

Abstract
Liver is one of the most common sites for metastases, which can occur on account of primary tumors from multiple sites of origin. Identifying the primary site of origin (PSO) of a metastasis can help in guiding therapeutic options for liver metastases. In this pilot study, we hypothesized that computer extracted handcrafted (HC) histomorphometric features can be utilized to identify the PSO of liver metastases. Cellular features, including tumor nuclei morphological and graph features as well as cytoplasm texture features, were extracted by computer algorithms from 175 slides (114 patients). The study comprised three experiments: (1) comparing and (2) fusing a machine learning (ML) model trained with HC pathomic features and deep learning (DL)‐based classifiers to predict site of origin; (3) identifying the section of the primary tumor from which metastases were derived. For experiment 1, we divided the cohort into training sets composed of primary and matched liver metastases [60 patients, 121 whole slide images (WSIs)], and a hold‐out validation set (54 patients, 54 WSIs) composed solely of liver metastases of known site of origin. Using the extracted HC features of the training set, a combination of supervised machine classifiers and unsupervised clustering was applied to identify the PSO. A random forest classifier achieved areas under the curve (AUCs) of 0.83, 0.64, 0.82, and 0.64 in classifying the metastatic tumor from colon, esophagus, breast, and pancreas on the validation set. The top features related to nuclear and peri‐nuclear shape and textural attributes. We also trained a DL network to serve as a direct comparison to our method. The DL model achieved AUCs for colon: 0.94, esophagus: 0.66, breast: 0.79, and pancreas: 0.67 in identifying PSO. A decision fusion‐based strategy was deployed to fuse the trained ML and DL classifiers and achieved slightly better results than ML or DL classifier alone (colon: 0.93, esophagus: 0.68, breast: 0.81, and pancreas: 0.69). For the third experiment, WSI‐level attention maps were also generated using a trained DL network to generate a composite feature similarity heat map between paired primaries and their associated metastases. Our experiments revealed that epithelium‐rich and moderately differentiated tumor regions of primary tumors were quantitatively similar to paired metastatic tumors. Our findings suggest that a combination of HC and DL features could potentially help identify the PSO for liver metastases while at the same time also potentially identify the spatial sites of origin for the metastases within primary tumors.
Keywords: digital pathology, machine learning, deep learning, quantitative histomorphometric image analysis
Introduction
A common diagnostic dilemma among surgical pathologists is to identify the primary site of origin (PSO) for a newly diagnosed metastatic adenocarcinoma [1]. Adenocarcinomas are the most common cancer subtype which metastasize to the liver [2, 3]. Since many currently successful therapies are tumor site specific, it is paramount to identify the PSO for metastatic disease [4]. Pathologists generally rely on morphological and immunohistochemical assessments to define or confirm a primary source for the metastasis [5, 6]. Based on pathology assessment alone, the site of origin for up to 20% of metastatic tumors to the liver, the majority being adenocarcinomas, cannot be identified [2]. With an ever‐increasing number of high‐quality antibodies for immunohistochemistry (IHC) as well as the increasing availability of molecular technologies [7, 8, 9], a majority of the metastatic tumors' site of origin can be identified [10] though at the expense of ever‐increasing costs and turnaround times which prohibit the adoption of these new technologies in low‐cost scenarios [5, 10]. These technologies are also tissue destructive, often leading to tissue block depletion before critical assays, such as assessment of biomarkers predictive of response to targeted therapies, can be performed. Thus, there is an unmet clinical need for tissue nondestructive and efficient approaches to identifying the PSO associated with a metastatic tumor.
With the increased availability of digitized whole slide images (WSIs) and computational power, computerized assessment of WSI including deep learning (DL) and handcrafted (HC) approaches has been widely used for disease characterization. Multiple studies have demonstrated the utility of HC features in defining molecular phenotype and correlating with clinical outcomes across different cancer types [11, 12, 13, 14] using histomorphometric features from both cancerous as well as adjacent noncancerous stromal and intratumoral inflammatory components [15, 16]. A DL‐based assistive tool developed by Lu et al [17] automatically identified the site of origin of tumors using only H&E‐stained histology slides. Although various algorithms have been deployed to improve the interpretability, the DL‐based approach's black‐box nature represents a challenge in explaining the rationale behind the classifier's decision process which also impedes quality control assessments, including identification of individual result inaccuracies – important for potential clinical implementation. Thus, an approach based on the decision fusion of HC features and DL models was developed to identify PSO.
In this study we sought to demonstrate the ability of histomorphologic features, specifically in so far as they relate to cellular features of nuclei shape and texture, spatial graph‐based feature [11, 18, 19], as well as cytoplasm texture from H&E‐stained WSIs, (1) to identify the site of origin of liver metastases; and (2) to explore the potential ‘subclone’ regions of primary tumors that show morphological similarities to their corresponding metastases, suggesting a link to tumor heterogeneity.
Methods
Dataset description
Primary adenocarcinomas along with their corresponding liver metastasis, and liver metastases without corresponding primary tumors from four sites of origin, breast (ductal type only), colon (standard /NOS type only), esophagus, and pancreas, were obtained from University Hospitals Cleveland Medical Center and digitized using Ventana iScan HT scanner at ×40 (0.25 μm per pixel) under an Institutional Review Board‐approved protocol (02‐13‐42C). We collected 180 WSIs from 118 patients; 5 WSIs from 4 patients were excluded due to impaired image quality or misclassification as liver metastasis, unpaired primary, leaving a final dataset of 175 WSIs from 114 patients with primary cancer (n = 60) or liver metastases (n = 115) (see supplementary material, Figure S1 and Table S1). These comprised 121 WSIs from 60 patients with matched primary and metastatic cancers and 54 WSIs from liver metastasis from patients whose primary origin was known. Tumor regions were manually identified by BM and JW. HistoQC [20], a digital image quality assessment tool, was then applied to the WSIs for artifact and pen‐marker detection. Patches with artifacts including but not limited to pen markings, blurriness, and bubbles were excluded from the analysis.
Methodological design
The final dataset consisted of 175 H&E‐stained WSIs (114 patients) with primary cancers, matched or unpaired liver metastases from the colon (59 images), esophagus (43 images), breast (36 images), and pancreas (37 images). A combination of supervised machine learning (ML)‐based classification and unsupervised clustering was then applied to identify the tumors' site of origination using the fore‐mentioned histomorphometric features from the acquired WSIs. A DL‐based model was trained to serve as a direct comparison to the HC‐based approach. We also fused the decisions generated by the trained ML and DL models together to evaluate whether this combined model could better identify the site of origin of liver metastases. Histomorphometric feature analysis of metastatic cancers was then compared with their initial primary to define the likely ‘clone of origin’ of the metastasis on a small number of primary‐metastasis pairs. An overview of the methodological design is illustrated in Figure 1.
Figure 1.
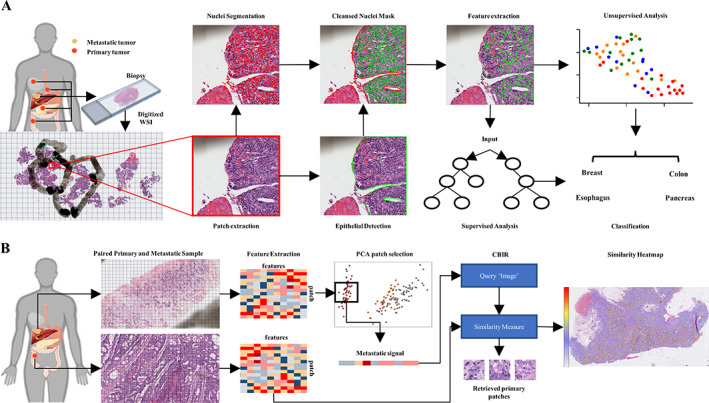
Illustration of workflow. (A) Biopsies were prepared and digitized into WSI at UHCMC. WSIs were dissected into 2,048 × 2,048 pixel2 patches based on tumor masks annotated by an experienced pathologist. Within the annotated tumor region, nuclei were segmented by the StarDist algorithm. The epithelial region was segmented by a pre‐established model. Cytoplasm masks were generated by dilating the nuclei masks. TILs and nuclei within the stromal region were removed from the nuclei mask. Nuclear and cytoplasm histomorphometric features were extracted. The features were then evaluated to identify the site of origin of each tumor via supervised classification and other unsupervised approaches. (B) Workflow for experiment 2. Four paired metastases and primary tumors were randomly selected (one pair for each organ site). To increase the resolution of the resulting heat map, WSIs were dissected into 256 × 256 pixel2 patches. Histomorphometric features were extracted for both primary and metastatic patches. Principal component ananlysis (PCA) was then applied to metastatic patches to select the most representative patches. Next, the most representative patches were averaged to acquire the metastatic signal, which was utilized as the Query Image for the CBIR classifier. The CBIR then calculated the Euclidean distance between each primary patch and the metastatic signal and retrieved patches with the lowest distance. The similarity was then calculated as the reciprocal of the distance to generate the final similarity heat map.
Image analysis and nuclear segmentation
StarDist, a DL‐based cell segmentation approach was deployed to segment nuclei from the individual image patches [21, 22]. Epithelial and stromal components within each patch were segmented by a pre‐trained DL model [12]. As the goal was also to assess histomorphometric image features associated with the cytoplasm, masks of tissue regions corresponding to cytoplasm were generated by first dilating the boundary associated with the nuclei masks, and then subtracting the nuclei masks from the dilated masks. Finally, tumor‐infiltrating lymphocytes (TILs) were detected using a pre‐trained ML model [15]. The TIL classifier, which was trained using seven features derived from texture, shape, and color attributes of the segmented nuclei, was deployed to classify the individual nuclei as TILs or non‐TILs [15]. Nuclei identified as TILs or as occurring within the stroma region were excluded from the final nuclei mask. A kernel density map was generated using the two‐dimensional (2D)‐PCA embedding of the extracted features. The top 20 percentile regions with highest estimated kernel density were chosen as the most representative samples for a given WSI [23]; these tiles were employed for the subsequent pathomic analysis (supplementary material, Figure S2).
Feature extraction
A total of 13 512 features from eight feature families were extracted from the segmented cancer nuclei. These eight feature families are: (1) nuclei shape and morphology; (2) nuclei Haralick texture; (3) cytoplasm Haralick texture; (4) cell cluster graphs (CCGs); (5) feature‐driven local cell graph (FeDeG); (6) local cellular diversity (CCM); (7) fractal dimension (FD); and (8) cell run length. Shape features included major/minor axis ratio, area, as well as invariant moment, Fourier descriptor, and length/width ratios to reflect the morphological differences among tumor nuclei originating from different organ sites. A total of 312 Haralick‐based texture features from within the nuclei and cytoplasm were captured (supplementary material, Table S3). CCGs were also constructed to capture basic shape features of tumor nuclei within the local neighborhood [13]. FeDeG [19] was calculated to investigate the interaction between different cell graphs. Furthermore, CCM [11] features were extracted to reflect the morphological differences among tumor nuclei originating within local CCGs from different organ sites. Full feature descriptions are included within the supplementary material, feature description section. Pearson correlation coefficient was calculated for features in the training sets (T1 and T2) and used to remove highly correlated features (correlation coefficient >0.95) in T1 and T2. These identified correlated features were subsequently removed from V1 and V2. To normalize the feature values, extracted features were min–max normalized separately in the training sets (T1 and T2) and hold‐out validation sets (V1 and V2).
Experimental design
Experiment 1: evaluating HC approaches for predicting site of origin
For experiment 1, unpaired liver metastases were selected as part of hold‐out validation set, V1 (54 WSIs: colon – 16; esophagus – 11; breast – 12; pancreas – 15; see supplementary material, Figure S1), while the remaining paired primary and liver metastases were used as the training set, T1. To prevent information leakage, images obtained from the same patient were not present within the training and validation set simultaneously. Three‐fold cross‐validation (CV) was used to train the classification models, . For each iteration, three sub‐training sets, , that consisted of two‐thirds of the original dataset were randomly selected from T1. The remaining cases were included in the corresponding validation set . Next, two feature selection methods were utilized to identify the top 10 most discriminative features out of the . The selected features were used to train three classification models, , for each iteration to identify the tumor's site of origin. Area under the receiver operating characteristic curve (AUC) on was calculated for each fold and iteration. The final AUC was reported as the average of AUC across each fold and iteration. Finally, the model with the highest averaged AUC, , was selected and evaluated on the hold‐out validation set.
Uniform Manifold Approximation and Projection (UMAP) [24] was applied to reduce the dimensionality of the selected features. The resulting low‐dimensional embedding of image patches was then subsequently clustered via hierarchical clustering. The violin plots of the top features identified via feature selection from each feature family were also plotted. Paired Student's t tests were invoked to identify whether features were distributed differently among the four organ sites. To further evaluate the features, a Content‐Based Image Retrieval (CBIR) model was evaluated on the hold‐out validation set, V1. Each WSI within V1 was represented by the selected top 10 features (supplementary material, Table S2). The algorithm loop through V1 with each time one WSI was selected as the query image. After that, the Euclidean distances between the query image and the remaining WSI within the V1 were calculated. Subsequently, the class of each query image was determined by the class of the closest WSI. The precision–recall (PR) curve and the area under PR curve (AUPRC) were also reported.
Experiment 2: integrating HC and DL approaches
A modified version of DenseNet [25] model, , was trained to classify the site of origin of tumor metastasis from colon, breast, esophagus, or pancreas. The customized DenseNet consisted of four dense blocks, where each dense block had two layers and a growth rate of k = 32. The model has 415 554 learnable parameters and accepts 256 × 256 pixel [2] image patches with a corresponding site of origin as input. The structure of the network is plotted in supplementary material, Figure S3. The training and validation process was performed using the same training and hold‐out validation set (T1 and V1) used by ML classifiers (supplementary material, Figure S1). Thirty percent of the training sets were randomly selected and used as test set to select the model with the best performance. The training patches were augmented by rotating each patch by 180°, randomly flipping the image horizontally or vertically, and assigning color or gray scale to the patches [26]. The classifier outputs from the individual patches from the same patient were averaged to obtain the per‐patient aggregated output. The network was trained on a RTX 2080Ti GPU for 100 epochs with CUDA 10.0 and cuDNN 7.6.5 optimized by Adam optimizer built into Pytorch and a fixed batch size of 128. The cross‐entropy error function was used. The model that achieved the lowest loss on the test set was selected as the trained model.
To harness the power of both DL features and histomorphologic features, a fused model, , was created by combining the decision made by and . After the probability of each WSI was generated by and , the model outputs were fused together into a single vector with each column representing the probability of current WSI originated from each organ site (colon, esophagus, breast, and pancreas). The final classification was given by identifying the highest probability within the final fused probability vector (supplementary material, Figure S4). The AUC of was reported and compared with the AUCs of and respectively.
Experiment 3 – identifying site of metastasis in the primary tumor
WSIs were partitioned into image patches of size of 256 × 256 pixel [2] at a magnification level of ×40. For this experiment, only nuclei texture and morphological features (a total of 376 features) were extracted. The total number of features was then reduced to 10 via Wilcoxon ranked sum test (WRST; 5 features corresponding to nuclear shape and 5 to nuclear texture, detailed information is listed in supplementary material, Table S2). The Euclidean distance between patch‐wise features from the metastases and their corresponding primary tumor regions for four selected cases (one tumor pair from each organ site) was calculated. The similarity was then calculated as the reciprocal of calculated distances for each patch. Finally, similarity heat maps were generated by stitching the patches together with the associated similarity distance and overlaying these measurements on the corresponding WSIs. To qualitatively evaluate the similarity heat map, regions of interests (ROIs), including focal regions of moderately or poorly differentiated carcinoma (colon) and epithelial or stromal rich regions, were visually identified by an experienced pathologist. We then calculate the averaged similarity within each identified ROI to reveal regional similarity of these epithelial components with the corresponding metastatic tumor.
In order to generate visual attention maps for the DL algorithm, new training and hold‐out validation sets, T2 and V2, were re‐generated with a patient train–test ratio of 75:38. This was done since the original training set V1 was comprised solely of metastasis cases. WSIs were randomly selected to enter the hold‐out validation set regardless of their primary or metastasis status (60 WSIs: colon – 14; esophagus – 12; breast – 15; pancreas – 19; primary cancers – 17; metastases – 43. See supplementary material, Figure S1), while the remaining cases were used for training. To generate the attention map, a DL model was trained to classify WSIs into primary or metastatic categories. A Guided Grad‐CAM [27] was deployed to generate the attention map revealing the metastatic activation map on four selected cases. The WSI‐level attention maps were generated by stitching attention maps at the patch level.
Experimental results
Experiment 1 – evaluating HC approaches for predicting site of origin
In general, the model trained using a combination of WRST and the random forest classifier had the best performance out of all combinations of feature ranking methods and classifiers using top 10 features selected from all feature families. Across different combinations of feature ranking methods and classifiers, the highest AUCs for colon: 0.80, esophagus: 0.74, breast: 0.67, and pancreas: 0.71 were achieved using a combination of WRST and the random forest classifier over the 100 iteration of CV on T1. Using WRST as the feature selection method, the performance of the models with the highest AUCs during the CV training phase was reported on V1 (Table 1). achieved high AUC in classifying tumors from the colon (0.83) and breast (0.82). The had an intermediate performance for esophagus (0.64) and pancreas (0.64).
Table 1.
Averaged AUC of ML classifiers trained using top HC features selected from all features over the training set.
| Training set | Validation set | |||||
|---|---|---|---|---|---|---|
| AUC | LDA | QDA | RF | ML | DL | ML + DL |
| Colon | 0.78 | 0.77 | 0.80 | 0.83 | 0.94 | 0.93 |
| Esophagus | 0.61 | 0.64 | 0.74 | 0.64 | 0.68 | 0.71 |
| Breast | 0.40 | 0.60 | 0.67 | 0.82 | 0.79 | 0.81 |
| Pancreas | 0.61 | 0.61 | 0.71 | 0.64 | 0.69 | 0.72 |
Based on the mean AUC over CV, models trained using RF + WRST were selected and evaluated on the hold‐out validation set. The result of the DL model as well as the fused model is also reported. The machine learning model with the highest performance on the training set is highlighted in bold.
LDA, linear discriminant analysis; QDA, quadratic discriminant analysis; RF, random forest.
To further demonstrate the utility of each HC feature family, the classifiers were trained using the top 10 features selected from each individual feature family and the AUCs evaluated on the V1 are reported in Table 2. CCM features demonstrated the highest classification value among all eight feature families, followed by Haralick texture features extracted from the nuclei. Across all feature families, the highest performance of the classifier was achieved on the colon tumors with an AUC of 0.85, using features corresponding to both basic nuclei shape and cytoplasm texture. The classifiers trained using FD features achieved the highest AUCs in classifying the esophageal and breast tumors (0.62 and 0.77, respectively). The highest AUC (0.78) in classifying pancreatic metastases was achieved using CCM features.
Table 2.
AUC of ML classifiers trained using top HC features in different feature families on validation set.
| WRST | Colon | Esophagus | Breast | Pancreas |
|---|---|---|---|---|
| Nuclei morphology | 0.85 | 0.51 | 0.65 | 0.55 |
| Cell run length | 0.59 | 0.59 | 0.53 | 0.71 |
| Cytoplasm texture | 0.85 | 0.57 | 0.70 | 0.52 |
| Fractal dimension | 0.76 | 0.62 | 0.77 | 0.51 |
| FeDeG | 0.75 | 0.55 | 0.45 | 0.62 |
| Nuclei texture | 0.79 | 0.59 | 0.76 | 0.62 |
| Local cell diversity | 0.77 | 0.55 | 0.71 | 0.78 |
The feature families that achieved the highest AUC on each organ site are highlighted in bold.
Figure 2A shows the 2D UMAP embedding of the validation set using the top 10 most selected features over 100 iterations of CV across all feature families. Tumors originating from breast, colon, esophagus, and pancreas were colored by green, red, blue, and orange, respectively. Subclusters from each organ site were visually identified and highlighted using bounding boxes with the corresponding color code. These subclusters could be readily identified for tumors from all organs within the 2D embedding. Since the UMAP embedding clearly illustrates the presence of two clusters of breast cancers (highlighted with a green bounding box in Figure 2A), we also acquired ER, PR, and HER2 status of these cancers, and plotted the 2D UMAP embedding with the corresponding molecular information. The resulting embedding is illustrated in Figure 2B and supplementary material, Figure S5A,B, with ‘+’ or ‘−’ signs representing tumors with positive or negative molecular status for ER, PR, or HER2, respectively. However, from the embedding it is clear that the breast tumors did not appear to cluster based off the molecular data. The hierarchical clustering‐based heat map using the embedding of the top 10 most selected features from all feature families is illustrated in Figure 3A. When setting the number of clusters at four, the only distinct cluster identified was the colon cluster, 14 of the 17 WSIs in the cluster correspond to colon cancer. Based on the 2D UMAP embedding, we performed an agglomerative clustering assessment by setting the number of clusters to be identified as four. The contour of each identified cluster was also plotted for better visual representation in Figure 3B. When comparing Figure 3B with Figure 2A, the colon cluster identified by the algorithm (red cluster in Figure 3B) mirrors the cluster identified visually (highlighted by bounding boxes in Figure 2A), while the orange cluster identified in Figure 3B contains a majority of the pancreatic WSIs within the hold‐out validation set. However, the agglomerative clustering failed to identify separate and distinct clusters for breast and esophageal tumors.
Figure 2.
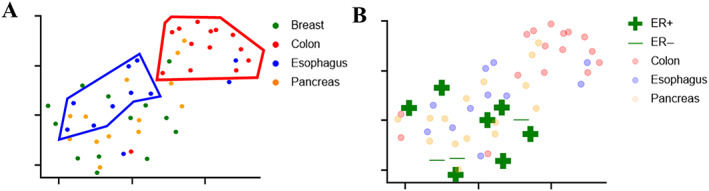
2D UMAP embedding. (A) Top 10 most selected features from all feature families. (B) ER status of breast cases. Tumors originated from breast, colon, esophagus, and pancreas are colored with green, red, blue, and orange respectively. Green ‘+’ or ‘−’ signs are used to represent positive or negative tumor with corresponding molecular status. To highlight the IHC status of breast metastases, metastases from other organ sites are lowlighted.
Figure 3.
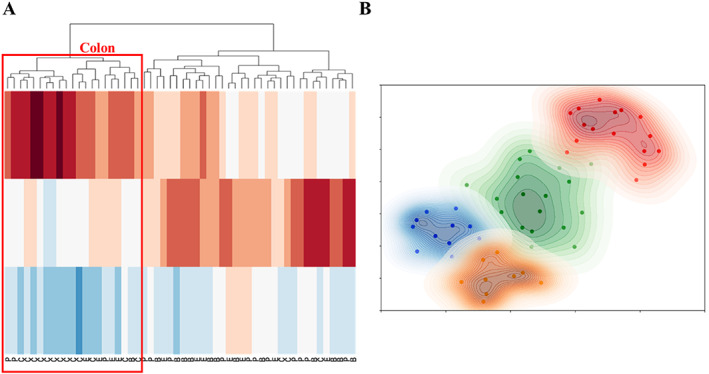
Hierarchical clustering of top 10 most selected feature from all feature families that generated maximum cluster separation. (A) Heat map of the 3D embedment with true class labels is shown on the bottom of the plot. (B) Agglomerative clustering analysis results when set the number of clusters to be identified into four. Color scheme for cancers as in Figure 2. B, breast; C, colon; E, esophagus; P, pancreas.
Supplementary material, Figure S6 shows the PR curve and AUPRC of the CBIR classifier built using the top 10 commonly selected features over 100 iterations of CV from all feature families (supplementary material, Figure S6A) and CCM (supplementary material, Figure S6B). For the usually best performed colon tumors, the CBIR classifier only yielded an AUPRC of 0.49 (all feature families) and 0.57 (CCM).
Figure 4 shows the violin plots of the most commonly selected feature over the 100 iterations of CV from all feature families (Figure 4A) and CCM (Figure 4B). The most commonly selected feature from all feature families was from FeDeG family [19], which emphasizes the local cell graph's mean area. Based on Student's t test, the colon tumors have a significantly different distribution when compared with pancreatic tumors. As for Figure 5B, the most commonly selected CCM feature characterizes the nuclei intensity range within the local cell graph. The t test demonstrated that the colon tumors had a different distribution when compared with tumors from other organ sites. However, breast, esophageal, and pancreatic tumors failed to show any significance in the distribution of the top selected feature. A more detailed description for the top 10 features and distribution of the top 3 features are shown in supplementary material, Table S2 and Figure S7 respectively.
Figure 4.
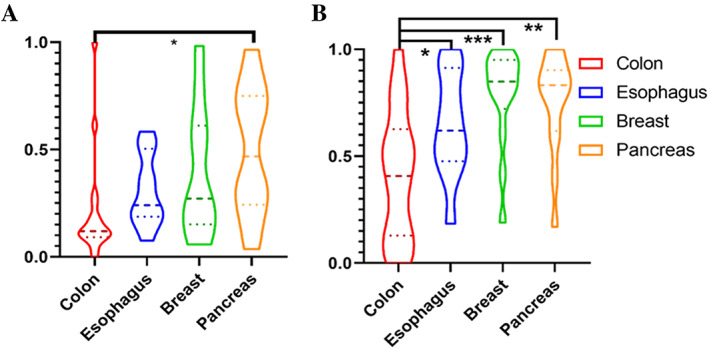
Violin plots of top feature selected (A) from all feature families and (B) from CCM. Student's t test was applied to the normalized feature. Organs with significant differences in the distribution of the most selected feature was labeled with *p < 0.05; **p < 0.01; or ***p < 0.001.
Figure 5.
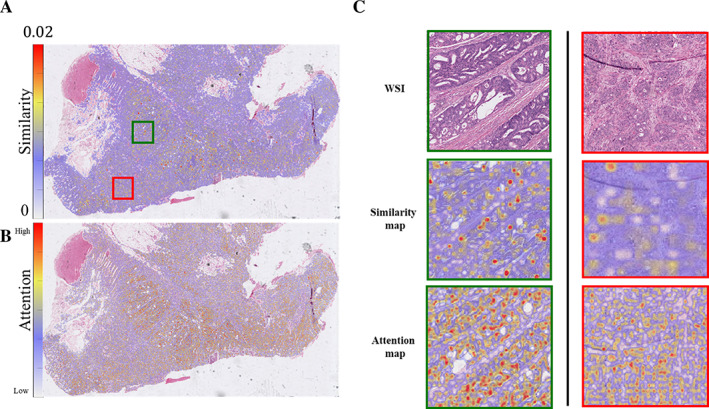
Similarity heat map of a selected colon case. Euclidean distances between the metastatic signal and patches from its corresponding primary tumor were calculated using the CBIR classifier. Similarity was then calculated as the reciprocal of the calculated distance and overlaid as a heat map (A). Guided Grad‐CAM attention map was generated by DL model to highlight important region to classify tumor as metastasis. ArI‐based pathology predicts origins of metastases from paired primary (B). The magnification of pathologist‐identified moderately differentiated (in green) and poorly differentiated (in red) regions is also plotted in panel (C). A patch with a cooler color represents lower similarity between the image patch and its corresponding metastatic slide.
Experiment 2 – integrating HC and DL approaches
In comparison with the performance of , the trained DL model, , achieved better performance on the classification of colon tumors (0.94), and comparable performance in identifying tumors from esophagus (0.68), breast (0.79), and pancreas (0.69). Using the decision fusion strategy described above, the fused model, , achieved a slightly better results than ML or DL classifier alone on the validation set (esophagus: 0.71, breast: 0.81, and pancreas: 0.72). had a comparable performance (AUC: 0.93) in identifying metastatic tumors originated from colon when comparing with (AUC: 0.94).
Experiment 3 – identifying site of metastasis from within a primary tumor
For four selected primary‐metastasis pairs (one pair for each organ site), the similarity between metastatic and corresponding primary tumors was calculated and overlaid on top of the original WSIs as heat maps. A representative case from a colon cancer primary is illustrated in Figure 5A. The trained DL model's performance on hold‐out validation is shown in supplementary material, Figure S8. The attention map corresponding to the metastases activation map as engendered by the DL model is illustrated in Figure 5B. A similarity map and an attention map of moderately and poorly differentiated regions are highlighted by green and red bounding boxes and in a higher resolution in Figure 5C. Patches from the primary and corresponding metastatic tumors identified to have the highest similarity are illustrated in supplementary material, Figure S9A. In a representative breast case (Figure 6 and supplementary material, Figure S9B), patches located within the regions of primary tumor with high epithelial content demonstrated a higher similarity with the metastatic tumor compared with patches of primary tumor located in regions with high stromal components. This observation is also consistent with the selected primary tumors from all four organ sites (supplementary material, Figures S10–S13). This observation aligns with our expectations and serves as a form of quality control for our methodology. Given that the features of metastatic tumors are extracted from epithelium, it is anticipated that these tumors would exhibit greater resemblance to epithelial‐rich primary tumors. In the similarity map illustrated in Figure 5C, poorly differentiated tumor cluster regions had lower similarity to the metastatic tumors compared with the moderately differentiated cluster regions in the same tumors (see also supplementary material, Figure S10). Also illustrated in supplementary material, Figure S9B are the top three primary patches with the highest similarity to metastatic regions, all of these image patches correspond to epithelial rich (stromal poor) tumor regions.
Figure 6.

Similarity heat map of a selected breast case. Euclidean distances between the metastatic signal and patches from its corresponding primary were calculated using the CBIR classifier. Similarity was then calculated as the reciprocal of the calculated distance and overlaid as a heat map (A). The magnification of the raw WSI, generated similarity heat map, as well as attention map are also plotted in panel (B). Patches with cooler colors in the similarity map represent lower similarity between these patches and the corresponding metastatic slide. The attention map highlighted region with higher importance to classify tumor as metastasis.
Discussion
Adenocarcinoma represents almost 75% of all liver metastasis cases with primary sources with the most common primary sites of origin being colon, breast, pancreas, and esophagus [2, 3]. However, the site of origin for approximately 4.1% of metastatic liver tumors and up to 15.6% of adenocarcinomas cannot be identified [2]. Despite advances in molecular techniques and IHC, the cost in both time and money as well as the tissue destructive nature of molecular assays represent technological limitations, especially in resource scarce and low‐ and middle‐income country settings. Computational approaches using digitized WSIs offer distinct advantages including low cost of implementation, scalability and, most importantly, non‐tissue destructive tissue assays using H&E images of routinely acquired diagnostic pathology slides. Given the need to define tumor sites of origin to direct cancer specific therapies, and to preserve what are often small tissue specimens for advanced molecular testing, digital pathomics technologies have a potentially important role to play in these patients.
Computational approaches using digitized WSIs offer distinct advantages including low cost of implementation, scalability and, most importantly, non‐tissue destructive tissue assays using H&E images of routinely acquired diagnostic pathology slides.
In this work, we constructed a dataset consisting of paired liver metastatic tumors and corresponding primary adenocarcinomas derived from four common PSO of liver metastases. Models were trained using either a HC‐ or DL‐based approach to identify PSO of liver metastases. By fusing the decisions generated by the HC and DL classifiers together, we also created a fused model. The trained model was then evaluated on a hold‐out validation set comprised solely of liver metastases without corresponding primary tumor. We demonstrated that the fused DL + HC classifier slightly outperformed classifiers trained via HC or DL approaches alone. This work demonstrates the feasibility of a combined computational pathology approach, which harnesses the strengths of both HC and DL for the identification of a site of origin in clinical samples of liver metastases for four common cancers.
Histomorphometric features have been widely used to risk‐stratify tumors and have yielded reliable performance in characterizing various cancer types [11, 12, 13, 15, 28, 29, 30]. This work extends the use of histomorphometric features to the identification of the origin of tumors. Among the various feature families considered in this work, CCM features yielded the highest classification value, followed by texture features of the nuclei. The most selected features were from the FeDeG family, features that reflect the spatial architecture of individual cancer cells. These CCM and FeDeG features likely reflect the morphological and glandular differences of nuclei with sites of origin. In addition, cytoplasmic texture further enabled discrimination between tumors of different origins. Despite substantial research demonstrating the value of TILs and carcinoma‐associated stroma (CAS) being prognostic for various cancer types [31, 32, 33, 34, 35], we intentionally excluded TILs and CAS from our analysis as these were not unique to certain primary tumors and no literature was identified to help support the case that these features are associated with PSO.
Additionally as part of a proof of concept demonstration, we were able to illustrate the utility of the HC approach to suggest the intratumoral site of origin of cancer metastases in a set of paired primary/metastatic tumors. While the approach needs to be validated via molecular‐based approaches, this approach could have the potential for defining cancer subclone populations with varying biological profiles directly from H&E slides.
The metastatic signal mapped back to the corresponding primary tumors implies that the metastatic tumor was more similar to moderately differentiated tumor regions as opposed to poorly differentiated regions. We also identified that the metastatic signal was more similar to the image patches of the primary tumor from the epithelium rich regions compared with stromal‐rich regions. We note that findings from experiment 3 are somewhat exploratory and preliminary and consequently we abstain from drawing too many conclusions from these findings. However, these initial results do suggest to the need to correlate these morphometric findings with spatial transcriptomics to get at the molecular underpinning of our observations.
The work of Lu et al [17] is the closest work related to our study. Both studies focused on identifying the origin of tumors using computational pathology. Lu et al utilized multi‐task, multi‐instance DL methods to predict the origin of metastases in 18 different cancers. Their model also deployed an attention‐based, multiple‐instance learning algorithm to identify those regions with higher importance. While our model had a lower performance when compared directly with the work of Lu et al, our approach also did not have the benefit of the large number of training cases employed in the Lu et al study. A benefit of our approach lies in that we demonstrated the utility and inclusion of interpretable image features, our preliminary results indicate that the combination of the interpretable image features with the DL classifier enables more accurate prediction compared with a DL or HC classifier alone.
Our work does have its limitations. The dataset size was a major limitation of this work, with only 175 cases from 114 patients. In experiment 2, we did not see a statistically significant improvement in the combination of the HC and DL approaches, this however may be on account of the limited sample size considered in this study. However given the complementarity of the two categories of approaches, we expect that on larger, powered datasets, fusion strategies, such as deep orthogonal fusion, could demonstrate superiority over either individual classifier [36]. The manual delineation of the tumor regions on the WSIs was yet another limitation of the study and we will look to apply automated approaches for tumor segmentation in future studies. Given the constraints of the 256 × 256 pixel [2] patch size, we primarily utilized features at the cytological level in our third experiment. We acknowledge that the inclusion of higher level features such as architectural or slide‐level ones could potentially enhance the accuracy of our results. Nevertheless, our choice of a smaller patch size was intentional, as it allows for the generation of more detailed heat maps. It is a conscious trade‐off we made to balance between detailed feature extraction and heat map resolution clarity.
An additional limitation of this study is the necessity to use biopsy samples for the majority of liver metastases analyzed as well as for a significant number of cancer primaries. The latter were chosen to avoid primary cancers that had undergone preoperative chemoradiation – an accepted practice in breast, pancreas, and esophageal oncologic practice. Intrinsically, biopsies provide less information than resection slides as the spatial and anatomical orientation were lost during the procedure. However, due to the scarcity of paired primary and metastatic tumor samples, it is impossible for us to exclusively use resection WSIs. Another limitation of this study is the absence of external, independent validation sets which restricts our ability to thoroughly test the generalizability of the trained models. Moreover, the investigation focused primarily on adenocarcinomas derived from four common sites of liver metastases. Future work will include a broadening of the scope of the study by including diverse cancer types, such as gastric adenocarcinoma and cholangiocellular carcinoma. Moreover, the use of external open image sources such as The Cancer Genome Atlas will be pursued to enhance the robustness and validity of the findings. By addressing these limitations, we aim to improve the predictive power and clinical utility of our pathomics approach.
In conclusion, we present an approach combining DL with HC histomorphometric features to identify the site of origin of metastatic liver tumors. Since our approach requires only H&E‐stained WSIs, it is tissue non‐destructive and cheaper compared with molecular assays. We also demonstrate that our approach can potentially be used to spatially identify the likely site of origin of a metastasis by direct comparison of matched primary‐metastasis pairs. Future work will validate these features on multi‐site, multi‐institutional datasets and extend the approach to additional tumor sites.
Author contributions statement
CC designed the workflow, trained the model, and analyzed the data. CL assisted with feature extraction. JW, BM and BM contributed to dataset annotation. CC wrote the manuscript with input from VV, CL, JW and AM. JW and AM conceived the original idea and supervised the project.
Supporting information
Figure S1. Dataset distribution of experiments 1, 2, and 3
Figure S2. Example of PCA patch selection
Figure S3. Workflow of DL model
Figure S4. Decision fusion strategy
Figure S5. 2D UMAP embedding of top 10 most selected features from all feature families
Figure S6. PR curve and AUPRC for CBIR
Figure S7. Identified top three features and representative patches
Figure S8. ROC of trained DL model on validation set in experiment 2
Figure S9. Top patch identified by CBIR
Figure S10. Regional similarity analysis – colon case
Figure S11. Regional similarity analysis – breast case
Figure S12. Regional similarity analysis – pancreas case
Figure S13. Regional similarity analysis – esophagus case
Figure S14. Example of patch analysis (cited only in Supplementary material)
Figure S15. Example of Delaunay triangulation graph (cited only in Supplementary material)
Figure S16. Example of nuclear Haralick texture features (cited only in Supplementary material)
Figure S17. Relationship between nuclei density and nFD (cited only in Supplementary material)
Figure S18. Example of CRL feature (cited only in Supplementary material)
Table S1. Clinical data for the dataset used
Table S2. Identified top 10 features and their explanations in experiment 1
Table S3. Identified top five features from nuclei shape and textures families
Table S4. Thirteen Haralick measurements of the co‐occurrence matrix (cited only in Supplementary material)
Acknowledgements
The research reported in this publication was supported by the National Cancer Institute under award numbers R01CA268287A1, U01CA269181, R01CA26820701A1, R01CA249992‐01A1, R01CA202752‐01A1, R01CA208236‐01A1, R01CA216579‐01A1, R01CA220581‐01A1, R01CA257612‐01A1, 1U01CA239055‐01, 1U01CA248226‐01, 1U54CA254566‐01, and 2P50CA150964; National Heart, Lung and Blood Institute 1R01HL15127701A1 and R01HL15807101A1; National Institute of Biomedical Imaging and Bioengineering 1R43EB028736‐01; National Center for Research Resources under award number 1 C06 RR12463‐01; VA Merit Review Award IBX004121A from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Service, the Office of the Assistant Secretary of Defense for Health Affairs, through the Breast Cancer Research Program (W81XWH‐19‐1‐0668), the Prostate Cancer Research Program (W81XWH‐20‐1‐0851), the Lung Cancer Research Program (W81XWH‐18‐1‐0440, W81XWH‐20‐1‐0595), the Peer Reviewed Cancer Research Program (W81XWH‐18‐1‐0404, W81XWH‐21‐1‐0345, W81XWH‐21‐1‐0160), the Kidney Precision Medicine Project (KPMP) Glue Grant and sponsored research agreements from Bristol Myers‐Squibb, Boehringer‐Ingelheim, Eli‐Lilly, and Astrazeneca. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the U.S. Department of Veterans Affairs, the Department of Defense, or the United States Government.
No conflicts of interest were declared.
Contributor Information
Chuheng Chen, Email: cche349@emory.edu.
Joseph Willis, Email: joseph.willis@case.edu.
Anant Madabhushi, Email: anantm@emory.edu.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Pavlidis N, Pentheroudakis G. Cancer of unknown primary site. Lancet 2012; 379: 1428–1435. [DOI] [PubMed] [Google Scholar]
- 2. de Ridder J, de Wilt JHW, Simmer F, et al. Incidence and origin of histologically confirmed liver metastases: an explorative case‐study of 23,154 patients. Oncotarget 2016; 7: 55368–55376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Horn SR, Stoltzfus KC, Lehrer EJ, et al. Epidemiology of liver metastases. Cancer Epidemiol 2020; 67: 101760. [DOI] [PubMed] [Google Scholar]
- 4. Vibert J, Pierron G, Benoist C, et al. Identification of tissue of origin and guided therapeutic applications in cancers of unknown primary using deep learning and RNA sequencing (TransCUPtomics). J Mol Diagn 2021; 23: 1380–1392. [DOI] [PubMed] [Google Scholar]
- 5. Amin K, El‐Rayes D, Snover D, et al. Patterns of immunohistochemistry utilization in metastases to the liver. Appl Immunohistochem Mol Morphol 2019; 27: 441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park JH, Kim JH. Pathologic differential diagnosis of metastatic carcinoma in the liver. Clin Mol Hepatol 2019; 25: 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Penson A, Camacho N, Zheng Y, et al. Development of genome‐derived tumor type prediction to inform clinical cancer care. JAMA Oncol 2020; 6: 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao Y, Pan Z, Namburi S, et al. CUP‐AI‐Dx: a tool for inferring cancer tissue of origin and molecular subtype using RNA gene‐expression data and artificial intelligence. EBioMedicine 2020; 61: 103030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen Y, Chu Q, Yin X, et al. TOD‐CUP: a gene expression rank‐based majority vote algorithm for tissue origin diagnosis of cancers of unknown primary. Brief Bioinform 2021; 22: 2106–2118. [DOI] [PubMed] [Google Scholar]
- 10. Rassy E, Pavlidis N. Progress in refining the clinical management of cancer of unknown primary in the molecular era. Nat Rev Clin Oncol 2020; 17: 541–554. [DOI] [PubMed] [Google Scholar]
- 11. Lu C, Lewis JS, Dupont WD, et al. An oral cavity squamous cell carcinoma quantitative histomorphometric‐based image classifier of nuclear morphology can risk stratify patients for disease‐specific survival. Mod Pathol 2017; 30: 1655–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koyuncu CF, Lu C, Bera K, et al. Computerized tumor multinucleation index (MuNI) is prognostic in p16+ oropharyngeal carcinoma: a multi‐site validation study. J Clin Invest 2021; 131: e145488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu C, Romo‐Bucheli D, Wang X, et al. Nuclear shape and orientation features from H&E images predict survival in early‐stage estrogen receptor‐positive breast cancers. Lab Invest 2018; 98: 1438–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee G, Sparks R, Ali S, et al. Co‐occurring gland angularity in localized subgraphs: predicting biochemical recurrence in intermediate‐risk prostate cancer patients. PLoS One 2014; 9: e97954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Corredor G, Wang X, Zhou Y, et al. Spatial architecture and arrangement of tumor‐infiltrating lymphocytes for predicting likelihood of recurrence in early‐stage non‐small cell lung cancer. Clin Cancer Res 2019; 25: 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H, Bera K, Toro P, et al. Collagen fiber orientation disorder from H&E images is prognostic for early stage breast cancer: clinical trial validation. NPJ Breast Cancer 2021; 7: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lu MY, Chen TY, Williamson DFK, et al. AI‐based pathology predicts origins for cancers of unknown primary. Nature 2021; 594: 106–110. [DOI] [PubMed] [Google Scholar]
- 18. Lewis JS, Tarabishy Y, Luo J, et al. Inter‐ and intra‐observer variability in the classification of extracapsular extension in p16 positive oropharyngeal squamous cell carcinoma nodal metastases. Oral Oncol 2015; 51: 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu C, Wang X, Prasanna P, et al. Feature driven local cell graph (FeDeG): predicting overall survival in early stage lung cancer. In: Medical Image Computing and Computer Assisted Intervention – MICCAI 2018. Lecture Notes in Computer Science, Frangi AF, Schnabel JA, Davatzikos C, et al. (Eds). Springer International Publishing: Cham, 2018; 407–416. 10.1007/978-3-030-00934-2_46. [DOI] [Google Scholar]
- 20. Janowczyk A, Zuo R, Gilmore H, et al. HistoQC: an open‐source quality control tool for digital pathology slides. JCO Clin Cancer Inform 2019; 3: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmidt U, Weigert M, Broaddus C, et al. Cell detection with star‐convex polygons. arXiv 2018; arXiv:180603535 Cs. 10.1007/978-3-030-00934-2_30 [DOI]
- 22. Weigert M, Schmidt U, Haase R, et al. Star‐convex polyhedra for 3D object detection and segmentation in microscopy. In 2020 IEEE Winter Conference on Applications of Computer Vision (WACV) , 2020; 3655–3662. 10.1109/WACV45572.2020.9093435 [DOI]
- 23. Ding R, Prasanna P, Corredor G, et al. Compactness measures of tumor infiltrating lymphocytes in lung adenocarcinoma are associated with overall patient survival and immune scores. In Proceedings of the SPIE, Volume 11320 , 2020. 10.1117/12.2549588 [DOI]
- 24. McInnes L, Healy J, Melville J. UMAP: uniform manifold approximation and projection for dimension reduction. arXiv 2018; arXiv:180203426 Cs Stat. [Accessed 29 June 2020]. Available from: http://arxiv.org/abs/1802.03426
- 25. Huang G, Liu Z, van der Maaten L, et al. Densely connected convolutional networks. arXiv 2018; arXiv:160806993 Cs. [Accessed 11 June 2020]. Available from: http://arxiv.org/abs/1608.06993
- 26. Perez L, Wang J. The effectiveness of data augmentation in image classification using deep learning. arXiv 2017; arXiv:1712.04621. 10.48550/arXiv.1712.04621 [DOI]
- 27. Selvaraju RR, Cogswell M, Das A, et al. Grad‐CAM: visual explanations from deep networks via gradient‐based localization. In 2017 IEEE International Conference on Computer Vision (ICCV) , 2017; 618–626. 10.1109/ICCV.2017.74 [DOI]
- 28. Li H, Whitney J, Bera K, et al. Quantitative nuclear histomorphometric features are predictive of Oncotype DX risk categories in ductal carcinoma in situ: preliminary findings. Breast Cancer Res 2019; 21: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang X, Janowczyk A, Zhou Y, et al. Prediction of recurrence in early stage non‐small cell lung cancer using computer extracted nuclear features from digital H&E images. Sci Rep 2017; 7: 13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fu Y, Jung AW, Torne RV, et al. Pan‐cancer computational histopathology reveals mutations, tumor composition and prognosis. Nat Cancer 2020; 1: 800–810. [DOI] [PubMed] [Google Scholar]
- 31. Wagner P, Koch M, Nummer D, et al. Detection and functional analysis of tumor infiltrating T‐lymphocytes (TIL) in liver metastases from colorectal cancer. Ann Surg Oncol 2008; 15: 2310–2317. [DOI] [PubMed] [Google Scholar]
- 32. Kroemer M, Turco C, Spehner L, et al. Investigation of the prognostic value of CD4 T cell subsets expanded from tumor‐infiltrating lymphocytes of colorectal cancer liver metastases. J Immunother Cancer 2020; 8: e001478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. El Bairi K, Haynes HR, Blackley E, et al. The tale of TILs in breast cancer: a report from The International Immuno‐Oncology Biomarker Working Group. NPJ Breast Cancer 2021; 7: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brown KM, Xue A, Smith RC, et al. Cancer‐associated stroma reveals prognostic biomarkers and novel insights into the tumour microenvironment of colorectal cancer and colorectal liver metastases. Cancer Med 2021; 11: 492–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lopez de Rodas M, Nagineni V, Ravi A, et al. Role of tumor infiltrating lymphocytes and spatial immune heterogeneity in sensitivity to PD‐1 axis blockers in non‐small cell lung cancer. J Immunother Cancer 2022; 10: e004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Braman N, Gordon JWH, Goossens ET, et al. Deep orthogonal fusion: multimodal prognostic biomarker discovery integrating radiology, pathology, genomic, and clinical data. arXiv 2021; arXiv:210700648 Cs Q‐Bio. [Accessed 9 April 2022]. Available from: http://arxiv.org/abs/2107.00648
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dataset distribution of experiments 1, 2, and 3
Figure S2. Example of PCA patch selection
Figure S3. Workflow of DL model
Figure S4. Decision fusion strategy
Figure S5. 2D UMAP embedding of top 10 most selected features from all feature families
Figure S6. PR curve and AUPRC for CBIR
Figure S7. Identified top three features and representative patches
Figure S8. ROC of trained DL model on validation set in experiment 2
Figure S9. Top patch identified by CBIR
Figure S10. Regional similarity analysis – colon case
Figure S11. Regional similarity analysis – breast case
Figure S12. Regional similarity analysis – pancreas case
Figure S13. Regional similarity analysis – esophagus case
Figure S14. Example of patch analysis (cited only in Supplementary material)
Figure S15. Example of Delaunay triangulation graph (cited only in Supplementary material)
Figure S16. Example of nuclear Haralick texture features (cited only in Supplementary material)
Figure S17. Relationship between nuclei density and nFD (cited only in Supplementary material)
Figure S18. Example of CRL feature (cited only in Supplementary material)
Table S1. Clinical data for the dataset used
Table S2. Identified top 10 features and their explanations in experiment 1
Table S3. Identified top five features from nuclei shape and textures families
Table S4. Thirteen Haralick measurements of the co‐occurrence matrix (cited only in Supplementary material)
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.