

Abstract
Granular cell tumors are rare tumors that develop from Schwann cells, which are glial cells surrounding neurons of the peripheral nervous system, which serve in the process of myelination. Granular cell tumors are rarely associated with the central nervous system in humans. In this report, we analyze a patient with granular cell tumor and review the current literature.
Keywords: granular cell tumors, Schwann cells, malignant giant cell, astrocytoma
Introduction
Granular cell tumors (GCTs) are soft tissue tumors [1], originating from Schwann cells, thereby possessing characteristic pathological findings. These can behave as either benign or malignant, affecting the gastrointestinal tract, skin, and oral cavity more frequently. However, granular cell tumors may occur in any part of the body [2]. They often are classified as benign, but it has been discovered that <2% of the total cases of granular cell tumors can be malignant [3, 4]. Certain genetic disorders, including Noonan syndrome and neurofibromatosis, may include GCTs as a clinical finding [5]. There are numerous types of GCTs, including diffusely infiltrative pattern granular cell tumor, epithelioid appearance granular cell tumor, spindle cell appearance Abrikossoff’s tumor, granular cell myoblastoma, granular cell nerve sheath tumor, granular cell schwannoma, and much more [6, 7]. Definitive management of GCTs is surgical resection [8]. These are usually small (typically 1–3 cm), painless, solitary, and can be asymptomatic initially in case of benign lesions. GCTs are slow growing and tend to have a good prognosis if diagnosed early in malignant lesions, whereas benign does not require any typical course of action.
Granular cell astrocytoma (GCA) is a rare type of malignant brain tumor and is usually positive for glial fibrillary acidic protein (GFAP), S100, CD68, and epithelial membrane antigen [9]. GCTs of the brain tend to be malignant, more frequent in females, and can remain asymptomatic until detected by a biopsy. GCAs are made up of massive astrocytic cells with plenty of eosinophilic granular cytoplasm, which react with the GFAP and S100 proteins. Except for the GCA, most GCTs of the central nervous system (CNS) and other body parts tend to be benign; yet, when malignant, these tumors are rare and often difficult to treat [10]. Generally, all GCAs occur in the cerebral hemispheres and are a type of infiltrative brain tumor, with most reported cases occurring in the suprasellar region. GCAs can be seen, although very rarely infratentorial within the cerebellum or spinal cord [11].
Case report
A 39-year-old male presented to the outpatient department with decreased word output, inability to complete sentences, and difficulty in reading. He had occasional headaches and vomiting. There were no episodes of seizures and no history of weakness of limbs. He did not have any other comorbidities and no similar history in the family.
MRI brain with contrast revealed a heterogeneous irregular rim of an enhancing lesion with a size of 3.6 × 3.4 × 2.4 cm appearing in the left frontal subcortical region, with mild perilesional edema extending into the corona radiata and external capsule (Fig. 1). Due to an increased intracranial pressure, there was a mild midline shift of 5.3 mm toward the right side. Moreover, there was no evidence of intralesional hemorrhage or calcification seen.
Figure 1.

MRI brain with contrast—a heterogeneous irregular rim of an enhancing lesion with a size of 3.6 × 3.4 × 2.4 cm appearing in the left frontal subcortical region, with mild perilesional edema extending into the corona radiata and external capsule.
A magnetic resonance spectroscopy (MRS) was performed and revealed an increased choline, considered as a marker of neoplasms, reduced NAA, and prominent lipid/lactate peaks. (Ch/NAA-4.0) (Ch/Cr-1.7) (Fig. 2).
Figure 2.

MRS showed an increased choline, reduced NAA, and prominent lipid/lactate peaks; (Ch/NAA-4.0) (Ch/Cr-1.7).
A tractography was also performed, showing destruction of the left superior longitudinal fasciculus, superior occipito-frontal fasciculus, and partial destruction of the left cortical spinal tract (Fig. 3). Gray and white matter differentiation was maintained and no abnormal signals were visualized in the rest of the cerebral hemispheres. There was subtle contrast enhancement.
Figure 3.

Tractography showing destruction of the left superior longitudinal fasciculus, superior occipito-frontal fasciculus, and partial destruction of the left cortical spinal tract.
In view of the location of the lesion and the involvement of the speech area, an awake craniotomy was planned for resection of the lesion. The goal of the surgery was maximal safe resection with function preservation. The patient was positioned in the supine position, with the patient’s head fixed with a three-pin holder (Fig. 4), and ultrasound was used to localize the lesion before opening the dura. The lesion was identified and microsurgical gross total resection (Fig. 5) was completed by using speech mapping techniques. Post-procedure, the patient had intact speech and had no new onset deficits.
Figure 4.
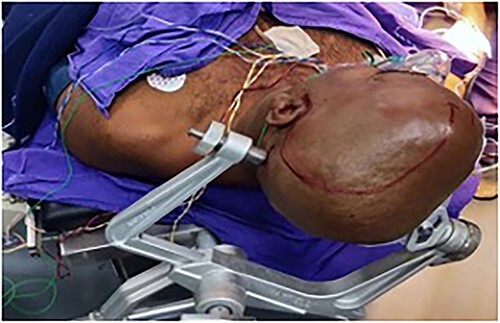
The patient was positioned in the supine position, with the head fixed with a three-pin holder.
Figure 5.
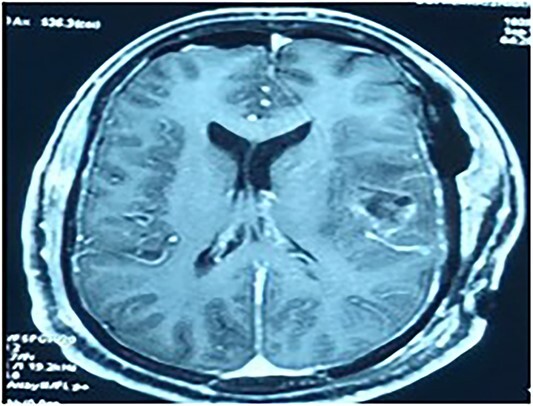
Contrast MRI depicting the tumor cavity and gross total resection.
The histopathology (Fig. 6) was reported as granular cell tumor. Immunohistochemistry also confirmed the same. Considering the nature of the lesion, the tumor board decided to give radiation to the tumor bed along with chemotherapy and follow-up imaging. The patient has completed radiotherapy and is tumor free at 4 years follow-up, without any deficits.
Figure 6.
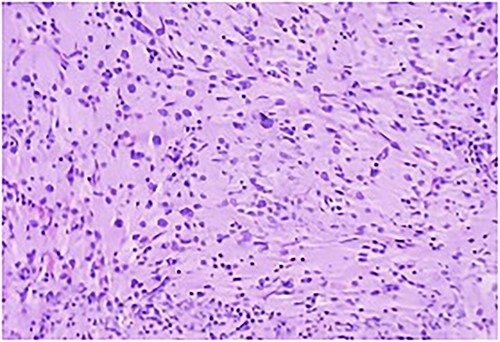
Section study shows a poorly defined mass composed of sheets of cells separated by thin collagenous bands; cells are round to polygonal with abundant eosinophilic cytoplasm with coarse granules; cell border is distinct at places with a syncytial pattern at some area; nuclei is large and vesicular.
Discussion
Granular cell tumor was determined to be originating from peripheral nerves due to its similar resemblance to peripheral nerves and to Schwann cells, which was shown by immunohistochemistry and electron microscopy [12]. According to their location, GCTs may have different cell sources and noticeably varied clinical symptoms. GCTs have been observed in a number of CNS regions. GCA, also known as cerebral GCT, is an uncommon variant of infiltrative astrocytoma that contains a significant number of granular cells. There have only been around 70 cases of GCA documented in the literature [10]. But unlike GCTs, which are often benign, GCAs frequently present as an aggressive glioblastoma. The cerebellum, pineal gland, and spinal cord have all been reported in a very small number of cases, but the cerebral hemispheres account for the majority of cases. Most hemispheres involve more than one lobe, with the parietal lobe appearing most frequently, followed by the frontal, temporal, and occipital lobes in that order [13, 14]. Patients with GCA are often between the age range of 27 and 83 (median age = 58 years). The male to female ratio among patients is 2:1, with a male predominance [12].
From a histological perspective, macrophages and granular tumor cells might be mistaken. Because of this, it is critical to distinguish GCA from a variety of reactive lesions, including multiple sclerosis, progressive multifocal leukoencephalopathy, and cerebral infarction [15]. In addition to considerable nuclear enlargement and atypia, GCAs frequently exhibit extensive eosinophilic nucleoli, characteristics that are not common in reactive CNS lesions in macrophages. To differentiate GCA from other disorders, it is also useful to look for a conventional infiltrating astrocytoma component and tumor cells that are bigger than macrophages [16].
Histologically, GCAs with WHO Grades II and III might display aggressive behavior. Tumors of Grade 2 were those that occasionally exhibited nuclear atypia, but lacked mitosis. But the presence of an uncommon mitosis does not transform the tumor into an anaplastic astrocytoma. Tumors classified as Grade 3 have hypercellularity, evident nuclear atypia, and noticeable mitotic activity [15]. The combination of conventional infiltrating astrocytoma and high-grade neoplastic findings in histology supports the diagnosis of GCA. Granular cell tumor histology demonstrates strong GFAP immunostaining, and its association with traditional astrocytoma suggests an astrocytic origin [10].
High-grade astrocytoma overlaps with genetic anomalies found in GCAs, which are not always specific. These include epidermal growth factor receptor amplification, homozygous deletion of CDKN2A, allelic losses at Loci 1p, 8p, 9p, 10q, 13q, 17p, 19q, and 22q, as well as methylation of the MGMT promoter [13]. Nearly all patients, including those with low-grade lesions, had loss of 9p and 10q. Several high-grade GCA cancers had TP53 mutations as well as simultaneous deletions of p14 and p16. Despite being a frequent occurrence in high-grade astrocytomas, epidermal growth factor receptor amplification was not seen in a small group of GCA [15].
Due to the aggressive nature of GCAs, most patients die within the first year of diagnosis. Patients exhibit symptoms such as new onset seizures, headache, nausea, impaired vision, disorientation, aphasia, and hemiparesis. Patients with GCAs were treated surgically and then postoperatively followed by chemotherapy and radiotherapy [14].
A few similar cases in the literature are described below (Table 1).
Table 1.
List of similar published articles.
| Study; year | Demographic features | Past history | Duration of illness | Investigations | Treatment given |
|---|---|---|---|---|---|
| A case of recurrent granular cell tumor; 2007 [8] | 16-year-old female | Previous history of granular cell tumor | 3-year history of a painless mass on the right side of her neck | Clinical examination: ~2 × 1 cm of a yellowish, solid, nodular mass was seen. Ultrasonography: to rule out metastases, cervical lymph nodes were examined. |
Under local anesthetic, the lesion was removed. Postoperative treatment: silicone blocks and topical steroid therapy were used. Follow up. |
| Granular cell tumors in the CNS; 2016 [10] | 29-year-old female | Amenorrhea | 12 months | MRI | Surgical: total resection, pterional approach. |
| Granular cell tumors in the CNS; 2016 [10] | 44-year-old male | None | MRI | Surgical: subtotal resection, trans-sphenoidal approach. | |
| Granular cell tumors in the CNS; 2016 [10] | 50-year-old female | Bilateral visual deficits | 3 years | MRI | Surgical: total resection, pterional approach. |
| Granular cell tumors in the CNS; 2016 [10] | 9-year-old male | Lumbodorsal pain | 2 months | MRI | Surgical: subtotal resection, laminectomy. |
| Granular cell tumors in the CNS; 2016 [10] | 12-year-old female | None | MRI | Surgical: subtotal resection, laminectomy. | |
| Granular cell tumors in the CNS; 2016 [10] | 25-year-old female | Epilepsy | 3 weeks | MRI | Surgical: total resection, left frontal craniotomy. |
| Granular cell tumors in the CNS; 2016 [10] | 45-year-old female | Facial hypesthesia | 2 years | MRI | Surgical: biopsy, transnasal approach. |
| Granular cell tumors in the CNS; 2016 [10] | 29-year-old male | Occipital pain | 1 year | MRI | Surgical: subtotal resection, far lateral craniotomy. |
| GCA of the pineal region; 2015 [11] | 16-year-old male | Parinaud syndrome, raised intracranial tension | 1 month | MRI: 3 × 3 × 4–cm iso-intensity lesion and homogeneous contrast enhancement | Surgical: subtotal resection, subtemporal approach, CSF shunt. |
| Granular cell variant meningioma; 2019 [12] | 49-year-old female | Unsteady walking | None | MRI: sized at 5.3 × 6.7 × 5.2 cm and situated in the right parietal and temporal area. | Surgical: total resection, craniotomy |
| GCA; 2018 [13] | 81-year-old female | Seizure and left-sided hemiparesis | None | CT: hypodensity in the right temporal lobe; MRI: subcortical white matter mass in the right temporal occipital region. | Surgical: total resection |
| Granular cell tumor of the neurohypophysis; 2018 [14] | 28-year-old male | Factor-VII deficiency | 2 years | Clinical examination: decrease facial and body hair; ophthalmological examination: bilateral optic atrophy; MRI: big suprasellar mass compressing pituitary and optic chiasm. | First: trans-sphenoidal endoscopy Second: surgical subtotal resection; transcranial approach. |
| GCA; 2012 [16] | 75-year-old male | 8-year history of hypertension and MI 5 years back | 3 months | CT and MRI: 6.5 cm mass seen in the frontal lobe and anterior genu of corpus callosum; Biopsy |
Conclusion
Granular cell tumors of the brain are extremely rare lesions. Majority of the GCTs in the CNS tend to be benign and only <2% of the tumors detected are malignant, referred as GCA. Almost all GCAs are found in the cerebral hemispheres having distinct morphologic features. Unfortunately, malignant granular cell tumors are associated with aggressive clinical behavior, often difficult to treat, and require expertise and precision.
Acknowledgements
The preprint of the manuscript is published in Authorea, with the DOI https://doi.org/10.22541/au.168396309.91269977/v1.
Contributor Information
Shyam Duvuru, Department of Neurosurgery, Apollo Specialty Hospitals, Tamil Nadu, India; Team Erevnites, Trivandrum, India.
Vivek Sanker, Team Erevnites, Trivandrum, India; Department of Neurosurgery, Trivandrum Medical College Hospital, Trivandrum, India.
Deepak Pandit, Team Erevnites, Trivandrum, India; Yerevan State Medical University, Armenia.
Sheezah Khan, Team Erevnites, Trivandrum, India; Yerevan State Medical University, Armenia.
Sara Alebrahim, Team Erevnites, Trivandrum, India; Faculty of Medicine, Mansoura University, MMPME.
Tirth Dave, Team Erevnites, Trivandrum, India; Bukovinian State Medical University, Chernivtsi, Ukraine.
Conflict of interest statement
None declared.
Funding
None declared.
Data availability
The data used to support the findings of this study are restricted by the ETHICS BOARD in order to protect PATIENT PRIVACY. Data are available from the corresponding author for researchers who meet the criteria for access to confidential data.
Author contributions
All authors contributed equally in drafting, editing, revising, and finalizing the case report.
Ethical approval
Ethical approval was not required for the case report as per the country’s guidelines.
Consent
Written informed consent was obtained from the patient to publish this report.
References
- 1. National Cancer Institute . NCI Dictionary of Cancer Terms. n.d.. Retrieved February 19, 2023, from https://www.cancer.gov/publications/dictionaries/cancer-terms/def/granular-cell-tumor.
- 2. Neelon D, Lannan F, Childs J. Granular Cell Tumor - Statpearls - NCBI Bookshelf. Granular Cell Tumor. n.d.. Retrieved February 19, 2023, from https://www.ncbi.nlm.nih.gov/books/NBK563150/. [PubMed]
- 3. Gunson DT. Granular Cell Tumour. DermNet. 2018. Retrieved February 19, 2023, from https://dermnetnz.org/topics/granular-cell-tumour.
- 4. Vladimir O Osipov, MD . Granular Cell Tumors. Practice Essentials, Etiology and Pathophysiology, Epidemiology. 2022. Retrieved February 19, 2023, from https://emedicine.medscape.com/article/282430-overview.
- 5. National Organization for Rare Disorders . Granular Cell Tumor. 2022. Retrieved February 24, 2023, from https://rarediseases.org/gard-rare-disease/granular-cell-tumor/.
- 6. Wikimedia Foundation . Book Sources. Wikipedia. n.d. Retrieved February 19, 2023, from https://en.wikipedia.org/wiki/Special:BookSources/978-1-4160-2999-1.
- 7. David G, Hicks MD, Lester SC, et al. Granular Cell Tumor. 2016. Granular Cell Tumor - an overview | ScienceDirect Topics. Retrieved February 19, 2023, from https://www.sciencedirect.com/topics/medicine-and-dentistry/granular-cell-tumor.
- 8. Yilmaz AD, Unlu RE, Orbay H, et al. Recurrent granular cell tumor: how to treat. J Craniofac Surg 2007. Retrieved February 19, 2023, from https://pubmed.ncbi.nlm.nih.gov/17912112/. [DOI] [PubMed] [Google Scholar]
- 9. Shi Y, Morgenstern N. Granular cell astrocytoma. Arch Pathol Lab Med 2008. Retrieved February 24, 2023, from https://pubmed.ncbi.nlm.nih.gov/19061297/. [DOI] [PubMed] [Google Scholar]
- 10. Li P, Yang Z, Wang Z, et al. Granular cell tumors in the central nervous system: a report on eight cases and a literature review. Br J Neurosurg 2016. Retrieved February 24, 2023, from https://pubmed.ncbi.nlm.nih.gov/27188824/. [DOI] [PubMed] [Google Scholar]
- 11. Asri ACE, Baallal H, Zoubeir Y, et al. Diagnosis and management challenge of a granular cell astrocytoma of the pineal region: case report. J Neurosurg Pediatr 2015. Retrieved February 24, 2023, from https://thejns.org/pediatrics/view/journals/j-neurosurg-pediatr/15/5/article-p506.xml. [DOI] [PubMed] [Google Scholar]
- 12. Gong C, Zhang Z. Granular cell variant of meningioma: a case report and review of the literature. Int J Clin Exp Pathol 2019. Retrieved February 24, 2023, from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6949636/. [PMC free article] [PubMed] [Google Scholar]
- 13. Rohan, Gupta A, Supriya Gupta B, Nathaniel Shapiro B, et al. Granular cell astrocytoma: case report. Hum Pathol Case Rep 2018. Retrieved February 24, 2023, from https://www.sciencedirect.com/science/article/pii/S2214330018300555. [Google Scholar]
- 14. Bello CT, Cipriano P, Henriques V, et al. Granular cell tumour of the neurohypophysis: an unusual cause of hypopituitarism. EDM Case Rep 2018. Retrieved February 24, 2023, from https://edm.bioscientifica.com/view/journals/edm/2018/1/EDM17-0178.xml. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi Y, Morgenstern N. Granular cell Astrocytoma [Internet]. Allen Press, 2008. [cited 2023Apr12]. Available from: https://meridian.allenpress.com/aplm/article/132/12/1946/63850/Granular-Cell-Astrocytoma [DOI] [PubMed] [Google Scholar]
- 16. Kim KH, Song JY, Choi CH, et al. Granular cell astrocytoma: report of a case. Korean J Pathol 2012;46:370. 10.4132/koreanjpathol.2012.46.4.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are restricted by the ETHICS BOARD in order to protect PATIENT PRIVACY. Data are available from the corresponding author for researchers who meet the criteria for access to confidential data.