
Figure 1.
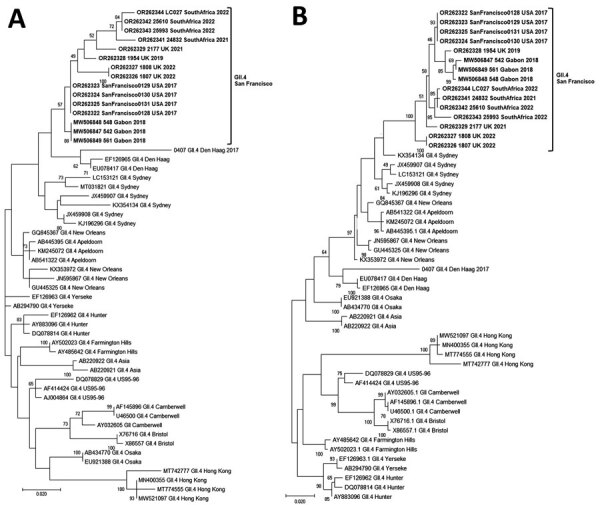
Phylogenetic trees of the emerging novel norovirus GII.4 strains on 3 continents. A) Common genotyping region C; 250 nt from the 5′ end of ORF2; B) complete VP1 aa sequences. Phylogenetic trees show novel GII.4 San Francisco strains and GII.4 variants, including recently identified clusters like GII.4 Hong Kong. Evolutionary analyses were conducted in MEGA X (https://www.megasoftware.net) using the maximum-likelihood method based on the Tamura-Nei model for the C region and Jones-Taylor-Thornton matrix-based model for VP1. We used a discrete gamma distribution to model evolutionary rate differences among sites; 5 categories γ parameter = 0.2174. Bootstrap (100) values are indicated at the nodes. Trees were drawn to scale. Scale bar represents nucleotide substitutions per site. ORF, open reading frame; VP1, viral protein 1.