

Abstract
Griscelli syndrome is a rare inherited autosomal recessive syndrome that causes immunodeficiency. Hemophagocytic lymphohistiocytosis (HLH), which is characterized by a high mortality rate, may develop because of Griscelli syndrome type 2 (GS2). We aimed to share our experience with the diagnosis and treatment methods of patients who developed HLH secondary to GS2. Patients with GS2 who were diagnosed and treated for HLH between 2017 and 2022 at the Cukurova University Division of Pediatric Allergy & Immunology and Division of Pediatric Hematology were included in the study. Microscopic examination of the hair shaft and next-generation sequencing for molecular genetic testing of RAB27A helped in the diagnosis of GS2. The first clinical presentation of 8 patients was HLH. One patient presented with CNS involvement and two patients presented with recurrent fever. Over 5 years, GS2 was diagnosed in 15 patients, of whom 11 (73.3%) developed HLH. The HLH-2004 protocol was used to treat these patients. Hematopoietic stem cell transplantation (HSCT) was performed in five patients who were matched with suitable donors. While all patients who underwent HSCT were alive, three patients who could not undergo HSCT because no donor could be found died. Deletion of CAAGC at nucleotides 514_518 in GS2 patients is associated with CNS involvement and a poor prognosis. HLH may be the first sign of presentation in patients with GS2. Although further research is needed, regardless of the conditioning regimen utilized, early HSCT remains the primary therapy option for preventing GS2-induced mortality in HLH.
Keywords: Griscelli syndrome, primary immunodeficiencies, hemophagocytic lymphohistiocytosis, hematopoietic stem cell transplantation
Introduction
Griscelli syndrome type-2 (GS2) is a rare autosomal recessive disorder associated with autosomal recessive mutations in RAB27A, causing immunodeficiency and phenotypically characterized by silver-gray hair and partial albinism [1]. Hemophagocytic lymphohistiocytosis (HLH) is a syndrome with a high mortality rate triggered by severe systemic hyperinflammation [2].
The RAB27A gene encodes RAB27A, a member of the GTPase family of proteins that controls organelle dynamics and vesicular transport in many tissues [3]. GS2 patients with the RAB27A mutation may develop HLH by overwhelming T-cell and macrophage activation and have a poor prognosis unless prompt intervention is initiated [4]. In cytotoxic T lymphocytes and NK cells, the RAB27A protein interacts with the Munc13-4 protein and synaptotagmin-like proteins (Slp1, Slp2a, and Slp3) as part of the complex responsible for binding lytic granules before excretion of perforin- and granzyme-containing granules to induce target cell apoptosis [5]. The absence of target cell death causes HLH due to lymphoproliferation and the resulting cytokine storm [5]. This inflammation causes various symptoms in the body [6].
Clinical and laboratory findings of HLH include fever, organomegaly, liver damage, consumption coagulopathy, hypertriglyceridemia, cytopenias, dermatological abnormalities, and increases in acute phase reactants (especially serum ferritin) [7].
In addition to systemic findings, neurological dysfunctions may develop in patients with GS2. The potential cause of neurological dysfunction could be lymphohistiocytic infiltration of the central nervous system (CNS) after HLH develops in GS2 [8]. However, there are hypotheses that CNS infiltration in GS2 may be directly related to the defect in the RAB27A protein due to the natural course of the disease [9].
The distribution of pigment-containing melanosomes in melanocytes is regulated by RAB27A, which coordinates the melanophilin/myosin Va motor complex and the SPIRE/FMN actin filament assembly complex at the initiation of melanin exocytosis [10]. In addition to its exocytotic roles at T cell receptors, RAB27A also has a role in binding cytosolic granules to the cell membrane after signal activation [11,12]. These mechanisms partially explain why oculocutaneous albinism and immunodeficiency are among the clinical features of GS2.
GS2 is a very rare syndrome, and its pathology and clinical features remain to be elucidated [13]. Because of consanguineous marriages in our region, rare diseases such as GS2 are not rare. For this purpose, we examined our patients’ clinics and treatment regimens and shared them with the literature.
Methods
We retrospectively analyzed pediatric patients with GS2 who were diagnosed and treated with HLH from 2017 to 2022 at Cukurova University, Division of Pediatric Allergy & Immunology, and Division of Pediatric Hematology. The diagnosis of GS2 was based on a microscopic examination of the hair shaft, clinical signs, and molecular genetic testing of the RAB27A gene using next-generation sequencing. Variant assessments were performed according to the American College of Medical Genetics and Genomics (ACMG) Criteria [14].
In 5 years, 15 patients presented with GS2, and 11 of them developed HLH. Eleven patients fulfilled at least 5 of the 8 diagnostic criteria of HLH 2004 [15]. Symptoms and laboratory parameters (white blood cell (WBC), absolute neutrophil count, absolute lymphocyte count, hemoglobin, platelet counts, lactate dehydrogenase (LDH), fibrinogen, ferritin, triglyceride, albumin, alanine transaminase (ALT), aspartate aminotransferase (AST), and C-reactive protein (CRP)) at the time of diagnosis of HLH and laboratory parameters in the second week of treatment were evaluated. The HLH 2004 protocol was used for the treatment. Hematopoietic stem cell transplantation (HSCT) was performed in patients with suitable donors. The ESID guidelines for HSCT were used as guidance for the conditioning regimen applied to patients [16].
Statistical analysis
Categorical variables are expressed as numbers and percentages, whereas continuous variables are summarized as mean, standard deviation, median, and minimum-maximum, where appropriate. The normality of the distribution for continuous variables was confirmed using the Shapiro-Wilk test.
To compare paired continuous variables (laboratory parameters at baseline and second week of treatment), paired-samples t-test or Wilcoxon signed rank test based on statistical hypotheses was used.
All analyses were performed using IBM SPSS Statistics Version 20.0 statistical software. The statistical level of significance for all tests was considered to be 0.05.
Results
Clinical features, laboratory parameters, and genetics
Eleven patients were enrolled in this study. Six were male and five were female. The mean age at diagnosis was 29.3 months (min. 3-max. 137). HLH was detected in 8 (72.7%) patients. Three (27%) patients presented with CNS involvement, and two (18.1%) patients presented with recurrent febrile diseases. The mean HLH development time was 35.5±6.2 months in patients without HLH at first presentation. At admission, 90% (n = 10) of the patients had cytopenia in at least two hematological series. Hypofibrinogenemia was detected in 54.5% (n = 6) patients. Ferritin elevation was detected in all patients, and 36.3% (n = 4) had high triglyceride levels. Hemophagocytosis in the bone marrow was documented in all patients. Hemaphagocytes were detected in the cerebrospinal fluid (CSF) samples of 1 patient (9%) (Figure 1A and 1B). The parents of patients 1, 3, 4, and 7 were cousins. There was no consanguinity between the other patients’ parents. The genetic mutation was RAB27A in 10 patients. All RAB27A mutations were homozygous. Nonsense mutations were detected in 63.6% (n = 7), missense mutations in 8.3% (n = 1), and large deletions in 8.3% (n = 1) of the patients (Details of clinical features, laboratory parameters, and genetic testing results are given in Table 1. The pathogenicity classifications are given in Table 2). All patients’ hair shafts were microscopically inspected, and irregular clumps of melanin pigment were detected (Figure 2A and 2B).
Figure 1.
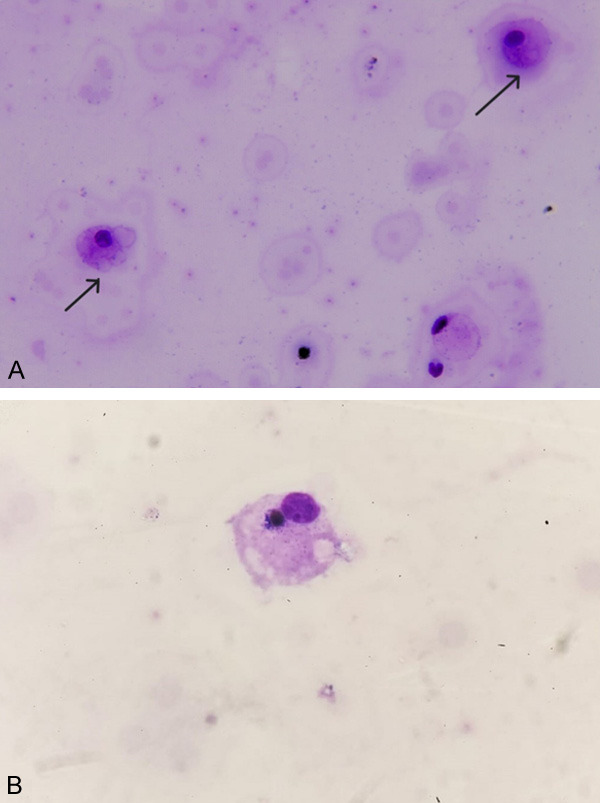
A. Histiocytes that have phagocytosed erythrocytes in the CSF smear prepared by the cytocentrifuge method (arrows). B. In the CSF smear prepared by the cytocentrifuge method, erythrocytes and lymphocyte-phagocytosed histiocytes are observed (×1000 May Grünwald Giemsa staining).
Table 1.
Clinical features, laboratory parameters, and mutations
| Patient | Gender | RAB27A mutation | Protein Change | Ethnicity | Age at diagnosis (month) | Time from diagnosis to HLH (month) | Symptoms at the time of diagnosis | CNS involvement | Time from diagnosis to CNS involvement | HSCT | Survival |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | ex2-5del | - | Turkish | 3 | - | HLH | N | - | P | S |
| 2 | F | ND | ND | Turkish | 60 | 2 | RFD | N | - | P | S |
| 3 | M | c.514_518del | p.Q172Nfs*2 | Turkish | 12 | 6 | HLH | N | - | P | S |
| 4 | F | c.550C>T | p.Arg148Ter | Turkish | 6 | - | HLH | N | - | P | S |
| 5 | M | c.514_518del | p.Q172Nfs*2 | Turkish | 3 | - | HLH | P | 1 | P | S |
| 6 | F | c.148delA | p.R50KfsX35 | Turkish | 137 | 108 | RFD | N | - | N | S |
| 7 | M | c.454G>C | p.A152P | Syrian | 4 | - | HLH | N | - | N | S |
| 8 | F | c.514_518del | p.Q172Nfs*2 | Turkish | 7 | - | HLH | N | - | N | E |
| 9 | M | c.514_518del | p.Q172Nfs*2 | Syrian | 12 | - | HLH | N | - | N | S |
| 10 | F | c.514_518del | p.Q172Nfs*2 | Syrian | 72 | 2 | CNS | P | 0 | N | E |
| 11 | M | c.514_518del | p.Q172Nfs*2 | Syrian | 7 | - | HLH | P | 12 | N | E |
M, Male; F, Female; HLH, Hemophagocytic Lymphohistiocytosis; HSCT, Hematopoietic stem cell transplantation; RFD, Recurrent febrile diseases; CNS, Central nervous system involvement; N, Negative; P, Positive; S, Survived; E, Exitus; ND, Not detected.
Table 2.
Pathogenicity classifications
| Patient | RAB27A mutation | Protein Change | ACMG classification |
|---|---|---|---|
| 3 | c.514_518del | p.Q172Nfs*2 | Pathogenic |
| 4 | c.550C>T | p.Arg148Ter | Pathogenic |
| 5 | c.514_518del | p.Q172Nfs*2 | Pathogenic |
| 6 | c.148delA | p.R50Kfs*35 | Likely Pathogenic |
| 7 | c.454G>C | p.A152P | VUS |
| 8 | c.514_518del | p.Q172Nfs*2 | Pathogenic |
| 9 | c.514_518del | p.Q172Nfs*2 | Pathogenic |
| 10 | c.514_518del | p.Q172Nfs*2 | Pathogenic |
| 11 | c.514_518del | p.Q172Nfs*2 | Pathogenic |
Figure 2.
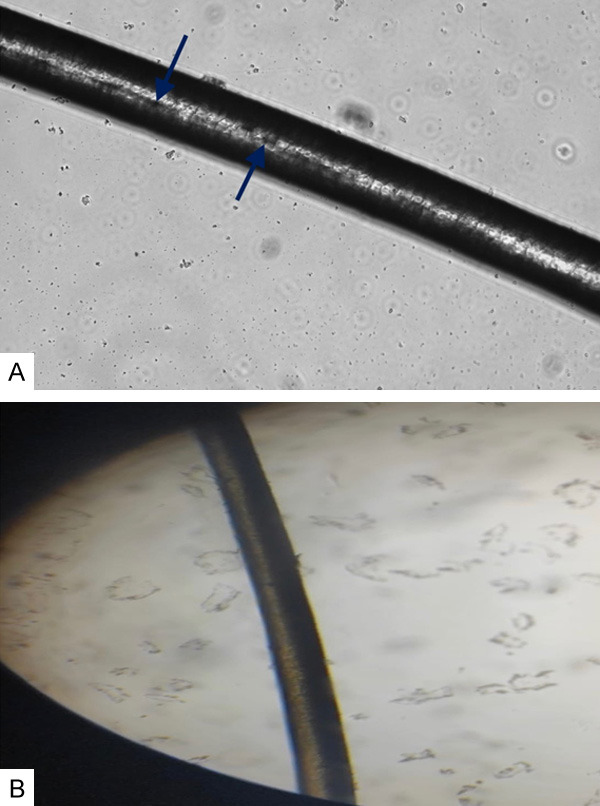
A. Clumps of melanin pigment in the patients’ hair shaft (arrows). B. Normal hair shaft.
Statistically significant decreases were found in ferritin (P<0.01) and CRP (P = 0.028) levels, and a significant increase in fibrinogen (P = 0.016) levels was detected in the second week of treatment (Table 3).
Table 3.
Pre- and post-treatment laboratory findings in patients
| Laboratory Parameters | Pre-Treatment | Post-Treatment | p |
|---|---|---|---|
| WBC (m/mm3) | 3500 (2171-12.700) | 5300 (2350-18.200) | 0.168b |
| Absolute Neutrophil Count | 1200 (300-8000) | 1900 (0-16.200) | 0.091b |
| Absolute Lymphocyte Count | 1770 (900-4600) | 2500 (600-4700) | 0.646b |
| Hemoglobin (g/dL) | 9.4±1.6 | 10.1±1.8 | 0.164a |
| Platelet (μL) | 88.000 (10.000-852.000) | 280.000 (57.000-387.000) | 0.062b |
| LDH (U/L) | 329 (200-483) | 281 (177-561) | 0.441b |
| Fibrinogen (mg/dl) | 129 (100-297) | 250 (118-291) | 0.016 b |
| Ferritin (ng/ml) | 1353±673 | 469±278 | <0.001 a |
| Triglycerides (mg/dl) | 375±166 | 255±137 | 0.110a |
| AST (U/L) | 35 (21-479) | 37 (13-241) | 0.350b |
| ALT (U/L) | 39±20 | 48±32 | 0.334a |
| Albumin (g/L) | 34.5±7 | 35.2±7 | 0.589a |
| CRP (mg/L) | 13.6 (1-79) | 5 (1-9) | 0.028 b |
Data were summarized as mean ± standard deviation or median (min-max).
paired-samples t-test;
Wilcoxon Signed Rank.
Treatment
Treatment was performed using the protocol for HLH-2004. During the active phase of HLH, all patients received intravenous immunoglobulin (IVIG) and corticosteroid treatments. Three patients with CNS involvement received intrathecal methotrexate therapy. Two of these patients underwent plasmapheresis therapy (Table 4).
Table 4.
HLH treatment
| Patient | Intrathecal Therapy | Other | HSCT |
|---|---|---|---|
| 1 | - | - | + |
| 2 | - | - | + |
| 3 | - | - | + |
| 4 | - | - | + |
| 5 | Methotrexate | - | + |
| 6 | - | - | - |
| 7 | - | - | - |
| 8 | - | - | - |
| 9 | - | - | - |
| 10 | Methotrexate | Plasmapheresis | - |
| 11 | Methotrexate | Plasmapheresis | - |
Two patients underwent allogeneic HSCT, two underwent haploidentical HSCT, and one underwent an unrelated donor HSCT. Four out of five patients showed remission after HSCT. Unrelated donor HSCT was performed for the second time in one patient because of a lack of engraftment. Remission was observed after the second transplantation. Two patients received treosulfan (14 mg/m2/day for 3 days) + fludarabine (30 mg/m2/day for 5 days) + thiotepa (10 mg/kg/day for 1 day) + anti-thymocyte globulin (10 mg/kg/day for 4 days). One patient received fludarabine (45 mg/m2/day for four days) + busulfan (3.5 mg/kg/day for four days) + anti-thymocyte globulin (5 mg/kg/day for three days). One patient received fludarabine (30 mg/m2/day for 5 days) and treosulfan (12 mg/m2/day for 3 days). In the first conditioning regimen of the patient whose engraftment could not be achieved in the first HSCT, fludarabine (30 mg/m2/day for 5 days) + treosulfan (14 mg/m2/day for 3 days) + alemtuzumab (0.2 mg/kg/day for 5 days) were administered. In the second HSCT, fludarabine (40 mg/m2/day for 4 days) + busulfan (4.9 mg/kg/day for 4 days) and anti-thymocyte globulin (10 mg/kg/day for 3 days) were administered. Four patients received cyclosporine and short-term methotrexate to prevent graft-versus-host disease (GVHD), and one patient who underwent haploidentical transplantation received cyclophosphamide and cyclosporine. After HSCT, the patients were followed for 23.3±5.2 months. There were no signs of acute or chronic GVHD. EBV and CMV PCR positivity was not detected in patients who underwent transplantation (Table 5).
Table 5.
Patients undergoing HSCT
| Patient | Age at HSCT (months) | Time from diagnosis to HSCT (months) | Conditioning regimen | Donor type | Source of stem cells | GVHD prophylaxis | AGVHD | CGVHD |
|---|---|---|---|---|---|---|---|---|
| 1 | 36 | 33 | Treo/Flu/Thio/ATG | Haploidentical Donor | PB | CsA | N | N |
| 2 | ||||||||
| 1*st HSCT | 65 | 5 | Flu/Treo/Alem | Matched UD | PB | CsA/MMF | N | N |
| 2*nd HSCT | 72 | 12 | Flu/Bu/ATG | Matched UD | PB | CsA/Mtx | N | N |
| 3 | 17 | 5 | Flu/Bu/ATG | Mismatched FD | BM | CsA/Mtx | N | N |
| 4 | 12 | 6 | Flu/Treo | Matched FD | BM | CsA/Mtx | N | N |
| 5 | 7 | 3 | Treo/Flu/Thio/ATG | Haploidentical Donor | PB | CsA/Cyc | N | N |
Treo, Treosulfan; Flu, Fludarabine; Thio, Thiotepa; Alem, Alemtuzumab; Bu, Busulfan; ATG, Anti-thymocyte globulin; CsA, Cyclosporine; Cyc, Cyclophosphamide; MMF, Mycophenolate; Mtx, Methotrexate; FD, Family Donor; UD, Unrelated Donor; PB, Peripheral Blood; BM, Bone Marrow; AGVHD, Acute GVHD; CGVHD, Chronic GVHD.
Patients with CNS involvement
Patient 5: The patient had a tonic-clonic seizure and strabismus in the first month of HLH treatment. Therefore, brain magnetic resonance imaging (MRI) was performed. MRI revealed gliotic changes in the periventricular white matter. HSCT was performed three months after the diagnosis; his complaints regressed after HSCT, and the patient had no seizures during follow-up. A control MRI was performed, and the result was normal.
Patient 10: The first complaints were tremors in the hands, an inability to walk, and difficulty swallowing. With these complaints, a brain MRI was performed. MRI revealed areas of contrast enhancement and many lesion foci with similar morphologies in both cerebral hemispheres (Figure 3A).
Figure 3.
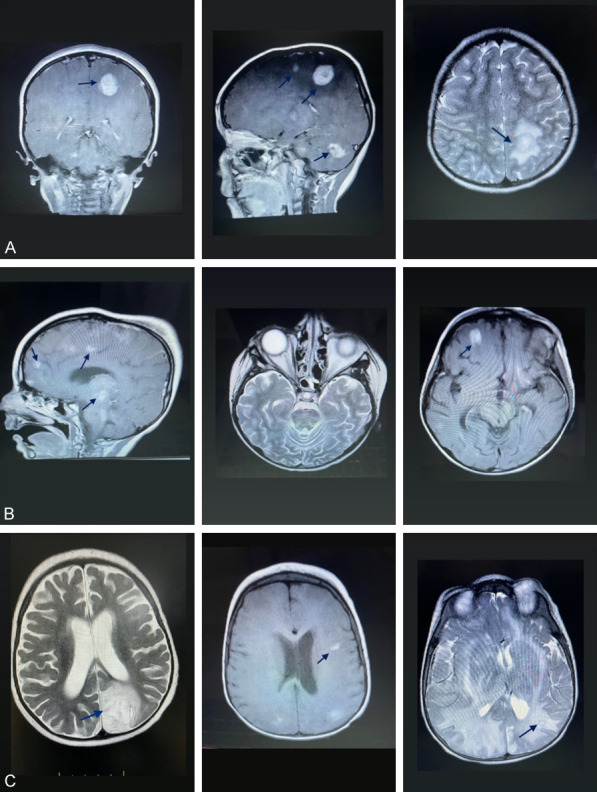
A. MRI of patient 10 at the time of admission (MRI showing nodular enhancing lesions in the contrast-enhanced weighted-T1 sequence, and a peripherally hypointense and centrally hyperintense lesion in T2A). B. Control MRI of patient 10 (T1A showing contrast-enhancing lesions in the frontal lobe and brainstem). C. MRI of patient 11 at admission (T2A showing a hyperintense lesion located in the parieto-occipital cortical and subcortical areas. Fluid-attenuated inversion recovery (FLAIR) showing increased signal in bilateral parieto-occipital white matter and left parietal subcortical lesion) (arrows).
The patient underwent a brain biopsy. The biopsy result was compatible with hemophagocytosis (Figure 4A and 4B). The HLH protocol was initiated. Radiotherapy was administered. Urinary and fecal incontinence developed during the first month of treatment, and speech difficulty occurred during the second month of treatment. Because of clinical worsening, a follow-up MRI was performed. In the control MRI, regression of previous lesions was observed (Figure 3B). Although treatment was continued, the patient developed respiratory failure and died in the 5th month of treatment.
Figure 4.
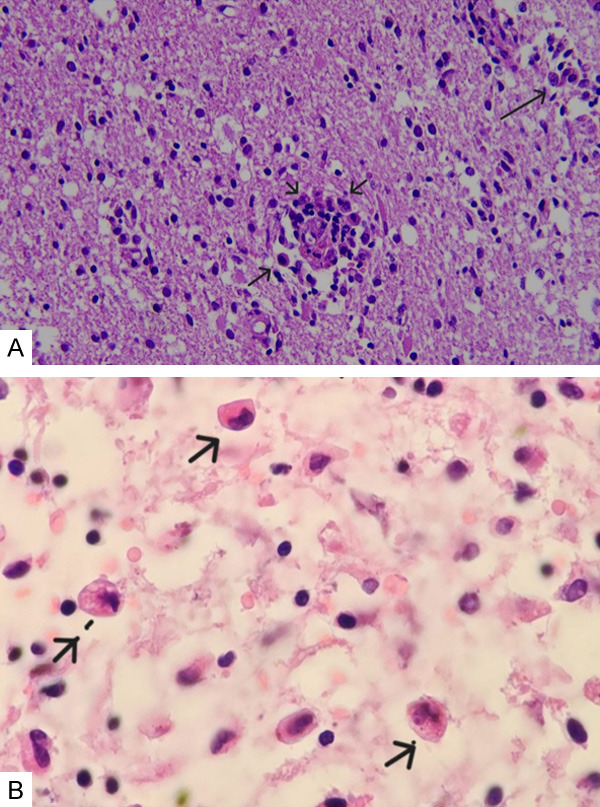
A, B. Numerous histiocytes are noted in the brain parenchyma, showing reactive gliosis. Erythrocytes and lymphocytes were observed in the cytoplasm of some histiocytes (×200 and ×1000 Hematoxylin & Eosin staining).
Patient 11: The patient presented with a seizure in the 12th month of HLH treatment. A brain MRI was performed. MRI revealed that signal pathologies extending to the cortical region were observed in the cerebral and cerebellar white matter and developing millimetric lesions (Figure 3C). The infection tests were normal, and infective causes were excluded. It was thought that the CNS involvement was due to HLH. While the patient continued his HLH treatment, he died one month after CNS involvement.
RAB27A mutations in patients with CNS involvement are shown in Figure 5.
Figure 5.

RAB27A mutation in patients 10 and 11 was shown in comparison with the control. Pathogen mutation data was shown with the help of the IgV program using the next-generation sequencing method.
Survival
In this retrospective analysis, 3 of 11 patients died. One patient died due to septic shock (Patient 8). The other two patients who died were patients with CNS involvement (Patient 10 and Patient 11).
Discussion
RAB27A is an important component of vesicular trafficking in various cell types, including blood leukocytes, platelets, and melanocytes [17]. Different expressions of this molecule in various tissues may affect the clinical symptoms of GS2 [17]. Siddiahgari et al. examined patients with silvery hair syndromes and reported that the first presenting finding in 3 of 4 cases of Griscelli syndrome was HLH [18]. Cetinkaya et al. examined patients who applied to the pediatric immunology clinic with HLH within 5 years and stated that most of them were diagnosed with GS2 [19]. Similarly, in our study, most patients’ first clinical manifestations were HLH.
In GS2, there may be a link between the beginning of the disease, its clinical course, and heredity. In a study reported by Russ et al., one of two sisters with the same genetic mutation of GS2 presented with CNS involvement, whereas the other’s first presenting complaint was systemic HLH [20]. Likewise, in our study, although the mutations of some patients were the same (Table 1), their presenting complaints were different.
According to a study by Mesache et al., the genetic variation in the RAB27A gene that results in the premature codon has very low cytotoxic activity [12]. Therefore, it has been suggested that genetic mutations in patients with GS2 can lead to variations in phenotype and genotype characteristics [12]. In addition, the study mentioned above [12], stated that the most reported RAB27A mutations associated with GS2 were nonsense or frameshift mutations. The loss of CAAGC between nucleotides 514_518 caused a frameshift mutation in 54.5% of patients (n = 6), which was the most prevalent mutation in our analysis.
In another study in which 9 patients were examined, CNS involvement developed in 4 patients, and it was stated that 2 of these patients had a genetic mutation c.514_518del [21]. This mutation was the same as that observed in patients with CNS involvement in our study. Moreover, the c.514_518del mutation was detected in 5 patients, 3 of whom died [21]. Likewise, our patients with this genetic change also died. In addition to the fact that it is the most common genetic alteration in our patients, we emphasize that c.514_518del may be associated with CNS involvement and a poor prognosis.
RAB27A is not expressed in neuronal cells; therefore, neurological findings in GS2 may be due to lymphohistiocytic infiltration [22]. The pathophysiology of infiltration of the central nervous system by hemophagocytes is unclear, but it is thought that a cytokine storm secreted by inflammatory cells crosses the blood-brain barrier and causes damage, a vasicentric process that usually results in meningeal infiltration [23]. Five cases with GS2 in a study were examined by Tesi et al., and notably, three of them had CNS involvement, and the patient who underwent early HSCT survived [24]. In a case reported by Messinger et al., HLH and respiratory failure occurred in a patient with GS2, the patient had CNS involvement, and the patient’s imaging and CSF were normal after HSCT [25]. In a case reported by Zhang et al., a 3-year-old child diagnosed with GS2 had neurological complaints such as loss of mental status and a decrease in physical strength, and a regression in these complaints was observed after HSCT [26]. HSCT is one of the most useful treatments for ensuring the regression of neurological symptoms and survival in patients with CNS involvement due to primary HLH [23]. However, studies on the best treatment method for CNS involvement due to GS2 have not yet been conducted. In our study, similar to the studies mentioned above [24-26], patients with CNS involvement had complaints of gait disturbance and seizures, and post-HSCT regression was observed in neurological symptoms. Although more detailed studies are needed on this subject, we suggest that HSCT is one of the most curative treatments for treating CNS involvement in patients with HLH due to GS2.
The most common c.514_518del mutation in our study may be the founder mutation in Turkey. However, predisposing to any founder effect based on the findings and literature knowledge is less likely due to the lack of population-specific databases. To speculate on a possible founder effect, more population-wide studies focused on haplotype analysis are recommended.
In a study conducted in India, ferritin was found to be one of the most important markers for assessing treatment response and activation in the pediatric population to treat HLH [27]. CRP levels are not required for a conclusive diagnosis of HLH [15]. CRP is, nevertheless, a prominent acute-phase protein whose concentration can rise in inflammatory situations [28]. When we evaluated our patients’ laboratory parameters before HLH treatment, we found that they all had high ferritin levels and almost half had low fibrinogen levels. A statistically significant decrease in ferritin and CRP levels, as well as a substantial rise in fibrinogen levels, two weeks after HLH treatment showed that these indicators can be used to assess early response to treatment.
There are three main types of conditioning regimens for allogeneic HSCT: myeloablative conditioning (MAC), non-myeloablative conditioning, and reduced-intensity conditioning (RIC) [29]. Overall survival increases with HSCT using the MAC regimen, but it mostly raises the risk of veno-occlusive disease and early death from transplant-related causes [30]. Although long-term complications and acute toxicity after transplantation are less prevalent with the RIC regimen, graft rejection is more likely [31]. Reduced toxicity regimens are preferred for lower rates of complications [32]. In a study by Kuskonmaz et al., ten patients with GS2 underwent HSCT, and the survival rate was 80% (8/10) [33]. In another study, Al-Mofareh et al. performed HSCT on 35 patients and found a mortality rate of 37.1% (13/35) after 87.7 months of follow-up [34]. Schmid et al. evaluated ten patients with GS2 who underwent HSCT, and the patients were followed for a mean of 5.2 years: seven survived and three died [35]. In a different study, HSCT was performed on five patients with GS2 using an HLA-matched related donor [36]. The patients were followed for 19 months, and 4 out of 5 of them survived [36]. In our study, a reduced toxicity regimen was administered to all patients who underwent HSCT. GVHD was not observed, and all patients continued their lives in remission. The optimal conditioning regimen for HSCT in patients with GS2 is yet to be defined. However, in the studies mentioned above [33-36] and in our study, it was revealed that regardless of the conditioning regimen, HSCT is the main treatment for survival and disease remission in GS2 patients who develop HLH. Furthermore, the fact that patients in our study had 100% survival and no GVHD development may be because the patients in our research had a shorter mean follow-up duration than the patients in the studies mentioned above.
According to the Genetic and Rare Diseases Information Center, approximately 150 cases of GS2 have been reported in the literature [13]. Fifteen cases of this rare disease are our patients. The genetic variants found in our patients showed that deletion of CAAGC at nucleotides 514_518 in GS2 patients is associated with CNS involvement and a poor prognosis. HLH may be the first sign of presentation in patients with GS2. Ferritin, fibrinogen, and CRP can be used as markers in the evaluation of treatment response. Although further research is needed, regardless of the conditioning regimen utilized, early HSCT remains the primary therapy option for preventing GS2-induced mortality in HLH.
Disclosure of conflict of interest
None.
References
- 1.Castaño-Jaramillo LM, Lugo-Reyes SO, Cruz Muñoz ME, Scheffler-Mendoza SC, Duran McKinster C, Yamazaki-Nakashimada MA, Espinosa-Padilla SE, Saez-de-Ocariz Gutierrez MDM. Diagnostic and therapeutic caveats in Griscelli syndrome. Scand J Immunol. 2021;93:e13034. doi: 10.1111/sji.13034. [DOI] [PubMed] [Google Scholar]
- 2.Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. 2020;135:1332–1343. doi: 10.1182/blood.2019000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohishi Y, Ammann S, Ziaee V, Strege K, Groß M, Amos CV, Shahrooei M, Ashournia P, Razaghian A, Griffiths GM, Ehl S, Fukuda M, Parvaneh N. Griscelli syndrome type 2 sine albinism: unraveling differential RAB27A effector engagement. Front Immunol. 2020;11:612977. doi: 10.3389/fimmu.2020.612977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zondag TCE, Torralba-Raga L, Van Laar JAM, Hermans MAW, Bouman A, Hollink IHIM, Van Hagen PM, Briggs DA, Hume AN, Bryceson YT. Novel RAB27A variant associated with late-onset hemophagocytic lymphohistiocytosis alters effector protein binding. J Clin Immunol. 2022;42:1685–1695. doi: 10.1007/s10875-022-01315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowman SL, Bi-Karchin J, Le L, Marks MS. The road to lysosome-related organelles: insights from Hermansky-Pudlak syndrome and other rare diseases. Traffic. 2019;20:404–435. doi: 10.1111/tra.12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esteban YM, de Jong JLO, Tesher MS. An overview of hemophagocytic lymphohistiocytosis. Pediatr Ann. 2017;46:e309–e313. doi: 10.3928/19382359-20170717-01. [DOI] [PubMed] [Google Scholar]
- 7.Ponnatt TS, Lilley CM, Mirza KM. Hemophagocytic lymphohistiocytosis. Arch Pathol Lab Med. 2022;146:507–519. doi: 10.5858/arpa.2020-0802-RA. [DOI] [PubMed] [Google Scholar]
- 8.Woodward KE, Shah RM, Benseler S, Wei XC, Ng D, Grossman J, Hahn C, Thomas MA, Wright NAM, Appendino JP. Considering immunologic and genetic evaluation for HLH in neuroinflammation: a case of Griscelli syndrome type 2 with neurological symptoms and a lack of albinism. Pediatr Blood Cancer. 2020;67:e28312. doi: 10.1002/pbc.28312. [DOI] [PubMed] [Google Scholar]
- 9.Panigrahi I, Suthar R, Rawat A, Behera B. Seizure as the presenting manifestation in Griscelli syndrome type 2. Pediatr Neurol. 2015;52:535–8. doi: 10.1016/j.pediatrneurol.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 10.Alzahofi N, Welz T, Robinson CL, Page EL, Briggs DA, Stainthorp AK, Reekes J, Elbe DA, Straub F, Kallemeijn WW, Tate EW, Goff PS, Sviderskaya EV, Cantero M, Montoliu L, Nedelec F, Miles AK, Bailly M, Kerkhoff E, Hume AN. Rab27a co-ordinates actin-dependent transport by controlling organelle-associated motors and track assembly proteins. Nat Commun. 2020;11:3495. doi: 10.1038/s41467-020-17212-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izumi T. In vivo roles of Rab27 and its effectors in exocytosis. Cell Struct Funct. 2021;46:79–94. doi: 10.1247/csf.21043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, Wulffraat N, Bianchi D, Fischer A, Le Deist F, de Saint Basile G. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25:173–176. doi: 10.1038/76024. [DOI] [PubMed] [Google Scholar]
- 13.The portal for rare diseases and orphan drugs. (2023, Nov 19) Griscelli syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=381&lng=EN.
- 14.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 16.European Society for Immunodeficiencies. (2023, Nov 19). EBMT/ESID guidelines for hematopoietic stem cell transplantation for primary immunodeficiencies. https://esid.org/content/download/
- 17.Zamani R, Shahkarami S, Rezaei N. Primary immunodeficiency associated with hypopigmentation: a differential diagnosis approach. Allergol Immunopathol (Madr) 2021;49:178–190. doi: 10.15586/aei.v49i2.61. [DOI] [PubMed] [Google Scholar]
- 18.Siddiahgari S, Soma SK, Penmetcha C, Vaddadi S, Bandi V, Lingappa L. Case series on silvery hair syndromes: single center experience. Indian J Dermatol. 2022;67:164–168. doi: 10.4103/ijd.IJD_723_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cetinkaya PG, Cagdas D, Gumruk F, Tezcan I. Hemophagocytic lymphohistiocytosis in patients with primary immunodeficiency. J Pediatr Hematol Oncol. 2020;42:e434–e439. doi: 10.1097/MPH.0000000000001803. [DOI] [PubMed] [Google Scholar]
- 20.Russ A, Mack J, Green-Murphy A, Occidental M, Mian A. Griscelli type 2 syndrome and hemophagocytic lymphohistiocytosis: sisters with the same mutation but different presentations. J Pediatr Hematol Oncol. 2019;41:473–477. doi: 10.1097/MPH.0000000000001522. [DOI] [PubMed] [Google Scholar]
- 21.Mamishi S, Modarressi MH, Pourakbari B, Tamizifar B, Mahjoub F, Fahimzad A, Alyasin S, Bemanian MH, Hamidiyeh AA, Fazlollahi MR, Ashrafi MR, Isaeian A, Khotaei G, Yeganeh M, Parvaneh N. Analysis of RAB27A gene in griscelli syndrome type 2: novel mutations including a deletion hotspot. J Clin Immunol. 2008;28:384–389. doi: 10.1007/s10875-008-9192-5. [DOI] [PubMed] [Google Scholar]
- 22.Krzewski K, Cullinane AR. Evidence for defective Rab GTPase-dependent cargo traffic in immune disorders. Exp Cell Res. 2013;319:2360–2367. doi: 10.1016/j.yexcr.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horne A, Wickström R, Jordan MB, Yeh EA, Naqvi A, Henter JI, Janka G. How to treat involvement of the central nervous system in hemophagocytic lymphohistiocytosis? Curr Treat Options Neurol. 2017;19:3. doi: 10.1007/s11940-017-0439-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tesi B, Rascon J, Chiang SCC, Burnyte B, Löfstedt A, Fasth A, Heizmann M, Juozapaite S, Kiudeliene R, Kvedaraite E, Miseviciene V, Muleviciene A, Müller ML, Nordenskjöld M, Matuzeviciene R, Samaitiene R, Speckmann C, Stankeviciene S, Zekas V, Voss M, Ehl S, Vaiciene-Magistris N, Henter JI, Meeths M, Bryceson YT. A RAB27A 5’ untranslated region structural variant associated with late-onset hemophagocytic lymphohistiocytosis and normal pigmentation. J Allergy Clin Immunol. 2018;142:317–321. e8. doi: 10.1016/j.jaci.2018.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Messinger YH, Pozos TC, Griffiths AG, Mize WA, Olson DR, Smith AR. Delayed diagnosis of Griscelli syndrome type 2 with compound heterozygote RAB27A variants presenting with pulmonary failure. Pediatr Hematol Oncol. 2021;38:593–601. doi: 10.1080/08880018.2021.1895925. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Q, Zhao YZ, Ma HH, Wang D, Zhang N, Li ZG, Zhang R. Successful rescue of a lethal Griscelli syndrome type 2 presenting with neurological involvement and hemophagocytic lymphohistiocytosis: a case report. BMC Pediatr. 2021;21:253. doi: 10.1186/s12887-021-02720-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basu S, Maji B, Barman S, Ghosh A. Hyperferritinemia in hemophagocytic lymphohistiocytosis: a single institution experience in pediatric patients. Indian J Clin Biochem. 2018;33:108–112. doi: 10.1007/s12291-017-0655-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pathak A, Agrawal A. Evolution of C-reactive protein. Front Immunol. 2019;10:943. doi: 10.3389/fimmu.2019.00943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lankester AC, Neven B, Mahlaoui N, von Asmuth EGJ, Courteille V, Alligon M, Albert MH, Serra IB, Bader P, Balashov D, Beier R, Bertrand Y, Blanche S, Bordon V, Bredius RG, Cant A, Cavazzana M, Diaz-de-Heredia C, Dogu F, Ehlert K, Entz-Werle N, Fasth A, Ferrua F, Ferster A, Formankova R, Friedrich W, Gonzalez-Vicent M, Gozdzik J, Gungor T, Hoenig M, Ikinciogullari A, Kalwak K, Kansoy S, Kupesiz A, Lanfranchi A, Lindemans CA, Meisel R, Michel G, Miranda NAA, Moraleda J, Moshous D, Pichler H, Rao K, Sedlacek P, Slatter M, Soncini E, Speckmann C, Sundin M, Toren A, Vettenranta K, Worth A, Yesilipek MA, Zecca M, Porta F, Schulz A, Veys P, Fischer A, Gennery AR. Hematopoietic cell transplantation in severe combined immunodeficiency: the SCETIDE 2006-2014 European cohort. J Allergy Clin Immunol. 2022;149:1744–1754. e8. doi: 10.1016/j.jaci.2021.10.017. [DOI] [PubMed] [Google Scholar]
- 30.Seo JJ. Hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis: recent advances and controversies. Blood Res. 2015;50:131–139. doi: 10.5045/br.2015.50.3.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiesa R, Veys P. Reduced-intensity conditioning for allogeneic stem cell transplant in primary immune deficiencies. Expert Rev Clin Immunol. 2012;8:255–266. doi: 10.1586/eci.12.9. quiz 267. [DOI] [PubMed] [Google Scholar]
- 32.Contreras CF, Long-Boyle JR, Shimano KA, Melton A, Kharbanda S, Dara J, Higham C, Huang JN, Cowan MJ, Dvorak CC. Reduced toxicity conditioning for nonmalignant hematopoietic cell transplants. Biol Blood Marrow Transplant. 2020;26:1646–1654. doi: 10.1016/j.bbmt.2020.06.004. [DOI] [PubMed] [Google Scholar]
- 33.Kuskonmaz B, Ayvaz D, Gokce M, Ozgur TT, Okur FV, Cetin M, Tezcan I, Uckan Cetinkaya D. Hematopoietic stem cell transplantation in children with Griscelli syndrome: a single-center experience. Pediatr Transplant. 2017;21 doi: 10.1111/petr.13040. [DOI] [PubMed] [Google Scholar]
- 34.Al-Mofareh M, Ayas M, Al-Seraihy A, Siddiqui K, Al-Jefri A, Ghemlas I, Alsaedi H, El-Solh H, Al-Sweedan S, Al-Saud B, Al-Mousa H, Al-Dhekri H, Arnaout R, Mohammed R, Al-Muhsen S, Al-Ahmari A. Hematopoietic stem cell transplantation in children with Griscelli syndrome type 2: a single-center report on 35 patients. Bone Marrow Transplant. 2020;55:2026–2034. doi: 10.1038/s41409-020-0885-6. [DOI] [PubMed] [Google Scholar]
- 35.Pachlopnik Schmid J, Moshous D, Boddaert N, Neven B, Dal Cortivo L, Tardieu M, Cavazzana-Calvo M, Blanche S, de Saint Basile G, Fischer A. Hematopoietic stem cell transplantation in Griscelli syndrome type 2: a single-center report on 10 patients. Blood. 2009;114:211–218. doi: 10.1182/blood-2009-02-207845. [DOI] [PubMed] [Google Scholar]
- 36.Hamidieh AA, Pourpak Z, Yari K, Fazlollahi MR, Hashemi S, Behfar M, Moin M, Ghavamzadeh A. Hematopoietic stem cell transplantation with a reduced-intensity conditioning regimen in pediatric patients with Griscelli syndrome type 2. Pediatr Transplant. 2013;17:487–491. doi: 10.1111/petr.12092. [DOI] [PubMed] [Google Scholar]