

Abstract
Background:
Erythropoiesis stimulating agents (ESAs) are the first-line therapy in patients with lower-risk myelodysplastic syndromes (LR-MDS). Some predictive factors for ESAs response have been identified. Type and number of somatic mutations have been associated with prognosis and response to therapies in MDS patients.
Objectives:
The objective was to evaluate the outcomes after ESAs in patients with LR-MDS and to address the potential predictive value of somatic mutations in ESAs-treated patients.
Design:
Multi-center retrospective study of a cohort of 722 patients with LR-MDS included in the SPRESAS (Spanish Registry of Erythropoietic Stimulating Agents Study) study. Retrospective analysis of 65 patients with next generation sequencing (NGS) data from diagnosis.
Methods:
ESAs’ efficacy and safety were evaluated in patients receiving ESAs and best supportive care (BSC). To assess the potential prognostic value of somatic mutations in erythroid response (ER) rate and outcome, NGS was performed in responders and non-responders.
Results:
ER rate for ESAs-treated patients was 65%. Serum erythropoietin (EPO) level <200 U/l was the only variable significantly associated with a higher ER rate (odds ratio, 2.45; p = 0.036). Median overall survival (OS) in patients treated with ESAs was 6.7 versus 3.1 years in patients receiving BSC (p < 0.001). From 65 patients with NGS data, 57 (87.7%) have at least one mutation. We observed a trend to a higher frequency of ER among patients with a lower number of mutated genes (40.4% in <3 mutated genes versus 22.2% in ⩾3; p = 0.170). The presence of ⩾3 mutated genes was also significantly associated with worse OS (hazard ratio, 2.8; p = 0.015), even in responders. A higher cumulative incidence of acute myeloid leukemia progression at 5 years was also observed in patients with ⩾3 mutated genes versus <3 (33.3% and 10.7%, respectively; p < 0.001).
Conclusion:
This large study confirms the beneficial effect of ESAs and the adverse effect of somatic mutations in patients with LR-MDS.
Keywords: erythropoiesis stimulating agents, lower-risk myelodysplastic syndromes, myelodysplastic syndromes, next generation sequencing, somatic mutations
Background
Lower-risk myelodysplastic syndromes (LR-MDS) patients usually present with cytopenias (mainly anemia), and absence of excess blasts or poor prognosis cytogenetic abnormalities. These patients have a substantially longer overall survival (OS) than higher-risk MDS (HR-MDS),1,2 but their expectancy and quality of life are compromised by the severity of anemia and the need of red blood cell (RBC) transfusions, with the consequent risk of iron overload, alloimmunization, and cardiovascular disease.
Most patients with LR-MDS and symptomatic anemia receive erythropoiesis stimulating agents (ESAs) as first line treatment. However, the probability of erythroid response (ER) and its duration are highly variable. Predictive factors for ESAs response include serum erythropoietin (EPO) level, severity of RBC transfusion dependency (RBC-TD), and International Prognostic Scoring System (IPSS)/IPSS Revised (IPSS-R) score.3–7 The absence of ER and a shorter duration of response have been associated with poor OS and higher risk of acute myeloid leukemia (AML) transformation. 8 Apart from lenalidomide in patients with deletion 5q or luspatercept in MDS with ring sideroblasts and SF3B1 mutations, no alternative treatment for most anemic LR-MDS patients refractory or intolerant to ESAs has been approved by regulatory agencies.
Several somatic mutations have been associated with poor prognosis in LR-MDS, like TP53, SRSF2, RUNX1, ASXL1, EZH2, or NRAS mutations, while only isolated SF3B1 mutations have been clearly associated with a good prognosis.9–12 Also, the presence of somatic mutations could influence the response to therapies in MDS patients.12–21 The main purposes of this study were to evaluate the outcomes after ESAs and to compare them with those obtained with the best supportive care (BSC) in a large series of anemic patients with LR-MDS from the Spanish group of MDS (GESMD), and to address the potential predictive value of somatic mutations in ESAs-treated patients.
Design
From April to December 2013, 959 patients with MDS from the RESMD (Spanish Registry of MDS) were included in the SPRESAS (Spanish Registry of Erythropoietic Stimulating Agents Study) study. Patients should have (1) de novo MDS, (2) hemoglobin level <11 g/dl, (3) lower-risk according to the IPSS (low and intermediate-1), (4) not have received any disease modifying treatment, (5) be diagnosed before December 2011, and (6) have sufficient available data. In total, 237 cases were excluded from the study (23 treated with disease modifying agents, 38 with secondary MDS, 21 with IPSS > intermediate-1 risk, 97 with hemoglobin level >11 g/dl, 31 diagnosed after December 2011, 3 progressing to HR-MDS, and 24 with incomplete data). The 722 remaining patients constituted the subject of this study (Table 1). The primary endpoint was to evaluate the efficacy of ESAs (ER rate, ER duration, OS length, and cumulative incidence of AML transformation). Second endpoints were to address the safety of ESAs treatment and to determinate the potential influence of somatic mutations on outcomes in ESAs-treated patients.
Table 1.
Main characteristics of ESA and BSC patients in SPRESAS study.
| Patients and disease characteristics | ESAs (n = 530) | Best supportive care (n = 192) | p Value |
|---|---|---|---|
| Age (median) [p25–p75] | 77 years [70–80] | 76 years [68–81] | 0.042 |
| Gender | 530 | 192 | 0.398 |
| Male | 280 (54%) | 111 (58%) | |
| Female | 240 (46%) | 81 (42%) | |
| WHO 2008 | 413 | 121 | <0.001 |
| RCUD | 48 (11.6%) | 13 (10.7%) | |
| RCMD | 182 (44.1%) | 42 (34.7%) | |
| RARS | 101 (24.4%) | 14 (11.6%) | |
| RAEB-1 | 18 (4.4%) | 19 (15.7%) | |
| RAEB-2 | 2 (0.5%) | 11 (10%) | |
| MDS NOS | 3 (0.7%) | 0 (0%) | |
| MDS with del(5q−) | 17 (4.1%) | 10 (8.3%) | |
| CMML | 33 (8%) | 10 (8.3%) | |
| MDS/MPN | 7 (1.7%) | 2 (1.7%) | |
| Not available | 117 | 71 | |
| IPSS | 484 | 185 | <0.001 |
| Low | 305 (63%) | 74 (40%) | |
| Intermediate-1 | 179 (37%) | 111 (60%) | |
| Not available | 46 | 7 | |
| IPSS-R | 484 | 185 | <0.001 |
| Very low | 130 (26.9%) | 18 (9.7%) | |
| Low | 285 (58.9%) | 94 (50.8%) | |
| Intermediate | 63 (13%) | 57 (30.8%) | |
| High | 6 (1.2%) | 16 (8.6%) | |
| Not available | 46 | 7 | |
| Hemoglobin (median) [p25–p75] | 10 g/dl [9–10] | 9 g/dl [8–10] | <0.001 |
| Neutrophils (median) [p25–p75] | 2.74 × 109/l [1.6–4.0] | 2.58 × 109/l [1.6–4.3] | 0.742 |
| Platelets (median) [p25–p75] | 214 × 109/l [131–299] | 198.5 × 109/l [110–315] | 0.384 |
| Bone marrow blasts (median) [p25–p75] | 1% [0–2] | 3% [1–5] | <0.001 |
| EPO (median) [p25–p75] | 59.5 U/l [29.0–114.2] | 142.5 U/l [42.8–372.8] | 0.003 |
| Ferritin (median) [p25–p75] | 315 ng/ml [142–555] | 376 ng/ml [159–813] | 0.009 |
| Transfusion dependency | 329 | 192 | <0.001 |
| Yes | 185 (56.2%) | 184 (95.8%) | |
| No | 144 (43.8%) | 8 (4.2%) | |
| Not available | 201 | 0 | |
| Cytogenetics IPSS-R | 484 | 185 | 0.227 |
| Very good | 28 (5.8%) | 5 (2.7%) | |
| Good | 418 (86.5%) | 163 (88.1%) | |
| Intermediate | 37 (7.2%) | 14 (7.6%) | |
| Poor | 3 (0.6%) | 2 (1.1%) | |
| Very poor | 0 (0%) | 1 (0.5%) | |
| Not available | 46 | 7 | |
| ECOG | 74 | 9 | 0.200 |
| <1 | 59 (79.7%) | 5 (55.6%) | |
| ⩾2 | 15 (20.3%) | 4 (45.4%) |
CMML, chronic myelomonocytic leukemia; ECOG, Eastern Cooperative Oncology Group; ESA, erythropoietic stimulating agents; IPSS, international prognostic scoring system; IPSS-R, IPSS revised; MDS-U, myelodysplastic syndrome unclassifiable; MDS/MPN, myelodysplastic syndrome/myeloproliferative neoplasm; NOS, not otherwise specified; RAEB, refractory anemia with excess blasts; RARS, refractory anemia with ringed sideroblasts; RCMD, refractory cytopenia with multilineage dysplasia; RCUD, refractory cytopenia with unilineage dysplasia; SC, supportive care; SPRESAS, Spanish Registry of Erythropoietic Stimulating Agents Study.
We also retrospectively analyzed data from 65 patients treated with ESAs from five participating centers and diagnosed between 1997 and 2011. Date of last follow-up was updated to August 2019. Only patients with at least 12 months of ER duration and non-responders were included in the analysis, based on the results of a prior study demonstrating similar outcomes between non-responders and responders with a shorter ER duration. 8
Methods
Definitions
ER rate was defined according to the International Working Group 2006 criteria. 22 OS was calculated from date of diagnosis to date of death from any cause or last follow up for living patients. Transformation to AML was defined as the presence of ⩾20% of blast in bone marrow (BM) or peripheral blood.
Mutations sub-analysis
We selected genomic DNA from BM mononucleated cells at diagnosis; only in one case we used peripheral blood. DNA extraction was performed with QIAgen (Qiagen©, Venlo, Netherlands) and quantification by fluorometry with Qubit 2.0 (Life Technologies©, Carlsbad, CA, USA). Detection of somatic mutations was performed by next generation sequencing (NGS), using a capture sequencing strategy with MiSeq platform (Illumina©, San Diego, CA, USA) and a 117 myeloid genes panel (Table 2) designed with DesignStudio software (Illumina©) using 3259 probes targeting 1740 regions, including exons, splicing regions, and UTR 3′/5′ regions. Only those variants located in exonic or splicing regions that cause a change in the protein sequence were considered; polymorphisms, defined as variants with a minor allele frequency in the healthy people higher than 1%, were also excluded.
Table 2.
117 genes NGS panel used in the study.
| ABL1, AEBP2, ARID2, ASXL1, ATRX, BCAS1, BCOR, BCORL1, BCR, BMI1, BRAF, CALR, CBFB, CBL, CBLB, CBLC, CD177, CDH13, CDH23, CDH3, CDK2, CDKN2A, CEBPA, CREBBP, CSF3R, CSNK1A1, CTCF, CTNNA1, CUX1, DNMT3A, UBR5, EGFR, EIF2AK2, ENG, EP300, ETV6, EZH2, FBXW7, FLT3, G3BP1, GATA1, GATA2, GCAT, GNAS, GNB1, HRAS, IDH1, IDH2, IKZF1, IL3, IRF1, JAK1, JAK2, JAK3, JARID2, JKAMP, KDM6A, KIT, KRAS, LUC7L2, MECOM, KMT2A, KMT2D, MPL, MTOR, NF1, NOTCH1, NPM1, NRAS, NR2F6, NTRK1, NUP98, PBRM1, PDGFRA, PDGFRB, PHF19, PHF6, PHLPP1, PTEN, PTPN1, PTPN11, RAD21, RARA, RET, RPS14, RUNX1, SALL4, SBDS, SETBP1, SETD2, SF1, SF3A1, SF3B1, SFPQ, SH2B3, SMC1A, SMC3, SPARC, SRSF2, STAG1, STAG2, SUZ12, TCL1B, TERC, TERT, TET2, TGM2, TIMM50, TNFAIP3, TP53, TYK2, U2AF1, UMODL1, USB1, WASF3, WT1, ZRSR2 |
NGS, next generation sequencing.
Sequencing was performed in two steps: first, capture and enrichment using a Nextera Rapid Capture Enrichment Custom methodology, starting from 50 ng of DNA from each sample; and second, generating clusters and sequencing with the MiSeq platform, using a 300 cycles reagent kit (obtaining an average coverage of 500–600 lectures). Minimum median coverage of sequenced exons was 50–100 reads with more than 10–20 mutated reads. IGV software (Broad Institute©, Cambridge, MA, USA) was used for the variant analysis. All variants were aligned with the reference genome (GRCh37/hg19). Variant calling and annotation were performed with VarScan (v2.4) (McDonnell Genome Institute, St. Louis, MO, USA), SAMTools v1.3.1 (Genome Research Limited, Hinxton, Saffron Walden, UK), and ANNOVAR (Center for Applied Genomics, Philadelphia, PA, USA). We used databases COSMIC and ClinVar and predictors SIFT, PolyPhen-2, and Mutation Taster to evaluate the oncogenic potential of variants. Variants with a variant allele frequency (VAF) <5% were validated re-sequencing amplicons with GS-Junior (Roche©, Basel, Switzerland) and those with a VAF >5% with Sanger method.
Statistical analysis
Comparisons of proportions and ranks in different variables were made with the Chi-square test and Fisher’s exact test. OS was analyzed by the Kaplan–Meier method and curves were compared by the log-rank test. A Cox multivariable model was used to adjust for clinical characteristics. For SPRESAS study, a landmark analysis was performed. Patients in the BSC arm were censored if they died before day 100 from diagnosis (the median time to ESAs treatment onset in our series). The cumulative incidence of AML progression (CI of AML) was calculated using competing risks and compared using the Gray test. A two-side 0.05 p value was considered as statistically significant. Calculations were performed using R (v3.1.1) (The R Foundation for Statistical Computing, Vienna, Austria), IBM SPSS v20.0 and v25.0 (IBM Corporation©, Armonk, NY, USA), and Xlstat version 2018.6 (Microsoft Corporation©, Redmond, WA, USA) statistical packages.
Equator network guideline accordance
This paper is in accordance with the Strengthening the Reporting of Observational Studies in Epidemiology statement 23 (Supplemental Material).
Results
SPRESAS study
Patients and disease characteristics
From the entire cohort of 722 patients, 530 were treated with ESAs and 192 managed only with BSC. Median follow up was 2.9 years (range, 0.02–25.5), 3.1 years for ESAs-treated group, and 2.4 years for BSC group (p = 0.002). Median age at diagnosis was 76 years (range, 19–101). Patients and disease characteristics from SPRESAS study are summarized in Table 1.
Outcome
ER in ESAs-treated patients
Data on ER were available in 478 patients, of whom 310 (64.8%) responded, with a median ER duration of 1.8 years (range, 0.23–10.4) in the 284 patients with ER duration available. Two-hundred sixty-five (93%) and 213 (75%) sustained the response at 6 and 12 months, respectively. No differences in ER rate and ER duration were observed among ESAs compounds: darbepoetin (n = 243), epoetin α (n = 24), epoetin β (n = 75), and others (n = 15). ER was similar in patients with serum EPO level <100 U/l (67.9%) and <200 U/l (66.6%).
Serum EPO level <200 U/l was the only variable independently associated with a higher ER rate in the multivariable analysis [odds ratio, 2.45; 95% confidence interval (CI): 1.06–5.64; p = 0.036] (Table 3).
Table 3.
Univariable and multivariable analysis for erythroid response (SPRESAS study).
| Variables | Univariable | Multivariable | ||||
|---|---|---|---|---|---|---|
| OR | p Value | OR | p Value | CI 95% | CI 95% | |
| Age (⩾77 versus <77 [ref]) | 0.780 | 0.196 | 1.629 | 0.157 | 0.828 | 3.204 |
| Sex (male versus female [ref]) | 1.088 | 0.663 | 1.296 | 0.450 | 0.661 | 2.540 |
| Transfusion dependency (yes versus no [ref]) | 1.655 | 0.033 | 1.898 | 0.087 | 0.911 | 3.953 |
| IPSS (intermediate-1 versus low [ref]) | 1.779 | 0.005 | 1.348 | 0.438 | 0.634 | 2.867 |
| Hemoglobin (continuous) | 0.785 | 0.003 | 1.104 | 0.502 | 0.827 | 1.473 |
| Hemoglobin (<10 versus ⩾10 g/dl [ref]) | 1.511 | 0.032 | ||||
| Neutrophils (<800 versus ⩾800 g/dl [ref]) | 1.160 | 0.767 | ||||
| Peripheral blood blasts (>0 versus 0 [ref]) | 2.929 | 0.045 | 1.332 | 0.740 | 0.240 | 7.244 |
| Bone marrow blasts (continuous) | 1.588 | 0.022 | 1.104 | 0.398 | 0.895 | 1.322 |
| Serum EPO (⩾200 versus <200 U/l [ref]) | 3.969 | <0.001 | 2.446 | 0.036 | 1.061 | 5.639 |
| Ferritin (continuous) | 1.001 | 0.001 | 1.000 | 0.429 | 0.999 | 1.000 |
| Cytogenetics (intermediate versus favorable [ref]) | 1.876 | 0.056 | ||||
| WHO 2008 (RARS versus RCUD + RCMD [ref]) | 0.859 | 0.574 | ||||
| RAEB | 2.323 | 0.106 | ||||
| MDS 5q− | 2.613 | 0.059 | ||||
| Others MDS | 2.821 | 0.008 | ||||
EPO, erythropoietin; IPSS, International Prognostic Scoring System; MDS, myelodysplastic syndrome; RAEB, refractory anemia with excess blasts; RARS, refractory anemia with ringed sideroblasts; RCMD, refractory cytopenia with multilineage dysplasia; RCUD, refractory cytopenia with unilineage dysplasia; SPRESAS, Spanish Registry of Erythropoietic Stimulating Agents Study.
Safety of ESAs treatment
Adverse events were reported in 10 patients, of which 3 were severe adverse events (thrombocytopenia, mucocutaneous bleeding, and traumatic fall). None of them were considered as treatment-related by the investigator.
Overall survival
Median OS from diagnosis was 6.7 years (95% CI, 5.5–7.2) for ESAs-treated patients and 3.1 years (95% CI, 2.6–4.4) for BSC patients (p < 0.001) (Figure 1). OS at 5 years was 58% and 37% for each group, respectively. Among ESAs-treated patients, median OS from ESAs onset was 6.8 years (95% CI, 5.7–not reached) and 3.1 years (95% CI, 2.4–3.8) for each group, respectively (p < 0.001) (Figure 2).
Figure 1.

Overall survival in SPRESAS study according to the management strategy: best supportive care versus ESAs treatment (landmark analysis).
ESA, erythropoiesis stimulating agents; SPRESAS, Spanish Registry of Erythropoietic Stimulating Agents Study.
Figure 2.

Overall survival from ESAs onset in SPRESAS study according to erythroid response.
ESA, erythropoiesis stimulating agents; SPRESAS, Spanish Registry of Erythropoietic Stimulating Agents Study.
Variables significantly associated with a better OS in the multivariable analysis were younger age (hazard ratio, 1.05; 95% CI, 1.01–1.04; p < 0.001), female gender (hazard ratio, 0.67; 95% CI, 0.53–0.85; p = 0.036), higher hemoglobin level (hazard ratio, 0.874; 95% CI, 0.804–0.951; p = 0.001), lower leukocyte count (hazard ratio, 1.02; 95% CI, 1.02–1.04; p = 0.003), low-risk IPSS (hazard ratio, 1.91; 95% CI, 1.51–2.41; p < 0.001), and ESAs treatment (hazard ratio, 0.65; 95% CI, 0.51–0.83; p < 0.001).
Non-responders had similar outcome to responders with ER duration <12 months and <6 months, with a median OS from ESAs onset and from 12 and 6 months after ESAs onset, respectively, of 3.09, 2.5 (p = 0.851), and 2.37 years (p = 0.238). Conversely, responders with ER duration ⩾12 and ⩾6 months had longer OS: 6.2 years (p < 0.001) and 6.5 years (p < 0.001), respectively.
CI of AML at 5 years from diagnosis was 18% for ESAs-treated patients and 20% for BSC patients (p = 0.350). According to response to ESAs, CI of AML was 13% for responders and 28% for non-responders (p < 0.001). Variables significantly associated with a lower CI of AML in the multivariable analysis were female gender (hazard ratio, 0.61; 95% CI, 0.40–0.92; p = 0.02), lower BM percentage of blasts (hazard ratio, 1.23; 95% CI, 1.13–1.33; p < 0.001), and low-risk IPSS (hazard ratio, 1.99; 95% CI, 1.20–3.30; p = 0.007). Among ESAs-treated patients, CI of AML at 5 years from ESAs onset was 20% in responders versus 37% in non-responders (p < 0.001).
Mutations sub-analysis
Patients and disease characteristics
Among 65 patients with available samples at diagnosis for NGS, 23 (35%) were ESAs-treated patients with ER duration ⩾12 months and 42 (65%) were non-responders. Among responders, 13 patients (57%) sustained ER until the date of last follow-up while 10 (43%) lost their response. Median ER duration was 4.6 years (range, 1–11.9). Median ESAs onset from diagnosis was 0.4 years (range, 0–14.1). There were 15 patients (23.1%) receiving granulocyte-colony stimulating factor (G-CSF) concomitant with ESAs.
The most frequent cytogenetic abnormality was del(5)(q13q31), that was present in five patients (7.7%), all of them categorized as MDS with del(5q−). Other cytogenetic abnormalities were: −Y and +8 in three patients, del(11q) in two patients and del(20q), +17, and inv(9) in one patient each abnormality. In one case del(7q) was detected by fluorescence in situ hybridation (FISH) but not by conventional karyotype.
Clinical characteristics of responders and non-responders are resumed in Table 4. Comparing patients from ESAs-treated cohort from SPRESAS registry with patients from mutations sub-analysis, we found that both groups were similar regarding most of variables.
Table 4.
Main characteristics of responders and non-responders in mutations sub-analysis.
| Patients and disease characteristics | Response, n = 23 (35.4%) | Non-response, n = 42 (64.6%) | p Value |
|---|---|---|---|
| Age (median) [p25–p75] | 76 years [71–83] | 76 years [65–80] | 0.422 |
| Gender | 23 | 76 | 0.200 |
| Male | 9 (39.1%) | 24 (57.1%) | |
| Female | 14 (60.9%) | 18 (42.9%) | |
| WHO 2008 | 21 | 41 | 0.042 |
| <5% blasts | 20 (95.2%) | 30 (73.2%) | |
| RCUD | 2 (9.5%) | 2 (4.9%) | |
| RARS | 9 (42.9%) | 9 (22%) | |
| RCMD | 8 (38.1%) | 17 (41.5%) | |
| MDS-U | 1 (4.35%) | 2 (4.9%) | |
| >5% blasts and CMML | 1 (4.8%) | 6 (14.5%) | |
| RAEB-1 | 0 (0%) | 1 (2.4%) | |
| RAEB-2 | 0 (0%) | 1 (2.4%) | |
| CMML | 1 (4.3%) | 3 (7.3%) | |
| Others MDS/MPN | 0 (0%) | 1 (2.4%) | |
| MDS with del(5q) | 0 (0%) | 5 (12.2%) | |
| Not available | 2 | 1 | |
| IPSS | 23 | 40 | 0.781 |
| Low | 17 (73.9%) | 28 (70%) | |
| Intermediate-1 | 6 (26.1%) | 12 (30%) | |
| Not available | 0 | 2 | |
| IPSS-R | 23 | 37 | 0.157 |
| Very low | 9 (39.1%) | 8 (20.5%) | |
| Low | 13 (56.5%) | 25 (64.1%) | |
| Intermediate | 1 (4.3%) | 6 (15.3%) | |
| Not available | 0 | 5 | |
| Hemoglobin (median) [p25–p75] | 9.8 g/dl [9.3–10.4] | 9.2 g/dl [8.1–10.1] | 0.033 |
| Hemoglobin | 23 | 42 | 0.188 |
| >8 g/dl | 21 (91.3%) | 32 (76.2%) | |
| <8 g/dl | 2 (8.7%) | 10 (23.8%) | |
| Platelets (median) [p25–p75] | 270 × 109/l [196–453] | 230 × 109/l [185–315] | 0.244 |
| Leucocytes (median) [p25–p75] | 5.76 × 109/l [3.9–8.4] | 4.62 × 109/l [3.4–6.6] | 0.144 |
| EPO (median) [p25–p75] | 47.9 U/l [22–118] | 110.5 U/l [41–172] | 0.027 |
| EPO 100 | 18 | 26 | 0.124 |
| <100 U/l | 13 (72.2%) | 12 (46.2%) | |
| ⩾100 U/l | 5 (21.7%) | 14 (33.3%) | |
| Not available | 5 | 16 | |
| EPO 200 | 18 | 26 | 0.211 |
| <200 U/l | 17 (94.4%) | 20 (76.9%) | |
| ⩾200 U/l | 1 (5.6%) | 6 (23.1%) | |
| Not available | 5 | 16 | |
| Ferritin (median) [p25–p75] | 449 ng/ml [256–621] | 368 ng/ml [p25–p75] | 0.652 |
| Ferritin | 18 | 35 | 1.000 |
| <500 ng/ml | 12 (66.7%) | 24 (68.6%) | |
| >500 ng/ml | 6 (33.3%) | 11 (31.4%) | |
| Not available | 5 | 7 | |
| Transfusion dependency | 23 | 42 | <0.001 |
| Yes | 11 (47.8%) | 39 (92.9%) | |
| No | 12 (52.5%) | 3 (7.1%) | |
| Cytogenetics | 23 | 39 | 0.034 |
| Normal | 21 (91.3%) | 26 (66.7%) | |
| Abnormal | 2 (8.7%) | 13 (33.3%) | |
| Not available | 0 | 3 | |
| Mutated genes | 23 | 42 | 0.248 |
| <3 | 19 (82.6%) | 28 (66.7%) | |
| ⩾3 | 4 (17.4%) | 14 (33.3%) |
CMML, chronic myelomonocytic leukemia; EPO, erythropoietin; IPSS, International Prognostic Scoring System; IPSS-R, IPPS Revised; MDS/MPN, myelodisplastic syndrome/myeloproliferative neoplasm; MDS-U, myelodysplastic syndrome unclassifiable; RAEB, refractory anemia with excess blasts; RARS, refractory anemia with ringed sideroblasts; RCMD, refractory cytopenia with multilineage dysplasia; RCUD, refractory cytopenia with unilineage dysplasia; WHO, World Health Organization.
Eight patients (12.3%) had no mutations while 57 patients (87.7%) had at least one mutation. Twenty patients (30.8%) had one mutated gene, 19 (29.2%) had two, 6 (9.2%) had three, 7 (10.8%) had four, and 5 patients had five mutated genes (7.7%). The median number of somatic mutations per patient was 2. Only three patients have two mutations in the same gene: two in SF3B1 and one in TP53. The median VAF was 34.6% (range, 2.7–92.7). For the statistical analysis, patients were divided in two groups: <3 and ⩾3 mutated genes.
Most frequently mutated genes were: SF3B1 (54.8%, 35 patients), DNMT3A (16.9%, 11 patients), TET2 (13.8%, 9 patients), SRSF2 (10.8%, seven patients), ZRSR2 (9.2%, 6 patients), ASXL1 (7.7%, 5 patients), IDH2 (7.7%, 5 patients), EZH2 (6.2%, 4 patients), STAG2 (6.2%, 4 patients), and TP53 (6.2%, 4 patients). Other frequently reported mutations in MDS were detected in <5% patients. There were 47 patients (72.3%) with mutations in splicing genes, being the most frequently mutated group; only three patients had two splicing genes simultaneously mutated: two with SF3B1 and SRSF2 and one with SF3B1 and ZRSR2. Mutations in TET2 and DNMT3A frequently co-occurred with other mutations: 8 out of 9 patients with TET2 mutations and 9 out of 11 patients with DNMT3A mutations presented mutations in splicing genes, mostly SF3B1 (6/9 and 9/11, respectively). Mutations in signaling pathways and transcription factors genes, described commonly associated with disease progression, were infrequent (16.9% and 26.2%, respectively) but nearly always associated to other mutations (9/11 patients with mutations in transcription genes and 17/17 with mutations in signaling genes). Particularly, from patients with mutations in signaling pathways genes, only 17.6% presented <3 mutated genes, as compared to patients with mutations in splicing genes, in which only 35.4% presented ⩾3 mutated genes (Figure 3).
Figure 3.

Patients with LR-MDS treated with ESAs: somatic mutations and cytogenetics.
ESA, erythropoiesis stimulating agents; LR-MDS, lower-risk myelodysplastic syndromes.
Outcome
ER and mutational status
ER was 40.4% among patients with <3 mutated genes and 22.2% in ⩾3 (p = 0.170) (Figure 4). The percentage of patients with <3 mutated genes was 82.6% among responders versus 66.7% among non-responders. No mutation proved to be clearly related to ER rate, however, we observed a trend toward a better response in patients with TET2 mutations versus TET2 wild-type patients: 66.7% (6/9 patients) versus 30.4% (p = 0.057). Variables related with ER rate in the univariable analysis were RBC-TD (p < 0.001), cytogenetics (p = 0.034), serum EPO level (p = 0.025), hemoglobin level (p = 0.032), and BM percentage of blasts (p = 0.015). None of them reached the statistical signification in the multivariable analysis.
Figure 4.

Erythroid response according to the number of mutated genes.
Only two variables were significantly associated with ER duration in the univariable analysis, RBC-TD (hazard ratio, 3.53; 95% CI, 1.04–12.0; p = 0.043), and ⩾3 mutated genes (hazard ratio, 4.37; 95% CI, 1.07–18.78; p = 0.047), but none of them retained their impact on multivariable analysis.
Overall survival
After a median follow-up in alive patients of 9.5 years (range, 2.7–17.6), 50 patients (76.9%) have died (14 responders and 36 non-responders), with a median OS from the diagnosis of 5.9 years (95% CI, 4.1–7.7) in the entire cohort, 8.0 years (95% CI, 7.0–9.0) in responders, and 4.4 years (95% CI, 2.18–6.69) in non-responders (p = 0.007) [Figure 5(a)]. In multivariable analysis variables independently associated with lower OS were older age (hazard ratio, 1.1; 95% CI, 1.05–1.15; p < 0.001), male gender (hazard ratio, 2.41; 95% CI, 1.15–5.10; p = 0.02), RBC-TD (hazard ratio, 3.49; 95% CI, 1.42–8.60; p = 0.007), intermediate-1 IPSS (hazard ratio, 2.83; 95% CI, 1.28–6.25; p = 0.01), and having ⩾3 mutated genes (hazard ratio, 2.8; 95% CI, 1.22–6.43; p = 0.015). Co-variables with a disproportionately high CI (WHO 2018 diagnosis, STAG2 mutations, GNAS mutations) were excluded from the multivariable analysis (Table 5). STAG2 mutations were always subclonal.
Figure 5.
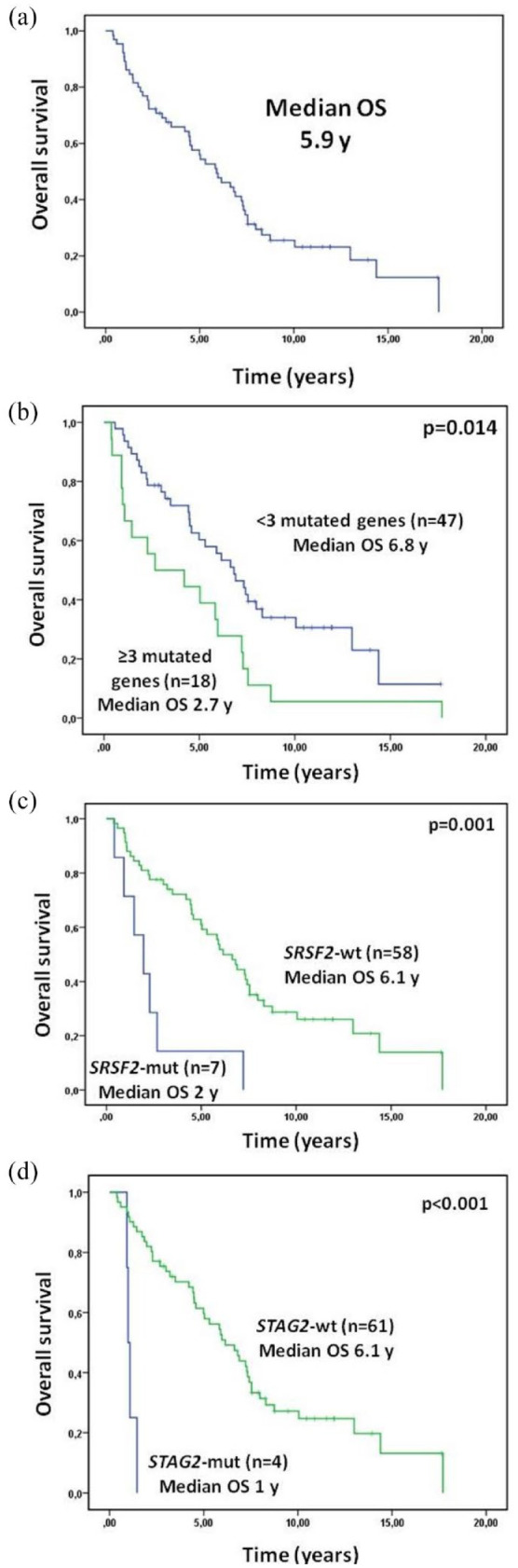
Overall survival: entire cohort (a) and according to the number of mutated genes (b), SRSF2 mutational status (c), and STAG2 mutational status (d).
Table 5.
Univariable and multivariable analysis for overall survival (mutations sub-analysis).
| Variable | Univariable | Multivariable | ||||
|---|---|---|---|---|---|---|
| HR | p Value | HR | p Value | CI 95% | CI 95% | |
| Age (continuous) | 1.077 | <0.001 | 1.102 | <0.001 | 1.053 | 1.153 |
| Sex (male versus female [ref]) | 1.869 | 0.036 | 2.419 | 0.020 | 1.148 | 5.096 |
| Transfusion dependency (yes versus not [ref]) | 2.791 | 0.012 | 3.490 | 0.007 | 1.417 | 8.599 |
| IPSS (int-1 versus low [ref]) | 2.297 | 0.010 | 2.827 | 0.010 | 1.279 | 6.251 |
| Hemoglobin (continuous) | 0.786 | 0.055 | ||||
| Hemoglobin (<8 versus ⩾8 g/dl [ref]) | 1.523 | 0.259 | ||||
| Leukocytes (continuous) | 1.023 | 0.225 | ||||
| Platelets (continuous) | 0.999 | 0.273 | ||||
| Bone marrow blasts (continuous) | 1.257 | 0.004 | 1.028 | 0.778 | 0.848 | 1.247 |
| Serum EPO (continuous) | 0.999 | 0.552 | ||||
| Serum EPO (⩾100 versus <100 U/l [ref]) | 0.846 | 0.631 | ||||
| Serum EPO (⩾200 versus <200 U/l [ref]) | 0.873 | 0.779 | ||||
| Ferritin (⩾500 versus <500 ng/ml [ref]) | 0.917 | 0.797 | ||||
| Cytogenetics (abnormal versus normal [ref]) | 1.969 | 0.045 | 1.371 | 0.421 | 0.635 | 2.956 |
| WHO 2008 classification (<5% blasts versus 5q− [ref]) | 0.552 | 0.276 | ||||
| ⩾5% blasts and CMML versus <5% blasts [ref] | 5.632 | 0.012 | ||||
| Mutated genes (⩾3 versus <3 [ref]) | 1.247 | 0.028 | 2.803 | 0.015 | 1.223 | 6.425 |
| SRSF2 (mutated versus wt [ref]) | 3.914 | 0.001 | 2.285 | 0.157 | 0.727 | 7.182 |
| STAG2 (mutated versus wt [ref])* | 11.665 | <0.001 | ||||
| GNAS (mutated versus wt [ref])* | 6.550 | 0.003 | ||||
CMML, chronic myelomonocytic leukemia; EPO, erythropoietin; IPSS, International Prognostic Scoring System; IPSS-R, IPSS Revised; WHO, World Health Organization.
STAG2 and GNAS were not included in the multivariable analysis because of a disproportionally high CI.
Patients with <3 mutated genes showed an improved outcome, with a median OS of 6.8 years and a 5-years OS of 60.3% versus 2.7 years and 4% in patients with ⩾3 mutated genes, respectively; p = 0.014) [Figure 5(b)]. Among responders, the higher number of mutated genes retained its unfavorable prognosis (5-years OS 75% for ⩾3 mutated genes versus 84% for <3 mutated genes; p = 0.012).
CI of AML
Eighteen patients (27.7%) transformed into AML, 3 responders, and 15 non-responders. CI of AML was 17.5% at 5 years (95% CI, 9.9–29.2), 10.7% in patients with <3 mutated genes (95% CI, 4.7–24.5), and 33.3% in patients with ⩾3 mutated genes (95% CI, 17.5–63.4; Gray test, p = 0.006) (Figure 6). Additionally, mutations in STAG2 gene were associated with an increased risk of AML at 5 years: 75% in mutated patients (95% CI, 42.6–1.3) versus 13.2% (95% CI, 6.9–25.2) in non-mutated (Gray test, p < 0.001) [Figure 6(c)].
Figure 6.
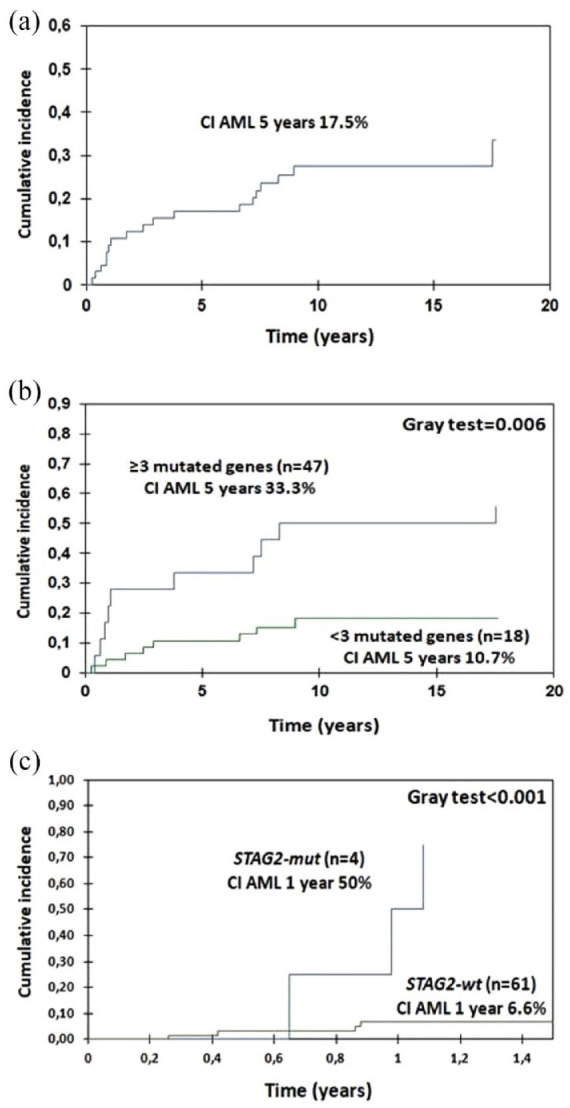
Cumulative incidence of acute myeloid leukemia: entire cohort (a), according to the number of mutated genes (b), and according to STAG2 mutational status (c).
Discussion
In the current study, we report the results of a large analysis on the use of ESAs in anemic LR-MDS patients. The proportion of patients responding to ESAs was 64.8%, similar to that observed in most previous retrospective studies, including an article by Messa with 1145 low-risk patients receiving ESAs versus BSC.3,6,7,24–27 Median duration of ER was 1.8 years, also close to previous reports. The only variable significantly associated with better ER rate in the multivariable analysis was a serum EPO level <200 U/l, validating the results of prior studies (500 U/l in Hellström-Lindberg score, 200 U/l in Park and Santini studies, and 100 U/l in Buckstein and Houston studies).3–6 This cut-off value has been also selected by the European Medicines Agency for the approval of ESAs in LR-MDS, based on the results from the clinical trial with epoetin-α performed by Fenaux. 28 We also confirmed that ESAs were safe and not related with AML progression. In addition to serum EPO level, both IPSS and IPSS-R and RBC-TD have been consistently associated with ER rate,3–6 but in our study these variables were only significant in the univariable analysis.
OS in our series was similar to previously reported. 21 We confirmed a higher OS among responders to ESAs, which has also been previously observed, although it was slightly higher in our study, probably due to a longer follow-up, a lower proportion of patients with RAEB treated with ESAs in our study (3.8%) comparing with others (19–38%) and the inclusion of patients with low and intermediate-1 IPSS exclusively, while other studies also included patients with intermediate-2 and high risk IPSS (11–24%).3,4,27,29 Among responders to ESAs, OS was inferior for patients with an ER duration lower than 6 and 12 months, confirming the data reported by Kelaidi et al., 8 who observed that ER <6 months was associated with a higher mortality and progression to AML and similar to the one seen in non-responders. By contrast, Messa did not observe any difference in OS between responders and non-responders. We argue that this could be due to that they did not differentiate patients according to ER duration. 21
AML transformation was similar in BSC and ESAs-treated patients; nevertheless, we observed that non-responders had a higher CI of AML (37% at 5 years), even higher than that observed in BSC-treated patients (20% at 5 years). The lower incidence of AML among responders could be explained by baseline disease characteristics and mutational profile.
Prevalence of somatic mutations was similar to that observed by Kosmider et al. 30 (87%) in their study about the impact of somatic mutations in LR-MDS patients receiving ESAs. In our study, however, SF3B1 mutations were more frequent (54.8% versus 40.5% in Kosmider’s study), maybe because we included more patients with low IPSS comparing to intermediate-1 (69% versus 45%, respectively). Most of patients have two or more mutations, suggesting that cooperation between mutations could be, almost partially, necessary for the phenotypic disease evolution. We observed that mutations in DNMT3A and TET2, typically related with clonal hematopoiesis, frequently coexisted with mutations in SF3B1 and other genes. We also found that mutations in signaling pathways and transcription factors gene were rare and nearly always associated with mutations in other genes. These data support the hypothesis that disease development and transformation to higher-risk forms are consequence of acquisition and accumulation of successive different mutations.
We observed that a higher number of mutated genes (⩾3) in LR-MDS patients tended to be associated with a lower rate of ER, although statistical signification was not reached, probably because of the limited sample size. Also, ER response duration seemed to be inversely related to the number of mutated genes. Thus, a higher genetic complexity could be associated with refractoriness and could be a predictive factor of response to ESA, similar to serum EPO, RBC-TD, or IPSS. In the same way, adverse impact of somatic mutations in OS was assessed, even among responders to ESAs, according with Kosmider et al. 30 Further studies including a higher number of patients are needed to confirm these findings.
Mutations in three genes were associated with a worse OS in univariable analysis: STAG2, GNAS, and SRSF2 [Figure 5(c) and (d)], all previously described as associated with poor prognosis and high risk features.31–35 Conversely, we did not find clear differences in OS regarding the presence of SF3B1 mutations. In any case, we cannot draw definitive conclusions about the role of isolated mutations, given the small sample size and the difficulty of isolating the role of each mutation separately. Patients with ⩾3 mutated genes also presented a higher incidence of transformation to AML, as well as those with mutations in STAG2. In fact, although only four patients had mutations in this gene, all of them had almost other three mutated genes; in all cases, STAG2 mutations were subclonal. These findings support the hypothesis that AML transformation is commonly caused by secondary or cooperative mutations, which emerged during disease evolution over a prior non-leukemic clone, according to a recently published paper from our group about the role of RAS pathway and cohesin complex mutations in AML evolution among LR-MDS patients. 35
Conclusion
To sum up, ESAs are a safe and efficient first-line treatment for symptomatic anemia in LR-MDS, and ER is associated with favorable outcome. A higher number of somatic mutations in these patients was associated with an adverse prognosis. The absence of response to ESAs, which could be partly related to an adverse mutational profile, might be considered as a warning signal for disease progression and a lower OS expectancy. To better clarify the impact of somatic mutation on the outcome after ESAs treatment in LR-MDS, it should be evaluated in larger series through an international cooperative study.
Supplemental Material
Supplemental material, sj-docx-1-tah-10.1177_20406207231218157 for Outcomes and effect of somatic mutations after erythropoiesis stimulating agents in patients with lower-risk myelodysplastic syndromes by Juan Carlos Caballero, Julio Dávila, María López-Pavía, Esperanza Such, Teresa Bernal, Fernando Ramos, Marisa Calabuig, Jesús María Hernández Sánchez, Helena Pomares, Mercedes Sánchez Barba, María Abáigar, Bernardo González, Brayan Merchán, Reyes Sancho-Tello, Marta Callejas, Carolina Muñoz-Novas, Carlos Cerveró, Guillermo Sanz, Jesús María Hernández Rivas and María Díez Campelo; in Therapeutic Advances in Hematology
Acknowledgments
None.
Footnotes
ORCID iD: Juan Carlos Caballero  https://orcid.org/0000-0001-5269-521X
https://orcid.org/0000-0001-5269-521X
Supplemental material: Supplemental material for this article is available online.
Contributor Information
Juan Carlos Caballero, Hematology Department, Hospital Clínico Universitario de Valladolid, Valladolid, Spain.
Julio Dávila, Hematology Department, Hospital Nuestra Señora de Sonsoles, Ávila, Spain.
María López-Pavía, Hematology Department, Hospital Universitario y Politécnico La Fe, Valencia, Spain; CIBERONC, Instituto de Salud Carlos III, Madrid, Spain; Universidad de Valencia, Valencia, Spain.
Esperanza Such, Hematology Department, Hospital Universitario y Politécnico La Fe, Valencia, Spain; CIBERONC, Instituto de Salud Carlos III, Madrid, Spain; Universidad de Valencia, Valencia, Spain.
Teresa Bernal, Hematology Department, Hospital Universitario Central de Asturias, Oviedo, Spain.
Fernando Ramos, Hematology Department, Hospital Universitario de León, León, Spain.
Marisa Calabuig, Hematology Department, Hospital Clínico Universitario de Valencia, Valencia, Spain.
Jesús María Hernández Sánchez, Instituto de Biología Molecular y Celular del Cáncer, Instituto Biosanitario de Salamanca, Centro de Investigación del Cáncer, USAL-CSIC, Salamanca, Spain.
Helena Pomares, Hematology Department, Hospital Universitario de Bellvitge-Hospital Duran I Reynals, Instituto Catalán de Oncología, L’Hospitalet del Llobregat, Spain.
Mercedes Sánchez Barba, Statistics Department, Universidad de Salamanca, Salamanca, Spain.
María Abáigar, Instituto de Biología Molecular y Celular del Cáncer, Instituto Biosanitario de Salamanca, Centro de Investigación del Cáncer, USAL-CSIC, Salamanca, Spain.
Bernardo González, Hematology Department, Hospital Universitario de Canarias, La Laguna, Spain.
Brayan Merchán, Hematology Department, Hospital Universitario Vall d’Hebron, Barcelona, Spain.
Reyes Sancho-Tello, Hematology Department, Hospital Arnau de Vilanova, Valencia, Spain.
Marta Callejas, Hematology Department, Hospital Universitario Príncipe de Asturias, Alcalá de Henares, Spain.
Carolina Muñoz-Novas, Hematology Department, Hospital Universitario Infanta Leonor, Madrid, Spain.
Carlos Cerveró, Hematology Department, Hospital Virgen de la Luz, Cuenca, Spain.
Guillermo Sanz, Hematology Department, Hospital Universitario y Politécnico La Fe, Valencia, Spain; CIBERONC, Instituto de Salud Carlos III, Madrid, Spain; Universidad de Valencia, Valencia, Spain.
Jesús María Hernández Rivas, Instituto de Biología Molecular y Celular del Cáncer, Instituto Biosanitario de Salamanca, Centro de Investigación del Cáncer, USAL-CSIC, Salamanca, Spain Hematology Department, Hospital Universitario de Salamanca, Salamanca, Spain Instituto Biosanitario de Salamanca, Salamanca, Spain; Hematology Department, Hospital Universitario de Salamanca, Salamanca, Spain Instituto Biosanitario de Salamanca, Salamanca, Spain; Instituto Biosanitario de Salamanca, Salamanca, Spain.
María Díez Campelo, Hematology Department, Hospital Universitario de Salamanca, Paseo de San Vicente 58-182, Salamanca 37007, Spain; Instituto Biosanitario de Salamanca, Salamanca, Spain.
Declarations
Ethics approval and consent to participate: The study was approved by Ethics Committee of Clinical Investigation from the University Hospital of Salamanca (30 April 2012) and was conducted according to the Declaration of Helsinki. Ref. CEIC E.O. 12/249. All patients provided written informed consent.
Consent for publication: Not applicable.
Author contributions: Juan Carlos Caballero: Conceptualization; Data curation; Formal analysis; Methodology; Writing – original draft; Writing – review & editing.
Julio Dávila: Conceptualization; Data curation; Formal analysis; Methodology; Writing – original draft; Writing – review & editing.
María López-Pavía: Data curation; Writing – review & editing.
Esperanza Such: Data curation; Writing – review & editing.
Teresa Bernal: Data curation; Writing – review & editing.
Fernando Ramos: Data curation; Writing – review & editing.
Marisa Calabuig: Data curation; Writing – review & editing.
Jesús María Hernández Sánchez: Investigation; Methodology; Writing – original draft.
Helena Pomares: Data curation; Writing – review & editing.
Mercedes Sánchez Barba: Formal analysis; Methodology; Writing – original draft.
María Abáigar: Investigation; Methodology; Writing – original draft.
Bernardo González: Data curation; Writing – review & editing.
Brayan Merchán: Data curation; Writing – review & editing.
Reyes Sancho-Tello: Data curation; Writing – review & editing.
Marta Callejas: Data curation; Writing – review & editing.
Carolina Muñoz-Novas: Data curation; Writing – review & editing.
Carlos Cerveró: Data curation; Writing – review & editing.
Guillermo Sanz: Supervision; Writing – review & editing.
Jesús María Hernández Rivas: Conceptualization; Investigation; Methodology; Supervision; Writing – review & editing.
María Díez Campelo: Conceptualization; Formal analysis; Methodology; Supervision; Writing – review & editing.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III, Ministerio de Sanidad: PI17/01741. Gerencia Regional de Salud de Castilla y León (Sacyl): GRS1349/A/16.
The authors declare that there is no conflict of interest.
Availability of data and material: Not applicable.
References
- 1. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89: 2079–2088. [PubMed] [Google Scholar]
- 2. Greenberg PL, Tuechler H, Schanz J, et al. Revised International Prognostic Scoring System for myelodysplastic syndromes. Blood 2012; 120: 2454–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Park S, Grabar S, Kelaidi C, et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood 2008; 111: 574–582. [DOI] [PubMed] [Google Scholar]
- 4. Santini V, Schemenau J, Levis A, et al. Can the revised IPSS predict response to erythropoietic-stimulating agents in patients with classical IPSS low or intermediate-1 MDS? Blood 2013; 122: 2286–2288. [DOI] [PubMed] [Google Scholar]
- 5. Houston BL, Jayakar J, Wells RA, et al. A predictive model of response to erythropoietin stimulating agents in myelodysplastic syndrome: from the Canadian MDS patient registry. Ann Hematol 2017; 96: 2025–2029. [DOI] [PubMed] [Google Scholar]
- 6. Buckstein R, Balleari E, Wells R, et al. ITACA: a new validated international erythropoietic stimulating agent-response score that further refines the predictive power of previous scoring systems: BUCKSTEIN et al. Am J Hematol 2017; 92: 1037–1046. [DOI] [PubMed] [Google Scholar]
- 7. Hellstrom-Lindberg E, Gulbrandsen N, Lindberg G, et al. A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin + granulocyte colony-stimulating factor: significant effects on quality of life. Br J Haematol 2003; 120: 1037–1046. [DOI] [PubMed] [Google Scholar]
- 8. Kelaidi C, Park S, Sapena R, et al. Long-term outcome of anemic lower-risk myelodysplastic syndromes without 5q deletion refractory to or relapsing after erythropoiesis-stimulating agents. Leukemia 2013; 27: 1283–1290. [DOI] [PubMed] [Google Scholar]
- 9. Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011; 364: 2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol 2012; 30: 3376–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malcovati L, Papaemmanuil E, Bowen DT, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 2011; 118: 6239–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jädersten M, Saft L, Smith A, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol 2011; 29: 1971–1979. [DOI] [PubMed] [Google Scholar]
- 13. Montalban-Bravo G, Takahashi K, Patel K, et al. Impact of the number of mutations in survival and response outcomes to hypomethylating agents in patients with myelodysplastic syndromes or myelodysplastic/myeloproliferative neoplasms. Oncotarget 2018; 9: 9714–9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Itzykson R, Kosmider O, Cluzeau T, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011; 25: 1147–1152. [DOI] [PubMed] [Google Scholar]
- 15. Bejar R, Lord A, Stevenson K, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014; 124: 2705–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Du M, Zhou F, Jin R, et al. Mutations in the DNA methylation pathway predict clinical efficacy to hypomethylating agents in myelodysplastic syndromes: a meta-analysis. Leuk Res 2019; 80: 11–18. [DOI] [PubMed] [Google Scholar]
- 17. Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med 2020; 382: 140–151. [DOI] [PubMed] [Google Scholar]
- 18. Bejar R, Stevenson KE, Caughey B, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol 2014; 32: 2691–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Della Porta MG, Gallì A, Bacigalupo A, et al. Clinical effects of driver somatic mutations on the outcomes of patients with myelodysplastic syndromes treated with allogeneic hematopoietic stem-cell transplantation. J Clin Oncol 2016; 34: 3627–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lindsley RC, Saber W, Mar BG, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med 2017; 376: 536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Messa E, Gioia D, Masiera E, et al. Effects of erythropoiesis-stimulating agents on overall survival of International Prognostic Scoring System low/intermediate-1 risk, transfusion-independent myelodysplastic syndrome patients: a cohort study. Haematologica 2019; 104: e4–e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheson BD. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006; 108: 419–425. [DOI] [PubMed] [Google Scholar]
- 23. Von Elm E, Altman DG, Egger M, et al.; STROBE Initiative. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet 2007; 370: 1453–1457. [DOI] [PubMed] [Google Scholar]
- 24. Negrin RS, Stein R, Vardiman J, et al. Treatment of the anemia of myelodysplastic syndromes using recombinant human granulocyte colony-stimulating factor in combination with erythropoietin. Blood 1993; 82: 737–743. [PubMed] [Google Scholar]
- 25. Jädersten M, Malcovati L, Dybedal I, et al. Erythropoietin and granulocyte-colony stimulating factor treatment associated with improved survival in myelodysplastic syndrome. J Clin Oncol 2008; 26: 3607–3613. [DOI] [PubMed] [Google Scholar]
- 26. Greenberg PL, Sun Z, Miller KB, et al. Treatment of myelodysplastic syndrome patients with erythropoietin with or without granulocyte colony-stimulating factor: results of a prospective randomized phase 3 trial by the Eastern Cooperative Oncology Group (E1996). Blood 2009; 114: 2393–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Musto P, Villani O, Martorelli MC, et al. Response to recombinant erythropoietin alpha, without the adjunct of granulocyte-colony stimulating factor, is associated with a longer survival in patients with transfusion-dependent myelodysplastic syndromes. Leuk Res 2010; 34: 981–985. [DOI] [PubMed] [Google Scholar]
- 28. Fenaux P, Santini V, Aloe Spiriti M, et al. A phase 3 randomized, placebo-controlled study assessing the efficacy and safety of epoetin-α in anemic patients with low-risk MDS. Leukemia 2018; 32: 2648–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park S, Fenaux P, Greenberg P, et al. Efficacy and safety of darbepoetin alpha in patients with myelodysplastic syndromes: a systematic review and meta-analysis. Br J Haematol 2016; 174: 730–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kosmider O, Passet M, Santini V, et al. Are somatic mutations predictive of response to erythropoiesis stimulating agents in lower risk myelodysplastic syndromes? Haematologica 2016; 101: e280–e283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zheng X, Zhan Z, Naren D, et al. Prognostic value of SRSF2 mutations in patients with de novo myelodysplastic syndromes: a meta-analysis. PLoS One 2017; 12: e0185053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thol F, Kade S, Schlarmann C, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012; 119: 3578–3584. [DOI] [PubMed] [Google Scholar]
- 33. Thota S, Viny AD, Makishima H, et al. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 2014; 124: 1790–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Di Giacomo D, Lema Fernandez AG, Pierini T, et al. The GNAS1 gene in myelodysplastic syndromes (MDS). Leuk Res 2014; 38: 804–807. [DOI] [PubMed] [Google Scholar]
- 35. Martín-Izquierdo M, Abáigar M, Hernández-Sánchez JM, et al. Co-occurrence of cohesin complex and Ras signaling mutations during progression from myelodysplastic syndromes to secondary acute myeloid leukemia. Haematologica 2021; 106: 2215–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-tah-10.1177_20406207231218157 for Outcomes and effect of somatic mutations after erythropoiesis stimulating agents in patients with lower-risk myelodysplastic syndromes by Juan Carlos Caballero, Julio Dávila, María López-Pavía, Esperanza Such, Teresa Bernal, Fernando Ramos, Marisa Calabuig, Jesús María Hernández Sánchez, Helena Pomares, Mercedes Sánchez Barba, María Abáigar, Bernardo González, Brayan Merchán, Reyes Sancho-Tello, Marta Callejas, Carolina Muñoz-Novas, Carlos Cerveró, Guillermo Sanz, Jesús María Hernández Rivas and María Díez Campelo; in Therapeutic Advances in Hematology