

Abstract
We have cloned the gene encoding RNase HII (RNase HIIPk) from the hyperthermophilic archaeon Pyrococcus kodakaraensis KOD1 by screening of a library for clones that suppressed the temperature-sensitive growth phenotype of an rnh mutant strain of Escherichia coli. This gene was expressed in an rnh mutant strain of E. coli, the recombinant enzyme was purified, and its biochemical properties were compared with those of E. coli RNases HI and HII. RNase HIIPk is composed of 228 amino acid residues (molecular weight, 25,799) and acts as a monomer. Its amino acid sequence showed little similarity to those of enzymes that are members of the RNase HI family of proteins but showed 40, 31, and 25% identities to those of Methanococcus jannaschii, Saccharomyces cerevisiae, and E. coli RNase HII proteins, respectively. The enzymatic activity was determined at 30°C and pH 8.0 by use of an M13 DNA-RNA hybrid as a substrate. Under these conditions, the most preferred metal ions were Co2+ for RNase HIIPk, Mn2+ for E. coli RNase HII, and Mg2+ for E. coli RNase HI. The specific activity of RNase HIIPk determined in the presence of the most preferred metal ion was 6.8-fold higher than that of E. coli RNase HII and 4.5-fold lower than that of E. coli RNase HI. Like E. coli RNase HI, RNase HIIPk and E. coli RNase HII cleave the RNA strand of an RNA-DNA hybrid endonucleolytically at the P-O3′ bond. In addition, these enzymes cleave oligomeric substrates in a similar manner. These results suggest that RNase HIIPk and E. coli RNases HI and HII are structurally and functionally related to one another.
RNase H degrades only the RNA strand of an RNA-DNA hybrid (6). The genes encoding two types of RNase H have been cloned from various organisms (3, 7, 9, 15, 17, 18, 21, 38, 40). The rnhA gene encodes RNase HI, and the rnhB gene encodes RNase HII. Interestingly, some organisms contain both of these genes. However, in contrast to the structures and functions of the RNase HI enzymes, which have been extensively studied (5, 13, 23, 24, 32), much less is known about those of the RNase HII enzymes. At present, no information on the distinctive enzymatic properties, the secondary and tertiary structures, and the types of amino acid residues involved in catalytic function and substrate binding is available for the RNase HII enzymes. Escherichia coli RNase HII has been shown to specifically hydrolyze the RNA strand of an RNA-DNA hybrid (15). The specific activity of this enzyme was reported to be 0.4% that of E. coli RNase HI under assay conditions that were optimal for E. coli RNase HI (15). However, most of the enzyme accumulated in E. coli cells in an insoluble form upon induction of the gene (rnhB) for overexpression, probably due to its poor solubility. This poor solubility hampers further biochemical and enzymatic characterization of the enzyme.
Computer analyses recently showed that the genes encoding RNase HII enzymes exist in the chromosomes of bacteria, archaea, and eucarya (30). Because the genomes of archaea obviously contain only genes homologous to the rnhB genes (2, 26), it seems likely that the RNase HII enzymes are more ubiquitously present in various organisms than are the RNase HI enzymes. However, most of these proteins are predicted from translation of DNA sequences. In addition to E. coli RNase HII, only RNase HII from Streptococcus pneumoniae has been shown to possess RNase H activity (40). Transformation with the plasmid containing the rnhB gene encoding S. pneumoniae RNase HII complemented the RNase H-dependent temperature-sensitive growth phenotype of an rnhA-recC double-mutant strain of E. coli, and the enzyme expressed in E. coli showed RNase H activity in a renaturation gel assay, suggesting that this enzyme is functional both in vivo and in vitro. It has been suggested that RNase HII [RNase H(35)] from Saccharomyces cerevisiae has RNase H activity (9). However, there is no evidence indicating that this protein has RNase H activity. Because the enzymes produced from hyperthermophiles are usually highly stable and because this property not only facilitates the production of recombinant enzymes in E. coli in an amount sufficient for biochemical characterization but also facilitates the crystallization of the enzymes for X-ray analyses, we decided to clone the rnhB gene from the hyperthermophilic archaeon Pyrococcus kodakaraensis KOD1.
P. kodakaraensis KOD1, which was previously designated Pyrococcus sp. strain KOD1, was isolated from a solfatara at a wharf on Kodakara Island, Kagoshima, Japan (33). The growth temperature of this strain ranges from 65 to 100°C, and the optimal temperature is 95°C. Here we report that the rnhB gene was cloned from P. kodakaraensis KOD1 by its ability to complement the temperature-sensitive growth phenotype of E. coli MIC3001, with rnh-339::cat and recB270(Ts) mutations (16). A comparison of the biochemical properties of recombinant RNase HII from P. kodakaraensis (RNase HIIPk) with those of E. coli RNases HI and HII suggests that RNases HI and HII are structurally and functionally related, despite their poor sequence similarity.
MATERIALS AND METHODS
Cells and plasmids.
rnhA mutant strains E. coli MIC3001 and MIC3009 (16) and plasmid pMIC2721 for the overproduction of E. coli RNase HII (15) were kindly donated by M. Itaya. The genotype of E. coli MIC3009 is F− supE44 supF58 lacY1 or Δ(lacIZY)6 trpR55 galK2 galT22 metB1 hsdR14(rK− mK+) rnh-339::cat. E. coli MIC3001 has an additional recB270(Ts) mutation. Competent cells of E. coli HB101 [F− hsdS20(rB− mB−) recA13 ara-13 proA2 lacY1 galK2 rpsL20 (Smr) xyl-5 mtl-1 supE44 λ−] and plasmids pBR322 and pUC18 were from Takara Shuzo Co., Ltd. E. coli BL21 (DE3) [F− ompT hsdSB(rB− mB−) gal (λcI857 ind1 Sam7 nin5 lacUV5-T7 gene 1) dcm(DE3)] was from Novagen. Plasmid pJLA503 was from Medac Gentechnologie. E. coli cells were grown in Luria-Bertani medium (31) containing 100 mg of ampicillin per liter.
Materials.
[γ-32P]ATP (>5,000 Ci/mmol) and [α-32P]ddATP (>5,000 Ci/mmol) were from Amersham. Crotalus durissus phosphodiesterase was from Boehringer Mannheim Biochemicals. Lysyl endopeptidase was from Wako Pure Chemical Industries. Restriction and modifying enzymes were from either Toyobo Co., Ltd., or Takara Shuzo Co.
General DNA manipulations.
Genomic DNA was prepared from a Sarkosyl lysate of P. kodakaraensis KOD1 cells as described previously (14). Digestion of DNA with restriction enzymes, analysis of DNA fragments by agarose gel electrophoresis, transformation by a calcium chloride procedure, and small-scale preparation of plasmid DNA by an alkaline lysis method were performed as described previously (36).
Construction and screening of a KOD1 DNA library.
Genomic DNA of P. kodakaraensis KOD1 was completely digested with BamHI or HindIII, and the resultant DNA fragments were ligated to the alkaline phosphatase-treated BamHI or HindIII site of pBR322. The resultant plasmids were used to transform E. coli MIC3001. Colonies were grown on Luria-Bertani medium plates containing 100 mg of ampicillin per liter at both 30 and 42°C. Usually, after 1 to 2 days of incubation, colonies are detected on the plate incubated at 30°C but not on that incubated at 42°C, unless the gene contained in the plasmid functionally complements the rnhA or recB mutation of E. coli MIC3001. Plasmid DNAs were isolated from colonies grown at 42°C and used to transform E. coli MIC3001 again. Cloned DNAs which repetitively complemented the temperature-sensitive growth phenotype of E. coli MIC3001 were selected as positive.
DNA sequence determination.
A 3-kbp BamHI fragment cloned into pBR322 was digested with HindIII and NruI, and the resultant 1.2-kbp HindIII-BamHI fragment and 1-kbp NruI-HindIII fragment were ligated to the large BamHI-HindIII and SmaI-HindIII fragments of pUC18, respectively. By use of the resultant plasmids as templates, the DNA sequences of these fragments were determined by the dideoxy chain termination method with fluorescent dye terminators and a Prism 310 DNA sequencer (PE Applied Biosystems).
Overproduction and purification.
The open reading frame (ORF) for the rnhB gene (rnhBPk) was amplified by PCR with a set of forward (5′-TTAGGAGGTGAACATATGAAGATAGCGGGCATTGACGAGGC) and reverse (5′-GCGCGGTCGACTCGCGGCTGCCAGTTTTGCAT) primers, where the underlined bases show the positions of the NdeI (for the forward primer) and SalI (for the reverse primer) cleavage sites. PCR was performed in 25 cycles with a GeneAmp 2400 PCR system (The Perkin-Elmer Corp.) and a GeneAmp kit (The Perkin-Elmer, Corp.) in accordance with the procedures recommended by the supplier. All oligodeoxyribonucleotides were synthesized by Sawady Technology Co., Ltd. The resultant 700-bp NdeI-SalI fragment was ligated into plasmid pJLA503 to construct pJAL700K. An overproducing strain was constructed by transforming E. coli MIC3009 with pJAL700K. Overproduction was accomplished as described previously for E. coli RNase HI (25). Cells were then harvested by centrifugation at 6,000 × g for 10 min and subjected to purification procedures.
All purification procedures, except for heat treatment, were carried out at 4°C. Cells were suspended in 10 mM Tris-HCl (pH 7.5) containing 1 mM EDTA (TE buffer), disrupted by sonication with a model 450 Sonifier (Branson Ultrasonic Corp.), and centrifuged at 15,000 × g for 30 min. The supernatants were incubated at 90°C for 15 min to precipitate most of the proteins derived from host cells and centrifuged at 30,000 × g for 30 min. The supernatants were pooled, and the enzyme was precipitated at 70% saturation with ammonium sulfate to remove nucleic acids. The precipitates were collected by centrifugation at 10,000 × g for 20 min, dissolved in TE buffer, and applied to a DE-52 column equilibrated with the same buffer. The enzyme was eluted from the column by linearly increasing the NaCl concentration from 0 to 0.5 M. Fractions containing the enzyme were combined and applied to a HiLoad 16/60 Superdex 200 pg gel filtration column (Pharmacia) (1.6 by 60 cm) equilibrated with TE buffer containing 0.1 M NaCl. Fractions containing the pure protein were combined and used for further analyses. Overproduction and purification of E. coli RNase HII were performed as described previously (15). The purities of the enzymes were analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) on a 12% polyacrylamide gel (29), followed by staining with Coomassie brilliant blue. For estimation of the molecular weight of the enzyme by gel filtration, bovine serum albumin, ovalbumin, chymotrypsinogen, and RNase A were used as standard proteins.
Enzymatic activity.
The RNase H activity was determined at 30°C for 15 min by measuring the radioactivity of the acid-soluble digestion product from the substrate, the 3H-labeled M13 DNA-RNA hybrid, as previously described (25). The buffer was 10 mM Tris-HCl (pH 8.0) containing 50 mM NaCl, 1 mM 2-mercaptoethanol, 10 μg of bovine serum albumin per ml, and 10 mM CoCl2 (for RNase HIIPk), 10 mM MgCl2 (for E. coli RNase HI), or 10 mM MnCl2 (for E. coli RNase HII). One unit was defined as the amount of enzyme producing 1 μmol of acid-soluble material/min at 30°C. The specific activity was defined as the enzymatic activity per milligram of protein.
Cleavage of oligomeric RNA-DNA and DNA-RNA-DNA–DNA substrates.
The 29-base DNA-RNA-DNA (5′-AATAGAGAAAAAGaaaaAAGATGGCAAAG-3′), in which DNA and RNA are represented by uppercase and lowercase letters, respectively, and the 29-base DNA which is complementary to this 29-base DNA-RNA-DNA were kindly donated by ID Biomedical Corp. The 12-base RNA (5′-cggagaugacgg-3′) was chemically synthesized by Toray Research Center Co., Ltd. The 12-base DNA which is complementary to the 12-base RNA and the 13-base DNAs with the sequences of 5′-AATAGAGAAAAAG-3′ (DNA1) and 5′-AAGATGGCAAAGA-3′ (DNA2), in which the underlined sequences represent the 5′- and 3′-terminal sequences of the 29-base DNA-RNA-DNA, respectively, were synthesized by Sawady Technology Co. Three types of hybrid duplexes, in which the 12-base RNA was 32P labeled at the 5′ end or the 29-base DNA-RNA-DNA was 32P labeled at the 5′ end or the 3′ end, were constructed. Because the 29-base DNA-RNA-DNA was 32P labeled at the 3′ end by attaching [α-32P]ddATP by use of terminal deoxynucleotidyltransferase (36), the hybrid duplex in which the 29-base DNA-RNA-DNA was 32P labeled at the 3′ end was constructed by hybridizing the 30-base DNA-RNA-DNA to the 29-base DNA. These hybrid duplexes (1.0 μM) were prepared by hybridizing 32P-labeled 12-base RNA, 29-base DNA-RNA-DNA, or 30-base DNA-RNA-DNA with 1.5 molar equivalents of DNA oligomers.
Hydrolysis of the substrate was carried out as described for the M13 DNA-RNA hybrid. The hydrolysates were separated on a 20% polyacrylamide gel containing 7 M urea and were analyzed with Instant Imager (Packard). When the hybrid duplex in which RNA or DNA-RNA-DNA was 32P labeled at the 5′ end was used as a substrate, the hydrolysates were identified by comparing their migrations on the gel with those of oligonucleotides generated by partial digestion of 32P-labeled 12-base RNA or 29-base DNA-RNA-DNA with snake venom phosphodiesterase (19) or that of 32P-labeled DNA1. When the hybrid duplex in which the 29-base DNA-RNA-DNA was 32P-labeled at the 3′ end was used as a substrate, the hydrolysates were identified by comparing their migrations on the gel with that of 32P-labeled DNA2.
Protein concentration.
The protein concentration was determined from the UV absorption, with A2880.1% values of 0.63 for RNase HIIPk and 0.56 for E. coli RNase HII. These values were calculated by use of an ɛ of 1,576 M−1 cm−1 for Tyr and an ɛ of 5,225 M−1 cm−1 for Trp at 280 nm (10).
Analyses for primary structure.
Digestion of RNase HIIPk with lysyl endopeptidase and separation of the peptides by reverse-phase high-performance liquid chromatography were carried out as described previously (25). The NH2-terminal amino acid sequence was determined with a model 491 protein sequencer (PE Applied Biosystems). The molecular weight of the peptide was determined with an on-line mass spectrometer (LCQ; Finnigan MAT) by electrospray ionization. Mass spectral data were processed with Bioworks software (Finnigan MAT).
Nucleotide sequence accession number.
The nucleotide sequence reported in this paper has been deposited in the DDBJ database under accession no. AB012613.
RESULTS
Cloning of the gene encoding RNase H from P. kodakaraensis KOD1.
E. coli MIC3001 with the rnh-339::cat and recB270(Ts) mutations has an RNase H-dependent temperature-sensitive growth phenotype (16). We used this strain to isolate the DNA fragment containing the genes encoding RNase H enzymes from P. kodakaraenis KOD1 by a complementation assay. If KOD1 RNase H enzymes were expressed in E. coli and exhibited RNase H activities, MIC3001 transformants with the plasmids containing the genes coding for them could form colonies at a nonpermissive temperature, 42°C. Of 5 × 104 MIC3001 transformants with the plasmid libraries containing either the BamHI or HindIII fragments of the KOD1 genome, 11 transformants could grow at 42°C. Restriction enzyme analyses indicated that all of the plasmids isolated from these transformants were identical to one another and contained a 3-kbp BamHI fragment (data not shown).
DNA sequence analysis.
A plasmid that complements the temperature-sensitive growth phenotype of MIC3001 does not necessarily contain an rnh gene, because other genes may be able to complement the rnhA or recB mutation of this strain. Generally, a gel renaturation assay (4) has been used to identify positive clones from those obtained by the complementation method. However, in this experiment, we obtained only one transformant that could grow at a nonpermissive temperature. Therefore, we did not analyze this transformant for its RNase H activity by a gel renaturation assay. Instead, we analyzed the DNA sequence of the plasmid isolated from this transformant.
Because the 3-kbp BamHI fragment of the KOD1 genome contains an ORF encoding the amino acid sequence homologous to that of E. coli RNase HII, we designated this ORF and the encoded protein rnhBPk and RNase HIIPk, respectively. A putative ribosome binding site (5′-AGGAGGTG-3′), which is complementary to the 3′-terminal sequence of 16S rRNA from KOD1 (3′-UCCUCCAC-5′) (33), is located 6 nucleotides upstream from the initiation codon of the ORF. The protein is composed of 228 amino acid residues and has a calculated molecular weight of 25,799 and an isoelectric point of 5.43. A HindIII site is located ∼150 bp downstream from the initiation codon. This is the reason why we could not isolate the DNA fragment containing the rnhB gene from HindIII libraries.
Amino acid sequence comparison.
The amino acid sequence of RNase HIIPk was compared with those of its homologues from archaea, eucarya, and bacteria (Fig. 1). The sequences of the RNase HII homologues from Archaeoglobus fulgidus, Methanobacterium thermoautotrophicum, and Methanococcus jannaschii, to which RNase H function has been putatively ascribed, represent those of archaeal RNase HII enzymes. The sequences of S. cerevisiae RNase HII and the putative RNase HII from Caenorhabditis elegans represent those of eucaryotic RNase HII enzymes. The sequences of E. coli RNase HII and the putative RNase HII from Bacillus subtilis represent those of prokaryotic RNase HII enzymes. RNase HIIPk showed amino acid sequence identities of 42% to A. fulgidus RNase HII, 40% to M. thermoautotrophicum RNase HII or M. jannaschii RNase HII, 31% to S. cerevisiae RNase HII or C. elegans RNase HII, 25% to E. coli RNase HII, and 26% to B. subtilis RNase HII. Three motifs (motifs I to III) which are highly conserved in various RNase HII sequences (40) were also conserved in the RNase HIIPk sequence (Fig. 1). When the archaeal and eucaryotic RNase HII sequences were compared, an additional motif (motif IV) was observed (Fig. 1).
FIG. 1.

Alignment of RNase HII sequences. The amino acid sequences of RNase HII enzymes from bacteria, archaea, and eucarya are compared. Gaps are denoted by dashes. Amino acid residues which are conserved in at least three different enzymes are highlighted in black. When two types of amino acid residues were conserved at a given position, the most conserved one was selected; if they were conserved equally, the residue conserved in the RNase HIIPk sequence was selected; if they were conserved equally but either one of them was not conserved in the RNase HIIPk sequence, one of them was just selected randomly. Numbers represent the positions of the amino acid residues, which start from the initiator methionine for each enzyme. The ranges for the four sequence motifs which are well conserved in the RNase HII sequences are shown above the sequences. The sources of RNase HII enzymes were as follows: KOD1, P. kodakaraensis KOD1 (DDBJ accession no. AB012613); Afu, A. fulgidus (GenBank accession no. AE001062); Mth, M. thermoautotrophicum (GenBank accession no. AE000875); Mja, M. jannaschii (GenBank accession no. U67470); Sce, S. cerevisiae (GenBank accession no. Z71348, ORF YNL072w); Cel, C. elegans (GenBank accession no. Z66524, product T13H5.2); Eco, E. coli (Swiss-Prot code no. P10442); and Bsu, B. subtilis (EMBL accession no. 299112).
Overexpression and purification of RNase HIIPk.
Upon induction, recombinant RNase HIIPk accumulated in cells as the most abundant protein (Fig. 2). The level of production of the enzyme was estimated to be about 25 mg/liter of culture. RNase HIIPk was recovered in a soluble form after sonication lysis and purified to homogeneity by three procedures, with a yield of ∼20% (Fig. 2). Approximately 5 mg of the enzyme was purified from 1 liter of culture. The molecular weight of the enzyme was estimated to be 28,000 from SDS-PAGE and 33,000 from gel filtration chromatography (data not shown). These results suggest that RNase HIIPk exists in a monomeric form.
FIG. 2.
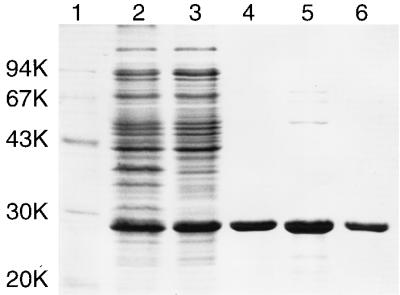
SDS-PAGE of RNase HIIPk in each purification step. Samples were subjected to SDS–12% PAGE and stained with Coomassie brilliant blue. Lane 1, low-molecular-weight marker kit (Pharmacia LKB Biotechnology) containing phosphorylase b, albumin, ovalbumin, carbonic anhydrase, trypsin inhibitor, and α-lactalbumin; lane 2, whole-cell lysate of E. coli MIC3009 harboring plasmid pJAL700K; lane 3, soluble fraction extracted from E. coli MIC3009 harboring plasmid pJAL700K; lane 4, soluble fraction obtained after heat treatment; lane 5, fractions pooled after DE-52 column chromatography; lane 6, fractions pooled after Superdex 200 pg column chromatography. Numbers beside the gel represent the molecular weights of individual standard proteins (in thousands [K]).
Amino acid sequence analyses.
Because the molecular weight of recombinant RNase HIIPk estimated from SDS-PAGE and gel filtration chromatography was slightly higher than the calculated one, we analyzed the enzyme by mass spectroscopy as well. The molecular weight of the enzyme determined by mass spectroscopy was 25,577, which is lower than the calculated one by 222. To examine whether a few NH2- or COOH-terminal amino acid residues are truncated in this enzyme, the NH2- and COOH-terminal amino acid sequences of the enzyme were determined as described in Materials and Methods. The results showed that the COOH terminus is Arg226 instead of Val228, indicating that recombinant RNase HIIPk is composed of 226 amino acid residues.
Enzymatic activity of RNase HIIPk.
It has been reported that E. coli RNase HI prefers the Mg2+ ion for activity and exhibits 2 to 3% maximal activity in the presence of the Mn2+ ion (1). It has also been reported that E. coli RNase HII requires the Mg2+ ion for activity and exhibits 0.4% of the enzymatic activity of E. coli RNase HI under conditions which are optimal for E. coli RNase HI (15). However, it remains to be determined whether E. coli RNase HII exhibits activity in the presence of metal ions other than Mg2+. Therefore, we determined the activities of these enzymes, as well as that of RNase HIIPk, in the presence of 10 mM MgCl2, MnCl2, BaCl2, CaCl2, CoCl2, CuSO4, FeCl2, FeCl3, NiCl2, SrCl2, and ZnCl2. The results are summarized in Table 1. As expected, E. coli RNase HI exhibited little activity in the presence of metal ions other than Mg2+ and Mn2+. Likewise, E. coli RNase HII exhibited activity only in the presence of the Mg2+ or Mn2+ ion. However, unlike E. coli RNase HI, E. coli RNase HII preferred the Mn2+ ion for activity. In contrast to these E. coli enzymes, RNase HIIPk showed a unique metal ion preference for activity. It exhibited activity in the presence of the Mg2+, Mn2+, Co2+, and Ni2+ ions but did not exhibit activity in the presence of other metal ions. The most preferred metal ion for RNase HIIPk was the Co2+ ion, but the Mg2+, Mn2+, and Ni2+ ions could be substituted for the Co2+ ion without seriously affecting enzymatic activity. These results indicate that the E. coli RNase HI and HII activities are dependent on the Mg2+ and Mn2+ ions, respectively, but that the RNase HIIPk activity is not necessarily dependent on a specific divalent cation. Instead, RNase HIIPk exhibits rather broad metal ion specificities. Thus, at pH 8.0, the specific activity of RNase HIIPk determined in the presence of 10 mM CoCl2 was shown to be 6.8-fold higher than that of E. coli RNase HII determined in the presence of 10 mM MnCl2 and 4.5-fold lower than that of E. coli RNase HI determined in the presence of 10 mM MgCl2.
TABLE 1.
Comparison of the specific activities of the enzymesa
| Enzyme | Metal ion | Sp act (U/mg) | Relative activity (%) |
|---|---|---|---|
| RNase HIIPk | CoCl2 | 2.1 | 100 |
| MgCl2 | 1.1 | 52.3 | |
| MnCl2 | 0.64 | 30.5 | |
| NiCl2 | 0.59 | 28.1 | |
| E. coli RNase HII | MnCl2 | 0.31 | 14.8 |
| MgCl2 | 0.001 | 0.05 | |
| E. coli RNase HI | MgCl2 | 9.5 | 452 |
| MnCl2 | 0.29 | 13.8 |
Hydrolysis of the M13 DNA-RNA hybrid with an enzyme was carried out at 30°C for 15 min with 10 mM Tris-HCl (pH 8.0) containing 50 mM NaCl, 1 mM 2-mercaptoethanol, 10 μg of bovine serum albumin per ml, and 10 mM MgCl2, BaCl2, CaCl2, CoCl2, CuSO4, FeCl2, FeCl3, MnCl2, NiCl2, SrCl2, or ZnCl2. Because E. coli RNase HI and RNase HII exhibited little activity (specific activity, <0.0005 U/mg) in the presence of 10 mM BaCl2, CaCl2, CoCl2, CuSO4, FeCl2 FeCl3, NiCl2, SrCl2, or ZnCl2, these data are not listed. Likewise, because RNase HIIPk exhibited little activity (specific activity, <0.0005 U/mg) in the presence of BaCl2, CaCl2, CuSO4, FeCl2, FeCl3, SrCl2, or ZnCl2, these data are not listed as well. Errors, which represented the 67% confidence limits, were all at or below ± 30% of the values reported.
The dependence of RNase HIIPk activity on the metal ion concentration was analyzed for Mg2+, Mn2+, Co2+, and Ni2+. As shown in Fig. 3, the concentrations of the metal ions which gave 50% maximal activity were ∼5 mM for Co2+ and Mg2+, ∼2 mM for Ni2+, and <1 mM for Mn2+. These results suggest that the Mn2+ ion binds most strongly to the enzyme and that the Co2+ and Mg2+ ions bind most weakly to the enzyme. The dependence of RNase HIIPk activity on the concentration of NaCl or KCl was also analyzed. The results are summarized in Table 2. These salts do not seriously affect enzymatic activity at concentrations lower than 100 mM but are inhibitory for activity at concentrations higher than 400 mM for NaCl and 200 mM for KCl.
FIG. 3.

Effect of metal ion concentrations on RNase HIIPk activity. Enzymatic activity was determined with 10 mM Tris-HCl (pH 8.0) containing 50 mM NaCl, 1 mM 2-mercaptoethanol, 10 μg of bovine serum albumin per ml, and various concentrations of MgCl2 (closed circle), MnCl2 (open square), CoCl2 (open circle), or NiCl2 (closed square).
TABLE 2.
Effect of NaCl and KCl on RNase HIIPk activitya
| Metal ion | Concn (mM) | Relative activity (%) |
|---|---|---|
| NaCl | 0 | 100 |
| 50 | 109 | |
| 100 | 95 | |
| 200 | 89 | |
| 400 | 46 | |
| KCl | 0 | 100 |
| 50 | 100 | |
| 100 | 81 | |
| 200 | 52 | |
| 400 | 10 |
Hydrolysis of the M13 DNA-RNA hybrid with an enzyme was carried out at 30°C for 15 min with 10 mM Tris-HCl (pH 8.0) containing 50 mM NaCl, 1 mM 2-mercaptoethanol, 10 μg of bovine serum albumin per ml, 10 mM of MgCl2, and various concentrations of NaCl or KCl.
The pH dependence of RNase HIIPk activity was not analyzed, because the solubility of the Co2+ ion dramatically decreased as the pH increased beyond 9.0. However, when the pH dependence was analyzed in the presence of the Mg2+ ion, the enzyme showed a pH optimum at pH 9.5 (data not shown), indicating that RNase HIIPk expresses activity at an alkaline pH, like E. coli RNase HI. The temperature dependence of RNase HIIPk activity was not analyzed either, because the substrate is unstable at high temperatures. We determined the enzymatic activity at 30°C, at which all of the substrates used in this experiment, including a 12-bp RNA-DNA substrate, are stable.
Cleavage of oligomeric substrates.
To examine whether RNase HIIPk hydrolyzes oligomeric substrates, we cleaved the 12-bp RNA-DNA substrate and the 29-bp DNA-RNA-DNA–DNA substrate, both of which were 32P labeled at the 5′ ends, with the enzyme. The results are shown in Fig. 4A. The enzyme cleaved the 12-bp RNA-DNA substrate at multiple sites, but most preferably at a9-c10, whereas it specifically cleaved the 29-bp DNA-RNA-DNA–DNA substrate at the phosphodiester bond between the third and fourth adenosines (a16-a17). The specific cleavage of this DNA-RNA-DNA–DNA substrate at a16-a17 with RNase HIIPk was further confirmed by use of the substrate in which the 29-base DNA-RNA-DNA was 32P labeled at the 3′ end (data not shown). We analyzed the E. coli RNase HI and E. coli RNase HII activities by using these substrates as well. The results are summarized in Fig. 4B. E. coli RNase HI cleaved the 12-bp RNA-DNA substrate in a manner similar to that of RNase HIIPk. It has already been reported that E. coli RNase HI cleaves the same substrate most preferably at a9-c10 (20). In addition, like RNase HIIPk E. coli RNase HI cleaved the 29-bp DNA-RNA-DNA–DNA substrate at a unique position, although it cleaved the substrate in the middle of the tetraadenosines (a15-a16). This result is consistent with the previous one that E. coli RNase HI specifically cleaves the 20-base DNA-RNA-DNA, which consists of tetraribonucleotides flanked on either side by 7- or 9-base DNA, only in the middle of the tetraribonucleotide in the presence of the cDNA strand (12). E. coli RNase HII cleaved these substrates in basically the same manner as RNase HIIPk (data not shown). Both RNase HII enzymes cleaved neither the 12-base RNA nor the 29-base DNA-RNA-DNA, suggesting that these enzymes cannot cleave single-stranded RNA or DNA.
FIG. 4.

Cleavage of oligomeric substrates by RNase HIIPk. (A) Autoradiograph of cleavage reactions. Hydrolysis of the 5′-end-labeled 12-base RNA hybridized to the 12-base DNA (a) and the 5′-end-labeled 29-base DNA-RNA-DNA hybridized to the 29-base DNA (b) with RNase HIIPk was carried out at 30°C for 15 min, and the hydrolysates were separated on a 20% polyacrylamide gel containing 7 M urea as described in Materials and Methods. The concentration of the substrate was 1.0 μM. (a) Lane 1, partial digest of the 12-base RNA with snake venom phosphodiesterase; lane 2, untreated substrate; lane 3, hydrolysate with 0.8 ng of the enzyme; lane 4, hydrolysate with 1.6 ng of the enzyme; lane 5, hydrolysate with 16 ng of the enzyme. (b) Lane 1, partial digest of the 29-base DNA-RNA-DNA with snake venom phosphodiesterase; lane 2, 5′-end-labeled 13-base DNA with a sequence which is identical to residues 1 to 13 of the 29-base DNA-RNA-DNA; lane 3, untreated substrate; lane 4, hydrolysate with 0.8 ng of the enzyme; lane 5, hydrolysate with 1.6 ng of the enzyme; lane 6, hydrolysate with 16 ng of the enzyme. (B) Sites and extents of cleavage by RNase H. Sites of cleavage of the 12-bp RNA-DNA hybrid with RNase HIIPk (a) and E. coli RNase HI (b) and those of the 29-bp DNA-RNA-DNA–DNA substrate with RNase HIIPk (c) and E. coli RNase HI (d) are shown by arrows. Differences in the sizes of the arrows reflect the relative cleavage intensities at the indicated positions. The sites of cleavage of these substrates with E. coli RNase HII were almost identical to those with RNase HIIPk. For both panel A and panel B, deoxyribonucleotides are shown by uppercase letters, and ribonucleotides are shown by lowercase letters.
Heat inactivation.
The stability of RNase HIIPk against irreversible heat inactivation was analyzed by incubating the enzyme solution (0.1 μg/ml) in 10 mM Tris-HCl (pH 7.5) containing 0.1 M NaCl, 1 mM EDTA, 10% glycerol, and 0.1 mg of bovine serum albumin per ml at 90°C. For comparative purposes, the stability of E. coli RNase HII against irreversible heat inactivation was analyzed by incubating the enzyme solution (1 μg/ml) in the same buffer at 70°C. At appropriate intervals, an aliquot of the enzyme solution was withdrawn, chilled on ice, and analyzed for remaining activity. As shown in Fig. 5, RNase HIIPk was fairly stable against irreversible heat inactivation at 90°C, whereas E. coli RNase HII rapidly lost activity even at 70°C, with a half-life of 9 min.
FIG. 5.

Stability against irreversible heat inactivation. RNase HIIPk (closed circles) and E. coli RNase HII (open circles) were incubated at 90 and 70°C, respectively, in 10 mM Tris-HCl (pH 7.5) containing 0.1 M NaCl, 1 mM EDTA, 10% glycerol, and 0.1 mg of bovine serum albumin per ml. At appropriate intervals, an aliquot was withdrawn and examined for remaining activity. The concentrations of the enzymes were 0.1 μg/ml for RNase HIIPk and 1 μg/ml for E. coli RNase HII. Enzymatic activity was determined at 30°C for 15 min with 10 mM Tris-HCl containing 10 mM MgCl2 (for RNase HIIPk) or MnCl2 (for E. coli RNase HII), 50 mM NaCl, 1 mM 2-mercaptoethanol, and 10 μg of bovine serum albumin per ml and with the M13 DNA-RNA hybrid as a substrate.
DISCUSSION
Classification of RNase H families.
In this report, we showed that RNase HIIPk, which may represent archaeal RNase HII enzymes, has RNase H activity both in vivo and in vitro. This result supports a previous proposal that RNase H enzymes can be classified into two families, RNase HI and RNase HII, based on the difference in the amino acid sequences (40). However, similarities in the substrate cleavage patterns and the requirements for enzymatic activity between RNase HIIPk and E. coli RNase HI suggest that RNase HI and HI enzymes are functionally and structurally related. It has been reported that Asp10, Glu48, Asp70, and Asp134 form the active site of E. coli RNase HI, to which the catalytically essential Mg2+ ion binds (24). Three or four acidic residues may form the active site of RNase HII as well, because several acidic amino acid residues (Asp7, Glu8, Asp40, Asp105, Asp135, and Asp153 for RNase HIIPk are fully conserved in RNase HII sequences (Fig. 1). We are currently undertaking site-directed mutagenesis and X-ray crystallographic studies to find an answer to the question of whether RNase HI and HII enzymes form a superfamily.
Purification of recombinant RNase HIIPk.
In contrast to E. coli RNase HII, RNase HIIPk accumulates in a soluble form in cells which overproduce the enzyme. A comparison of the hydropathy profiles of these enzymes suggests that E. coli RNase HII is more hydrophobic than RNase HIIPk (data not shown), especially at the COOH-terminal region. E. coli RNase HII has the sequence ALGTCVLILV at positions 193 to 202, whereas RNase HIIPk has the sequence GWKTLKKIAE at the corresponding region (positions 197 to 206). Such a difference in hydrophobicity may alter the solubilities of these enzymes.
Overproduction of RNase HIIPk in E. coli and purification of the recombinant enzyme yielded a truncated enzyme in which the COOH-terminal two residues were eliminated. The COOH-terminal region of RNase HIIPk probably assumes a disordered structure; therefore, the peptide bond between Arg226 and Lys227 is cleaved by a protease from E. coli, such as OmpT protease, which is known to specifically cleave the peptide bond between two basic amino acid residues (37). Partial digestion with trypsin or chymotrypsin also produced a truncated protein in which the COOH-terminal 10 to 15 residues were eliminated (34a).
Metal ion specificity.
RNase HIIPk shows a relatively broad metal ion specificity. In addition, other enzymes from KOD1, such as a DNase which is associated with a RecA homologue (35) and glycerol kinase (28), show broad metal ion specificity. Therefore, this broad metal ion specificity may be characteristic of the enzymes isolated from hyperthermophilic archaea. Because hyperthermophilic archaea have been thought to retain traces of early life forms and to produce enzymes which may represent prototypes in the same protein family, the difference in metal ion specificity between an archaeal enzyme and a bacterial or eucaryotic enzyme may reflect the difference in the contents of various metal ions in the soil or sea between the primitive earth and the modern earth.
Substrate specificity.
Good agreement in the autoradiograms of the cleavage reactions for the 12-bp RNA-DNA substrate between RNase HIIPk and E. coli RNase HI strongly suggests that, like E. coli RNase HI, RNase HIIPk cleaves the P-O3′ bond of the RNA endonucleolytically. In addition, the findings that RNase HIIPk cleaves the 12-bp RNA-DNA substrate at multiple sites but cannot cleave the 12-base RNA, the 29-base DNA-RNA-DNA, or the DNA strand of the 29-bp DNA-RNA-DNA–DNA substrates clearly indicate that the enzyme does not cleave single-stranded DNA, single-stranded RNA, or double-stranded DNA. Because RNase HIIPk has a sequence specificity similar to that of E. coli RNase HII and it has been shown that E. coli RNase HII does not cleave double-stranded RNA (15), it is highly likely that RNase HIIPk cleaves the RNA strand only when it is hybridized to the DNA strand.
Recently, the enzymatically active subunit of human RNase HI was suggested to be a member of the RNase HII family of proteins (9). A human RNase HI from erythroleukemic cells can cleave DNA-RNA junctions when a DNA-RNA-DNA–DNA hybrid in which a single ribonucleotide is flanked on either side by DNA is used as a substrate (8). This DNA-RNA junction cannot be cleaved by E. coli RNase HI (12). The 29-base DNA-RNA-DNA has two DNA-RNA junctions: DNA-RNA and RNA-DNA junctions from 5′ to 3′. Because we used two types of 29-bp DNA-RNA-DNA–DNA substrates, those which were 32P labeled at the 5′ end or the 3′ end, it is clear that RNase HIIPk cannot cleave either the DNA-RNA junction or the RNA-DNA junction. However, RNase HIIPk and E. coli RNase HII can cleave the 29-bp DNA-RNA-DNA–DNA substrate at the 5′ end of the last ribonucleotide at the RNA-DNA junction, whereas E. coli RNase HI cannot cleave it at this site. It has been reported that calf thymus E. coli RNase HI cannot cleave it at this site. It has been reported that calf thymus RNase HI shows junction RNase activity, which is responsible for cleavage at the 5′ end of the last ribonucleotide at the RNA-DNA junction of an Okazaki substrate regardless of whether or not it forms an RNA-DNA heteroduplex structure (34). RNase HIIPk and E. coli RNase HII may have such a junction RNase activity and thereby facilitate the elimination of RNA from an Okazaki substrate.
Stability of RNase HIIPk.
RNase HIIPk was shown to be remarkably more stable against irreversible heat inactivation than E. coli RNase HII. We previously showed that E. coli RNase HI almost fully lost activity after incubation at 70°C for 10 min (22). Therefore, there is no doubt that RNase HIIPk is more stable than E. coli RNase HI as well. It has been reported that an increase in the number of ion pairs or ion-pair networks and a decrease in the volume of cavities are responsible for the hyperstability of the enzymes from hyperthermophilic archaea (11, 27, 39). It would be informative to examine whether RNase HIIPk is stabilized by similar mechanisms.
ACKNOWLEDGMENTS
We thank M. Itaya, Mitsubishi Kasei Institute of Life Sciences, for providing E. coli MIC3001 and MIC3009 and plasmid pMIC2721 and ID Biomedical Corp. for providing the DNA-RNA-DNA probe for the RNase H assay. We also thank N. Ohtani and N. Hirano for technical assistance.
This work was supported in part by a grant-in-aid for scientific research (08455382) from the Ministry of Education, Science and Culture of Japan.
REFERENCES
- 1.Berkower I, Leis J, Hurwitz J. Isolation and characterization of an endonuclease from Escherichia coli specific for ribonucleic acid in ribonucleic acid · deoxyribonucleic acid hybrid structure. J Biol Chem. 1973;248:5914–5921. [PubMed] [Google Scholar]
- 2.Bult C J, et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 3.Campbell A G, Ray D S. Functional complementation of an Escherichia coli ribonuclease H mutation by a cloned genomic fragment from trypanosomatid Crithidia fasciculata. Proc Natl Acad Sci USA. 1993;90:9350–9354. doi: 10.1073/pnas.90.20.9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carl P L, Bloom L, Crouch R J. Isolation and mapping of a mutation in Escherichia coli with altered levels of ribonuclease H. J Bacteriol. 1980;144:28–35. doi: 10.1128/jb.144.1.28-35.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crouch R J. Ribonuclease H: from discovery to 3D structure. New Biol. 1990;2:771–777. [PubMed] [Google Scholar]
- 6.Crouch R J, Dirksen M-L. Ribonucleases H. In: Linn S M, Roberts R J, editors. Nuclease. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1982. pp. 211–241. [Google Scholar]
- 7.Dawes S S, Crouch R J, Morris S L, Mizrahi V. Cloning, sequence analysis, overproduction in Escherichia coli and enzymatic characterization of the RNase HI from Mycobacterium smegmatis. Gene. 1995;165:71–75. doi: 10.1016/0378-1119(95)00523-9. [DOI] [PubMed] [Google Scholar]
- 8.Eder P S, Walder J A. Ribonuclease H from K562 human erythroleukemia cells: purification, characterization, and substrate specificity. J Biol Chem. 1991;266:6472–6479. [PubMed] [Google Scholar]
- 9.Frank P, Braunshofer-Reiter C, Wintersberger U. Yeast RNase H(35) is the counterpart of the mammalian RNase HI, and is evolutionarily related to prokaryotic RNase HII. FEBS Lett. 1998;421:23–26. doi: 10.1016/s0014-5793(97)01528-7. [DOI] [PubMed] [Google Scholar]
- 10.Goodwin T W, Morton R A. The spectrophotometric determination of tyrosine and tryptophan in proteins. Biochem J. 1946;40:628–632. doi: 10.1042/bj0400628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hennig M, Darimont B, Sterner R, Kirschner K, Jansonius J N. A structure of indole-3-glycerol phosphate synthase from the hyperthermophile Sulfolobus solfataricus: possible determinants of protein stability. Structure. 1995;3:1295–1306. doi: 10.1016/s0969-2126(01)00267-2. [DOI] [PubMed] [Google Scholar]
- 12.Hogrefe H H, Hogrefe R I, Walder R Y, Walder J A. Kinetic analysis of Escherichia coli RNase H using DNA-RNA-DNA/DNA substrates. J Biol Chem. 1990;265:5561–5566. [PubMed] [Google Scholar]
- 13.Hostomsky Z, Hostomska Z, Matthews D A. Ribonucleases H. In: Linn S M, Roberts R J, editors. Nuclease. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1993. pp. 341–376. [Google Scholar]
- 14.Imanaka T, Tanaka T, Tsunekawa H, Aiba S. Cloning of the genes for penicillinase, penP and penI, of Bacillus licheniformis in some vector plasmids and their expression in Escherichia coli, Bacillus subtilis, and Bacillus licheniformis. J Bacteriol. 1981;147:776–786. doi: 10.1128/jb.147.3.776-786.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itaya M. Isolation and characterization of a second RNase H (RNase HII) of Escherichia coli K-12 encoded by the rnhB gene. Proc Natl Acad Sci USA. 1990;87:8587–8591. doi: 10.1073/pnas.87.21.8587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Itaya M, Crouch R J. A combination of RNase H (rnh) and recBCD or sbcB mutations in Escherichia coli K12 adversely affects growth. Mol Gen Genet. 1991;227:424–432. doi: 10.1007/BF00273933. [DOI] [PubMed] [Google Scholar]
- 17.Itaya M, Kondo K. Molecular cloning of a ribonuclease H (RNase HI) gene from an extreme thermophile Thermus thermophilus HB8: a thermostable RNase H can functionally replace the Escherichia coli enzyme in vivo. Nucleic Acids Res. 1991;19:4443–4449. doi: 10.1093/nar/19.16.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itaya M, McKelvin D, Chatterjie S K, Crouch R J. Selective cloning of genes encoding RNase H from Salmonella typhimurium, Saccharomyces cerevisiae and Escherichia coli rnh mutants. Mol Gen Genet. 1991;227:438–445. doi: 10.1007/BF00273935. [DOI] [PubMed] [Google Scholar]
- 19.Jay E, Bambara R, Padmanabham P, Wu R. DNA sequence analysis: a general, simple and rapid method for sequencing large oligodeoxyribonucleotide fragments by mapping. Nucleic Acids Res. 1974;1:331–353. doi: 10.1093/nar/1.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanaya E, Kanaya S. Kinetic analysis of E. coli ribonuclease HI using oligomeric DNA/RNA substrates suggests an alternative mechanism for the interaction between the enzyme and substrate. Eur J Biochem. 1995;231:557–562. doi: 10.1111/j.1432-1033.1995.0557d.x. [DOI] [PubMed] [Google Scholar]
- 21.Kanaya S, Crouch R J. DNA sequence of the gene coding for Escherichia coli ribonuclease H. J Biol Chem. 1983;258:1276–1281. [PubMed] [Google Scholar]
- 22.Kanaya S, Itaya M. Expression, purification, and characterization of a recombinant ribonuclease H from Thermus thermophilus HB8. J Biol Chem. 1992;267:10184–10192. [PubMed] [Google Scholar]
- 23.Kanaya S, Ikehara M. Structure and function of ribonuclease HI from Escherichia coli. In: Eckstein F, Lilley M J, editors. Nucleic acids and molecular biology. Vol. 7. Berlin, Germany: Springer-Verlag KG; 1993. pp. 285–302. [Google Scholar]
- 24.Kanaya S, Ikehara M. Functions and structures of ribonuclease H enzymes. In: Biswas B B, Roy S, editors. Subcellular biochemistry. 24. Proteins: structure, function, and engineering. New York, N.Y: Plenum Press; 1995. pp. 377–422. [DOI] [PubMed] [Google Scholar]
- 25.Kanaya S, Katsuda C, Kimura S, Nakai T, Kitakuni E, Nakamura H, Katayanagi K, Morikawa K, Ikehara M. Stabilization of Escherichia coli ribonuclease H by introduction of an artificial disulfide bond. J Biol Chem. 1991;266:6038–6044. [PubMed] [Google Scholar]
- 26.Klenk H P, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- 27.Knapp S, de Vos W M, Rice D, Ladenstein R. Crystal structure of glutamate dehydrogenase from the hyperthermophilic eubacterium Thermotoga maritima at 3.0 Å resolution. J Mol Biol. 1997;267:916–932. doi: 10.1006/jmbi.1996.0900. [DOI] [PubMed] [Google Scholar]
- 28.Koga, Y., M. Morikawa, M. Haruki, H. Nakamura, T. Imanaka, and S. Kanaya. Thermostable glycerol kinase from a hyperthermophilic archaeon: gene cloning and characterization of the recombinant enzyme. Protein Eng., in press. [DOI] [PubMed]
- 29.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 30.Mian I S. Comparative sequence analysis of ribonucleases HII, III, II, PH and D. Nucleic Acids Res. 1997;25:3187–3195. doi: 10.1093/nar/25.16.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller J H. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. p. 433. [Google Scholar]
- 32.Morikawa K, Katayanagi K. RNase H: three-dimensional structure and function. Bull Inst Pasteur Paris. 1992;90:71–82. [Google Scholar]
- 33.Morikawa M, Izawa Y, Rashid N, Hoaki T, Imanaka T. Purification and characterization of a thermostable thiol protease from a newly isolated hyperthermophilic Pyrococcus sp. Appl Environ Microbiol. 1994;60:4559–4566. doi: 10.1128/aem.60.12.4559-4566.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murante R S, Henricksen L A, Bambara R A. Junction ribonuclease: an activity in Okazaki fragment processing. Proc Natl Acad Sci USA. 1998;95:2244–2249. doi: 10.1073/pnas.95.5.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34a.Muroya, A. Unpublished data.
- 35.Rashid N, Morikawa M, Nagahisa K, Kanaya S, Imanaka T. Characterization of a RecA/RAD51 homologue from a hyperthermophilic archaeon Pyrococcus sp. KOD1. Nucleic Acids Res. 1997;25:719–726. doi: 10.1093/nar/25.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 37.Sugimura K, Nishihara T. Purification, characterization, and primary structure of Escherichia coli protease VII with specificity for paired basic residues: identity of protease VII and OmpT. J Bacteriol. 1988;170:5625–5632. doi: 10.1128/jb.170.12.5625-5632.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H, Lima W F, Crooke S T. Molecular cloning and expression of cDNA for human RNase H. Antisense Nucleic Acid Drug Dev. 1998;8:53–61. doi: 10.1089/oli.1.1998.8.53. [DOI] [PubMed] [Google Scholar]
- 39.Yip K S P, Stillman T J, Britton K L, Artymiuk P J, Baker B J, Sedelnikova S E, Engel P C, Pasquo A, Chiaraluce R, Consalvi V, Scandurra R, Rice D W. The structure of Pyrococcus furiosus glutamate dehydrogenase reveals a key role for ion-pair networks in maintaining enzyme stability at extreme temperatures. Structure. 1995;3:1147–1158. doi: 10.1016/s0969-2126(01)00251-9. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y-B, Ayalew S, Lackes S A. The rnhB gene encoding RNase HII of Streptococcus pneumoniae and evidence of conserved motifs in eucaryotic genes. J Bacteriol. 1997;179:3828–3836. doi: 10.1128/jb.179.12.3828-3836.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]