
Fig. 2 |. Resistance in ALK+ lung cancer and therapeutic interventions.
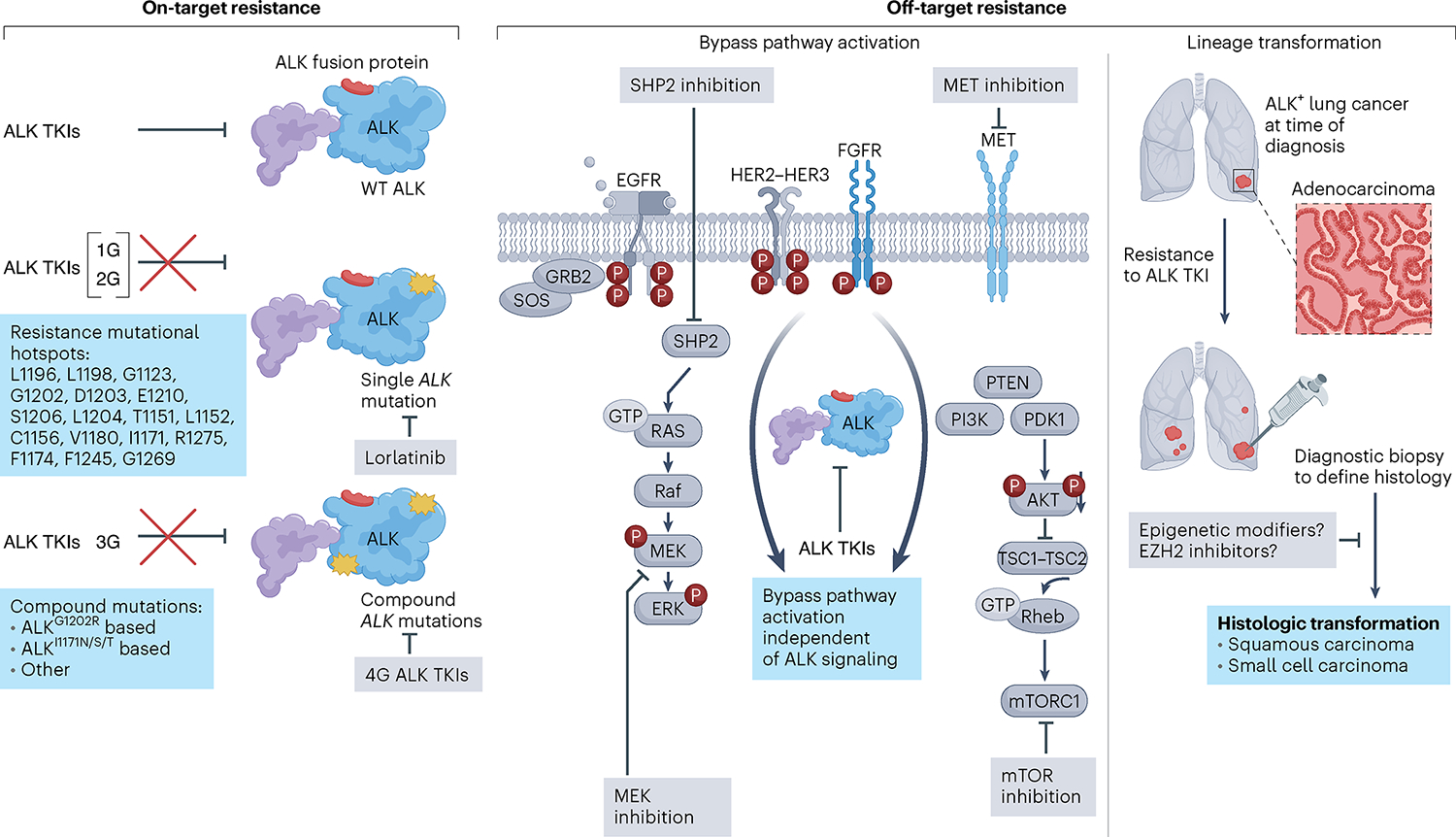
Resistance to ALK TKIs occurs through three main mechanisms. Left, on-target resistance is mediated by mutations in the ALK tyrosine kinase domain, which disrupt TKI binding to ALK, rendering tumor cells insensitive to ALK inhibition. ALK residues involved in ALK TKI resistance are listed. Single ALK mutations are most common after first- or second-generation ALK TKIs, while compound mutations are most common after sequential use of early-generation inhibitors culminating with a third-generation inhibitor, lorlatinib. This stepwise accumulation of ALK mutations confers resistance to ALK TKIs, with fourth-generation (4G) ALK TKIs designed to target compound mutations that are refractory to current FDA-approved ALK inhibitors. Middle, off-target resistance is mediated by bypass signaling activation or lineage transformation. Bypass pathway activation can occur through genetic mechanisms (amplifications, activation mutations, structural alterations) and non-genetic mechanisms (receptor hyperactivation), resulting in activation of signaling pathways that bypass ALK dependency. Rational combinations of ALK plus bypass pathway inhibition are being evaluated and are depicted in gray boxes. Right, lineage transformation is another off-target resistance mechanism that can lead to ALK TKI insensitivity. Diagnostic biopsies to define histology are necessary to select histology-specific chemotherapy regimens in squamous cell- or small cell-transformed tumors. Studies are underway to determine whether histologic changes are reversible and whether epigenetic modifiers may resensitize tumor cells to ALK inhibition. GRB2, growth factor receptor-bound protein 2; PTEN, phosphatase and tensin homolog; Rheb, Ras homolog enriched in brain; SOS, son of sevenless; SHP2, SH2 containing protein tyrosine phosphatase-2; TSC, tuberous sclerosis proteins 1 and 2; WT, wild type.