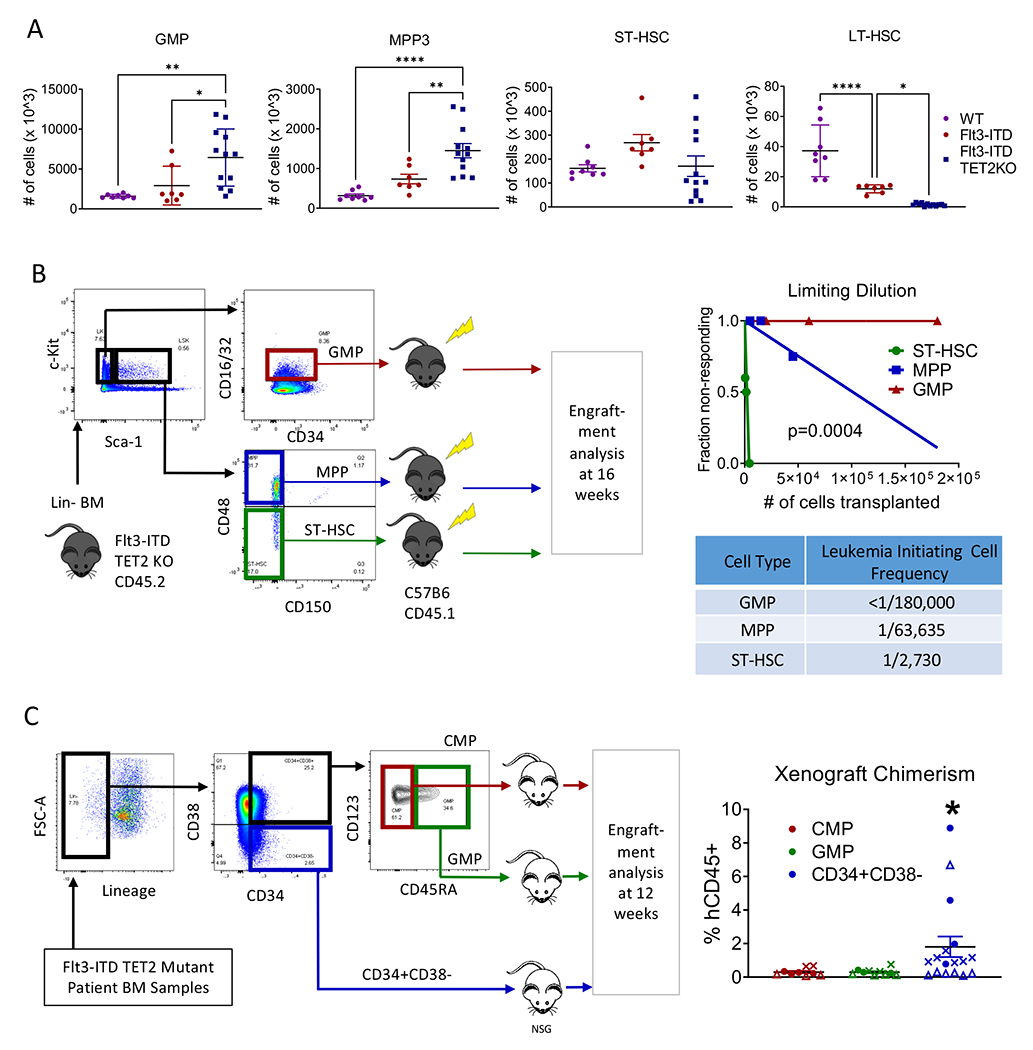
(A) Bone marrow frequencies of GMP (far left), MPP (left-center), ST-HSC (right-center), and LT-HSC (far right) in Flt3-ITD vs Flt3-ITD TET2-KO mice. (B) FACS sorting and transplantation scheme for limiting dilution transplants of sorted populations to identify leukemia initiating populations (left), limiting dilution plot of cell numbers transplanted (right-upper), and calculated leukemia initiating cell frequency for each population of transplanted cells (right-lower). (C) FACS sorting and xenotransplantation scheme for Flt3-ITD TET2 mutant human AML patient samples transplanted into NSG mice (left) and engraftment of the transplanted populations in NSG mouse xenografts (right). Each symbol represents a unique patient sample. Significance values: *p<0.05; **p<0.01; ****p<0.0001. Results represent mean ± SEM of multiple replicates.
To identify LSC-containing subpopulation(s) in human Flt3-ITD TET2-mutated AML samples, we transplanted FACS-sorted HSPC populations into NSG mice (Figure 2C, left). As with the mouse model, phenotypic LT-HSC were almost absent in all Flt3-ITD TET2 mutant samples analyzed (data not shown), consistent with previous reports,(23) and were not transplanted. We observed engraftment of human CD45+CD33+ cells following transplantation of primitive CD34+CD38− populations, but not committed progenitor populations (Figure 2C, right). Therefore the LSC-containing population in this mouse model recapitulates that of patients with similar mutations.
Flt3-ITD AML alters the BM microenvironment and increases CXCL12 expression in CAR cells
As CXCL12/CXCR4 signaling is essential for normal HSC function(9, 10, 24) and may contribute to AML progression and drug resistance(25), we interrogated our Flt3-ITD TET2-KO AML model for CXCR4 and CXCL12 expression. Expression of CXCR4 was increased in GMP, MPP and ST-HSC from Flt3-ITD TET2-KO vs. Flt3-ITD alone mice (Figure 3A). Flow cytometry analysis of stromal cell subsets revealed increased endothelial cell (EC, CD45-Ter119-CD31+) and CXCL12-abundant reticular cell (CAR, CD45-Ter119-CD31-VECadherin-CD51+Sca1−)(26) numbers and frequencies in Flt3-ITD TET2-KO compared with Flt3-ITD mice, whereas periarteriolar cells (PAC, CD45-Ter119-CD31-VECadherin-CD51+Sca1+)(27) were unaltered (Figure S3A–B). Since Mx-1 cre has activity in BM MSCs and perivascular cells(28), to ensure that changes in stromal populations were not related to stromal TET2 deletion, we assessed frequencies, numbers, and CXCL12 expression of stromal populations in CXCL12-GFP-knock-in reporter mice transplanted with WT or AML BM (Figure 3B). The frequency of EC and CAR (Figure 3C, left), and absolute number of CAR (Figure S3C), were increased in mice engrafted with Flt3-ITD TET2-KO cells. The percent and absolute number of CXCL12 expressing CAR was increased in Flt3-ITD TET2-KO AML engrafted mice (Figure 3C, right, Figure S3D). The observed increase in CXCR4-expressing HSPC and CXCL12-expressing CAR cells supported further investigation of the role of BMM CXCL12 expression in supporting Flt3-ITD TET2-KO AML HSPC.
Figure 3. Flt3-ITD AML alters the BM microenvironment and increases CXCL12 expression in CAR cells.
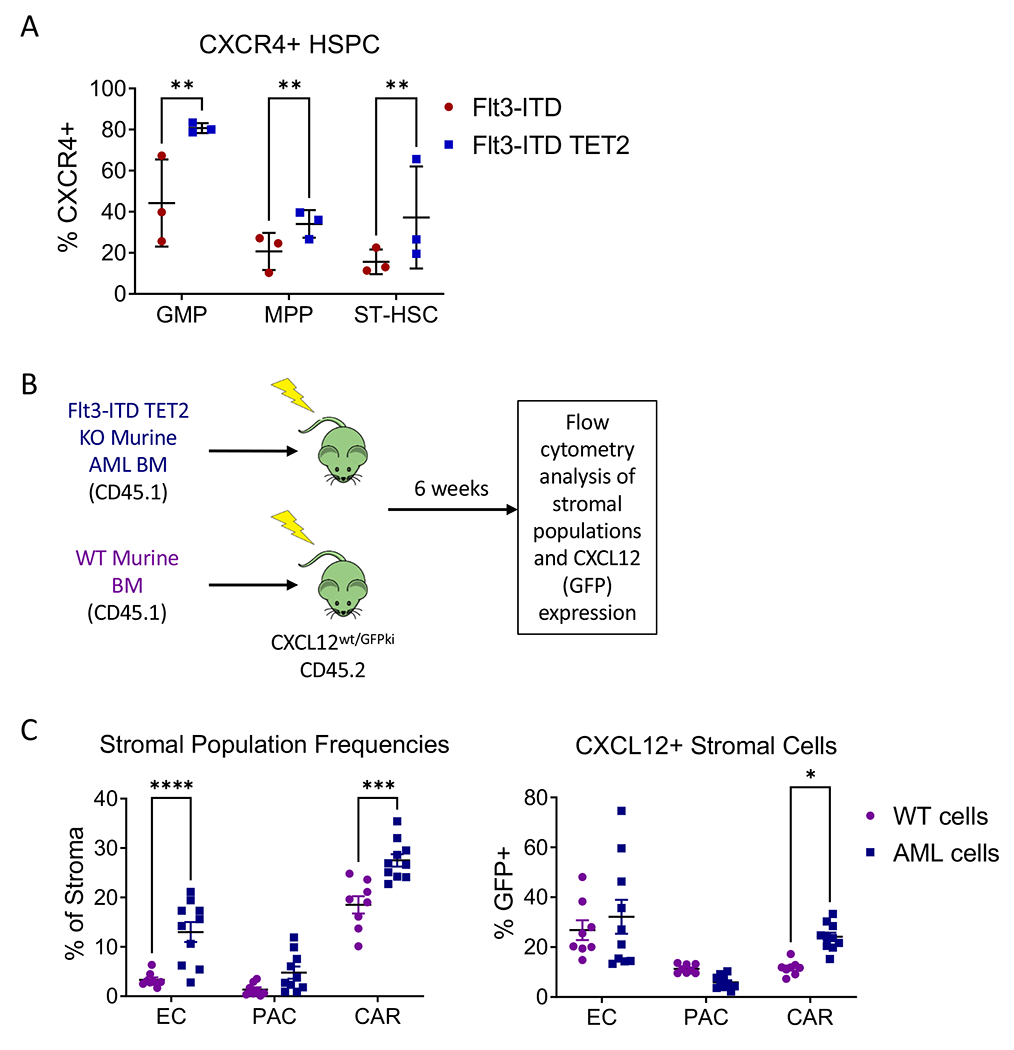
(A) CXCR4 positivity in BM HSPC from Flt3-ITD vs Flt3-ITD TET2-KO mice. (B) Transplant scheme for CXCL12-GFP mice. (C) Frequencies of endothelial cells, PAC, and CAR cells within CD45-Ter119-population (left) and percentage GFP (CXCL12) positivity in endothelial, PAC, and CAR cell populations (right) in CXCL12-GFP mice transplanted with WT vs. Flt3-ITD TET2-KO AML cells. Significance values: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ns, not significant. Results represent mean ± SEM of multiple replicates.
CXCL12 KO reduces AML disease burden
To assess the role of BMM CXCL12 in AML progression, we transplanted BM from Flt3-ITD TET2-KO AML mice into CXCL12flox/flox UBC-ERT2-cre mice treated with tamoxifen to induce CXCL12 deletion (CXCL12-KO) or tamoxifen-treated control CXCL12flox/flox cre-negative littermates (CXCL12WT, Figure 4A). Our previous studies show that Tamoxifen-treated UBC-ERT2-cre mice exhibit complete inhibition of CXCL12 mRNA expression as detected by Q-PCR(29). We observed reduced PB WBC counts (Figure 4B, left), moderate reduction in dysplastic neutrophils and increase in mature neutrophils (Figure S4A), increased RBC counts (Figure 4B, center), and reduced spleen weight (Figure 4B, right), in CXCL12-KO vs. CXCL12-WT AML mice at 8 weeks post-transplantation. A trend towards reduced BM cellularity (Figure 4C, left), and a reduction in BM GMP, but not other HSPC populations, was seen in CXCL12-KO vs. CXCL12-WT AML mice (Figure 4C, center and right). However, CXCL12 deletion did not result in significant change in survival of mice transplanted with Flt3-ITD AML BM (Figure S4B).
Figure 4. CXCL12 KO reduces AML disease burden.
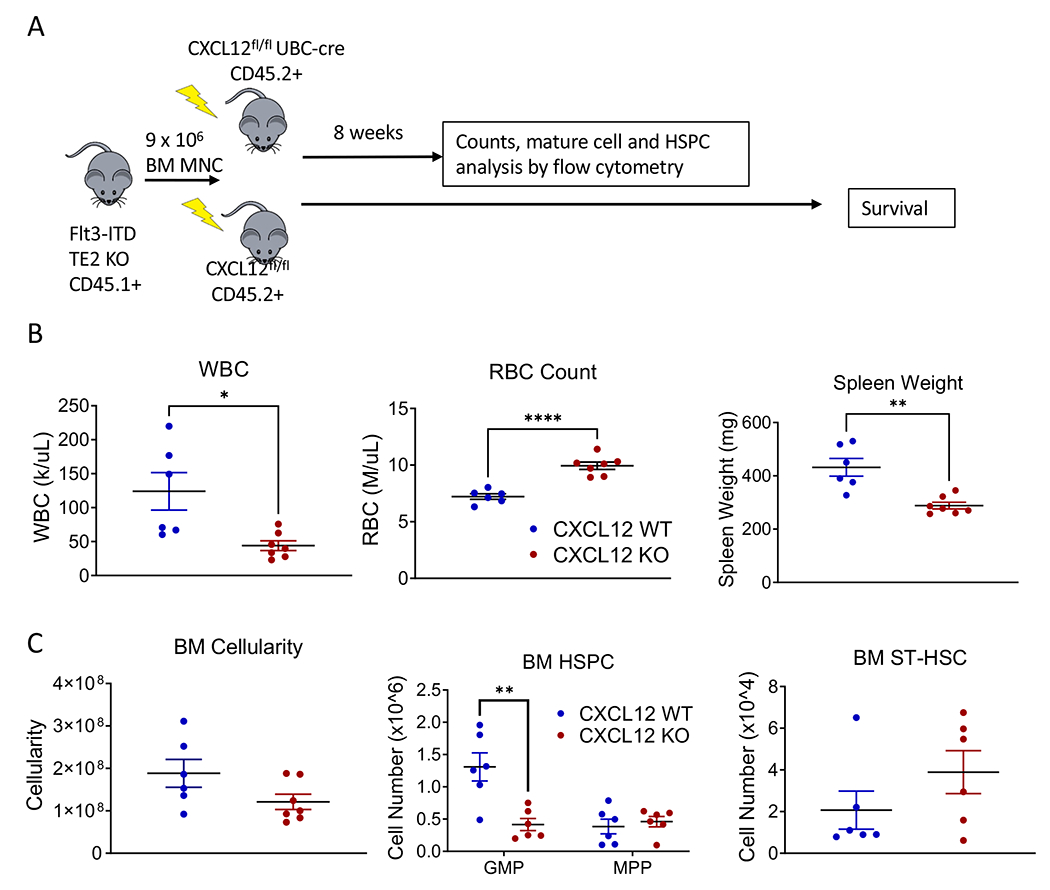
(A) Experimental design for transplanting AML cells into CXCL12 flox/flox UBC-cre mice to assess the effect of global CXCL12 KO on AML development. (B) WBC counts (left), RBC counts (center), and spleen weights (right) in CXCL12-WT vs. CXCL12-KO mice transplanted with AML cells. (C) BM cellularity (left), BM HSPC numbers (center), and BM ST-HSC (right) in CXCL12-WT vs. CXCL12-KO mice transplanted with AML cells. Significance values: *p<0.05; **p<0.01. Results represent mean ± SEM of multiple replicates.
To determine effects of CXCL12 KO on quizartinib response, we transplanted murine Flt3-ITD TET2-KO AML BM into CXCL12-KO or CXCL12-WT mice, and four weeks post-transplantation started 28-days treatment with quizartinib (10 mg/kg) or vehicle control (Figure 5A). Vehicle- and TKI-treated CXCL12-WT mice showed increased WBC counts after 4 weeks, while vehicle- and TKI-treated CXCL12-KO mice showed significantly reduced WBC counts (Figure 5B, left). PB c-Kit+ cells were decreased (Figure S5B) and RBC counts were increased (Figure S5A) in vehicle- and TKI-treated CXCL12-KO mice. Spleen weight was significantly reduced in vehicle and TKI-treated CXCL12-KO mice (Figure 5B, right). BM GMP were reduced by CXCL12 KO, quizartinib or both, but MPP and ST-HSC were unaffected (Figure 5C). Therefore, CXCL12 KO reduced leukemic burden in Flt3-ITD TET2-KO AML mice and quizartinib showed modest additional benefit in CXCL12-KO mice.
Figure 5. Effect of CXCL12 KO modestly on quizartinib response.
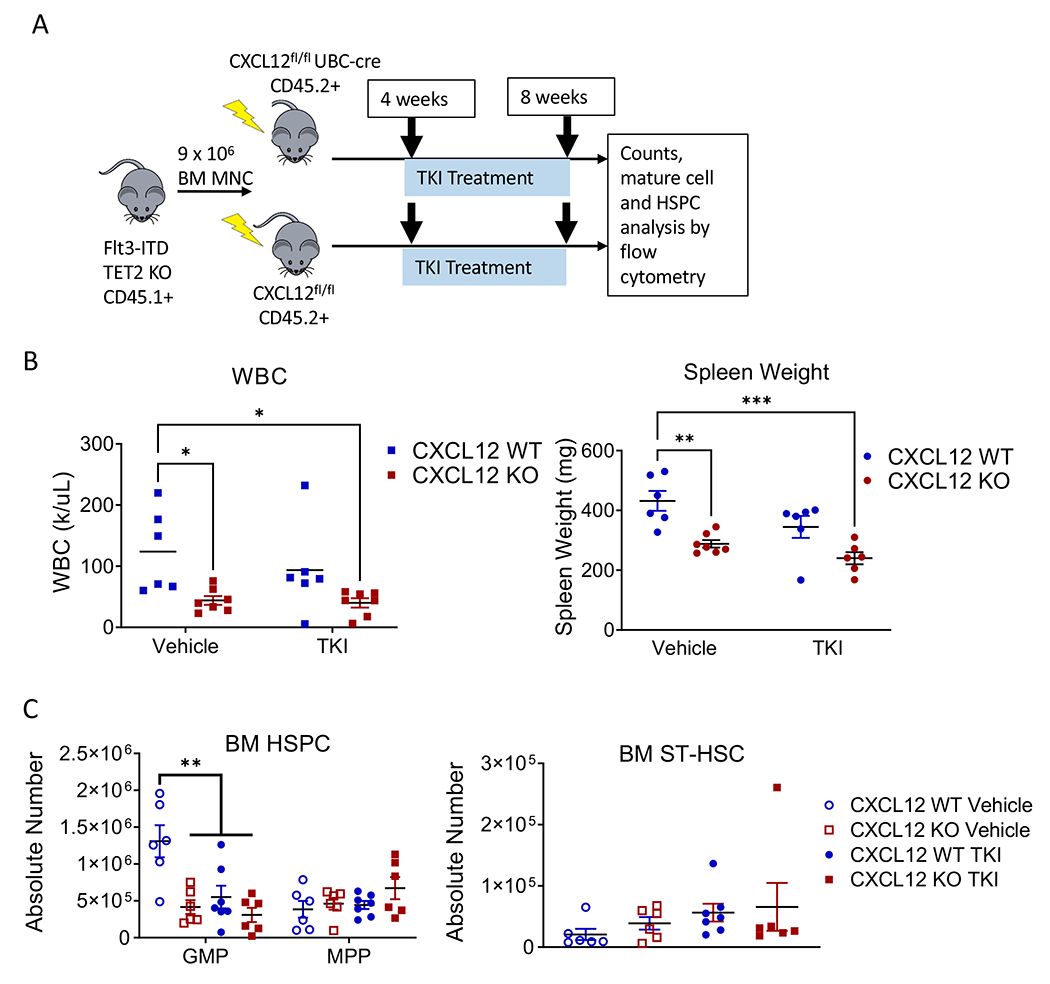
(A) Experimental design for single-agent quizartinib (AC220) treatment in CXCL12-WT and CXCL12-KO AML mice. (B) WBC counts (left) and spleen weights (right) and (C) HSPC (left) and ST-HSC numbers (right) in BM of CXCL12-WT or CXCL12-KO mice transplanted with AML BM and treated with vehicle control or single-agent quizartinib. Significance values: *p<0.05; **p<0.01; ***p<0.001. Results represent mean ± SEM of multiple replicates.
CXCL12 KO enhances response to combination chemotherapy plus quizartinib
To assess effects of CXCL12 KO on AML response to combination of quizartinib with chemotherapy, we transplanted BM from Flt3-ITD TET2-KO AML mice into CXCL12-KO mice and CXCL12-WT littermate controls, and four weeks post-transplantation began treatment with cytarabine, doxorubicin, and quizartinib (Figure S6A). Blood was taken after 2 and 3 weeks of treatment, and mice were sacrificed and BM analyzed immediately upon completion of 3 weeks of quizartinib treatment. Vehicle-treated mice had increased WBC counts after 2 weeks, whereas combination-treated mice had stable counts. However, after 3 weeks, combination-treated CXCL12-WT (WT-Rx) mice developed increased WBC whereas CXCL12-KO combination-treated (KO-Rx) mice maintained stable counts (Figure 6A, left). Immature c-Kit+ (Figure 6A, center) and LSK cells (Figure S6B) were decreased in PB of KO-Rx but not WT-Rx mice, whereas Gr-1high mature myeloid cells were increased (Figure 6A, right). Spleen weights (Figure 6B, left) and spleen LSK cells (Figure S6C) were reduced in KO-Rx mice. BM immature c-Kit+ (Figure S6D) and LSK cells (Figure 6B, center) were increased in vehicle-treated CXCL12-KO (KO-Veh) mice, and decreased in KO-Rx mice compared to KO-Veh mice, whereas mature myeloid cells were increased in KO-Rx mice (Figure S6E). ST-HSC numbers were decreased in KO-Rx compared to KO-Veh mice (Figure 6B, right). BM CXCR4+ expression was increased in KO-Veh mice (Figure S6F). The percentage of LSK within CXCR4+ BM cells showed a trend toward increase in WT-Rx mice, but was significantly decreased in KO-Rx mice (Figure S6G). These observations suggest that CXCL12 KO enhances targeting of CXCR4-expressing LSC by chemo+TKI.
Figure 6. CXCL12 KO enhances response to combination chemotherapy plus quizartinib.
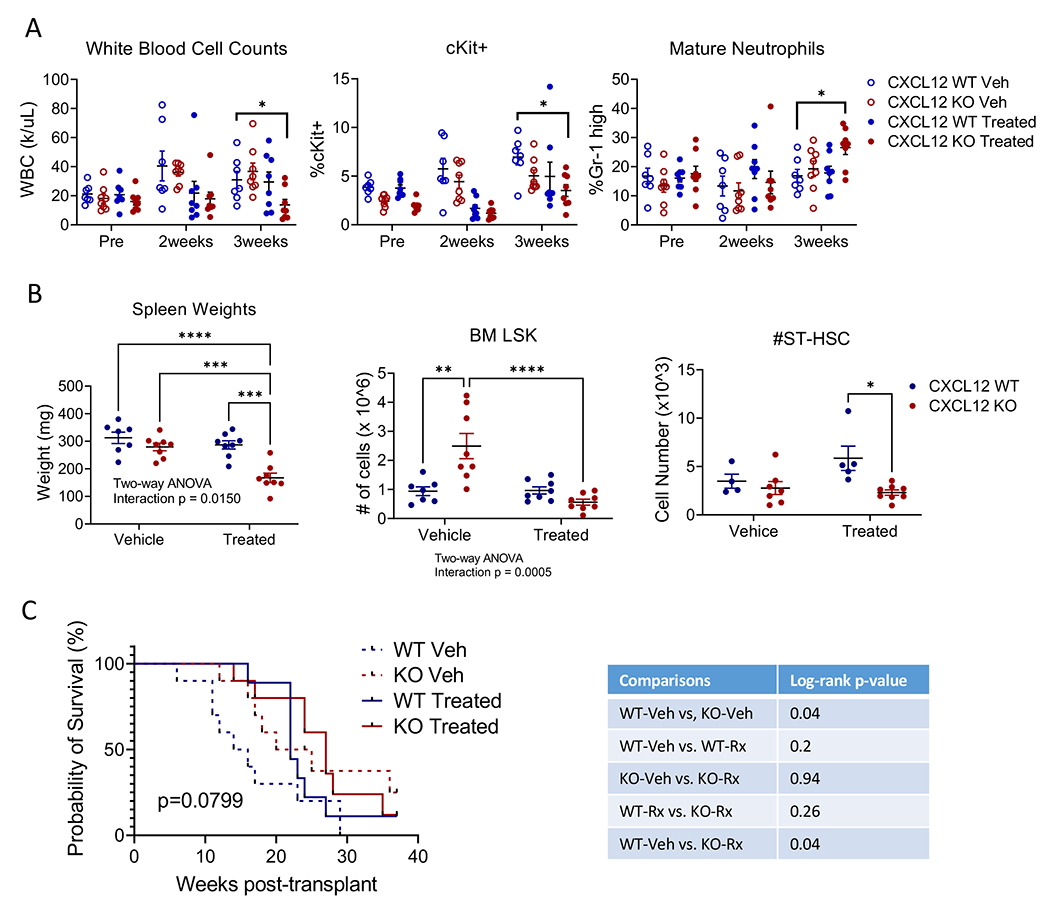
(A) WBC counts (left), %c-Kit+ cells (center), and %Gr-1 high mature neutrophils in PB (right), and (B) spleen weights (left), BM LSK (center) and BM ST-HSC (right) in CXCL12-WT or CXCL12-KO mice treated with vehicle control or combination chemotherapy plus quizartinib. (C) Kaplan-Meier survival analysis of secondary transplants of 2 x 10^6 BM MNC from control, KO-Veh, WT-Rx, or KO-Rx mice into WT recipient mice. Significance values: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. Results represent mean ± SEM of multiple replicates.
We also performed secondary transplantation of BM obtained upon treatment completion into lethally-irradiated WT mice to assess long-term engrafting and leukemia generating capacity (Figure 6C). Recipients of BM from WT-Rx mice show an initial survival benefit compared to WT-Veh recipients, but then rapidly decline, suggesting that treatment may deplete cells with short-term but not long-term engraftment capacity. Recipients of both KO-Veh and KO-Rx BM cells showed significant improvement in survival compared to WT-Veh recipients. We observed similar survival of WT-Rx recipients and KO-Rx recipients. However, unlike WT-Rx recipients, KO-Rx recipients showed a significant survival advantage over WT-Veh recipients. These observations suggest that treatment with CXCL12 KO was superior to treatment without CXCL12 KO.
Changes in gene expression in leukemia stem cells following CXCL12 KO and combination chemotherapy plus TKI
We performed RNA-sequencing to compare differentially-expressed genes (DEG) in BM ST-HSC selected from WT-Veh (Control), KO-Veh, WT-Rx, and KO-Rx mice (Figure S7A). Gene set enrichment analysis (GSEA) revealed enrichment of AML and oxidative phosphorylation-related gene signatures in WT-Rx ST-HSC, extracellular matrix receptor interaction-related gene signatures in KO-Veh ST-HSC, and extracellular matrix receptor interaction and hematopoietic cell lineage gene signatures in KO-Rx ST-HSC vs. Control ST-HSC (Figure S7B). Interestingly, comparison of genes uniquely upregulated in KO-Rx vs. Control ST-HSC, revealed that the top enriched gene sets (other than riboflavin metabolism and porphyrin and chlorophyll metabolism) were driven by upregulation of MAPK11 (p38 MAPKβ) and IL-10 (Figure 7A) gene expression. We also found that all comparisons showed downregulation of several genes related to p38 and ERK MAPK signaling (Figure 7B). These observations led us to further investigate the role of MAPK signaling in treatment response and resistance with or without CXCL12KO.
Figure 7. Changes in gene expression in leukemia stem cells following CXCL12 KO and combination chemotherapy plus TKI.
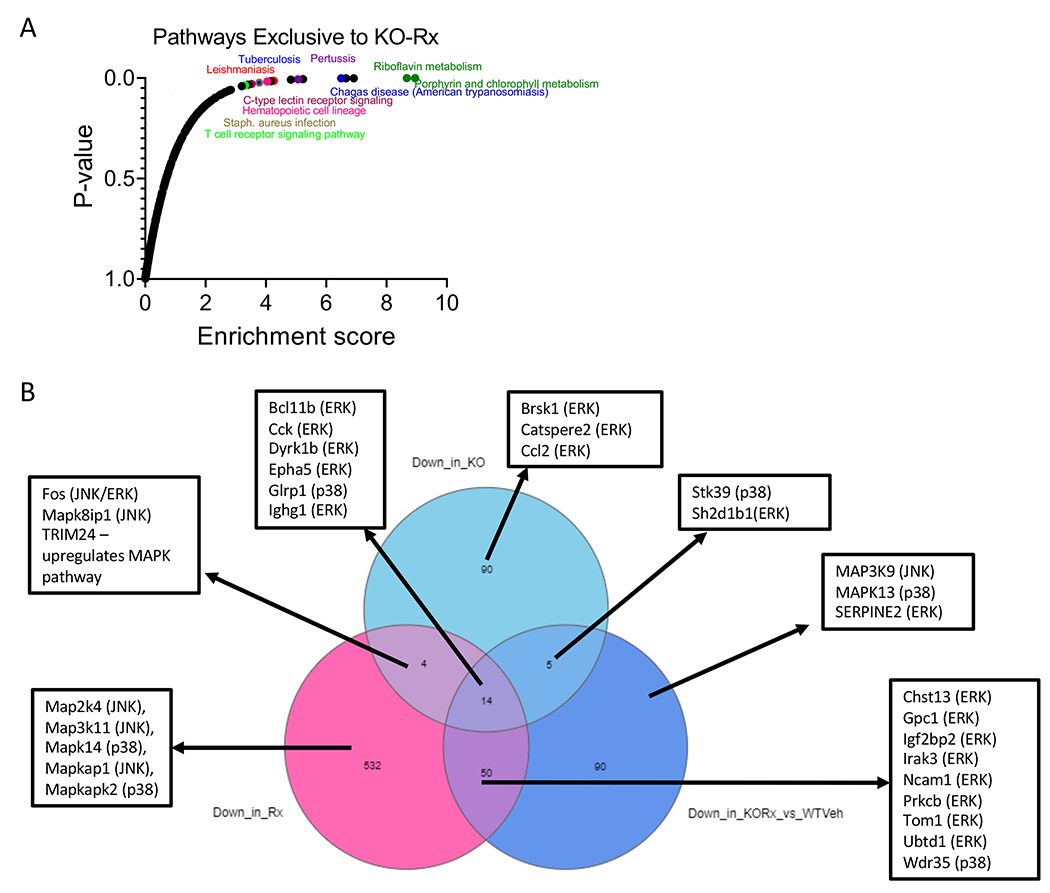
(A) Pathways exclusively upregulated in KO-Rx vs. Control. (B) DEG downregulated in KO-Veh vs Control, WT-Rx vs. Control, and/or KO-Rx vs. Control. Lists represent selected DEG related to MAPK signaling, with the relevant class of MAPK (ERK, p38, or JNK) indicated in parentheses.
p38 signaling contributes to AML LSK resistance to chemo + TKI
To confirm changes in MAPK signaling, we utilized intracellular flow cytometry to measure phosphorylated p38 and ERK in ST-HSC from Control, WT-Rx, KO-Veh, and KO-Rx mice following PMA stimulation. Phospho-p38 was reduced, but not significantly, in ST-HSC from WT-Rx and KO-Veh and significantly decreased, but not eliminated, in KO-Rx mice (Figure 8A). Phospho-ERK showed a trend toward reduction in ST-HSC from KO-Rx mice (Figure 8A). These observations are consistent with overall decrease in MAPK-related genes but upregulation of MAPK11 in KO-Rx mice (Figure 7B).
Figure 8. p38 signaling contributes to AML LSK resistance to chemo + TKI.
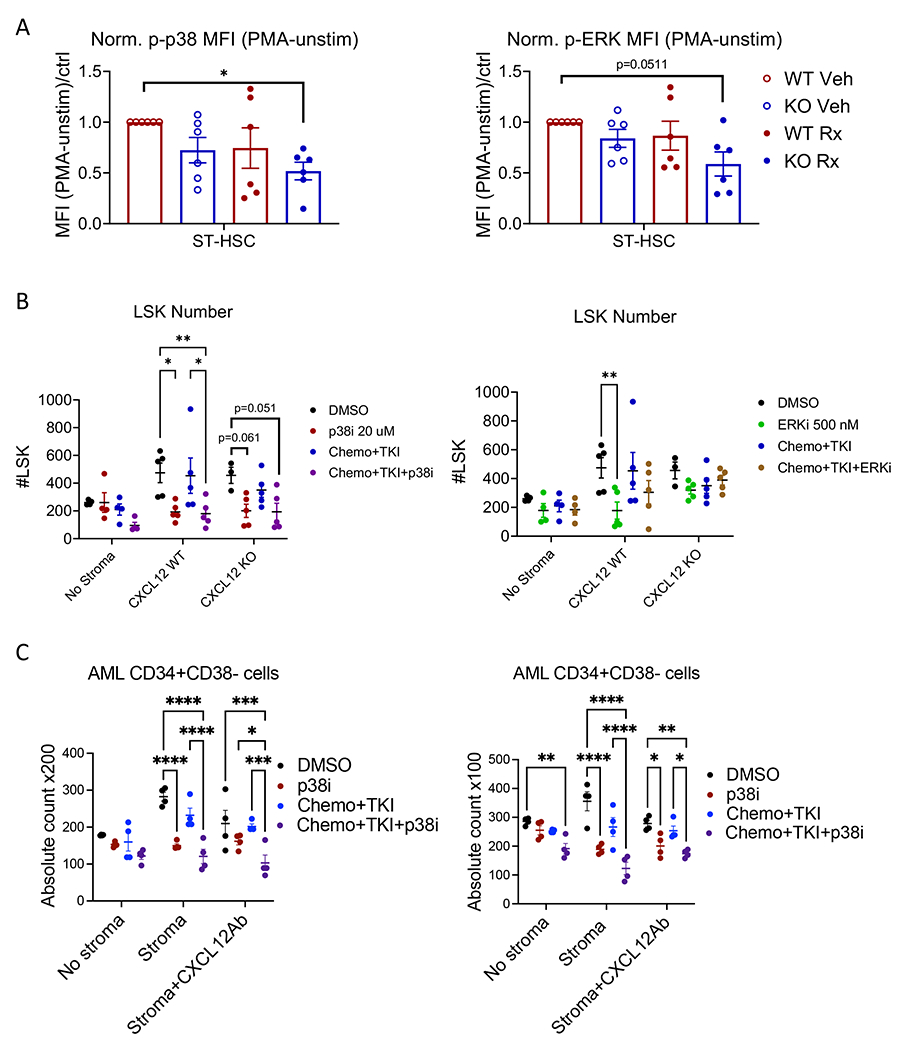
(A) Normalized mean fluorescence intensity – (MFI with PMA stimulation minus MFI without stimulation)/WT-Veh control – of phosphorylated p38 (left) and phosphorylated ERK (right) in BM cells from CXCL12-WT and CXCL12-KO mice treated with combination chemotherapy plus quizartinib (Rx) or vehicle as shown in Figure 6. (B) Total number of live AML LSK cells following co-culture with or without CXCL12-WT and CXCL12-KO stromal cells and treatment with DMSO (vehicle), p38i, chemo+TKI, or chemo+TKI+p38i (left); or vehicle, ERKi, chemo+TKI, or chemo+TKI+ERKi (right) for 3 days; MSC=mesenchymal stem cells; Chemo+TKI=combination chemotherapy plus quizartinib; p38i= losmapimod; and ERKi=ulixertinib. (C) Total number of live human AML CD34+CD38− cells following treatment of AML CD34+ cells with DMSO (vehicle), p38i, chemo+TKI, or chemo+TKI+p38i, with or without co-culture with stromal cells from the same patient, and with or without addition of CXCL12 blocking antibody. Results from two different AML samples, AML395 (left) and AML172 (right) are shown. Significance values: *p<0.05; **p<0.01, ***p<0.001, ****p<0.0001. Results represent mean ± SEM of multiple replicates.
We next evaluated the effect of inhibition of p38 and ERK signaling on AML LSC growth and response to chemo+TKI. FACS-sorted AML LSK were cultured with or without BM-derived MSC from CXCL12-WT or CXCL12-KO mice (Figure S8A). IC10 concentrations for doxorubicin, cytarabine and AC220, and IC20 concentrations for losmapimod, a p38 MAPK inhibitor (p38i), and ulixertinib, an ERK inhibitor (ERKi) were determined. Cultures were treated with 0.1% DMSO (vehicle control); doxorubicin, cytarabine, and AC220 (chemo+TKI, IC10); p38i (20 μM); ERKi (500 nM); chemo+TKI+p38i; or chemo+TKI+ERKi. Total AML LSK and CD45+ cell numbers were evaluated after 3 days.
AML CD45+ cell numbers were enhanced by MSC co-culture, but reduced by CXCL12 KO. Both p38i and ERKi reduced CD45+ cells co-cultured with CXCL12-WT or KO MSC (Figure S8B–C). Chemo+TKI reduced CD45+ cell numbers following culture with CXCL12-KO MSC but not CXCL12-WT MSC, consistent with in vivo studies. Chemo+TKI+p38i and chemo+TKI+ERKi both led to greater reduction in CD45+ cells than chemo+TKI with CXCL12-WT MSC. Chemo+TKI+p38i had significantly greater effect on AML CD45+ cells vs. p38i alone with CXCL12-WT but not CXCL12-KO MSC. In contrast, chemo+TKI+ERKi had similar effects as ERKi alone. We conclude that both p38 and ERK signaling contribute to MSC support of AML CD45+ cell growth, and that p38 signaling contributes to CXCL12-mediated protection from chemo+TKI.
Both p38i and ERKi significantly reduced LSK cell numbers following coculture with WT-MSC (Figure 8B). There was a trend toward significant reduction of LSK by p38i, but not ERKi, following coculture with CXCL12-KO MSC (Figure 8B). Treatment with chemo+TKI+p38i significantly reduced LSK numbers vs. chemo+TKI on CXCL12-WT but not CXCL12-KO MSC. There was a trend towards significant reduction in LSK cells cultured on CXCL12-KO MSC with chemo+TKI+p38i compared to controls. On the other hand, chemo+TKI+ERKi did not further reduce LSK numbers, with WT-MSC or CXCL12-KO MSC. These results show that both p38 and ERK signaling contribute to LSC maintenance by MSC. p38 has both CXCL12-dependent CXCL12-independent effects. They further show that p38, but not ERK, signaling contributes to AML LSC resistance to chemo+TKI both with and without CXCL12. p38 inhibition in combination with chemo+TKI leads to greater inhibition of AML LSC than CXCL12 KO alone, and could be even more effective than CXCL12 KO in sensitizing Flt3-ITD AML LSC to chemotherapy and Flt3 inhibition.
Finally we evaluated the effect of inhibition of p38 signaling on human AML LSC growth and response to chemo+TKI (Figure 8C). AML CD34+ were cultured with or without BM-derived MSC from the same patient, and with or without an anti-CXCL12 blocking antibody. Cells were exposed to 0.1% DMSO (vehicle control); chemo+TKI (IC10); p38i (20 μM) and chemo+TKI+p38i. AML CD34+CD38− cell numbers were evaluated after 3 days. MSC co-culture significantly enhanced total CD34+CD38− cell numbers, which were significantly reduced by CXCL12 blockade. p38i significantly reduced CD34+CD38− cell numbers when co-cultured with MSC. Treatment with chemo+TKI+p38i significantly reduced CD34+CD38− cell numbers vs. chemo+TKI following co-culture on stroma, with and without CXCL12 blockade. Similar results were obtained for cells with the CD34+CD38−CD45RA+ phenotype that has been associated with primitive AML LSC.(23) These results indicate that p38 contributes to MSC-mediated support of human AML LSC and that p38 inhibition in combination with chemo+TKI leads to greater inhibition of AML LSC than CXCL12 KO or chemo+TKI alone.
Discussion:
While Flt3-TKI show promise in treating Flt3-ITD-mutant AML, their efficacy remains limited. Clinical trials have combined Flt3 inhibitors with standard chemotherapy(3, 30) and hypomethylating agents (HMA)(31–33), but more rationally designed combination therapies are needed to effectively target Flt3-ITD LSC. Here we characterized the LSC population in a new GEMM of Flt3-ITD AML, and correlated with LSC populations in AML patients with similar mutations. Unlike some other AMLs where LSC are present in phenotypic GMP populations(34–36), LSC in this AML subtype have a more primitive phenotype(14). We observed upregulation of CXCR4 in LSC, and increased CAR cell numbers and CXCL12 expression in BM in this FLT3-ITD AML model. Inhibition of LSC interactions with the BMM by CXCL12 deletion reduced leukemic burden and increased LSC sensitivity to combined chemotherapy and Flt3 inhibition. Importantly, we identify p38 signaling as a targetable mechanism underlying microenvironmental support of FLT3-ITD LSC.
The CXCL12-CXCR4 axis is recognized as a potential mediator of AML progression and drug resistance(25, 37, 38). MSC from mice transplanted with AML upregulate CXCL12 expression(39). High CXCR4 expression is associated with poorer prognosis in AML, and CXCR4 expression is significantly higher in Flt3-ITD AML and contributes to poor prognosis(40). CXCR4 inhibitors tested in preclinical studies for the treatment of AML induced LSC mobilization and improved response to chemotherapy. However, clinical trials have not yet shown clear benefit over chemotherapy alone(25). In addition to CXCR4, CXCL12 also signals through CXCR7(41), so CXCR4 inhibition may not inhibit the full spectrum of CXCL12 signaling. Since CXCL12 expression has also been detected in AML cells(42), our CXCL12-targeting approach allows us to specifically block CXCL12 mediated LSC-BMM interactions.
Our studies identify a role for p38 in AML LSC maintenance and therapy resistance in the Flt3-ITD AML mouse model. Previous reports indicate that CXCL12 can activate ERK signaling.(43) Persistent ERK activation may contribute to reduced sensitivity to quizartinib of AML cell lines cultured on stroma.(44) Mutations in the Ras/MAPK pathway are an important mechanism of acquired resistance to the Flt3 inhibitor gilteritinib(45). The role of p38 MAPK signaling in Flt3-ITD AML is less well-explored. Forced expression of Flt3-ITD preferentially activates p38 MAPK.(46) BM stromal cells activate TGF-β-p38-ALDH2 signaling in AML cells to induce therapy resistance(47). p38 blockade enhances AML responsiveness to 5-fluorouracil in AML cell lines(48) and synergizes with the BCL2 inhibitor Venetoclax in ex vivo inhibition of primary AML cells not selected for Flt3 mutation status(49). However, the role of p38 MAPK in Flt3-ITD AML LSC maintenance and resistance to therapy remains unstudied. Our studies support a role for p38 signaling in human and murine Flt3-ITD AML LSC maintenance and for CXCL12-dependent and independent BMM-mediated resistance to chemo+TKI. p38 inhibition in combination with chemo+TKI leads to enhanced inhibition of AML LSC compared to CXCL12 KO indicating that p38 activity may be a critical vulnerability that could be exploited for therapy. These results provide a strong rationale for further therapeutic testing of p38 inhibitor containing combinations and investigation of mechanisms underlying p38 activation and mediation of resistance in LSC. Our results also support a role for ERK signaling in Flt3-ITD AML, but did not show a combinatorial effect of ERK inhibition with chemotherapy and Flt3 inhibition or with CXCL12 deletion.
Overall, the current study has characterized a new GEMM of Flt3-ITD AML and its LSC population, and identified a novel approach to target the Flt3-ITD AML LSC population. A recent phase I clinical trial(50) demonstrated favorable safety and tolerability of losmapimod at clinically relevant doses in facioscapulohumeral muscular dystrophy and a phase II study is underway. Our findings support further study of combining p38 inhibition with chemotherapy and Flt3 inhibition in Flt3-ITD AML.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health grant R01 number CA1772447 to R.B. N.A. was supported in part by UAB Cell and Molecular Biology T32 Training Grant T32GM008111.
The authors would like to thank: AML patients at UAB for the generous donation of their tissue samples; Drs. Douglas Hurst, Amjad Javed and Christopher Klug for helpful scientific discussions and comments; Maya Robinson and Amanda Mullen-Clem for procurement and processing of human samples. HudsonAlpha Institute for Biotechnology Sequencing Facility, Huntsville, Alabama for performing RNA sequencing; the UAB Comprehensive Flow Cytometry Core for providing help with FACS sorting; and the UAB Animal Resource Program for maintaining mouse colonies. N.R.A. is a PhD candidate at the University of Alabama at Birmingham. This work is submitted in partial fulfillment of the requirement for the PhD.
Footnotes
The authors declare no competing financial interests.
References
- 1.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, Reinhardt D, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood. 2006;108(12):3654–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017;377(5):454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med. 2019;381(18):1728–40. [DOI] [PubMed] [Google Scholar]
- 5.Hanekamp D, Cloos J, Schuurhuis GJ. Leukemic stem cells: identification and clinical application. Int J Hematol. 2017;105(5):549–57. [DOI] [PubMed] [Google Scholar]
- 6.Chopra M, Bohlander SK. The cell of origin and the leukemia stem cell in acute myeloid leukemia. Genes Chromosomes Cancer. 2019;58(12):850–8. [DOI] [PubMed] [Google Scholar]
- 7.Niu J, Peng D, Liu L. Drug Resistance Mechanisms of Acute Myeloid Leukemia Stem Cells. Front Oncol. 2022;12:896426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang A, Zhong H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology. 2018;23(10):729–39. [DOI] [PubMed] [Google Scholar]
- 9.Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495(7440):227–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495(7440):231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell. 2015;27(6):755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agarwal P, Isringhausen S, Li H, Paterson AJ, He J, Gomariz A, et al. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell. 2019;24(5):769–84 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee BH, Williams IR, Anastasiadou E, Boulton CL, Joseph SW, Amaral SM, et al. FLT3 internal tandem duplication mutations induce myeloproliferative or lymphoid disease in a transgenic mouse model. Oncogene. 2005;24(53):7882–92. [DOI] [PubMed] [Google Scholar]
- 14.Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. 2015;27(4):502–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyer SE, Qin T, Muench DE, Masuda K, Venkatasubramanian M, Orr E, et al. DNMT3A Haploinsufficiency Transforms FLT3ITD Myeloproliferative Disease into a Rapid, Spontaneous, and Fully Penetrant Acute Myeloid Leukemia. Cancer Discov. 2016;6(5):501–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ostrander EL, Kramer AC, Mallaney C, Celik H, Koh WK, Fairchild J, et al. Divergent Effects of Dnmt3a and Tet2 Mutations on Hematopoietic Progenitor Cell Fitness. Stem Cell Reports. 2020;14(4):551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen C, Liu Y, Lu C, Cross JR, Morris JPt, Shroff AS, et al. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. 2013;27(18):1974–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–9. [DOI] [PubMed] [Google Scholar]
- 20.Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, et al. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1(1):113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20(6):707–18. [DOI] [PubMed] [Google Scholar]
- 22.Chu SH, Heiser D, Li L, Kaplan I, Collector M, Huso D, et al. FLT3-ITD knockin impairs hematopoietic stem cell quiescence/homeostasis, leading to myeloproliferative neoplasm. Cell Stem Cell. 2012;11(3):346–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goardon N, Marchi E, Atzberger A, Quek L, Schuh A, Soneji S, et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19(1):138–52. [DOI] [PubMed] [Google Scholar]
- 24.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–88. [DOI] [PubMed] [Google Scholar]
- 25.Ladikou EE, Chevassut T, Pepper CJ, Pepper AG. Dissecting the role of the CXCL12/CXCR4 axis in acute myeloid leukaemia. Br J Haematol. 2020;189(5):815–25. [DOI] [PubMed] [Google Scholar]
- 26.Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33(3):387–99. [DOI] [PubMed] [Google Scholar]
- 27.Helbling PM, Pineiro-Yanez E, Gerosa R, Boettcher S, Al-Shahrour F, Manz MG, et al. Global Transcriptomic Profiling of the Bone Marrow Stromal Microenvironment during Postnatal Development, Aging, and Inflammation. Cell Rep. 2019;29(10):3313–30 e4. [DOI] [PubMed] [Google Scholar]
- 28.Joseph C, Quach JM, Walkley CR, Lane SW, Lo Celso C, Purton LE. Deciphering hematopoietic stem cells in their niches: a critical appraisal of genetic models, lineage tracing, and imaging strategies. Cell Stem Cell. 2013;13(5):520–33. [DOI] [PubMed] [Google Scholar]
- 29.Ramakrishnan R, Pena-Martinez P, Agarwal P, Rodriguez-Zabala M, Chapellier M, Hogberg C, et al. CXCR4 Signaling Has a CXCL12-Independent Essential Role in Murine MLL-AF9-Driven Acute Myeloid Leukemia. Cell Rep. 2020;31(8):107684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altman JK, Foran JM, Pratz KW, Trone D, Cortes JE, Tallman MS. Phase 1 study of quizartinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia. Am J Hematol. 2018;93(2):213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ravandi F, Alattar ML, Grunwald MR, Rudek MA, Rajkhowa T, Richie MA, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121(23):4655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swaminathan M, Kantarjian HM, Levis M, Guerra V, Borthakur G, Alvarado Y, et al. A phase I/II study of the combination of quizartinib with azacitidine or low-dose cytarabine for the treatment of patients with acute myeloid leukemia and myelodysplastic syndrome. Haematologica. 2021;106(8):2121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strati P, Kantarjian H, Ravandi F, Nazha A, Borthakur G, Daver N, et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am J Hematol. 2015;90(4):276–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heuser M, Yun H, Berg T, Yung E, Argiropoulos B, Kuchenbauer F, et al. Cell of origin in AML: susceptibility to MN1-induced transformation is regulated by the MEIS1/AbdB-like HOX protein complex. Cancer Cell. 2011;20(1):39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442(7104):818–22. [DOI] [PubMed] [Google Scholar]
- 36.Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17(24):3029–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho BS, Kim HJ, Konopleva M. Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: from bench to bedside. Korean J Intern Med. 2017;32(2):248–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su L, Hu Z, Yang YG. Role of CXCR4 in the progression and therapy of acute leukaemia. Cell Prolif. 2021;54(7):e13076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou HS, Carter BZ, Andreeff M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol Med. 2016;13(2):248–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rombouts EJ, Pavic B, Lowenberg B, Ploemacher RE. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood. 2004;104(2):550–7. [DOI] [PubMed] [Google Scholar]
- 41.Singh AK, Arya RK, Trivedi AK, Sanyal S, Baral R, Dormond O, et al. Chemokine receptor trio: CXCR3, CXCR4 and CXCR7 crosstalk via CXCL11 and CXCL12. Cytokine Growth Factor Rev. 2013;24(1):41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64(8):2817–24. [DOI] [PubMed] [Google Scholar]
- 43.Zaitseva L, Murray MY, Shafat MS, Lawes MJ, MacEwan DJ, Bowles KM, et al. Ibrutinib inhibits SDF1/CXCR4 mediated migration in AML. Oncotarget. 2014;5(20):9930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim BR, Jung SH, Han AR, Park G, Kim HJ, Yuan B, et al. CXCR4 Inhibition Enhances Efficacy of FLT3 Inhibitors in FLT3-Mutated AML Augmented by Suppressed TGF-b Signaling. Cancers (Basel). 2020;12(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith CC, Levis MJ, Perl AE, Hill JE, Rosales M, Bahceci E. Molecular profile of FLT3-mutated relapsed/refractory patients with AML in the phase 3 ADMIRAL study of gilteritinib. Blood Adv. 2022;6(7):2144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Odgerel T, Kikuchi J, Wada T, Shimizu R, Kano Y, Furukawa Y. MSK1 activation in acute myeloid leukemia cells with FLT3 mutations. Leukemia. 2010;24(5):1087–90. [DOI] [PubMed] [Google Scholar]
- 47.Yuan B, El Dana F, Ly S, Yan Y, Ruvolo V, Shpall EJ, et al. Bone marrow stromal cells induce an ALDH+ stem cell-like phenotype and enhance therapy resistance in AML through a TGF-beta-p38-ALDH2 pathway. PLoS One. 2020;15(11):e0242809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matou-Nasri S, Najdi M, AlSaud NA, Alhaidan Y, Al-Eidi H, Alatar G, et al. Blockade of p38 MAPK overcomes AML stem cell line KG1a resistance to 5-Fluorouridine and the impact on miRNA profiling. PLoS One. 2022;17(5):e0267855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurtz SE, Eide CA, Kaempf A, Long N, Bottomly D, Nikolova O, et al. Associating drug sensitivity with differentiation status identifies effective combinations for acute myeloid leukemia. Blood Adv. 2022;6(10):3062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mellion ML, Ronco L, Berends CL, Pagan L, Brooks S, van Esdonk MJ, et al. Phase 1 clinical trial of losmapimod in facioscapulohumeral dystrophy: Safety, tolerability, pharmacokinetics, and target engagement. Br J Clin Pharmacol. 2021;87(12):4658–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during during the current study are available from the corresponding author on reasonable request.