

Abstract
Purpose:
Non-muscle myosin II (NMII) complexes are master regulators of actin dynamics that play essential roles during embryogenesis with vertebrates possessing three NMII heavy chain genes, MYH9, MYH10, and MYH14. As opposed to MYH9 and MYH14, no recognizable disorder has been associated with MYH10. We sought to define the clinical characteristics and molecular mechanism of a novel autosomal dominant disorder related to MYH10.
Methods:
An international collaboration identified the patient cohort. CAS9-mediated knockout cell models were used to explore the mechanism of disease pathogenesis.
Results:
We identified a cohort of 16 individuals with heterozygous MYH10 variants presenting with a broad spectrum of neurodevelopmental disorders and variable congenital anomalies that affect most organ systems and are recapitulated in animal models of altered MYH10 activity. Variants are typically de novo missense changes with clustering observed in the motor domain. MYH10 knockout cells demonstrate defects in primary ciliogenesis and reduced ciliary length with impaired Hedgehog signaling. MYH10 variant overexpression produced a dominant-negative effect on ciliary length.
Conclusions:
These data present a novel genetic cause of isolated and syndromic neurodevelopmental disorders related to heterozygous variants in the MYH10 gene with implications for disrupted primary cilia length control and altered Hedgehog signaling in disease pathogenesis.
Keywords: MYH10, non-muscle myosin, primary cilia, hedgehog, neurodevelopmental disability, hypertelorism
Introduction
Non-muscle myosin II (NMII) motor complexes are master regulators of actin cytoskeletal dynamics that are implicated in cell division, migration, polarity, morphology, adhesion, and other processes.1 NMII crosslinks actin into bipolar filaments and generates a sliding motion via ATP-hydrolysis to generate contractile force that exerts tension on the actomyosin network.1 NMII complexes are hexameric with pairs of heavy chain, regulatory light chain, and essential light chains. The NMII heavy chains (NMHCII) possess an N-terminal Mg2+-ATPase actin-binding motor domain and a C-terminal coiled-coil domain involved in dimerization and other interactions. In vertebrates, three NMHCII genes encoded by MYH9, MYH10, and MYH14 exhibit divergent enzymatic activity, subcellular localization, and cellular roles.1 Several human disorders have been associated with variants in NMHCII genes. Heterozygous MYH9 variants cause macrothrombocytopenia and granulocyte inclusions with or without nephritis or sensorineural hearing loss syndrome (OMIM #155199), while heterozygous MYH14 variants cause peripheral neuropathy, myopathy, hoarseness, and hearing loss syndrome (OMIM #614369). However, there is no recognizable disorder associated with MYH10.
Mouse models of altered MYH10 activity exhibit a broad range of defects in several organ systems. Global MYH10 knockout causes embryonic/peri-natal lethality due to heart failure with structural cardiac defects including septal defects and abnormal aortic positioning.2 In the developing brain, MYH10 deficiency results in severe hydrocephalus.3 In the post-natal brain, MYH10 regulates actin dynamics to support long-term potentiation and synapse formation in the hippocampus and amygdala.4,5 MYH10 mouse models also show congenital lung, renal, and ureteral abnormalities.6,7
A mouse knock-in p.(R709C) missense model demonstrates additional phenotypes such as cerebellar hypoplasia and neuronal migration defects.8 R709 is required for the MYH10 Mg2+-ATPase activity and the p.(R709C) model demonstrates additional phenotypes that are not present in global MYH10 knockout including body wall anomalies (omphalocele, diaphragmatic hernia, ectopia cordis) and palatal defects, implicating a dominant-negative effect of this missense product in these tissues.9,10 Of note, the p.(R709C) variant was annotated using transcript XM_017314835.3 that lacks exons 5 (30bp) and 16 (63bp) and hereafter will be referenced as p.(R740C) based on the full-length mouse transcript (XM_006534489.4).
There are several candidate MYH10 variants reported in the literature that show several phenotypes consistent with the mouse data. A de novo nonsense variant p.(R867X) was reported in a fetal case of hydrocephalus with ventriculomegaly and acqueductal stenosis.11 Another nonsense variant (p.E908X) was associated with IUGR, unilateral multicystic dysplastic kidney, congenital diaphragmatic hernia, cryptorchidism, hip dysplasia, severe neurodevelopmental disability, microcephaly, strabismus, and nystagmus.12 The missense variant p.(R280C) was identified in a young girl with severe developmental delay, intractable epilepsy, and microcephaly.13 There are several additional candidate de novo and inherited missense and nonsense/frameshift variants identified in large cohorts of individuals with neurodevelopmental/neuropsychiatric disorders, autism spectrum disorder, congenital heart disease, and congenital diaphragmatic hernia.14–22
Herein, we describe 16 additional individuals with heterozygous variants in the MYH10 gene presenting with a spectrum of neurodevelopmental disorders and variable congenital anomalies. We also provide evidence for MYH10 in the regulation of primary ciliary length and proper Hedgehog signal transduction as part of the mechanism of disease pathogenesis.
Materials and Methods
MYH10 variant assessment
All variants correspond to the MYH10 reference sequence NM_001256012.3. The model of human MYH10 (UniProt P35580) was generated with YASARA homology modeling, merging PDB structures 4pd3, 6bih, 4cgk, 4h5y, and 4dvy. Vertebrate MYH10 sequences were extracted from NCBI ortholog, aligned with ClustalW2, and conservation called as previously laid out. Sequences for MYH10 human paralogs (Q7Z406, P35749, P35579, Q9Y2K3, A7E2Y1, P13533, P12883, Q9UKX3, P11055, P13535, Q9Y623, P12882, Q9UKX2) were also assessed for conservation. MYH10 variants were extracted from gnomAD, Geno2MP, ClinVar, and SFARI followed by assessing predicted changes using Provean, SIFT, PolyPhen2, Align-GVGD, and our functional conservation. A compiled variant score was generated by calculating the functional tool assessment (max 9) x human paralog conservation x 21-codon linear motif calculation / number of amino acids used throughout the MYH10 evolution.
Cell culture, transfections, and siRNA knockdown
NIH/3T3 cells were cultured per standard protocols. Lipofectamine 3000 (ThermoFisher) was used for DNA transfections and Lipofectamine RNAiMAX reagent (TheroFisher) for siRNA. Dicer-substrate short interfering RNA (dsiRNA) against mouse MYH10 (Integrated DNA Technologies mm.Ri.Myh10.13.1) was used at 10 nM concentration.
qPCR quantitation of HH target genes
Total RNA was isolated using RNeasy Mini kit (Qiagen). Reverse transcription and qPCR was performed using High Capacity cDNA Reverse Transcription kit (ThermoFisher), PowerUp SYBR Green Master Mix (ThermoFisher), and run on the QuantStudio 7 Flex RT-PCR System (ThermoFisher). Relative expression was normalized to GAPDH. Primers are listed in supplemental information.
Site-directed mutagenesis
The human MYH10 cDNA was cloned into the pcDNA3 vector and included a C-terminal HA tag. Patient-specific missense variants were generated using the QuickChange Lightening Site Directed Mutagenesis Kit (Agilent).
Generating MYH10 knockout and overexpression cell lines
CAS9 gene editing was accomplished in NIH/3T3 cells using co-transfection of two different guide RNAs cloned into eSpCas9(1.1) in addition to an EGFP expression vector (pCIG). Single GFP positive cells were sorted via flow cytometry after 48 hours for clonal expansion and screening. A single guide RNA was used to target human MYH10 in RPE-1 cells via a similar strategy. Wildtype cell lines with normal MYH10 protein expression and native MYH10 sequence were isolated through the same targeting experiment. Polyclonal stable cell lines were generated via transfection of linearized plasmid and selected via G418 treatment. Monoclonal stable lines were expanded after single-cell plating in 96-well plates.
Western Blot Analysis
Western blot analysis was performed essentially as previously described.23 Antibodies include: rabbit anti-NMHCIIB (BioLegend; 1:1000), rabbit anti-NMHCIIA (BioLegend; 1:1000), mouse HRP anti-beta actin (Abcam, ab49900; 1:10000), mouse anti-GAPDH (Invitrogen, AM4300, 1:10000), and mouse anti-HA (Cell Signaling, 3274, 1:2000).
Luciferase assays
Luciferase reporter assays were performed as previously described using the ptcΔ136-GL3 luciferase reporter.23 Cells were plated and transfected 24 hours later with ptcΔ136-GL3, pSV-β-galactosidase (transfection control, Promega), and the indicated constructs (siRNA transfected 24 hours later). Cells were grown to confluence for 48 hours, placed in low serum media to stimulate primary ciliogenesis, and provided with HH pathway stimulation as indicated. Luciferase (Luciferase Assay System kit, Promega) and beta-galactosidase (beta-Galactosidase Assay Reagent, ThermoFisher) activity were measured after 48 hours.
Immunofluorescence
Immunofluorescent analysis was performed per standard protocol. Fixation included 4% paraformaldehyde and for cilia staining was preceded by treatment with −20°C methanol (100%). Primary antibodies include rabbit anti-NMHCIIB (BioLegend; 1:2000), mouse anti-acetylated tubulin (Sigma, T7451, 1:3000), rabbit anti-ARL13B (ProteinTech, 17711–1, 1:3000), rabbit anti-HA (CellSignaling, #3724, 1:2000). Actin was visualized using Phalloidin-iFluor 488 (Abcam, 1:3000). AlexaFluor secondary antibodies (ThermoFisher) were used for visualization (1:500). Images were captured on a Zeiss Laser Scanning Confocal Microscope 880 with Fast Airyascan.
Results
Clinical features of MYH10 cohort
We present 16 individuals (including 1 fetal case) harboring heterozygous variants in the MYH10 gene associated with a spectrum of neurodevelopmental disorders and variable congenital anomalies. Individuals were identified through an international collaboration facilitated by GeneMatcher24 and all variants were identified by exome sequencing. Table S1 describes the variant data, demographics, and growth parameters of the cohort. Growth deficits include short stature (<3%ile, 6/15), low body weight (<10%ile, 5/15), and microcephaly (<3%ile, 3/15, Table S1). As a unifying finding, we observe a spectrum of neurodevelopmental disorders ranging from mild impairment to profound global developmental delays and intellectual disability (Table 1). This includes motor delays (12/15), expressive language delay (12/15), intellectual disability (6/15), and autism spectrum disorder (4/15; Table 2).
Table 1.
Summary of phenotypic and variant data from individuals with heterozygous MYH10 variants.
| Ind# | Variant | Inh | Developmental Neurologic | Neuroimaging | Cardiac | Pulmonary/Airway | GI | Ocular | GU | Musculoskeletal |
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| 1 | c.2219G>A; p.(Arg740Gln) | De novo | GDD | MRI: supratentorial volume loss, hypoplastic dysmorphic corpus callosum, cerebellar hypoplasia, absent septum pellucidum, dilated 4th ventricle | VSD, ASD, right aortic arch | Chronic respiratory failure, tracheostomy/ventilator dependent, interstitial lung disease (PIG), bronchomalacia, alveolar maldev. | Omphalocele GJ-tube | Negative | Left renal cyst | Left hip dysplasia, sternal cleft |
| 2 | c.2219G>A; p.(Arg740Gln) | De novo | GDD, borderline ID, manipulative behavior, llimited body perception, immature conflict resolution, hypotonia | US: partial agenesis of corpus callosum, dilated lateral ventricles | Bicuspid AV, PFO, dilated ascending aorta, thickened TV/MV, membranous ventricular septum | Chronic respiratory failure, restrictive lung disease | Omphalocele, hiatal hernia, bilateral inguinal hernias | Hyperopia | Right pelviectasis, streak gonads | Scoliosis, dysplastic thorax |
| 3 | c.111G>T; p.(Trp37Cys) | Maternal | Motor delay, learning difficulties, spasticity, LE dystonia | MRI: normal | No echocardiogram | Negative | Constipation | Myopia | Negative | Negative |
| 4 | c.111G>T; p.(Trp37Cys) | N/A | Mild learning disability | No imaging | No echocardiogram | Negative | Negative | Negative | Negative | Negative |
| 5 | c.4529_4588dup; p.(Arg1510_Ala1529dup) | De novo | GDD, hypotonia | No imaging | Pulmonic stenosis | Negative | Constipation, chokes with feeds | Congenital ptosis | Shawl scrotum | Negative |
| 6 | c.1183C>A; p.(Leu395Ile) (mosaic) | De novo | GDD, mild ID ASD | MRI: normal | Normal echocardiogram | Negative | Negative | Congenital ptosis, intermittent exotropia, myopia | Negative | Pectus excavatum |
| 7 | c.2320G>A; p.(Gly774Ser) | De novo | ASD, ADHD spasticity | MRI: T2 hyperintensities throughout white matter, cystic abnormality of pituitary | Small PFO, episode of SVT w/ pericardial effusion | Negative | Negative | Negative | Negative | Negative |
| 8 | c.2555G>A; p.(Arg852Gln) | De novo | GDD, ASD, mild ID | No imaging | No echocardiogram | Negative | Negative | Negative | Negative | Negative |
| 9 | c.2201G>A; p.(Gly734Asp) | De novo | GDD, ID, seizures, hypotonia | MRI: cerebellar hypoplasia | No echocardiogram | Negative | GERD | Astigmatism | Negative | Right genu valgum, s/p hemiepiphysiodesis |
| 10 | c.5219A>G; p.(Glu1740Gly) | De novo | GDD, ID, seizures | MRI: Agenesis of corpus callosum, polymicrogyria, PNH | No echocardiogram | Episodic wheezing, respiratory distress | Constipation, GERD | Left chorioretinal lacunae | Negative | Negative |
| 11 | c.5519A>G; p.(Lys1840Arg) | De novo | Severe GDD, hypotonia, spasticity seizures, CVI | MRI: Hypoplastic corpus callosum, abnormal signal in periventricular white matter | Normal echocardiogram | Negative | G-tube | Negative | Negative | Arthrogryposis |
| 12 | c.2554C>T; p.(Arg852Trp) | De novo | GDD, hearing loss | MRI: ventriculomegaly, IVH | PDA, PFO, fetal heart block | Chronic lung disease, subglottic stenosis, OSA | Aspiration | Lateral rectus weakness | Pelviectasis, cryptorchidism | Cleft lip and palate |
| 13 | c.2036 G>A; p.(Gly679Asp) | De novo | ASD, hypotonia | No imaging | VSD, ASD, PDA, bicuspid AV, hypoplastic aortic arch | Negative | Constipation | Congenital ptosis | Negative | Negative |
| 14 | c.2209G>A; p.(Glu737Lys) | De novo | GDD, hypotonia | MRI: Agenesis of corpus callosum | VSD | Negative | Negative | Negative | Horseshoe kidneys, VUR | Negative |
| 15 | c.1220_1221del; p.(Leu407Hisfs*33) | De novo | GDD, ID, hypotonia | No imaging | Normal echocardiogram | Recurrent sinusitis | Negative | Negative | Retractile testes | Negative |
| 16 | c.2002del; p.(Thr668Glnfs*28) | De novo | N/A (fetal case) | Fetal imaging: Agenesis of corpus callosum | N/A | N/A | N/A | N/A | N/A | N/A |
This table demonstrates the variant data and phenotypic data by system for each individual identified with a heterozygous MYH10 variant.
denotes that the individual is deceased.
indicates prenatal demise.
Abbreviations: Ind individual, yo years old, mo months old, w weeks gestation, NDD neurodevelopmental disability, ID intellectual disability, ASD autism spectrum disorder, ADHD attention deficit hyperactivity disorder, GI gastrointestinal, GU genitourinary, VSD ventricular septal defect, ASD atrial septal defect, AV aortic valve, PFO patent foramen ovale, TV tricuspid valve, MV mitral valve, LE lower extremity, SVT supraventricular tachycardia, GERD gastro esophageal reflux disease, CVI cortical visual impairment, PNH periventricular nodular heterotopia, PDA patent ductus arteriosus, OSA obstructive sleep apnea, VUR vesicoureteral reflux.
Table 2.
Broad multisystem manifestations are observed in the MYH10-related disorder.
| Neurodevelopmental | |||
|---|---|---|---|
|
| |||
| HPO | Phenotype | # Ind (N=15) | % |
|
| |||
| Overall | 15 | 100.0% | |
| HP:0002194 | Motor delay | 12 | 80.0% |
| HP:0000750 | Delayed speech and language development | 12 | 80.0% |
| HP:0001249 | Intellectual disability | 6 | 40.0% |
| HP:0000717 | Autism spectrum disorder | 4 | 26.7% |
| HP:0001290 | Hypotonia | 8 | 53.3% |
| HP:0001257 | Spasticity | 3 | 20.0% |
| HP:0001250 | Seizures | 3 | 20.0% |
| Neuroimaging | |||
|
| |||
| HPO | Phenotype | # Ind (N=11) | % |
|
| |||
| Overall | 9 | 81.8% | |
| HP:0001273 | Abnormality of the corpus callosum | 6 | 54.5% |
| HP:0001274 | Agenesis of the corpus callosum | 4 | 36.4% |
| HP:0002079 | Hypoplasia of the corpus callosum | 2 | 18.2% |
| HP:0002119 | Ventriculomegaly | 3 | 27.3% |
| HP:0002500 | Abnormality of cerebral white matter | 2 | 18.2% |
| HP:0001321 | Cerebellar hypoplasia | 2 | 18.2% |
| HP:0001331 | Absent septum pellucidum | 1 | 9.1% |
| HP:0002126 | Polymicrogyria | 1 | 9.1% |
| HP:0002282 | Periventricular nodular heterotopia | 1 | 9.1% |
| Cardiac | |||
|
| |||
| HPO | Phenotype | # Ind (N=10) | % |
|
| |||
| Overall | 7 | 70.0% | |
| HP:0001629 | Ventricular septal defect | 3 | 30.0% |
| HP:0001655 | Patent foramen ovale | 3 | 30.0% |
| HP:0001631 | Atrial septal defect | 2 | 20.0% |
| HP:0001643 | Patent ductus arteriosis | 2 | 20.0% |
| HP:0001647 | Bicuspid aortic valve | 2 | 20.0% |
| HP:0001642 | Pulmonic stenosis | 1 | 10.0% |
| HP:0001702 | Abnormality of the tricuspid valve | 1 | 10.0% |
| HP:0012020 | Right aortic arch | 1 | 10.0% |
| HP:0012304 | Hypoplastic aortic arch | 1 | 10.0% |
| HP:0005111 | Ascending aorta dilation | 1 | 10.0% |
| Gastrointestinal | |||
|
| |||
| HPO | Phenotype | # Ind (N=15) | % |
|
| |||
| Overall | 8 | 53.3% | |
| HP:0002019 | Constipation | 4 | 26.7% |
| HP:0011471 | Enteral tube dependent | 2 | 13.3% |
| HP:0001539 | Omphalocele | 2 | 13.3% |
| HP:0002036 | Hiatal hernia | 1 | 6.7% |
| HP:0000023 | Inguinal hernia | 1 | 6.7% |
| Pulmonary/airway | |||
|
| |||
| HPO | Phenotype | # Ind (N=15) | % |
|
| |||
| Overall | 4 | 26.7% | |
| HP:0006530 | Interstitial lung disease | 1 | 6.7% |
| HP:0002091 | Restrictive lung disease | 1 | 6.7% |
| HP:0006528 | Chronic lung disease | 1 | 6.7% |
| HP:0030828 | Recurrent wheezing | 1 | 6.7% |
| HP:0002780 | Bronchomalacia | 1 | 6.7% |
| Ocular | |||
|
| |||
| HPO | Phenotype | # Ind (N=15) | % |
|
| |||
| Overall | 8 | 53.3% | |
| HP:0000539 | Abnormality of refraction | 4 | 26.7% |
| HP:0000508 | Ptosis | 3 | 20.0% |
| HP:0007858 | Unilateral chorioretinal lacunae | 1 | 6.7% |
The phenotypes presented in the MYH10 cohort are listed by system and defined by human phenotype ontology (HPO) terminology. Note that N=15 for neurodevelopmental, gastrointestinal, pulmonary/airway, and ocular as the fetal case was not included. Neuroimaging data is derived from 11 individuals for whom data was available. Cardiac defects are described from 10 individuals who underwent echocardiogram.
Neurologic manifestations include hypotonia (8/15), spasticity (3/15), and epilepsy (3/15) (Tables 1 and 2). Neuroimaging was available for 11 probands with findings of corpus callosum abnormalities (4/11 with agenesis, 2/11 with hypoplasia of the corpus callosum), ventriculomegaly (3/11), cerebellar hypoplasia (2/11), and white matter lesions of unclear etiology (2/11; Tables 1 and 2). There is one report of an absent septum pellucidum and focal polymicrogyria (Table 1).
Echocardiograms were performed in 10 individuals that identified septal defects including ventricular septal defects (VSD, 3/10), atrial septal defects (ASD, 3/10), and patent foramen ovale (PFO, 3/10; Tables 1 and 2). Several valve anomalies are also observed including bicuspid aortic valve (2/10), and one case each of pulmonic stenosis and tricuspid valve thickening (Tables 1 and 2). Aortic abnormalities are also present including right aortic arch, hypoplastic aortic arch, and ascending aorta dilation (Tables 1 and 2).
Gastrointestinal issues include constipation (4/15) and feeding difficulties requiring enteral tube placement (2/15; Tables 1 and 2). Omphalocele was present in 2/16 individuals who share the same heterozygous de novo p.(R740Q) missense variant (Table 1). Individual 1 was diagnosed with interstitial lung disease requiring tracheostomy and home ventilation. A lung biopsy showed pulmonary alveolar maldevelopment, pulmonary interstitial glycogenosis, and pulmonary arterial hypertensive remodeling. Individual 2 had severe restrictive lung disease due to profound scoliosis and a highly dysplastic thorax (Figure 1A–B) and died from respiratory failure at 22-years-old. Other musculoskeletal findings include instances of hip dysplasia, cleft lip/palate, arthrogryposis, and genu valgum (Table 1).
Figure 1. Examples of dysmorphology observed in individuals with MYH10 variants.
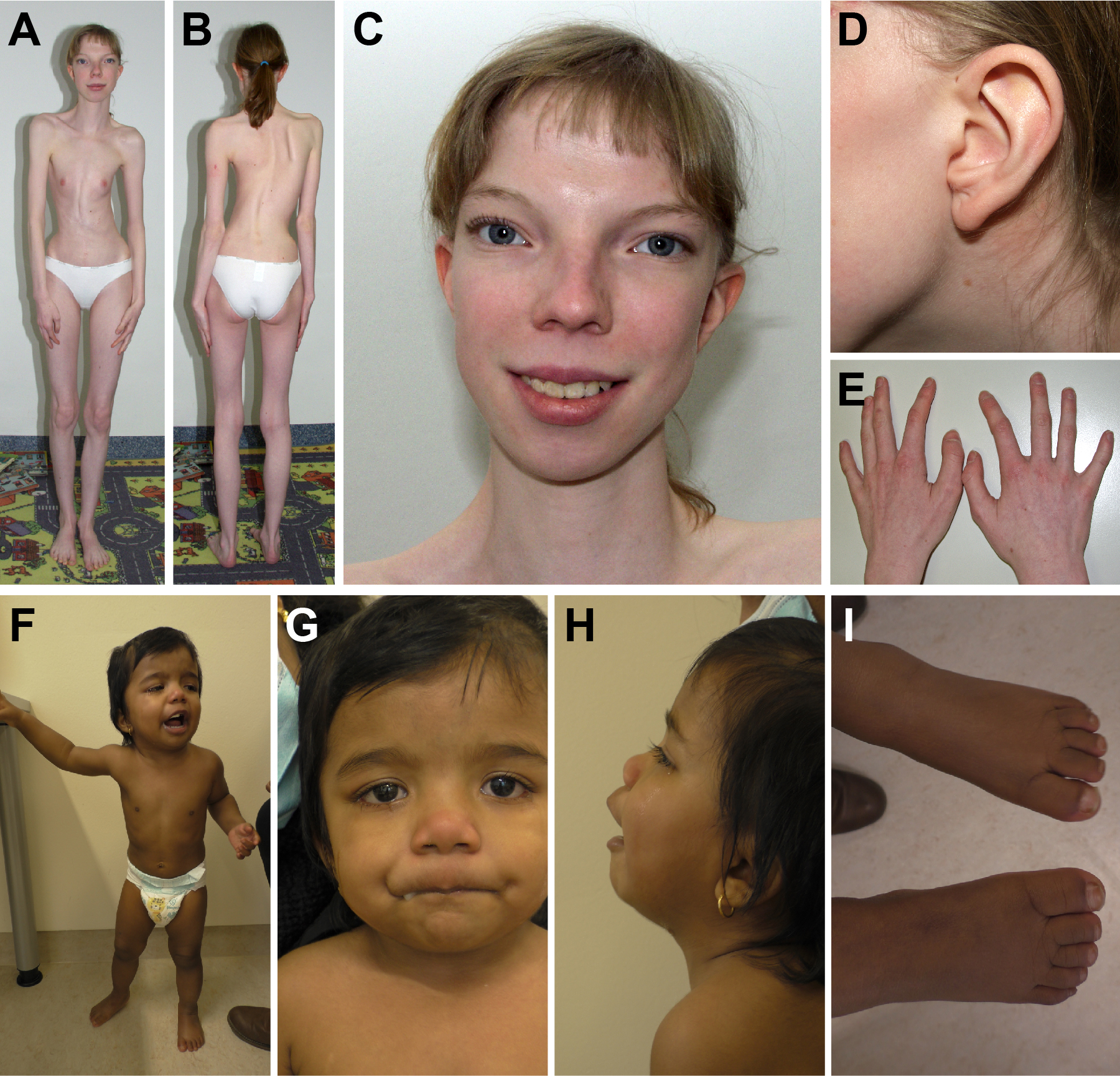
(A-E) Images obtained from individual 2 at age 19 years. Note the highly dysplastic thorax and scoliosis in (A,B), significant hypertelorism (C), preauricular pit (D), and 5th finger clinodactyly (E). (F-I) Images obtained from individual 14 at age 2 years demonstrating hypertelorism (G), broad nasal root (G), upturned nares (G), depressed nasal bridge (H), low set ears (H), broad toes (I), and 5th toe clinodactyly (I).
Ocular issues were identified including abnormalities of refraction (4/15), congenital ptosis (3/15), and one instance of unilateral chorioretinal lacunae (Tables 1 and 2). With respect to the genitourinary system, there are instances of renal cysts, pelviectasis, horseshoe kidney, vesicoureteral reflux, streak gonads, and cryptorchidism (Tables 1 and 2). Table S2 details the dysmorphology reported for each proband with hypertelorism being the most common finding (7/16, Figure 1, Table S2). Review of the previously published case reports and MYH10 candidate variants identified in large-scale cohorts demonstrates significant overlap between the phenotypes presented in our cohort (Figure S1).
Heterozygous variants identified in cohort
Fourteen unique heterozygous variants were identified with 11 missense variants, two frameshift variants, and one in-frame duplication (Table 1). Most cases are de novo (14/16), but we do observe inheritance from a mildly affected mother. All variants are rare and absent from the gnomAD database.25 Data from gnomAD show that MYH10 is intolerant of predicted loss-of-function (LOF) variants with a pLI score of 1, an observed/expected LOF variant ratio of 0.10 (CI 0.07–0.17), and a LOEUF score of 0.17.25 There is also strong constraint towards missense variants with an observed/expected ratio of 0.58 (CI 0.55–0.62; Z=5.01).25 There are two individuals with MYH10 variants and a second pathogenic variant (individuals 9 and 15; Table S1). Individual 10 presents with signs of mosaic Aicardi syndrome with corpus callosum agenesis, focal polymicrogyria, two small periventricular nodular heterotopias, and unilateral chorioretinal lacunae.
Figure 2A depicts the MYH10 protein topology including the N-terminal SH3 domain, the catalytic Mg2+ATPase motor domain, and a C-terminal myosin tail involved in protein-protein interactions. Variant mapping shows a broad distribution affecting all three functional domains (Figure 2A). A hot spot emerges in the motor domain including the critical R740 residue that is required for the Mg2+-ATPase activity (Figure 2A).9 In fact, the recurrent p.(R740Q) variant affects the same residue as the MYH10 p.(R740C) mouse knock-in model that develops similar phenotypes including omphalocele, brain anomalies, and cardiac defects as described above (Figure 2B).8 We also identify two missense variants affecting R852 [p.(R852W) and p.(R852Q)]. All affected residues show complete conservation across vertebrate species (Figure 2B). Alignment of the MYH10, MYH9, and MYH14 amino acid sequences show overlap between MYH10 variants and those defined as pathogenic in the MYH9- and MYH14-related disorders (Figure 2C). This includes the p.(R740Q) variant affecting the same amino acid as pathogenic variants in MYH9 [p.(R702C), p.(R702S), p.(R702H)] and MYH14 [p.(R767S)]. Similarly, p.(W37C) corresponds to the MYH9 p.(W33C) and p.(W33R) variants.
Figure 2. Mapping of MYH10 variants, structural modeling, and pathogenic predictions support deleterious effects of the identified missense variants.
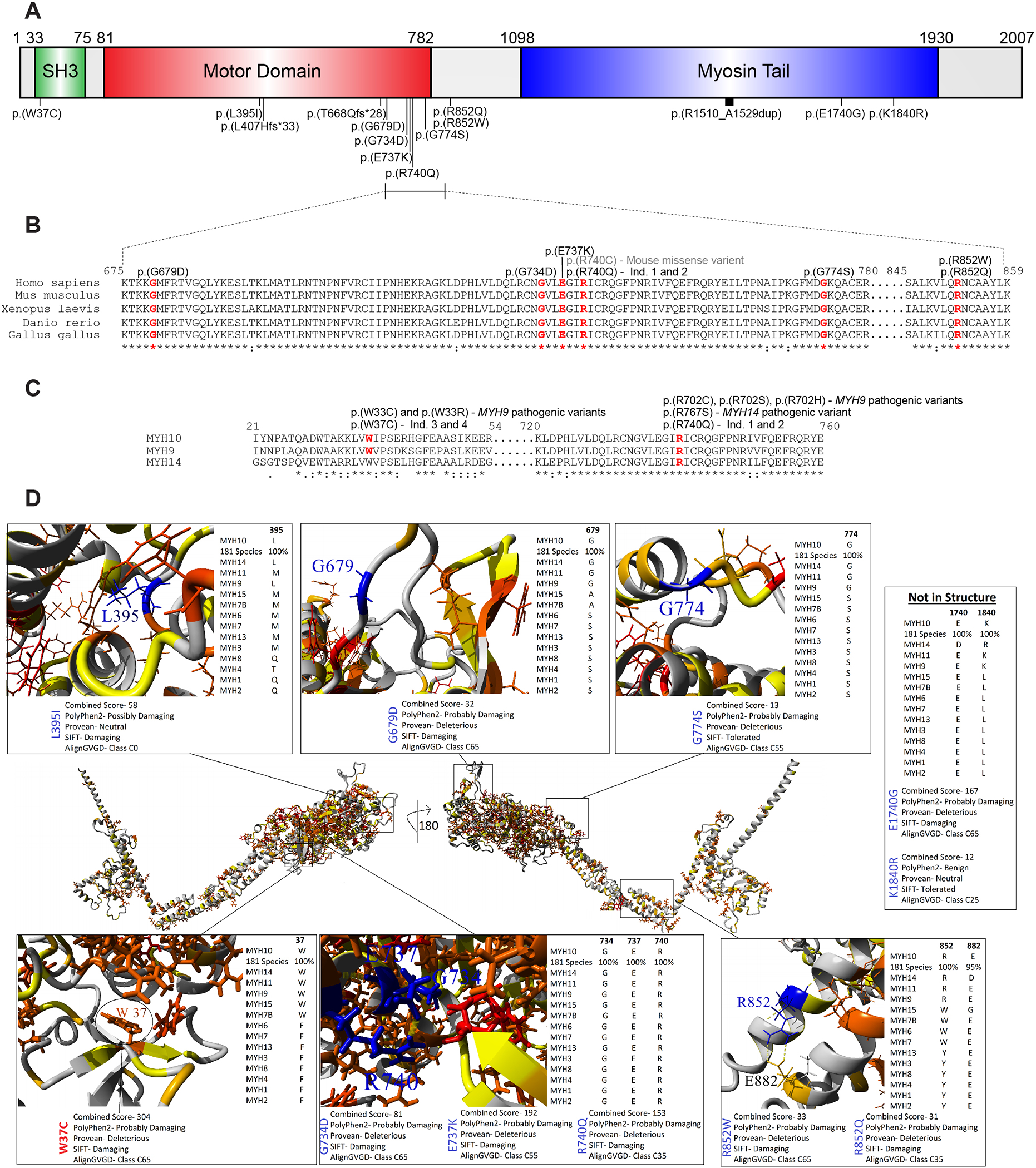
(A) Schematic of MYH10 protein structure demarcating the SH3, motor, and myosin tail domains. Variants are mapped to protein structure. All variants reported using transcript NM_001256012.3. (B) Alignment of vertebrate MYH10 protein sequences across species showing high conservation within the ‘hot spot’ region. Affected residues are delineated in red. Note the overlap between the p.(R740Q) variant identified in the cohort and the p.(R740C) missense variant described in the mouse knock-in model. (C) Alignment of human NMHCII proteins including MYH10, MYH9, and MYH14. Residues affected by missense variants are denoted in red. Note that the p.(W37C) and p.(R740Q) variants affect analogous residues that when affected are defined as pathogenic in the MYH9- and MYH14-related disorders. (D) In the middle is a model of MYH10 (numbering based on NM_001256012.3 / P35580–4) amino acids 1–1363 with a 180-degree rotation of the y-axis. The amino acid color is based on paralog conservation with 13 other human proteins, such that red is functionally conserved 100%, orange conserved functionally, and yellow some signs of conservation. On the outside are the zoom in view of each variant, often labeled in blue. For each variant is shown below the image the predictions of damage from various tools and to the right the conservation in other human paralog members in addition to the 181 vertebrate MYH10 sequences.
Variant impact analysis based on structural and evolutionary insights
A compiled analysis of MYH10 variants from this cohort, gnomAD, Geno2MP, ClinVar, and SFARI were assessed using an amino acid knowledgebase generated with structural insights, UniProt annotations, evolution of 181 vertebrate species, 14 human myosin paralog conservation, and multiple functional prediction scores. The VarSome variant data aggregation tool was used to perform additional functional predictions (Figure S2).26 Amino acid W37 is conserved as an aromatic amino acid throughout the myosin family, with 100% conservation within MYH10 evolution, damaging predictions with all functional tools, and is found within a hydrophobic core of the protein. L395 is hydrophobic in most myosin paralogs, is 100% conserved within MYH10 vertebrate evolution, resides within a hydrophobic core of the protein, and has damaging predictions with most tools (Figures 2D and S2). Variants G679 and G774 are conserved as a flexible (G, A or S) amino acid throughout the myosin family, with 100% conservation within MYH10 vertebrate evolution, damaging predictions with nearly all functional tools, and are within a flexible loop region of the protein. The amino acids G734, E737, and R740 are all conserved throughout the myosin family and MYH10 evolution, packed within the critical region of the motor domain, and have strong damaging/pathogenic predictions with most tools (Figure S2).
A crystal structure of the MYH10 motor domain in the rigor-like state reveals a complex interaction network in the environment of R740.27 This includes a critical salt bridge between R740 and E737 that forms the core of this interaction network.27 R740 and E737 also interact with D514 and N488, respectively, to further stabilize this structure.27 Hydrogen bonding between N733 and Q484 was also identified in this region, which is notable given the previously published p.(Q484E) missense variant (Figure S1).17,27 The two variants at R852 form a known salt bridge with E882, which are both highly conserved and strong damaging predictions with the majority of prediction tools. The E1740 and K1840 fall outside of known MYH10 structures but are conserved throughout MYH10 evolution and within the closely related myosin paralogs. The p.(E1740G) variant has strong pathogenic predictions while p.(K1840) has mostly tolerated/benign predictions (Figures 2 and S2).
One limitation of computational predictions involves the use of previously annotated variant data that may bias pathogenic predictions. Consequently, a new machine-learning model known as ‘Evolutionary model of variant effect’, or EVE, was developed that informs pathogenicity based on evolutionary information of sequence variation across species.28 This model outperforms prior computational approaches and data generated from high-throughput experiments.28 Similarly, the novel VARITY tool was also developed.29 Using both EVE and VARITY, pathogenic predictions are reached for variants p.(W37C), p.(G679D), p.(G734D), p.(E737K), p.(R740Q), p.(R852Q), p.(R852W), and p.(E1740G) (Figure S3). VARITY predicted pathogenicity for p.(G774S) while EVE yielded an uncertain result. Both tools predicted uncertain impact for p.(L395I) and benign variation for p.(K1840R) (Figure S3).
MYH10 is involved in primary ciliogenesis and ciliary length control
MYH10 has previously been implicated in the biogenesis of primary cilia, which are critical sensory organelles required to transduce developmental signals during embryogenesis. This includes Hedgehog signaling, a vital cell signaling pathway required for proper tissue patterning.30 There are two independent reports demonstrating a profound defect in primary cilia biogenesis resulting from MYH10 siRNA knockdown.31,32 However, conflicting data exists from high-throughput screens for modulators of ciliogenesis.33 Many features in the cohort including corpus callosum defects, cerebellar hypoplasia, cardiac septal defects, omphalocele, cleft palate, hypertelorism, absent septum pellucidum, skeletal dysplasia, and neuronal migration defects can be observed in mouse models and human diseases related to alterations in primary cilia and the Hedgehog signaling pathway.30,34
To investigate the role of MYH10 in primary cilia and HH signaling, we generated MYH10 knockout NIH/3T3 cells, which form primary cilia, express the necessary components to initiate a dynamic and sensitive HH response, and are widely used as a model for investigating primary cilia and HH signaling.35 We targeted the MYH10 locus using CAS9-mediated gene editing with two guide RNAs targeting exon 1 (Figure S4). Two independent knockout clones, KO-1 and KO-2, and one wildtype clone (WT-1) were chosen for analysis in comparison to the parental NIH/3T3 line. Sanger sequencing and western blot confirm successful MYH10 targeting, albeit with a residual faint band in knockout cells (Figures S4 and S5A). MYH9 levels were unaffected demonstrating the specificity of gene editing (Figure S5A). Immunofluorescent analysis shows co-localization of MYH10 and actin (phalloidin) in a striated pattern in the NIH/3T3 and WT-1 lines that was absent in line KO-1 (Figure S5B–M). We did not observe any gross changes to F-actin intensity or distribution (Figure S5B–M).
To investigate the role of MYH10 in primary ciliogenesis, cells were grown to confluence and then placed in low serum media for 24 hours to stimulate primary ciliogenesis that were labeled with acetylated tubulin and ARL13B. All cell lines tested were capable of generating primary cilia (Figure 3A–L). Of note, non-ciliary acetylated tubulin staining is significantly increased in knockout cells compared to controls (Figure 3A–D, S5N–U). MYH10 knockout lines showed a reduction in primary ciliogenesis when compared to NIH/3T3 and WT-1 cells, albeit with reduced magnitude compared to previously published siRNA data (Figure 3M).31,32
Figure 3. MYH10 knockout cells exhibit defects in primary ciliogenesis and ciliary length with evidence for a dominant-negative effect of patient-specific variants.
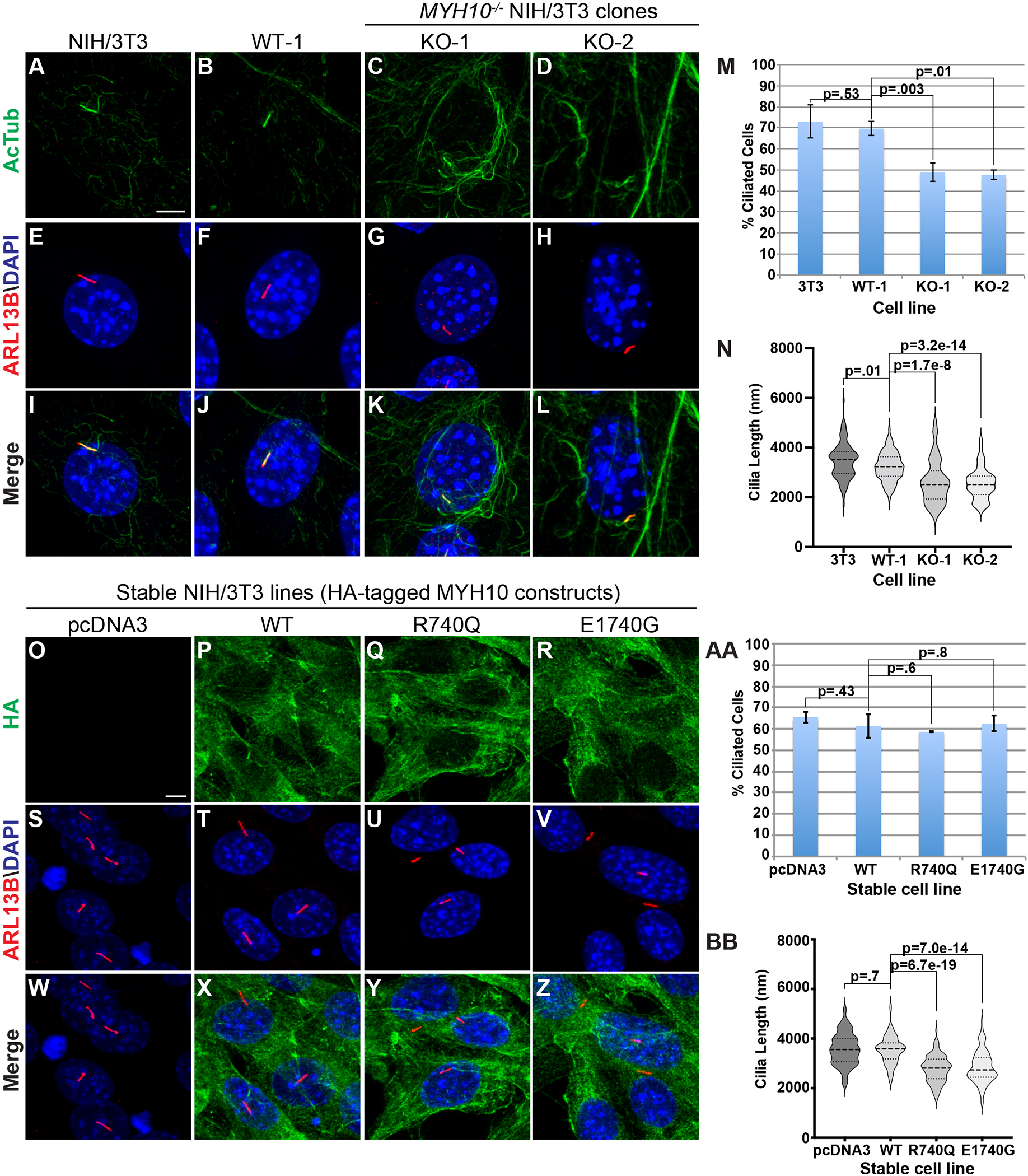
(A-L) Immunofluorescent analysis of primary cilia labeled with acetylated tubulin (green, A-D, I-L) and ARL13B (red, E-L) in parental NIH/3T3 (A, E, I), WT-1 (B, F, J), KO-1 (C, G, K), and KO-2 (D, H, L) lines. Nuclei labeled with DAPI (blue, E-L). Scale bar in A (5 um). (M) Analysis of primary ciliogenesis in control and knockout NIH/3T3 cell lines. The percent of cells with primary cilia are expressed between lines. (N) Violin plots of cilia length measurements between control and knockout NIH/3T3 cell lines. (U) Analysis of the percentage of cells with primary cilia in control and knockout RPE-1 cell lines. (O-Z) Immunofluorescent analysis of primary cilia in NIH/3T3 cells with stable overexpression of empty vector (pcDNA3; O, S, W), HA-tagged wildtype MYH10 (WT; P, T, X), HA-tagged p.(R740Q) (Q, U, Y), and HA-tagged p.(E1740G) (R, V, Z) and stained with antibodies against HA (green; O-R, W-Z) and ARL13B (red; S-Z). Nuclei are labeled with DAPI (blue; S, Z). Scale bar in O (5 um). (AA) Quantitation of the percent of ciliated cells between stable cell lines. (BB) Violin plots of primary cilia length measurements between stable NIH/3T3 cell lines. For ciliogenesis quantification, data was collected from 3 random fields of view (>50 cells/field of view) from maximum intensity projections of Z-stack images. Each experiment independently repeated at least N=3. For ciliary length quantitation, 100 cilia were measured from maximal intensity projected Z-stacks for each condition and each experiment was independently repeated at least N=3.
Previous data shows that pharmacologic or genetic manipulation of actin dynamics controls ciliary length and appropriate HH pathway responsiveness.33,36 Given the role of MYH10 to regulate actin dynamics, we measured primary cilia length in control and knockout cell lines. Violin plots demonstrate a small, albeit significant (p=0.01), decrease in ciliary length between parental NIH/3T3 cells (median 3469 nm) and line WT-1 (median 3232 nm; Figure 3N). Intriguingly, primary cilia length was significantly reduced in lines KO-1 and KO-2 when compared to control cells (KO-1 and KO-2 median length 2619 nm and 2536 nm, respectively (Figure 3N). This ~20% reduction in ciliary length is similar to the impact of other actin modulators.33,36
Impact of altered primary ciliogenesis on Hedgheog signaling
To investigate the functional consequences of altered primary cilia, we investigated HH pathway activity in NIH/3T3-derived cell lines by comparing HH target gene induction after treatment with conditioned media containing the bioactive N-terminal fragment of the Sonic Hedgehog (NSHH) ligand. NIH/3T3 and WT-1 cells demonstrate a robust induction of the HH target genes GLI1 and PTCH1 in response to NSHH-ligand stimulation (Figures S6A–B). By contrast, we observe a profoundly reduced response to NSHH ligand in the KO-1 and KO-2 NIH/3T3 lines (Figures S6A–B). We observe similar results via a HH-responsive luciferase reporter (Figure S6C). Moreover, siRNA knockdown of MYH10 also leads to a reduction in HH target gene induction and luciferase reporter activity (Figure S7). To determine the level at which MYH10 impacts HH signal transduction, we stimulated HH signaling downstream of HH ligand. Ligand binging to the HH receptor, Patched-1 (PTCH1), relieves inhibition of the key HH activator, Smoothened (SMO), leading to processing of GLI transcription to impact HH target gene expression (Figure S6C). Knockout cells failed to robustly respond to HH activation with a direct chemical SMO agonist (SAG) or with transfection of a constitutively active oncogenic SMO construct (SMOM2) compared to wildtype cells (Figure S6D–E). By contrast, expression of the GLI1 transcription factor produced robust reporter expression in both wildtype and knockout cells (Figure S6F). An intermediate phenotype with respect to primary ciliogenesis, shortened ciliary length, increased tubulin acetylation, and disrupted HH signaling is also observed in the MYH10 LOW-1 line (Figures S8 and S9). A second wildtype line also showed normal primary cilia and response to Hedgehog pathway activation (Figure S10). These data demonstrate that MYH10 mediates HH signaling at the level of, or downstream of SMO activation and upstream of GLI transcription factor processing, which are both dependent on primary cilia.30
We next attempted to rescue these deficits in primary ciliary length and HH pathway activity through re-expression of the human MYH10 cDNA. Unexpectedly, we observed several lower molecular weight bands after MYH10 expression in knockout cell lines (Figure S11A). This antibody recognizes the C-terminus of MYH10; thus, these fragments likely reflect N-terminal cleavage into the motor domain that may produce a dominant-negative effect on HH signaling. Consequently, MYH10 expression in line KO-1 was unable to rescue HH signaling defects (Figure S11B). Expression of a C-terminal HA-tagged MYH10 construct in control cells was found mostly in the full-length form, albeit with some cleavage (Figures S11C). Transient expression of several missense variants in NIH/3T3 cells lead to similar levels of expression (Figure S11C). Moreover, all variants interrogated demonstrate normal co-localization with actin based on phalloidin staining (Figure S12).
MYH10 variants exert a dominant-negative effect on primary ciliary length
To explore dominant-negative effects of MYH10 variation, we generated NIH/3T3 cells with stable overexpression of empty vector (pcDNA3), wildtype MYH10, or patient-specific variants. A C-terminal HA tag aided to distinguish endogenous and exogenous MYH10 protein. All lines demonstrated a similar distribution of MYH10 protein expression via HA staining (Figure S13A). Stable expression of wildtype MYH10 did not influence ciliary length; however, expression of p.(G734D), p.(E737K), p.(R740Q), and p.(E1740G) produced shortened primary cilia (Figure S13B). This was particularly pronounced in high-expressing cells (Figure S13C–T). We generated monoclonal stable cell lines and successfully identified high-expressing lines for p.(R740Q) and p.(E1740G) (Figure S13U). Primary cilia were significantly shortened in p.(R740Q) and p.(E1740G) lines compared to controls (Figure 3O–BB). Interestingly, there was no difference in primary ciliogenesis between these lines and no impact on HH signaling (Figure 3AA, data not shown). Overall, these data demonstrate a dominant-negative effect of patient-specific variants on primary ciliary length.
MYH10 loss impacts primary ciliogenesis in human RPE-1 cells.
We also derived MYH10−/− human retinal pigmented epithelial cells (RPE-1) via CAS9-mediated engineering (Figure S14). Intriguingly, we observe a significant reduction of primary ciliogenesis and ciliary length (RPE-1 median 6025 nm; WT-1 median 5947 nm; KO-1 median 4325 nm; KO-2 median 4401) resulting from MYH10 knockout (Figures S14E–R). RPE-1 cells did not produce a consistent response to HH pathway activation (data not shown). These data demonstrate that MYH10 plays a highly conserved role in primary ciliogenesis and ciliary length control.
Discussion
The data presented here defines a novel autosomal dominant cause of neurodevelopmental disorders and congenital anomalies caused by heterozygous MYH10 variants. The phenotypes presented are reflected in MYH10 mouse models. This includes CNS anomalies such as ventriculomegaly and cerebellar hypoplasia observed in our cohort and those in the literature in addition to cardiac septal defects.2 There is striking phenotypic overlap between both individuals with the p.(R740Q) variant and the mouse knock-in model of the p.(R740C) allele including omphalocele, CNS anomalies, and cardiac defects.8,10 Moreover, p.(R740C) mice also develop cleft palate and congenital diaphragmatic hernia, both of which are observed in our data and previously published cases.10,12,37 Combined MYH9/10 loss also disrupts ureteral development, which is interesting to note in light of the genitourinary features of the cohort.7 The pulmonary alveolar maldevelopment observed in individual 1 is reminiscent of the lung developmental defects resulting from MYH10 loss.6 MYH10-depdendent hippocampal circuit maturation, synaptic plasticity, and memory consolidation may help to explain the varying degrees of neurodevelopmental disorders observed.4,5 Additionally, the phenotypes and variable expressivity comports with existing case reports.14–22 Individual 10 presents with mosaic features of Aicardi syndrome and this proband’s p.(E1740G) variant exerts a dominant-negative effect on primary ciliary length. This may reflect a dual diagnosis; however, it is interesting to consider the role of altered actomyosin dynamics in the etiology of Acairdi syndrome, which has no clear genetic correlates.
All variants presented are rare and affect highly conserved amino acids. MYH10 exhibits significant constraint against missense and LOF variants.25 We observe clustering near and inclusive of the critical arginine residue (R740) in the protein’s motor domain. We also observe recurrent variation at R740 and R852 in unrelated families. MYH10 missense variants affect homologous residues that when mutated in the MYH9- and MYH14-related disorders are defined as pathogenic. Moreover, MYH10 structural modeling, deep conservation analysis, and functional predictions support a deleterious effect of all variants, with the exception of p.(K1840R). Structural analysis also identified variation at residues involved in key intermolecular interactions including R740, E737, R852, and Q484.27 These data strongly support the pathogenicity of variants related to the phenotypes in the cohort. Using ACMG/AMP variant classification guidelines,38 pathogenic or likely pathogenic predictions are reached for all variants except for p.(W37C) (Table S3); that this variant affects an analogous residue as a pathogenic variant in the MYH9-relaetd disorder strongly supports pathogenicity.
MYH10 variants are expected to have multifactorial and context dependent consequences. The p.(R740C) missense mouse model is instructive given that the complex phenotypes reflect both LOF and dominant-negative effects in a tissue-specific manner.10 In our cohort, missense variants may demonstrate reduced enzymatic activity compromising motor function while maintaining actin-scaffolding abilities as previously shown for variation at R740.9,10 We also show that several variants can produce a dominant-negative effect on ciliary length. Similarly, both LOF and dominant-negative mechanisms are implicated in the MYH9-related disorder.39
That the stable MYH10 patient-specific variant cell lines affect primary ciliary length, but do not impact HH signaling is interesting and is likely related to the wildtype MYH10 background of these cells. These data also suggest that MYH10 knockout may produce defects above and beyond the observed ciliary structural defects, such as trafficking of key HH pathway components through the primary cilia. Previous data show a defect in the trafficking of SMO and IFT88 resulting from MYH10 siRNA knockdown.31,32,33 Additional studies will investigate these key trafficking events in MYH10 knockout cells.
The pleiotropic effects of MYH10 may implicate alterations in a common set of signaling pathways. Our data demonstrate that MYH10 plays a role not only in primary ciliogenesis, but also in ciliary length control that is conserved between species and in cells of distinct developmental origin.31–33 This effect on ciliary length is consistent with other studies implicating altered actin dynamics in cilia length alterations.33,36 We confirm a functional ciliary defect based on a profound impairment of MYH10 knockout NIH/3T3 cells to respond to HH signals. Interestingly, many of the phenotypes observed in the MYH10 cohort overlap with anomalies observed in both humans and mice with alterations in primary cilia and HH signal transduction.30,34 It is not yet clear whether this disorder reflects a primary ciliopathy given that its effects are likely indirect via altered actomyosin dynamics since MYH10 has been excluded as a ciliary-localized protein.31,32 Moreover, HH signaling and altered primary cilia are likely one of several mechanisms underlying the phenotypes observed in the MYH10-related disorder.
The proteolytic cleavage observed after MYH10 expression in knockout cell lines is important to consider. That MYH10 protein expression in wildtype cells does not lead to significant cleavage implies a regulated event. One possibility involves changes in regulatory and essential light chain expression. Knockdown of the regulatory light chains MYL9/12A/12B in NIH/3T3 cells destabilizes MYH9 and MYH10.40 A reciprocal regulatory loop may exist and could impede MYH10 re-expression due to lower light chain expression. MYH10 also undergoes alternative splicing to generate four different products that demonstrate unique enzymatic activities and expression patterns.9 Future studies will explore the distinct roles of MYH10 isoforms in different cellular contexts.41
Overall, these data provide evidence for a unifying autosomal dominant disorder caused by heterozygous variants in the MYH10 gene. Future directions will include direct analysis of patient samples, CAS9-mediated variant knock-in cellular models, and targeted mouse knock-in models to delineate further the biochemical, cellular, and phenotypic impact of the identified variants that will likely reveal both overlapping and distinct mechanisms of action in a tissue-specific manner.
Supplementary Material
Acknowledgements
We would like to thank the patients and families for contributing to this study and to the providers who help to care for these families. We also thank Dr. Benjamin L. Allen (University of Michigan) for providing several resources for this study. This research was supported in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health (J.P.). Sequencing and analysis of individual #8 was provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 and in part by National Human Genome Research Institute grant R01 HG009141. W.B.D. is supported by R01 NS050375 and W.B.D. and E.H.S. are supported by R01 NS058721. Research reported in this publication was also supported by the National Institute Of Mental Health of the National Institutes of Health under Award Number U01MH119689. This work was also supported by a grant from SFARI (704413; E.B.R) and from the Stanley Center for Psychiatric Research at the Broad Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Ethics Declaration
All procedures within this study were performed in accordance with the Helsinki Declaration. The study was reviewed by the Boston Children’s Hospital Institutional Review Board protocol IRB-P00031211 (main IRB) in additional to approval by the ethics committees and the institutional review boards (IRB) from each contributing center. Written informed consent was obtained from all individuals undergoing exome sequencing and additional written consents were obtained for those contributing photographs.
Data Availability
All compiled analysis and data with respect to MYH10 evolutionary and variant modeling can be found at https://doi.org/10.6084/m9.figshare.16654690.v1
References
- 1.Newell-Litwa KA, Horwitz R, Lamers ML. Non-muscle myosin II in disease: mechanisms and therapeutic opportunities. Dis Model Mech. 2015;8(12):1495–1515. doi: 10.1242/dmm.022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tullio AN, Accili D, Ferrans VJ, et al. Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc Natl Acad Sci USA. 1997;94(23):12407–12412. doi: 10.1073/pnas.94.23.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tullio AN, Bridgman PC, Tresser NJ, et al. Structural abnormalities develop in the brain after ablation of the gene encoding nonmuscle myosin II-B heavy chain. J Comp Neurol. 2001;433(1):62–74. doi: 10.1002/cne.1125. [DOI] [PubMed] [Google Scholar]
- 4.Ozkan ED, Aceti M, Creson TK, et al. Input-specific regulation of hippocampal circuit maturation by non-muscle myosin IIB. Journal of neurochemistry. 2015;134(3):429–444. doi: 10.1111/jnc.13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gavin CF, Rubio MD, Young E, Miller C, Rumbaugh G. Myosin II motor activity in the lateral amygdala is required for fear memory consolidation. Learn Mem. 2012;19(1):9–14. doi: 10.1101/lm.024042.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim H-T, Yin W, Jin Y-J, et al. Myh10 deficiency leads to defective extracellular matrix remodeling and pulmonary disease. Nature Communications. 2018;9(1):4600. doi: 10.1038/s41467-018-06833-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haque F, Kaku Y, Fujimura S, Ohmori T, Adelstein RS, Nishinakamura R. Non-muscle myosin II deletion in the developing kidney causes ureter-bladder misconnection and apical extrusion of the nephric duct lineage epithelia. Dev Biol. 2017;427(1):121–130. doi: 10.1016/j.ydbio.2017.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma X, Kawamoto S, Hara Y, Adelstein RS. A point mutation in the motor domain of nonmuscle myosin II-B impairs migration of distinct groups of neurons. Mol Biol Cell. 2004;15(6):2568–2579. doi: 10.1091/mbc.e03-11-0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim K-Y, Kovács M, Kawamoto S, Sellers JR, Adelstein RS. Disease-associated mutations and alternative splicing alter the enzymatic and motile activity of nonmuscle myosins II-B and II-C. J Biol Chem. 2005;280(24):22769–22775. doi: 10.1074/jbc.M503488200. [DOI] [PubMed] [Google Scholar]
- 10.Ma X, Adelstein RS. A point mutation in Myh10 causes major defects in heart development and body wall closure. Circ Cardiovasc Genet. 2014;7(3):257–265. doi: 10.1161/CIRCGENETICS.113.000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petrovski S, Aggarwal V, Giordano JL, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393(10173):758–767. doi: 10.1016/S0140-6736(18)32042-7. [DOI] [PubMed] [Google Scholar]
- 12.Tuzovic L, Yu L, Zeng W, et al. A human de novo mutation in MYH10phenocopies the loss of function mutation in mice. Rare Diseases. 2014;1(1):e26144. doi: 10.1182/blood-2004-06-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamdan FF, Srour M, Capo-Chichi J-M, et al. De novo mutations in moderate or severe intellectual disability. PLOS Genetics. 2014;10(10):e1004772. doi: 10.1371/journal.pgen.1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iossifov I, O’Roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kosmicki JA, Samocha KE, Howrigan DP, et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat Genet. 2017;49(4):504–510. doi: 10.1038/ng.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Satterstrom FK, Kosmicki JA, Wang J, et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 2020;180(3):568–584.e23. doi: 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Cai T, Jiang Y, et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Molecular psychiatry. 2016;21(2):290–297. doi: 10.1038/mp.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aspromonte MC, Bellini M, Gasparini A, et al. Characterization of intellectual disability and autism comorbidity through gene panel sequencing. Hum Mutat. 2019;40(9):1346–1363. doi: 10.1002/humu.23822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carraro M, Monzon AM, Chiricosta L, et al. Assessment of patient clinical descriptions and pathogenic variants from gene panel sequences in the CAGI-5 intellectual disability challenge. Hum Mutat. 2019;40(9):1330–1345. doi: 10.1002/humu.23823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin SC, Homsy J, Zaidi S, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49(11):1593–1601. doi: 10.1038/ng.3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Roak BJ, Vives L, Girirajan S, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiao L, Wynn J, Yu L, et al. Likely damaging de novo variants in congenital diaphragmatic hernia patients are associated with worse clinical outcomes. Genet Med. 2020;22(12):2020–2028. doi: 10.1038/s41436-020-0908-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holtz AM, Peterson KA, Nishi Y, et al. Essential role for ligand-dependent feedback antagonism of vertebrate hedgehog signaling by PTCH1, PTCH2 and HHIP1 during neural patterning. Development. 2013;140(16):3423–3434. doi: 10.1242/dev.095083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35(11):1978–1980. doi: 10.1093/bioinformatics/bty897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Münnich S, Pathan-Chhatbar S, Manstein DJ. Crystal structure of the rigor-like human non-muscle myosin-2 motor domain. FEBS Lett. 2014;588(24):4754–4760. doi: 10.1016/j.febslet.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Frazer J, Notin P, Dias M, et al. Disease variant prediction with deep generative models of evolutionary data. Nature. 2021;599(7883):91–95. doi: 10.1038/s41586-021-04043-8. [DOI] [PubMed] [Google Scholar]
- 29.Wu Y, Li R, Sun S, Weile J, Roth FP. Improved pathogenicity prediction for rare human missense variants. Am J Hum Genet. 2021;108(10):1891–1906. doi: 10.1016/j.ajhg.2021.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kopinke D, Norris AM, Mukhopadhyay S. Developmental and regenerative paradigms of cilia regulated hedgehog signaling. Semin Cell Dev Biol. 2021;110:89–103. doi: 10.1016/j.semcdb.2020.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rao Y, Hao R, Wang B, Yao T-P. A Mec17-Myosin II Effector Axis Coordinates Microtubule Acetylation and Actin Dynamics to Control Primary Cilium Biogenesis. PLoS ONE. 2014;9(12):e114087. doi: 10.1371/journal.pone.0114087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong H, Kim J, Kim J. Myosin heavy chain 10 (MYH10) is required for centriole migration during the biogenesis of primary cilia. Biochemical and biophysical research communications. 2015;461(1):180–185. doi: 10.1016/j.bbrc.2015.04.028. [DOI] [PubMed] [Google Scholar]
- 33.Kim J, Lee JE, Heynen-Genel S, et al. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464(7291):1048–1051. doi: 10.1038/nature08895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elliott KH, Brugmann SA. Sending mixed signals: Cilia-dependent signaling during development and disease. Dev Biol. 2019;447(1):28–41. doi: 10.1016/j.ydbio.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breslow DK, Hoogendoorn S, Kopp AR, et al. A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat Genet. 2018;50(3):460–471. doi: 10.1038/s41588-018-0054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drummond ML, Li M, Tarapore E, et al. Actin polymerization controls cilia-mediated signaling. J Cell Biol. 2018;217(9):3255–3266. doi: 10.1083/jcb.201703196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiao L, Wynn J, Yu L, et al. Likely damaging de novo variants in congenital diaphragmatic hernia patients are associated with worse clinical outcomes. Genet Med. 2020;22(12):2020–2028. doi: 10.1038/s41436-020-0908-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. In: Vol 17. 2015:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pecci A, Canobbio I, Balduini A, et al. Pathogenetic mechanisms of hematological abnormalities of patients with MYH9 mutations. Human Molecular Genetics. 2005;14(21):3169–3178. doi: 10.1093/hmg/ddi344. [DOI] [PubMed] [Google Scholar]
- 40.Park I, Han C, Jin S, et al. Myosin regulatory light chains are required to maintain the stability of myosin II and cellular integrity. Biochem J. 2011;434(1):171–180. doi: 10.1042/BJ20101473. [DOI] [PubMed] [Google Scholar]
- 41.Ma X, Kawamoto S, Uribe J, Adelstein RS. Function of the neuron-specific alternatively spliced isoforms of nonmuscle myosin II-B during mouse brain development. Mol Biol Cell. 2006;17(5):2138–2149. doi: 10.1091/mbc.e05-10-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All compiled analysis and data with respect to MYH10 evolutionary and variant modeling can be found at https://doi.org/10.6084/m9.figshare.16654690.v1