

Abstract
It is estimated that Trichomonas vaginalis affects an astonishing 3.9% of the world’s population, and while many of those infected are asymptomatic, progression of the disease can lead to serious health problems. Currently, the nitroimidazoles constitute the only drug class approved to treat trichomoniasis in the United States, which makes the spread of drug resistance a realistic concern. We developed a new image-based, high-throughput, high-content assay for testing natural products (purified compounds and extracts) for antitrichomonal activity. Applying this assay system to a library of fungal natural product extracts led to the identification of three general classes of natural product inhibitors that exhibited moderate to strong activities against T. vaginalis: anthraquinones, xanthone-anthraquinone heterodimers, and decalin-linked tetramic-acid-containing metabolites. The tetramate natural products emerged as the most promising candidate molecules with pyrrolocin A (51) exhibiting potent activity against the parasite (EC50 = 60 nM), yet this metabolite showed limited toxicity to mammalian cell lines (selectivity index values of 100 and 167 versus 3T3 fibroblast and Ect1 normal cervical cells, respectively). The imaging-based assay system is a powerful tool for the bioassay-guided purification of single-component antitrichomonal biomolecules from complex natural product mixtures.
Keywords: Trichomonas vaginalis, natural products, fungi, quinones, tetramic acid
Trichomonas vaginalis is the most frequently encountered sexually-transmitted parasite in the United States.1 Those infected by the parasite exhibit an increased risk for (i) HIV infection, (ii) development of cervical cancer, and (iii) infertility, as well as certain adverse pregnancy outcomes including (iv) miscarriage and (v) low-preterm birth weight.2 Current clinical treatment options are limited to antiprotozoal nitroimidazoles (i.e., metronidazole and tinidazole), which still offer appreciably high levels of clinical efficacy (>90% of patients are cleared of the infection after a single course of treatment).3–5 However, concerns have arisen that in addition to several noted side effects (e.g., nausea, vomiting, and others), the nitroimidazoles may possess carcinogenic and mutagenic properties.6–7 This has led to worry about nitroimidazole treatment during pregnancy, as well as some apprehension about their use in the general population.6 A rather troubling problem, which came to light based on surveillance data collected from patients infected with T. vaginalis and who received standard courses of nitroimidazole treatment, is the high rate of infection recurrence (5–31% in women).5 While a portion of disease recurrence is attributable to further contact with untreated sexual partners, data suggests that a yet unknown proportion of T. vaginalis cases has the capacity to develop into persistent infectious states, which raises concerns about the clinical limitations of nitroimidazole therapies.8 Even more concerning, nitroimidazole resistance has been detected in up to 9.6% of clinical T. vaginalis isolates, raising alarm that nitroimidazoles might become clinically less effective.9–10 Considering the noted safety concerns and troubling clinical observations, there is a need for new disease management options that extend beyond the nitroimidazoles for treating T. vaginalis infections.
An examination of the research literature has revealed surprisingly sparse information concerning efforts to identify new classes of antiprotozoal molecules active against T. vaginalis, with only a small fraction of those endeavors dealing with natural products.11–12 A handful of prior studies had focused exclusively on testing plant extracts,13–16 but revealed little about the molecules responsible for their inhibitory effects. In a few instances, chemically-driven investigations of extracts have yielded a handful of secondary metabolites possessing activity against T. vaginalis including berberine,17 (−)-usnic acid,18 emodin,19 hamycin,20 hederagin,21 and mulinolic acid;22 however, these compounds suffer from poor potency and lack selectivity compared to metronidazole. Although the limited scope of reports pertaining to the discovery of new leads to fight T. vaginalis infections is worrisome, it also signals the potential that exists to identify new bioactive compounds for development into clinically useful agents. This expectation is reasonable, especially for natural products, which have long served as an outstanding resource for the discovery of new antiinfectious agents including some of the topmost antiparasitic compounds in current clinical use (e.g., avermectins, artemisinin, and more).23–24
Aside from the scarcity of natural-product-focused drug discovery efforts concentrating on inhibiting of T. vaginalis, there are relatively few reports of vetted assay methods that are amenable to the systematic screening and identification of compounds that inhibit this unique parasite. Most of the reported methods had employed manual microscopy-based approaches for cell enumeration25–26 or used metabolically-activated dyes such as resazurin as indicators of parasite viability.25–26 With this in mind, we set about developing an assay system that (i) was able to reliably identify samples containing antitrichomonal compound(s), (ii) was amenable to high-throughput sample testing, (iii) could accurately distinguish between complete versus partial inhibition of T. vaginalis, and (iv) was applicable for use involving a wide variety of sample types including pure compounds and natural product extracts. Herein we report the results of our assay development program, as well as describe new natural product scaffolds that exhibit promising inhibitory activities against T. vaginalis.
RESULTS AND DISCUSSION
Development of an Assay for Detecting T. vaginalis Inhibitors.
Resazurin-based colorimetric assays are widely used in bioactive compound screening campaigns to identify substances that inhibit cell viability and proliferation under aerobic and anaerobic conditions.27 This dye and its H2O soluble salts are readily reduced by NADH to yield the red, fluorescent product resorufin. Despite its reported use for the identification of T. vaginalis inhibitors, we were surprised to discover that, in our hands, this reagent was rather insensitive for the detection of reasonably large numbers of viable cells. We observed that under anaerobic assay conditions, a population of ~10,000 trichomonads per well in a 96-well microtiter plate was needed as a threshold to consistently signal a positive response for the presence of live T. vaginalis. Given that we established an inoculum of 40,000 trichomonads per well as an optimal starting point for assays, it meant that compounds affording modest (~75%) parasite-kill rates would be indistinguishable from more potent and potentially better agents (Figure 1). Therefore, we set about designing a new assay system that was more sensitive, as well as able to handle a wide-range of complex test substances (e.g., pure compounds and extracts containing colored or UV-active natural products).
Figure 1.

Comparison of detection methods, resazurin fluorescence assay (red squares) and the newly developed imaging-based assay (green triangles, two fields per well), for measuring live versus dead trichomonads. The imaging method’s limit of detection was ~1,000 trichomonads per well. In comparison, the resazurin assay detection limit was ~10,000 organisms per well.
One method deemed to be a promising alternative to the resazurin assay was high-content imaging.28 We speculated that this approach would offer a more responsive tool for evaluating the live/dead-status of individual trichomonads within sample populations. However, we quickly recognized that live trichomonads were not fully compatible with imaging-based detection methods; the rapid movements of the flagellated parasites caused the cells to severely blur even with a reasonably fast exposure time (10 ms). Accordingly, we identified a method to fix the trichomonads with glutaraldehyde followed by the application of a dual cell-staining system consisting of acridine orange (cell-permeable DNA stain) and propidium iodide (DNA stain that is not permeant to live cells) for live/dead determination (Figure 2). Initial tests conducted using the Operetta (PerkinElmer) high-content imaging system revealed that this assay tool could detect as few as one live or dead trichomonad per image field and was robust (Z-factor of 0.92). Moreover, this type of detection system was amenable to high-throughput screening of T. vaginalis viability, especially with UV/VIS-active natural products since the stored image files (i.e., two image fields recorded per well) could be manually inspected to identify potential false-positive and false-negative results. Additionally, DMSO was determined to be an acceptable vehicle for compound testing since a concentration of 1% by volume appeared to have no detrimental impact on the viability of T. vaginalis cells.
Figure 2.
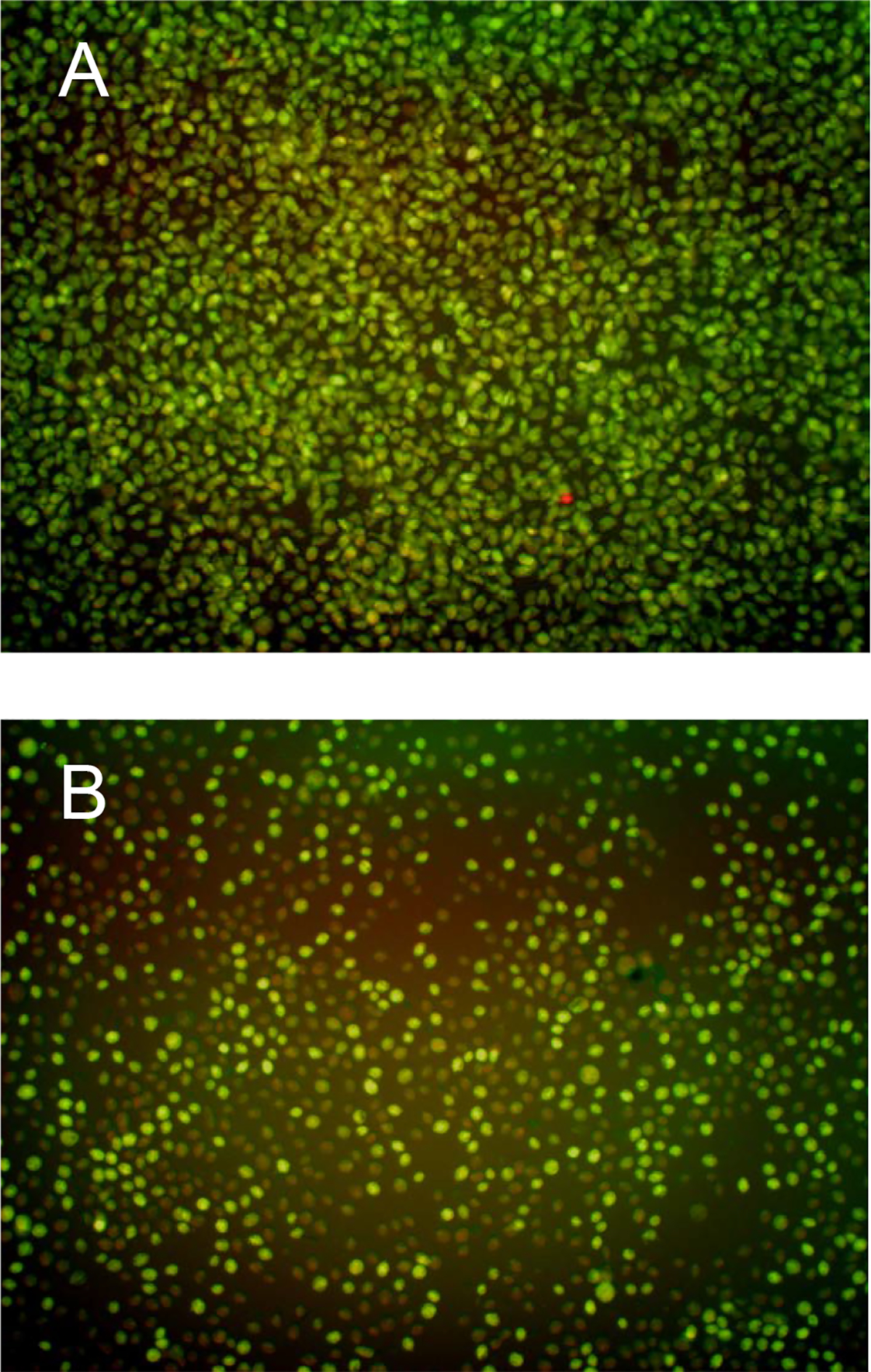
(A) Sample image produced with the Operetta showing the effects of fixing and staining (0.5% glutaraldehyde, 2.5 μM propidium iodide, and 2.5 μM acridine orange) a confluent T. vaginalis culture at 17 h. (B) Sample image showing the effects of a fungal extract causing partial inhibition of T. vaginalis at 17 h. Note the fewer number of cells, the rounded appearance of the remaining cells, as well as the large number of rust colored cells (the rust color is due to the overlay of green and red emission channels) indicating that a majority of the remaining T. vaginalis cells are dead or dying.
Testing Purified Natural Products.
To examine the probative capabilities of the new T. vaginalis imaging-based assay, a focused library of chemically diverse natural products was tested. The compound library consisted of 430 metabolites sourced by our research team from marine organisms, plants, bacteria, and fungi. An initial high-dose concentration (100 μM) was chosen to afford the opportunity to evaluate several challenging screening scenarios, especially those involving potential false-positives brought about by compound precipitation and UV/VIS-interference. The imaging-based assay proved invaluable since all the data files corresponding to potential hits (i.e., compounds that provided inhibition exceeding that afforded by 25 μM metronidazole) were immediately available for inspection to confirm that the number of live trichomonads had been reduced. This test revealed that 64 of the compounds we tested exhibited activities that were equal to or better than the positive control. The large number of active samples was not unexpected since our pure compound library was largely populated by bioactive substances that had previously demonstrated cytotoxicity toward mammalian cells and/or inhibited the proliferation of various microorganisms. To refine the list of natural products further, the hit compounds were screened at 25 μM, which resulted in a subset of 9 compounds that exhibited activities comparable to 25 μM metronidazole (Figure S1, Supporting Information). These results also reinforced the need for a mammalian cell-based cytotoxicity counter screen to aid in the elimination of undesirable non-specific cellular toxins. Accordingly, the 9 hits were tested over a wide-range of concentrations extending two-orders of magnitude (0.25–25 μM) against both T. vaginalis and a normal mouse fibroblast (NIH/3T3) cell line. With these data in hand, the EC50 values for all compounds against both the parasite and mammalian cells were estimated. The EC50 values were used to calculate a selectivity index (SI) value [SI3T3 = (EC50 for 3T3 cells)/(EC50 for T. vaginalis)] for each of the bioactive substances (Figure 3). While the SI values revealed that most of the pure natural products were equipotent inhibitors of trichomonads and mammalian cells, one noted exception was 2-bromoascididemin, which exhibited modest selectivity (SI3T3 = 14). Thus, we established that the new assay was amenable to the testing of structurally diverse natural products across a wide range of concentrations. Furthermore, when coupled with a mammalian cell cytotoxicity screen, it could reveal a substance’s ability to selectively inhibit the target parasite.
Figure 3.
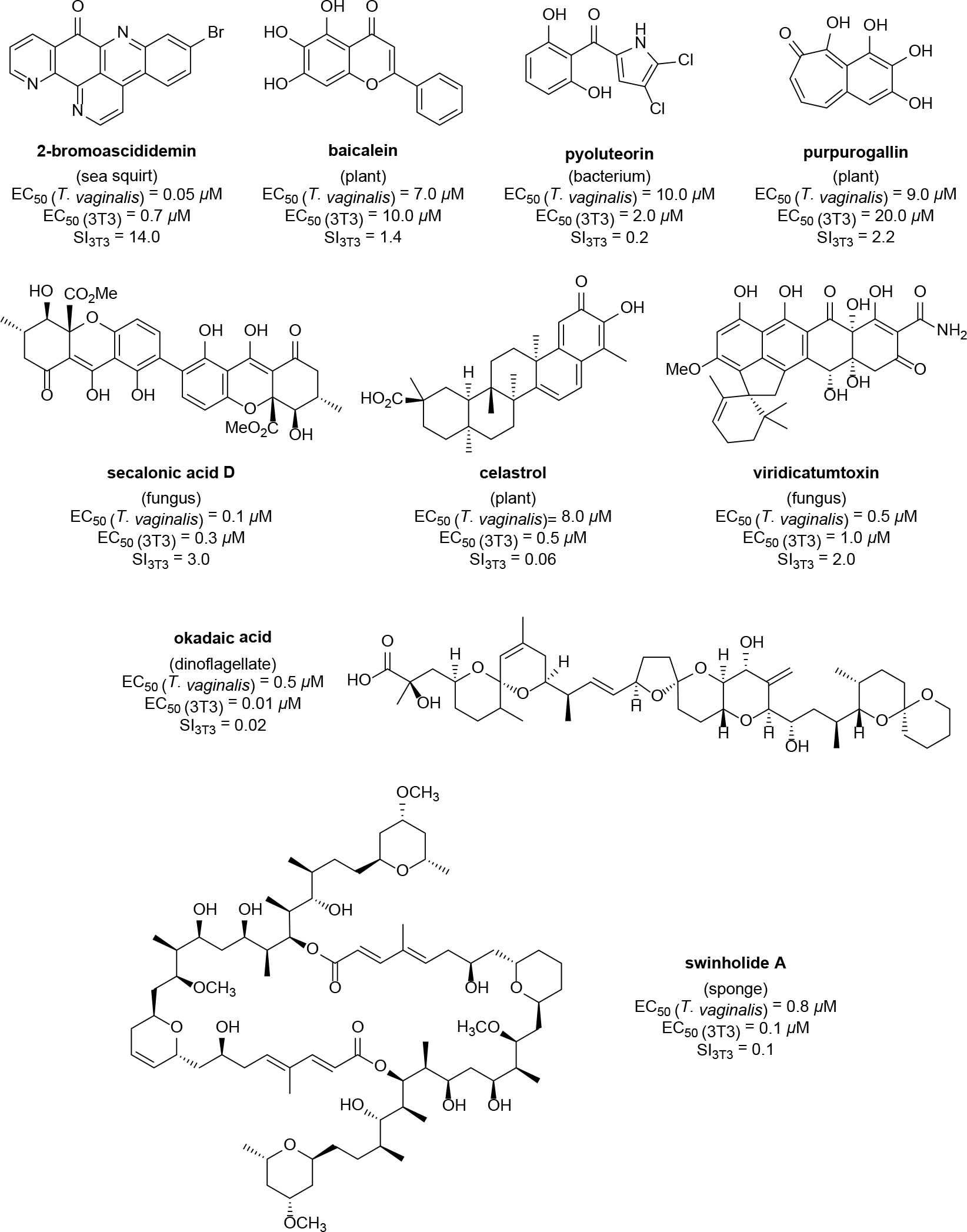
Results from screening 430 purified natural products against T. vaginalis in the imaging-based parasite viability assay. The structures of the 9 most active metabolites are illustrated along with their EC50 values against both the parasite and a mammalian cell line (3T3 fibroblasts). Based on those data, a SI3T3 value was calculated for each compound. Whereas most of the compounds exhibited near equipotent activities against both targets, 2-bromoascididemin exhibited a SI3T3 value of 14.0, which was the first semiselective natural product detected using this screening system.
Testing Fungal Natural Product Extracts.
The University of Oklahoma, Natural Products Discovery Group has generated a library of over 50,000 fungal extracts. Each extract consists of the organic residue obtained after a fungal isolate was cultured in small scale for 3 weeks on Cheerios breakfast cereal, extracted with EtOAc, and partitioned against H2O. For testing purposes, a subset of 1,748 samples was selected for examination in the high-content-imaging assay system. For the first stage of testing, we determined that it was most efficient to screen extracts in duplicate at a single concentration (15 μg/mL). This provided 111 ‘hits’ that inhibited the viability of T. vaginalis at levels equal to or better than the inhibition achieved using 25 μM metronidazole. These ‘hit’ extracts were subjected to a second stage of testing over a range of concentrations (0.15–15 μg/mL), which yielded 71 samples that retained potent activity and generated sigmoidal concentration-response curves. During this period in the assay development process, we considered the value of introducing a second mammalian cell type for selectivity evaluation since fibroblasts cells, although capable of vigorous in vitro growth that made them simple to test, might not offer a level of cytotoxin sensitivity or exhibit cellular properties typically associated with cells at the sites of T. vaginalis infections. Thus, Ect1/E6E7 normal-type cervical cells were chosen to provide a second point of reference for selectivity testing. Accordingly, the EC50 value for each extract’s parasite-inhibitory activity, as well as its cytotoxic effects toward Ect1/E6E7 cells were determined and the data were plotted as illustrated in Figure 4. This data visualization method provided a simple graphical means to identify samples that offered high levels of parasite inhibition yet exhibited limited toxicity toward mammalian cells. Thus, we proceeded to prioritize samples appearing in the upper right quadrant of the graph since those samples offered the best starting points for new bioactive compound discovery. Based on those data, three fungi responsible for generating potent and selective extracts were selected for follow-up chemical investigation including: two Fusarium spp. isolates (plate 55 well F11; SIEct1 = 36 and plate 72 well E6; SI3T3 ≥2.7) and a Humicola sp. isolate (plate 78 well A3; SI3T3 = 25).
Figure 4.

Prioritization scheme for selecting potent and selective bioactive fungal extracts. After an initial pool of 1,748 fungal extracts was tested, 71 of samples were identified as being more active than 25 μM metronidazole and exhibited sigmoidal concentration-response curves against T. vaginalis. The fungi responsible for generating the three active samples indicated in the chart by arrows were selected for bioassay-guided fractionation studies and the bioassay results for the resulting natural products are described in this communication.
Bioassay-Guided Purification and Testing of Natural Products from Fusarium sp. Isolate A.
An extract prepared from Fusarium sp. isolate A was subjected to bioassay-guided fractionation using silica gel vacuum liquid chromatography (VLC), HP20SS VLC, and C18 HPLC chromatography yielding three quinone-containing natural products: fusarubin (1),29 javanicin (2),30 and solaniol (3)31 (compounds were identified based on comparisons of experimental LC-ESIMS and NMR data to the published values)32–33 (Figure 5). The purified metabolites exhibited a range of potencies against T. vaginalis with 1 and 2 being the most potent (EC50 values of 2.5 μM and 1.3 μM, respectively), while 3 showed greatly reduced activity (EC50 = 40 μM). Metabolite 1 stood apart from the other natural products with a SIEct1 = 30, which greatly exceeded the SI values determined for 2 (SIEct1 = 3.1) and 3 (SIEct1 = 0.4) (Figure 5). These data initially suggested that simple quinone-containing scaffolds like 1 might serve as a starting point for the identification of structural analogues that offered improved selectivity and potency.
Figure 5.
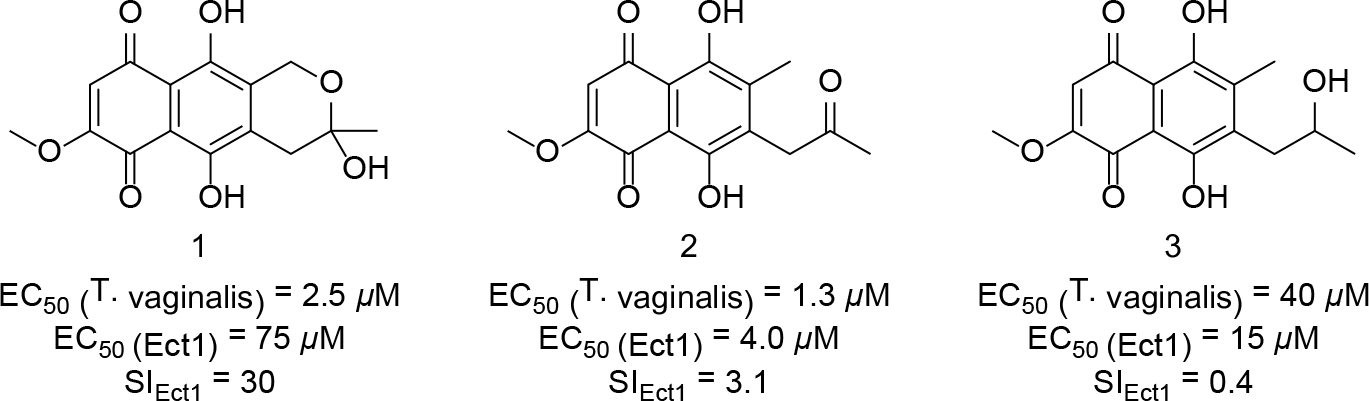
Structures of metabolites 1–3 from F. solani. EC50 values for each compound against T. vaginalis and Ect1 cells are provided in the figure along with their calculated SIEct1 values.
Exploring the Bioactivities for Analogues of Compound 1.
Based on the activities of natural products 1–3, we proceeded to test 42 additional natural and synthetic compounds (compounds 4–45, Table S1, Supporting Information) that shared many key structural elements with the metabolites obtained from Fusarium sp. isolate A to determine if compounds could be identified that afforded improved potency and selectivity. Although many of the tested molecules exhibited low-to-mid-micromolar inhibition of T. vaginalis viability, none of those compounds proved to be as selective as metabolite 1 (other compounds exhibited SIEct1 values ≤4.5) (Figure 6). Results of the bioactivity assessment are summarized in Table S1 (Supporting Information). Notably, atovaquone (37),34 a commercially available antiparasitic drug (used to treat some infections caused by Babesia spp., Plasmodium spp., and Toxoplasma spp.),35 exhibited only modest activity and a poor SIEct1 value of <0.1, suggesting that this compound did not offer a reasonable-level of crossover inhibition against T. vaginalis. Given that none of the tested compounds afforded more favorable activity compared to 1 (i.e., no improvement in potency or SI values), as well as redox-related concerns (e.g., potential for quinones to react with nucleophilic amino acid residues, as well as a notorious history of problems in a variety of assays),36 we became concerned that this scaffold might not provide a suitable opportunity for further development. To examine if redox activity might partially explain the activity of 1 and related compounds, we devised a test in which the potencies of compound 1, 2-methoxynaphthazarin (9), and metronidazole were assessed under three atmospheric conditions with different oxygen levels (i.e., aerobic, microaerophillic, and anaerobic). The results of those tests revealed that the activities of 1 and 9 both decreased as oxygen was introduced into the test culture conditions (Figure 7). Curiously, compounds 1 and 9 differed in another respect; 1 was only able to afford the partial elimination of trichomonads under strict anaerobic condition, whereas 9 was able to provide for the complete elimination of the parasite in an anaerobic atmosphere. In contrast, metronidazole maintained consistent potency across all the tested atmospheric conditions (Figure 7). Given the expected heterogeneity of oxygen levels across the various body sites infected by T. vaginalis, we abandoned further pursuit of 1 and its analogues in favor of other, more promising compounds.
Figure 6.
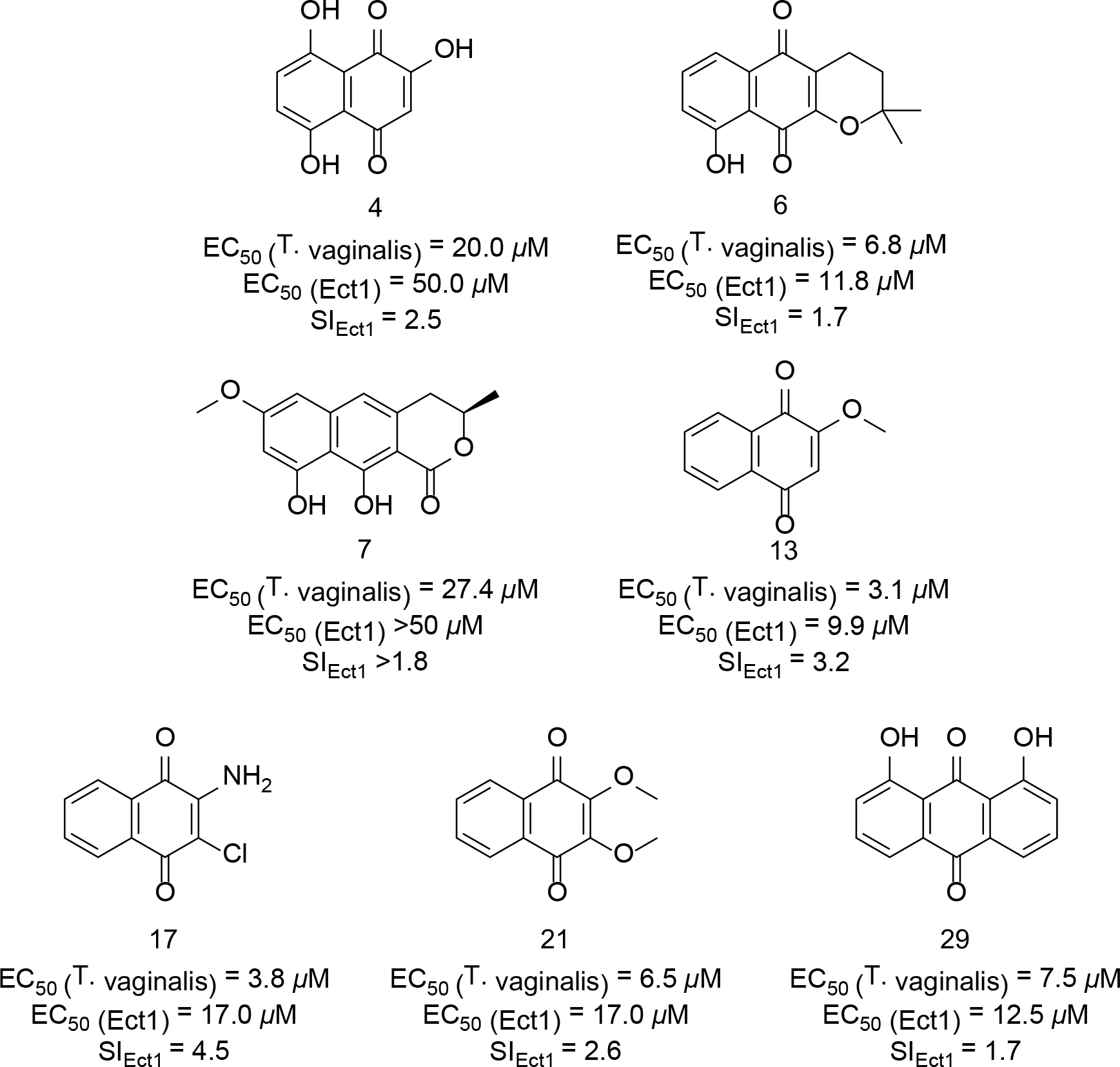
Structures and biological activity of synthetic and commercially available quinone-containing compounds that exhibited SIEct1 >1. EC50 values for each compound against T. vaginalis and Ect1 cells are provided in the figure along with their calculated SIEct1 values.
Figure 7.

Testing the effects of varying oxygen levels on the antitrichomonal activities of fusarubin (1), 2-methoxynaphthazarin (9), and metronidazole using the imaging assay [data are expressed as the mean of two visual fields per well performed in triplicate ± SD; DMSO (vehicle) did not exceed 1% by volume].
Bioassay-Guided Purification and Testing of Natural Products from a Humicola sp. Isolate.
The extract prepared from the Humicola sp. isolate was subjected to bioassay-guided fractionation using silica gel vacuum liquid chromatography (VLC), HP20SS VLC, and C18 HPLC purification steps to afford the xanthone-anthraquinone heterodimer xanthoquinodin A1 (46), which was identified by comparisons of its experimental 1H and 13C NMR spectra, LC-ESIMS data, and specific rotation value to the published values.37 Compound 46 was tested for activity against T. vaginalis, as well as counter-screened against Ect1/E6E7 cells, which revealed the natural product had a favorable SIEct1 of ~20. Based on our prior experiences with redox-active compounds (vide supra), 46 was tested under aerobic, microaerophillic, and anaerobic conditions to determine how those changes would influence its activity. To our surprise, compound 46 maintained consistent potency under all experimental conditions (average EC50 = 3 μM) (Figure S28, Supporting Information), which suggested that this compound functioned in a manner that was different than 1 and its analogues.
Reduction of Xanthoquinodin A1.
While the activity of 46 was different compared to the simple quinones that we had tested, we were interested in determining how this compound would work if it were subjected to chemical reduction. For that reason, compound 46 was treated with sodium borohydride to provide 47 (Scheme 1). Analysis of the 13C NMR data for 47 revealed the loss of a carbonyl chemical shift relative to 46 with the concomitant addition of a new spin at δC 71.9. Follow-up HSQC and HMBC experiments [e.g., correlation from H-10’ (δH 5.04) → C-13 (δC 30.5)] and ROESY [e.g., cross-peak observed between H-10’ ↔ H-4’ (δH 6.47)] confirmed that the C-10’ carbonyl in 46 had been stereospecifically reduced to an alcohol yielding a single isomeric product. Testing revealed that the inhibitory activity of 47 against T. vaginalis (EC50 = 3 μM) was indistinguishable from 46 (Figure S8, Supporting Information). This indicated that unlike compounds 1–45, the activity of natural product 46 may not be as susceptible to redox-dependent processes.
Scheme 1.
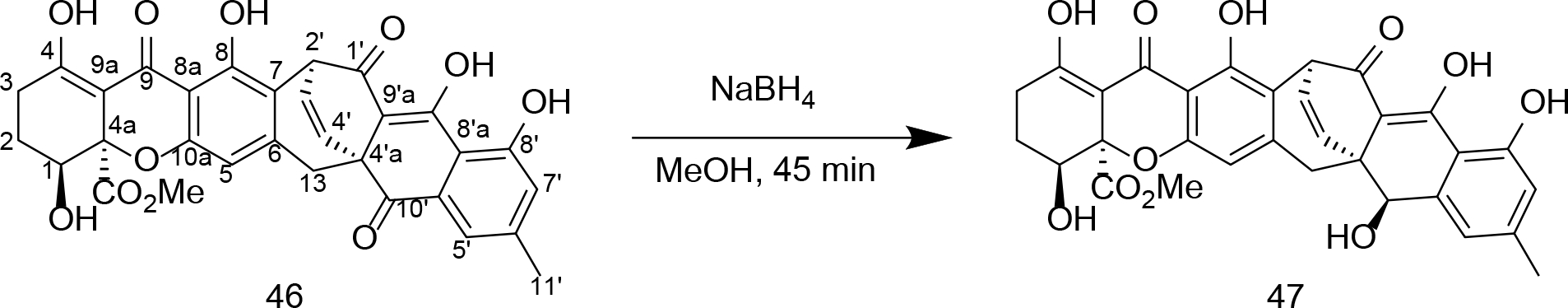
Structure of xanthoquinodin A1 (46) from the H. grisea isolate. The compound was subjected to NaBH4 reduction leading to the stereoselective generation of the new secondary-alcohol-containing product 47.
Establishing an Antibacterial Counter-Screen with Lactobacillus acidophilus.
While the antitrichomonal activity of 46 appeared promising, it was not apparent what effect this and other compounds we anticipated discovering would have on a person’s microflora. This was a concern because microbiome disruption can have deleterious consequences for patients following antibiotic treatment.38 For that reason, Lactobacillus acidophilus was chosen as a representative bacterium species because of its widespread occurrence in the human mouth, intestinal tract, and vagina.39 Unfortunately, compound 46 was determined to exhibit near equipotent activity against T. vaginalis and L. acidophilus (Figure 8). In contrast, metronidazole showed no inhibition of the bacterial indicator strain at the concentrations tested, although this compound does inhibit other bacteria.40 Consequently, 46 was dropped from further consideration given the negative impact that its antibacterial activity might have on the stability of a patient’s microflora.
Figure 8.
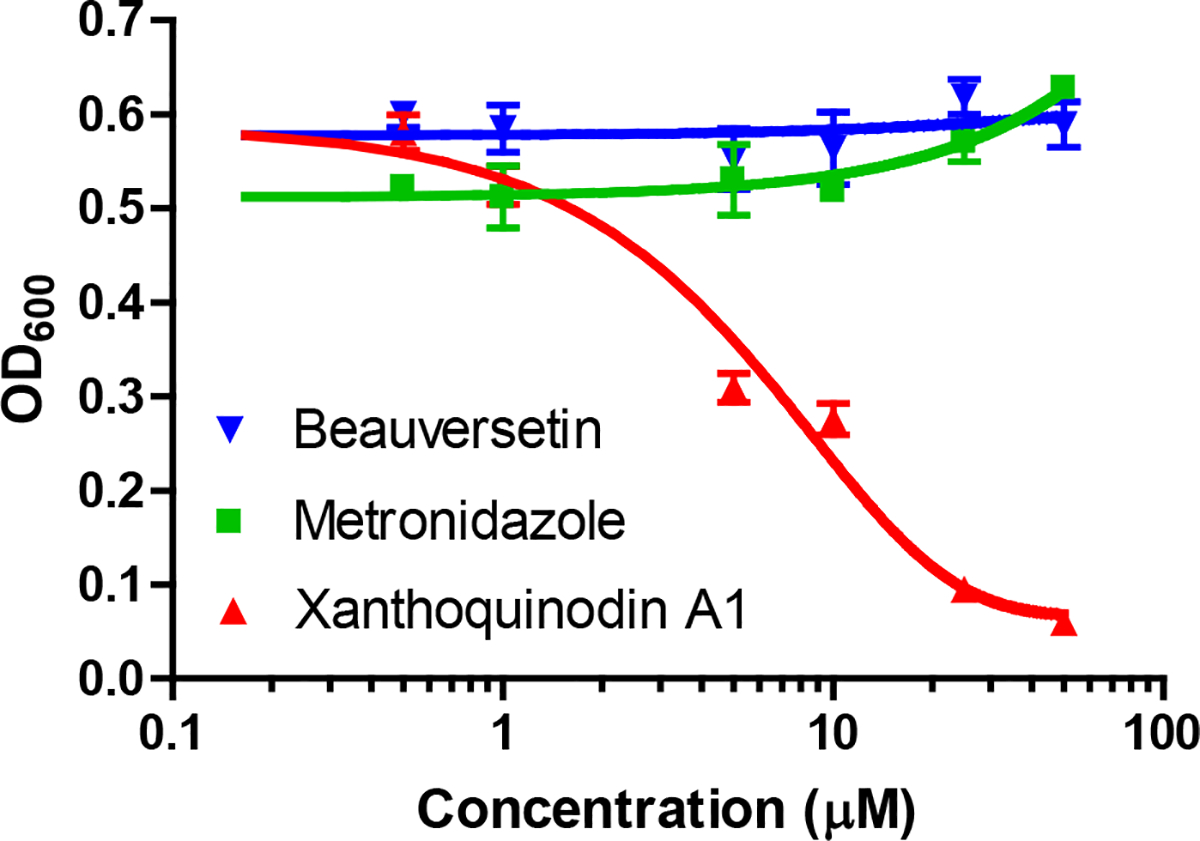
Effect of selected compounds [beauversetin (53), xanthoquinodin A1 (46), and metronidazole] on the growth and viability of L. acidophilus grown under anaerobic conditions. The tetramic-acid-containing compound (53) had no effect on the bacterium at the concentrations tested. In contrast, 46 exhibited significant inhibition of this important member of the vaginal microflora [data are expressed as the mean of triplicate experiments ± SD; DMSO (vehicle) did not exceed 1% by volume].
Bioassay-Guided Purification and Testing of Natural Products from Fusarium sp. Isolate B.
A crude extract prepared from Fusarium sp. isolate B was subjected to bioassay-guided fractionation using silica gel vacuum liquid chromatography (VLC), HP20SS VLC, and C18 HPLC to afford the compound equisetin (48). In the process of obtaining 48, 5’-epiequisetin (49) was also purified from the extract since its LC-ESIMS data had provided evidence that it was likely a structural analogue of the bioactive constituent. The purified compounds were identified by comparisons of their 1H and 13C NMR spectra and HRESIMS data to published values.41 The absolute configurations of the tetramic acid portions of both compounds were determined using a previously described oxidative bond-cleavage and LC-ESIMS-facilitated analysis methodology.42–43 Testing of the purified compounds revealed a stark difference in their respective antitrichomonal activities; whereas 48 exhibited rather potent inhibitory effects (EC50 = 3 μM) with good selectivity (SI3T3 = 33), 49 was inactive at concentrations up to 25 μM. An examination of both metabolites revealed that the remarkable difference in their biological activities could be traced to the epimerization of a single stereocenter (5’S in 48 and 5’R in 49). Taking these data into consideration, as well as noting that 48 was non-toxic to L. acidophilus at concentrations up to 50 μM (Table 3), there was ample reason to believe that further exploration of this family of natural products had the potential to provide additional bioactive analogues.
Table 3.
Bioassay results for the tetramic-acid-containing metabolites 48–56 including compound sources, as well as EC50 and SI index values
| Assay (EC50 Data in μM) |
Selectivity Index Value (SI) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Fungal Isolate Code | Isolate Identity from ITS | Compound | T. vaginalis | 3T3 Mouse Fibroblast | Ect1 Normal Cervical | L. acidophilus | SI3T3 | SIEct1 | SIL. acid |
|
| |||||||||
| Miller-1 Cz-3 | Fusarium sp. isolate B | equisetin (48) | 3.0 | 100 | 35 | >50 | 33 | 12 | >17 |
| 5’-epiequisetin (49) | Inactive | nt | nt | nt | nt | nt | nt | ||
|
| |||||||||
| Miller-26 SEA-3 | Fusarium sp. isolate C | trichosetin (50) | 2.5 | 30 | 34 | >50 | 12 | 14 | >20 |
|
| |||||||||
| KY6863 TV8-3 | Penicillium sp. | pyrrolocin A (51) | 0.060 | 6 | 10 | >50 | 100 | 167 | >833 |
| 5’-epipyrrolocin A (52) | Inactive | nt | nt | nt | nt | nt | nt | ||
|
| |||||||||
| Column L5 SEA-1 | Alternaria sp. | beauversetin (53) | 0.80 | 35 | 35 | >50 | 44 | 44 | >62 |
| 5’-epibeauversetin (54) | Inactive | nt | nt | nt | nt | nt | nt | ||
|
| |||||||||
| CA9310 TV8-3 | Phoma sp. | phomasetin (55) | 0.35 | 10 | 7 | >50 | 29 | 20 | >142 |
| 5’-epiphomasetin (56) | Inactive | nt | nt | nt | nt | nt | nt | ||
nt: not tested
To facilitate the identification of other natural products containing a tetramate linked to a trans-decalin system, we turned to our extensive library of fungal isolates and their associated chemical data. This family of natural products has been reported from a taxonomically diverse assemblage of fungi41–42, 44–46 and our records revealed that we had previously encountered these compounds from several fungal isolates in our collection. After considering the range of structural variation that we could anticipate from the fungal isolates available to us, 4 fungi were selected to serve as the subjects of a targeted (chemically-guided) purification process: (i) Fusarium sp. isolate C, (ii) Penicillium sp., (iii) Alternaria sp., and (iv) Phoma sp. P34E5. Those efforts led to the purification of 7 additional tetramic-acid-containing metabolites including trichosetin (50),45 pyrrolocin A (51),42 5’-epipyrrolocin A (52) (new), beauversetin (53),46 5’-epibeauversetin (54) (new), phomasetin (55),44 and 5’-epiphomasetin (56)44 (Figure 9 and Table 3). Whereas the previously reported tetramic acid metabolites were identified based on comparisons of their experimental versus published spectrometry and spectroscopy data, the new analogues became the foci of our structure determination efforts.
Figure 9.
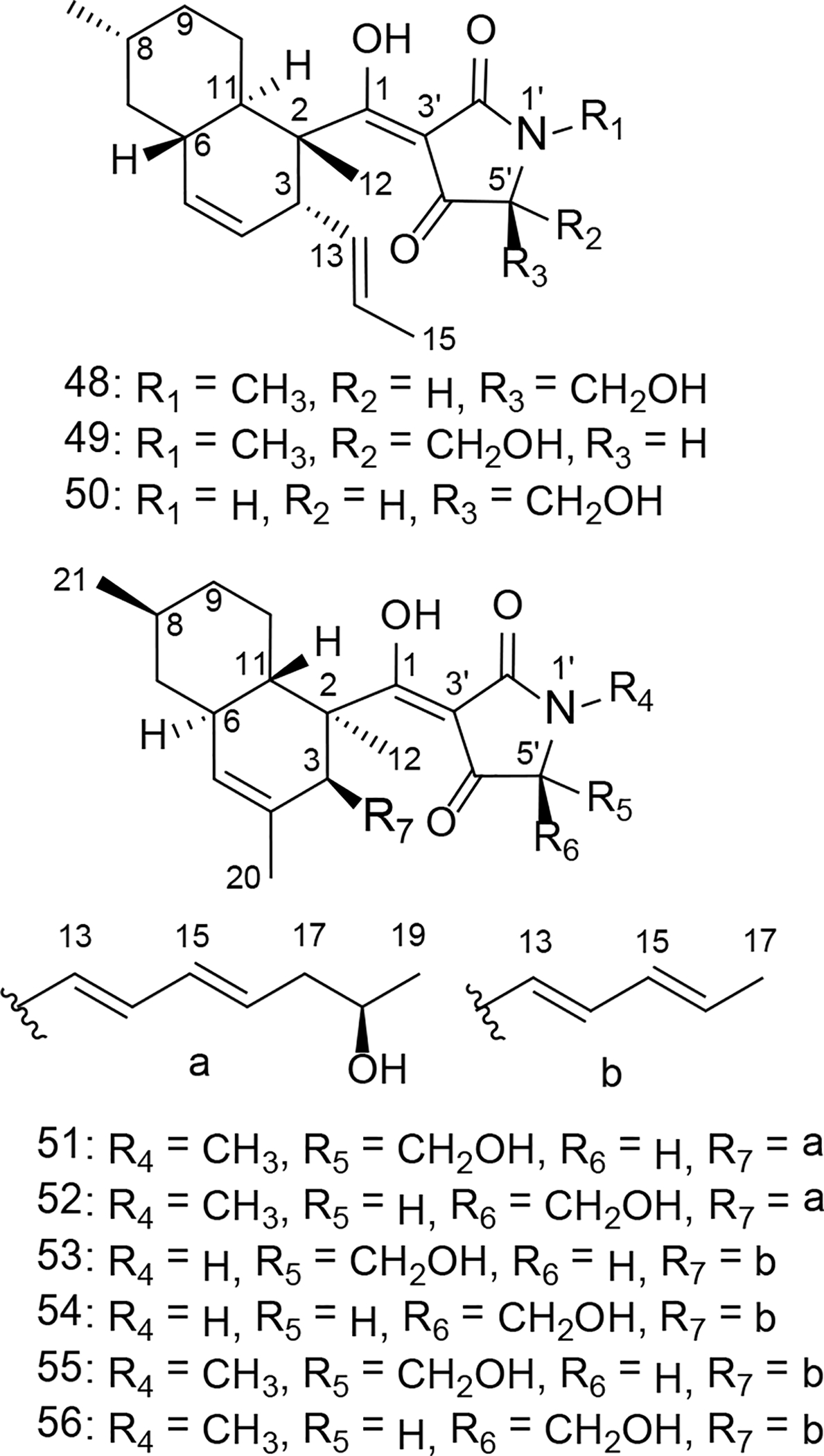
Structures of tetramic-acid-containing metabolites 48–49 from Fusarium sp. isolate B, 50 from Fusarium sp. isolate C, 51–52 from Penicillium sp., 53–54 from Alternaria sp., and 55–56 from Phoma sp. P34E5.
Compound 52 (purified from the Penicillium sp. isolate) was obtained as a pale yellow solid. Its molecular formula was deduced from HRESIMS data to be C27H39NO5 ([M-H]− ion at m/z 456.2747, calcd for 456.2755). The 1H and 13C NMR data (Tables 1 and 2) revealed the presence of eight olefinic carbons, an oxygenated methylene, an oxygenated methine, and 5 methyl groups. Comparison of the 1D NMR data of 52 with compound 51 (isolated from the same fungus) indicated that the only noteworthy difference in the 13C NMR data for these two molecules was that the carbon spin resonating at δC 68.1 in compound 51 now appeared at δC 67.4 in metabolite 52. Combining this information with the bond-line structure established for 52 (based on analyses of its 1H-1H COSY, HSQC, and HMBC data, Figure 10A), it was proposed that the new natural product was the 5’-epimer of 51.
Table 1.
1H NMR data of compounds 51–54 in DMSO-d6
|
δH (J in Hz) |
||||
|---|---|---|---|---|
| Position | 51a | 52a | 53b | 54b |
|
| ||||
| 3 | 3.15, m | 3.25, m | 3.20, m | 3.21, m |
| 5 | 5.19, s | 5.18, s | 5.19, s | 5.17, s |
| 6 | 1.79c | 1.78c | 1.79c | 1.78 c |
| 7ax | 0.82, m | 0.81, m | 0.80, m | 0.80, m |
| 7eq | 1.78c | 1.77c | 1.77c | 1.77 c |
| 8 | 1.49, m | 1.48, m | 1.48, m | 1.48, m |
| 9ax | 1.70, m | 1.70, m | 1.70, m | 1.70, m |
| 9eq | 1.01c | 1.00c | 1.01, m | 0.97, m |
| 10ax | 1.00c | 0.98c | 0.95, m | 0.94, m |
| 10eq | 1.89, m | 1.90, m | 1.93, m | 1.93, m |
| 11 | 1.57, m | 1.57, m | 1.54, m | 1.53, m |
| 12 | 1.33, s | 1.36, s | 1.33, s | 1.35, s |
| 13 | 5.22, m | 5.16, m | 5.15, m | 5.11, dd (10.4, 14.7) |
| 14 | 5.71, m | 5.79c | 5.72, dd (10.5, 14.7) | 5.78, dd (10.5, 14.7) |
| 15 | 5.88, m | 5.82c | 5.90, dd (10.5, 13.7) | 5.86, dd (10.5, 13.7) |
| 16 | 5.50, m | 5.46, m | 5.50, m | 5.46, m |
| 17 | 2.00, m | 1.99, m | 1.63, d (6.5) | 1.62, d (6.5) |
| 2.09, m | 2.08, m | |||
| 18 | 3.57, m | 3.57, m | 1.52, s | 1.51, s |
| 19 | 0.98, d (6.0) | 0.98, d (6.0) | 0.88, d (6.3) | 0.88, d (6.3) |
| 20 | 1.52, s | 1.52, s | ||
| 21 | 0.88, d (6.5) | 0.88, d (6.5) | ||
| 1’ | 9.25, s | 9.05, s | ||
| 5’ | 3.75, br s | 3.71c | 3.80, br s | 3.78, br s |
| 6’ | 3.81, d (11.0) | 3.81, m | 3.63, dd (2.1, 11.0) | 3.67, dd (2.1, 11.0) |
| 3.69, m | 3.72c | 3.59, m | 3.61, m | |
| N-Me | 2.92, s | 2.91, s | ||
Recorded at 500 MHz
Recorded at 400 MHz
Overlapped signals determined from HSQC correlations.
Table 2.
13C NMR data for compounds 51–54 in DMSO-d6 (100 MHz, δ in ppm)
|
δ
C
|
||||
|---|---|---|---|---|
| Position | 51 | 52 | 53 | 54 |
|
| ||||
| 1 | 195.9 | 196.5 | 199.0 | 199.0 |
| 2 | 49.0 | 49.6 | 49.2 | 49.5 |
| 3 | 49.2 | 48.6 | 48.5 | 48.1 |
| 4 | 131.3 | 131.9 | 131.4 | 131.6 |
| 5 | 126.4 | 126.1 | 126.0 | 125.7 |
| 6 | 39.1 | 39.2 | 38.7 | 38.7 |
| 7 | 42.4 | 42.5 | 42.1 | 42.2 |
| 8 | 33.3 | 33.4 | 33.0 | 33.0 |
| 9 | 35.8 | 35.9 | 35.5 | 35.5 |
| 10 | 28.1 | 28.1 | 27.6 | 27.7 |
| 11 | 39.4 | 39.9 | 39.3 | 39.3 |
| 12 | 14.0 | 14.2 | 13.4 | 13.5 |
| 13 | 130.9 | 131.4 | 130.5 | 130.6 |
| 14 | 132.7 | 133.0 | 132.1 | 132.5 |
| 15 | 131.8 | 132.0 | 131.1 | 131.6 |
| 16 | 130.9 | 130.5 | 128.0 | 127.7 |
| 17 | 42.7 | 42.8 | 17.8 | 17.9 |
| 18 | 66.3 | 66.3 | ||
| 19 | 23.5 | 23.5 | ||
| 20 | 22.6 | 22.7 | 22.2 | 22.3 |
| 21 | 22.8 | 22.9 | 22.5 | 22.5 |
| 2’ | 176.6 | 175.8 | 179.2 | 179.0 |
| 3’ | 100.9 | 101.6 | 99.4 | 99.8 |
| 4’ | 190.9 | 190.5 | 190.7 | 190.6 |
| 5’ | 68.1 | 67.4 | 63.2 | 62.9 |
| 6’ | 58.4 | 58.6 | 60.8 | 60.7 |
| N-Me | 27.2 | 27.3 | ||
Figure 10.
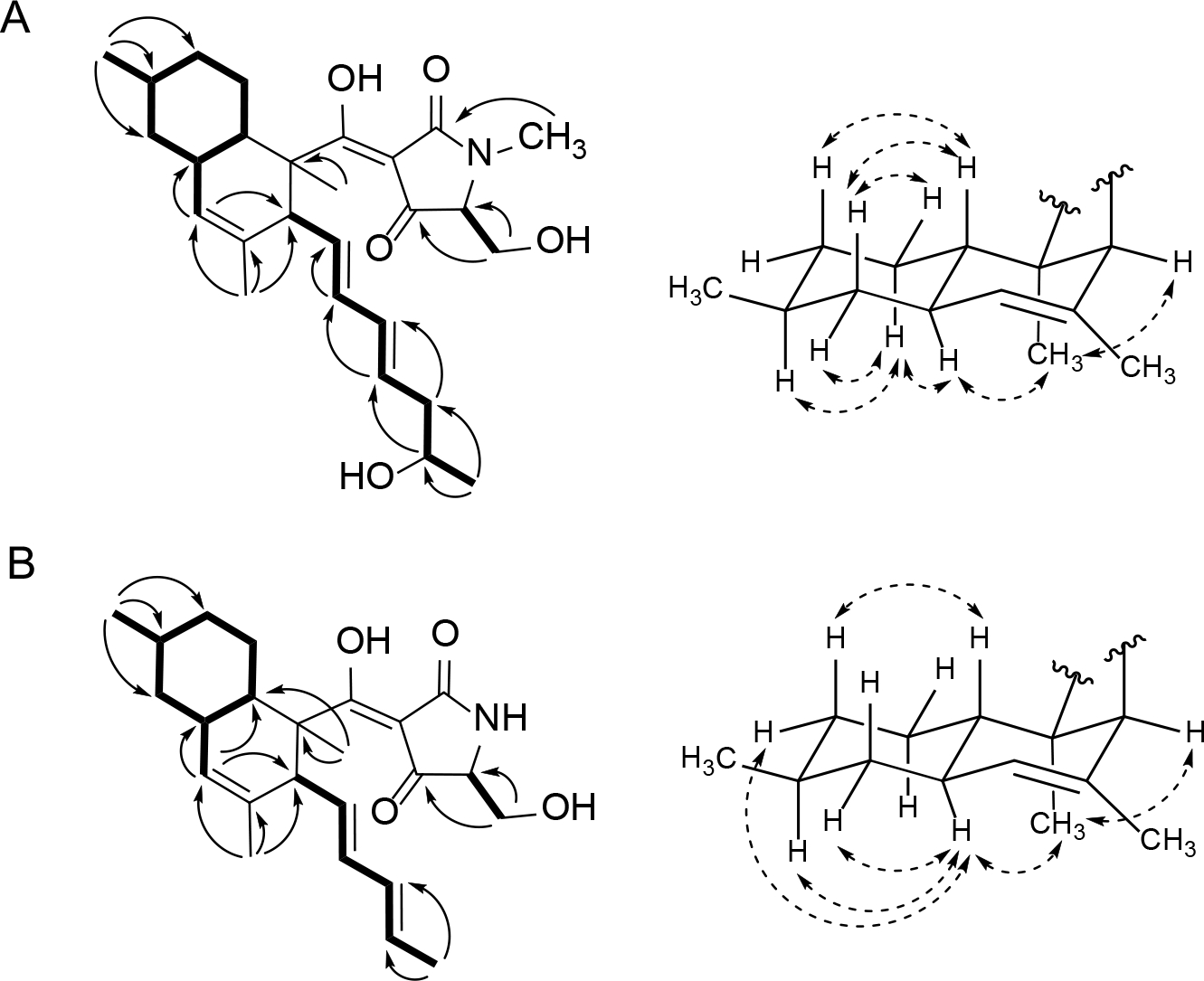
Key 1H-1H COSY (left side – bold bonds), 1H-13C HMBC (left side – single-headed arrows), 1H-1H ROESY (right side – double headed arrows) correlations for compounds 52 (A) and 54 (B).
Upon probing the stereochemical properties of compound 52, the relative configuration of the decalin system was affirmed based on 1H-1H ROESY correlations detected between the following sets of protons: Me-12 ↔ H-6 and H-3; H-10ax ↔ H-6, H-7eq, and H-8; as well as H-7ax ↔ H-10eq and H-11 (Figure 10A). The absolute configuration of this moiety was determined by comparing its CD spectrum with data obtained for similar metabolites. Specifically, the positive Cotton effects at 234 and 291 nm indicated that the absolute configuration of the decalin-ring portion of this structure was 2R,3S,6R,8S,11S (Figure 12, Figures S31 and S32, Supporting Information).42 The absolute configuration of the hydroxy group on the olefinic side chain was determined to be 18R based on an analysis employing the modified Mosher reaction (Figure 11).42, 47 The absolute configuration of C-5’ was determined by oxidative bond cleavage of the tetramic acid ring followed by acid hydrolysis to yield N-Me-serine.42–43 Marfey derivatization of the N-Me-serine was performed and the product was analyzed by LC-ESIMS together with the derivatized products generated from both N-Me-L- and D-serine standards. The N-Me-D-serine was made by racemization of commercially available N-Me-L-serine using acetic acid and salicylaldehyde as described previously.48–49 The resulting amino acid residue was determined to be N-Me-L-serine, thus, the absolute configuration of 52 was assigned as 2R,3S,6R,8S,11S,18R,5’S.
Figure 12.
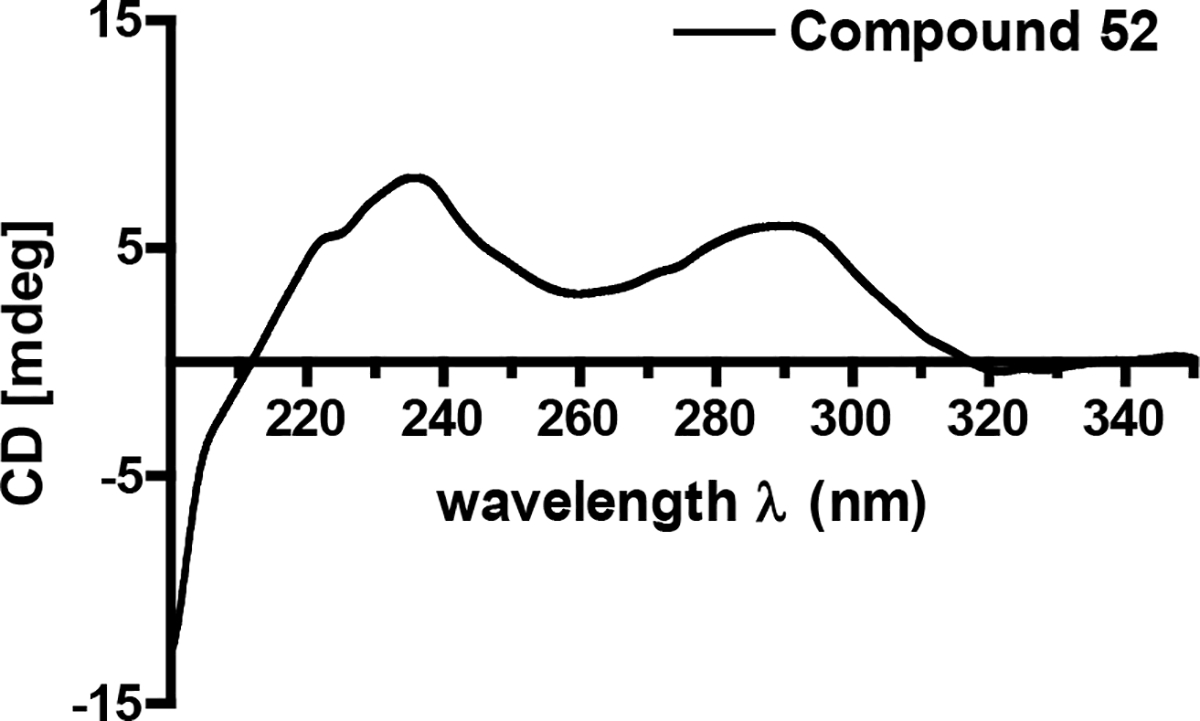
Circular dichroism (ECD) spectrum for compound 52.
Figure 11.
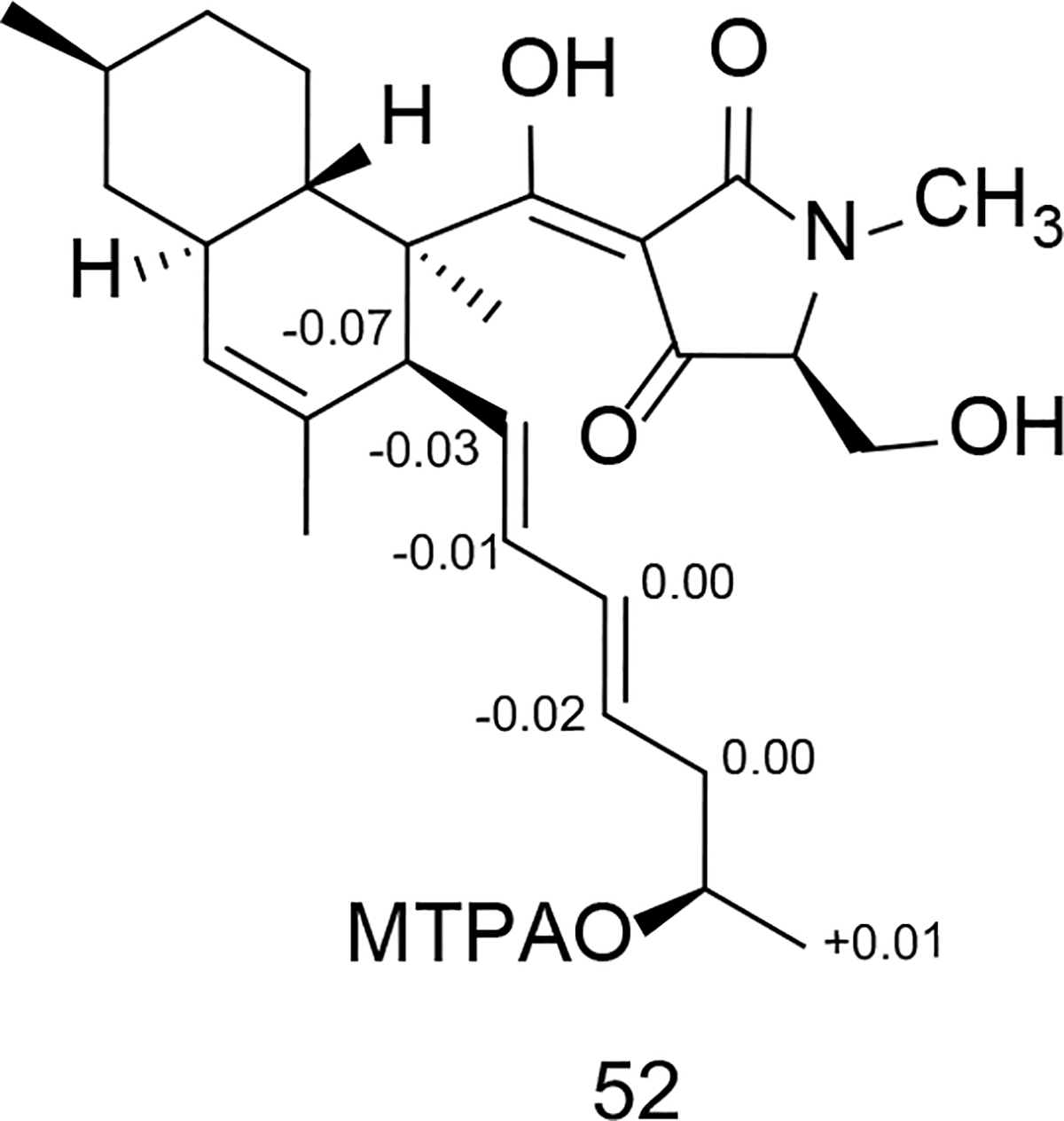
ΔδS-R values obtained in pyridine-d5 of the MTPA esters for compound 52.
Compound 54 was purified as a pale yellow solid. Its molecular formula was deduced as C24H33NO4 based on HRESIMS data. The 1H NMR spectrum of 54 (Table 1) showed thirty-one proton signals including 5 aromatic protons and 4 methyl spins. The 13C NMR spectrum (Table 2) contained 24 carbon spins, which were later classified according to their multiplicities using an HSQC experiment. Those data indicated that 54 was structurally similar to 52. Based on 2D NMR (1H-1H COSY, HSQC, and HMBC) data (Figure 10B), the bond-line structure of 54 was confirmed to be the same as metabolite 53, which indicated that this compound was likely to be another 5’-epimer.
The relative configuration of the decalin moiety was deduced as 2R*,3S*,6R*,8S*,11S* based on the key 1H-1H ROESY correlations between Me-12 ↔ H-3 and H-6, H-6 ↔ H-9eq, H-6 ↔ H-8, as well as H-11 ↔ H-7ax and H-9ax (Figure 10B). The positive Cotton effects recorded for 54 at 234 and 289 nm indicated that the absolute configuration of the decalin moiety in 54 was the same as that in compound 53 (Figure 13). Further analysis of key ROESY correlations including H-15 ↔ H-13 and Me-17 revealed both exocyclic olefins (Δ13 and Δ15) had trans geometries. Using the same oxidative bond cleavage and LC-ESIMS analysis method that had been applied to the tetramic acid moiety in metabolite 52, the absolute configurations of C-5’ stereocenters were determined as 5’R (53) and 5’S (54), respectively.42–43, 49 Thus, the absolute configuration of 54 was determined to be 2R,3S,6R,8S,11S,5’S.
Figure 13.
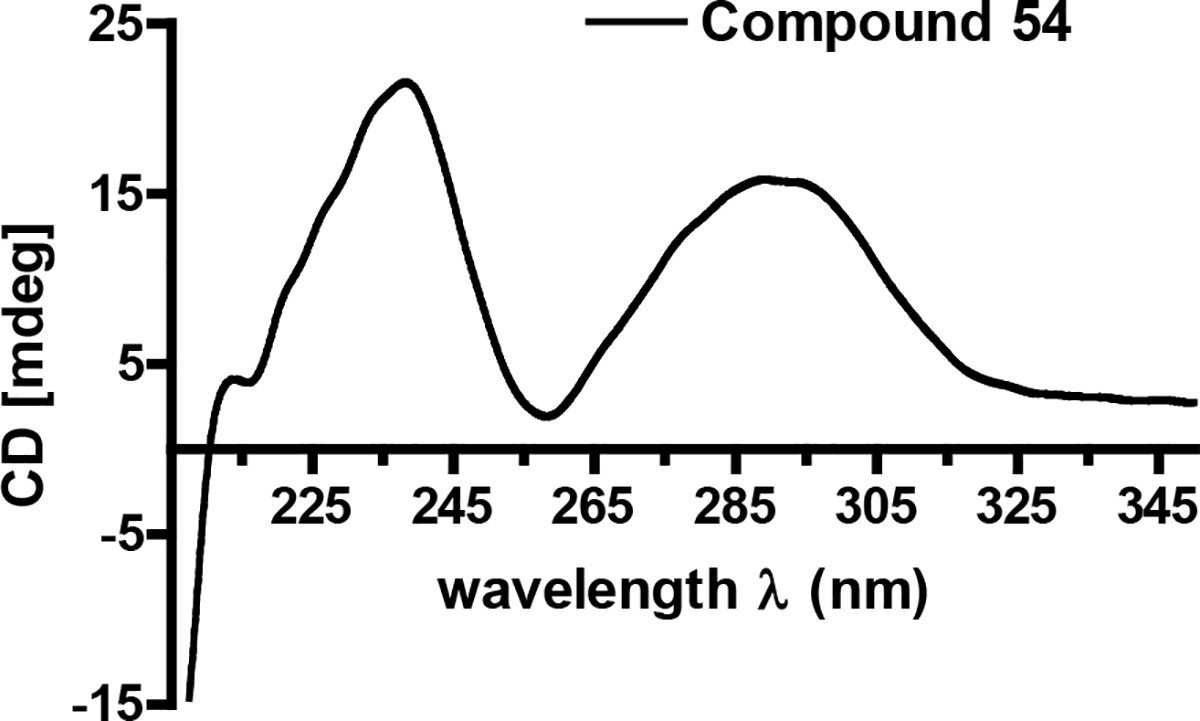
Circular dichroism (ECD) spectrum for compound 54.
Assessing the Biological Activities of the Tetramic-Acid-Containing Natural Products.
The antitrichomonal effects of the tetramic-acid-containing natural products were evaluated revealing an intriguing trend; whereas several of the compounds exhibited potent activities with EC50 values at or below single digit micromolar concentrations (i.e., 48, 50, 51, 53, and 55) their 5’-epimers were inactive even at concentrations as high as 25 μM (Table 3). The three most potent molecules, 51 (EC50 = 0.060 μM), 53 (EC50 = 0.80 μM), and 55 (EC50 = 0.35 μM), revealed that the tetramic-acid-containing molecules were among the most potent T. vaginalis inhibitors we have observed to date. To determine if the Trichomonas-inhibitory effects of the tetramic acid natural products were affected by the presence of molecular oxygen, metabolite 53 was tested under aerobic, microaerophilic, and anaerobic conditions (Figure S29, Supporting Information). Unlike many of the other compounds that we tested, 53 maintained potency under all the test atmospheric conditions exhibiting no statistically significant change in EC50 value, as well as affording the complete elimination of the parasite. Although some tetramic-acid-containing compounds have been reported to possess antibacterial activities,50–51 none of the compounds that inhibited T. vaginalis inhibited L. acidophilus at 50 μM (Figure 8). The selectivity of the tetramic-acid metabolites for T. vaginalis versus mammalian cells also appeared favorable (Table 3) with relatively large SI values for compound 51 (SI3T3 = 100, SIEct1 = 167). Based on these data, the tetramic-acid-containing natural products exhibited many promising characteristics that highlight their potential for further development as inhibitors of T. vaginalis.
CONCLUSIONS
A rapid and sensitive assay for the detection of T. vaginalis inhibitors was developed and used to test a variety of substances varying from pure compounds to complex fungal extracts. In the process of screening a portion of our fungal extract library, we detected and went on to purify several natural products that exhibited inhibitory activities against T. vaginalis and were relatively non-cytotoxic to human Ect1/E6E7 cells. In parallel to their PAINS-like qualities, quinone-containing compounds36, 52 were generally found to be problematical starting points for the development of new antitrichomonal scaffolds since their inhibitory properties were significantly influenced by the relative amounts of oxygen the trichomonads were exposed to throughout the assay period. Moreover, these types of rather promiscuous compounds exhibited antibacterial activities against L. acidophilus suggesting that their non-specific antibiosis effects could upset the microbiome structures of patients, thus creating new potential problems.
Based on the lessons learned from the quinones, we proceeded to test a second group of more promising tetramic-acid-containing natural products as inhibitors of T. vaginalis. A striking feature of their antitrichomonal effects was the decided difference in the biological activities of the 5’-epi-isomers, which generally showed no inhibitory effects against T. vaginalis. Compound 51 emerged as a rather promising metabolite based on its potent activity against the parasite (EC50 = 0.060 μM) and limited toxicity against both the human Ect1/E6E7 cervical cell line (SI = 167) and L. acidophilus (EC50 > 50 μM). Although our testing was limited to 9 tetramic-acid-containing natural products, the data suggested that modifications to the C-3 group of the trans-decalin system may provide additional opportunities for generating new analogues that offer even further improvements in potency and selectivity.
EXPERIMENTAL SECTION
General Experimental Procedures.
UV data were recorded on a Hewlett Packard 8452A diode array spectrophotometer. Optical rotation measurements were made on an AUTOPOL® III automatic polarimeter. The LC-ESIMS analyses were performed on a Shimadzu UFLC system with a quadrupole mass spectrometer using a Phenomenex Kinetex C18 column (3.0 mm × 75 mm, 2.6 μm) and MeCN-H2O (0.1% HCOOH) gradient solvent system. HRESIMS spectra were measured using an Agilent 6538 Ultra High Definition (UHD) Accurate-Mass Q-TOF system. NMR spectra were obtained on Varian spectrometers (500 MHz and 400 MHz for 1H, and 100 MHz for 13C) using DMSO-d6 and CDCl3 as solvents. Column chromatography was conducted using silica gel and HP20SS. HPLC was performed on a Waters System equipped with a 1525 binary HPLC pump coupled to a 2998 PDA detector with a Phenomenex Gemini C18 column (21.2 × 250 mm or 10 × 250 mm, 5 μm particle size).
Culture of Organisms.
Trichomonas vaginalis Donne (PRA-98) was purchased from the American Type Culture Collection (ATCC, Bethesda, MD). After some investigation, Keister’s Modified TYI-S33 medium53 (adjusted to pH 6.0 and without Diamond’s vitamin solution) was determined to be the optimum medium to support the growth of T. vaginalis. The medium was filter sterilized and aliquots stored frozen for later use. A notable difference in the growth of T. vaginalis was observed dependent upon the source from which the bovine bile obtained. The best growth of T. vaginalis was observed when Sigma B8381 bovine bile was used and it was adopted as the sole bovine bile source for all experiments. For culture maintenance and propagation, trichomonads were grown in a 37 °C incubator in sealed screw cap tubes. For microtiter plate assays, trichomonads were tested under several atmospheric conditions including standard atmospheric condition (aerobic atmosphere: ~21% O2, 0.04% CO2), candle jar (microaerophilic atmosphere: ~10% O2, 5% CO2), BD GasPak EZ Campy sachets (microaerophilic atmosphere: 6–16% O2, 2–10% CO2), and BD GasPak EZ Anaerobe sachets (anaerobic atmosphere: O2 at nondetectable level, >13% CO2). Unless specified, assays were carried out in conditions employing reduced levels of oxygen and increased levels of carbon dioxide (microaerophilic), which stimulates Trichomonas growth.54
NIH/3T3 (CRL-1658) mouse fibroblast and Ect1/E6E7 (CRL-2614) normal-type human ectocervical cell lines were purchased from ATCC. NIH/3T3 cells were maintained in Dulbecco’s Modified Eagle’s medium with 5% FetalClone III (Hyclone) and Ect1/E6E7 cells in keratinocyte-serum free medium (K-SFM, GIBCO-BRL 17005-042) or RPMI 1640 medium with 5% FetalClone III and supplemented with EGF (10 ng/mL Novoprotein C029).
Lactobacillus acidophilus (ATCC 4356) was purchased from ATCC. The bacterium was maintained and tested on MRS agar and broth at 37 °C under anaerobic conditions (BD GasPak EZ Anaerobe sachets).
Trichomonas vaginalis Assays.
For assays, a 250 μL aliquot of T. vaginalis cryopreserved in liquid N2 with 5% dimethyl sulfoxide (DMSO) of was thawed rapidly at 37 °C and put into a screw cap tube containing 12 mL of pre-warmed modified TYI-S-33 medium. The sample was incubated for 24 h at which point confluent growth of the parasite had occurred and live trichomonads were counted. A typical count yielded > 3.0×107 cells per tube. Stock cultures were diluted in fresh medium to achieve 40,000 trichomonads per 100 μL of medium in each well of a 96-well microtiter plate. Pin tools and pipets were used to dispense extracts and pure compounds dissolved in DMSO. Throughout the assay setup process, trichomonads were exposed to normal atmospheric conditions for not more than 30 min before experiments commenced. The trichomonads were determined to tolerate 1% DMSO with no detectable negative effects (no changes to the number of live trichomonads). Accordingly, all experiments were conducted so that the amount of DMSO did not exceed 1% by volume. All plates contained vehicle-only (DMSO) and positive (25 μM metronidazole) controls to ensure that the biological response of the trichomonads were consistent across assays using identical test conditions. For candle jar assays, plates were placed in a humidified candle jar, the candle was lit, and the jar was sealed before being placed in a 37 °C incubator for a 17 h incubation period. For assays using BD GasPak, the manufacturer’s instructions were followed as provided. After 17 h of incubation, microtiter plates were removed and 100 μL of room temperature fixation solution added to each well. The fixing solution was a PBS based solution developed in our lab that contained 1% glutaraldehyde, 5 μM propidium iodide, and 5 μM acridine orange (HCl salt). This solution was found to be stable at room temperature for several months and it lasted even longer when stored in a refrigerator. Plates treated with fixing solution were placed on an orbital shaker for 30 s to disperse clumps of cells and then moved to an incubator for 3 h for staining and fixation to occur. The plates were quantitatively imaged using the Perkin Elmer Operetta and analyzed using the Harmony 3.5.1 software package. Quantification involved the software identifying all trichomonads and subtracting live (green only) from dead (green and red) cells using a propidium iodide threshold of less than 6500 units; this threshold was determined to enable the assessment of membrane integrity and thus served as an indicator of live versus dead cells.
A traditional resazurin based assay employing a plate reader for live cell quantification was also performed.26 Trichomonads were grown in microtiter plates and at the end of experiments, 10 μL of a 0.1 mg/mL resazurin stock in PBS was added to each well. Plates were incubated at room temperature in the dark for 1 h. At the end of the incubation period, the plates were shaken and analyzed on a fluorescence plate reader (Tecan Infinite M200) with a 556 nm excitation wavelength and an emission of 590 nm.
Mammalian Cell Cytotoxicity Assays.
Mammalian cell assays were performed as described previously55 and viability determined by MTT assay56 or by a Calcein AM and Hoechst 33342 live cell area assay using an Operetta system. Assays were performed by adding to each well of the microtiter plate 5 μL of a 40 μM Calcein AM and 160 μM Hoechst 33342 stock solution prepared in DMSO that was diluted (1:5) in PBS buffer. After treatment, plates were incubated for 30 min before being analyzed on the Operetta system. Harmony software was used to calculate the live cell area by finding all Hoechst-labeled nuclei and the cells were assigned live or dead status based on their degree of green fluorescence. Live cells contain esterases that cleave the AM group from Calcein AM causing the cells to glow bright green.
Lactobacillus acidophilus Viability Assay.
Colonies of L. acidophilus were sampled with a loop and used to inoculate 10 mL of MRS broth and then vortexed. 100 μL aliquots were added to each well of a 96-well microtiter plate. Test compounds were dissolved in DMSO (0.5% final concentration DMSO) and added to the wells. Plates were read on a microplate reader at 600 nm to establish baseline absorbance (dispersion) values for each well, the plates were incubated at 37 °C for 18 h, and their contents analyzed on the microtiter plate reader at 600 nm to determine changes in their optical density values.
Purification of Secondary Metabolites 1–3.
A fungal isolate, Fusarium solani (internal strain designation Tree 400 EW+PNGP-3), identified based on its ribosomal internal transcribed spacer (ITS) sequence data (GenBank accession number MK424121), was obtained from a soil sample collected under a tree in the Oliver Wildlife Preserve in Norman, Oklahoma. This isolate was grown for 4 weeks on Cheerios breakfast cereal supplemented with a 0.3% sucrose solution with 0.005% chloramphenicol in three large mycobags (Unicorn Bags, Plano, TX) prior to being homogenized, extracted, and partitioned with EtOAc. The EtOAc soluble material (40.4 g) was subjected to silica gel vacuum liquid chromatography with elution steps of 1:1 hexanes/DCM, DCM, 10:1 DCM/MeOH, and MeOH, yielding 4 fractions. The 10:1 DCM/MeOH fraction (7.8 g) was subjected to HP20SS vacuum liquid chromatography and eluted with a step gradient of MeOH in H2O (30%, 50%, 70%, 90%, and 100%) and 1:1 DCM/MeOH, yielding a total of 6 fractions. Bioassay analysis of the resulting fractions indicated T. vaginalis-inhibitory activity was concentrated in the 90% MeOH fraction. This fraction was subjected to further bioassay-guided purification using preparative C18 HPLC (250 mm × 21.2 mm, 5 μm) with a MeOH-H2O gradient (30:70 to 100:0), followed by isocratic semi-preparative C18 HPLC (250 mm × 10 mm, 5 μm) with MeCN-H2O containing 0.1% HCOOH (50:50) to yield three known bioactive naphthoquinones [1 (1.5 mg), 2 (1.8 mg), and 3 (3.0 mg)].
Testing of Additional Commercially Available and Synthesized Quinones.
The structures of compounds 4–45 are shown in Table S1, Supporting Information. The identities of synthesized compounds were confirmed through 1H NMR, as well as by comparisons to additional spectroscopic data as reported in the literature.
Synthesis of Naphthopurpurin (8).
Compound 8 was synthesized according to a previously reported procedure.57 Specifically, 153 mg of naphthazarin (0.80 mmol) was dissolved in 45 mL of aqueous KOH (2 M). The solution was stirred vigorously and refluxed for 4 h. After cooling to ambient temperature, glacial acetic acid was used to neutralize the reaction mixture, resulting in a color change from purple to red. The reaction mixture was partitioned with 4 × 40 mL of chloroform. The aqueous phase was acidified with 5 mL of HCl (1 M) and partitioned with 4 × 50 mL of EtOAc. The organic phases were combined and the solvent was removed under vacuum to yield naphthopurpurin (130 mg, 78% yield). 1H NMR (400 MHz, CDCl3): 12.74 (s, 1H), 11.47 (s, 1H), 7.34 (d, J = 9.5 Hz, 1H), 7.21 (d, J = 9.5 Hz, 1H), 6.37 (s, 1H).
Synthesis of 2-Methoxynaphthazarin (9).
Compound 9 was synthesized according to a previously reported procedure.58 Specifically, 120 mg of naphthopurpurin (0.58 mmol) was dissolved in 5 mL of absolute MeOH and 80 μL of HCl (12 M). The solution was stirred and refluxed for 4 h, then brought back to room temperature. The product was purified from the reaction mixture by silica gel vacuum liquid chromatography using a stepwise gradient (hexanes, 3:1 hexanes/EtOAc, 2:1 hexanes/EtOAc, 1:1 DCM/MeOH, and MeOH). From the 3:1 hexanes/EtOAc fraction, 2-methoxynaphthazarin was obtained (17 mg, 13% yield). 1H NMR (400 MHz, CDCl3): 12.63 (s, 1H), 12.16 (s, 1H), 7.27 (d, J = 9.5 Hz, 1H), 7.20 (d, J = 9.5 Hz, 1H), 6.16 (s, 1H), 3.93 (s, 3H).
Synthesis of 1,4-Dihydro-5,8-dihydroxy-2-methyl-9,10-anthracenedione (10).
Compound 10 was synthesized according to a previously reported procedure.59 Specifically, 157 mg of naphthazarin (0.83 mmol) was dissolved in 5 mL glacial acetic acid. To this solution, 200 μL of isoprene (2.0 mmol) was added, and the mixture was stirred with heating to 70 °C for 48 h. The product was semi-purified from the reaction mixture by silica gel vacuum liquid chromatography, eluted with 25:1 hexanes/EtOAc, yielding a crude mixture of products. The mixture was dissolved in 5 mL of aqueous KOH (2 M) and stirred vigorously for 30 min. HCl (1 M) was added to acidify the solution, which was gravity filtered to yield 1,4-dihydro-5,8-dihydroxy-2-methyl-9,10-anthracenedione (38 mg, 18% yield). 1H NMR (400 MHz, CDCl3): 12.54 (s, 2H), 7.21 (s, 2H), 5.55 (m, 1H), 3.25 (m, 2H), 3.14 (m, 2H), 1.82 (s, 3H).
Synthesis of 1,4-Dihydro-2-methyl-9,10-anthracenedione (11).
Compound 11 was synthesized according to a previously reported procedure.59 Specifically, 187 mg of 1,4-naphthoquinone (1.2 mmol) was dissolved in 5 mL glacial acetic acid. To this solution, 200 μL of isoprene (2.0 mmol) was added, and the mixture was stirred with heating to 70 °C for 48 h. To the reaction mixture, 5 mL of H2O were added and the mixture was cooled to 0 °C. The mixture was gravity filtered to yield a crude mixture of products. The mixture was dissolved in 5 mL of aqueous KOH (2 M) and stirred vigorously for 15 min. The solution was acidified with 1 mL of HCl (12 M). The resulting solution was gravity filtered to yield 1,4-dihydro-2-methyl-9,10-anthracenedione (95 mg, 35% yield). 1H NMR (400 MHz, CDCl3): 8.07 (m, 2H), 7.69 (m, 2H), 5.54 (m, 1H), 3.23 (m, 2H), 3.12 (m, 2H), 1.80 (s, 3H).
Purification of Secondary Metabolite 46.
A fungal isolate, Humicola grisea (internal strain designation Mystery-9 SEA-2), was identified based on its ribosomal internal transcribed spacer (ITS) sequence data (GenBank accession number MK424122). The fungus was grown for 4 weeks on Cheerios breakfast cereal supplemented with a 0.3% sucrose solution containing 0.005% chloramphenicol in three large mycobags (Unicorn Bags, Plano, TX) prior to being homogenized, extracted, and partitioned with EtOAc. The EtOAc soluble material (57.5 g) was subjected to silica gel vacuum liquid chromatography (VLC) with elution steps of 1:1 hexanes/DCM, DCM, 10:1 DCM/MeOH and MeOH, yielding 4 fractions. The 10:1 DCM/MeOH fraction (8.0 g) was subjected to HP20SS VLC and eluted with a step gradient of MeOH in H2O (30%, 50%, 70%, 90%, and 100%) and 1:1 DCM/MeOH, yielding a total of 6 fractions. Bioassay analysis of the resulting fractions revealed the T. vaginalis-inhibitory activity was concentrated in the MeOH fraction from HP20SS VLC. The MeOH fraction was subjected to further bioassay-guided fractionation using preparative C18 HPLC (250 mm × 21.2 mm, 5 μm) with a MeOH-H2O gradient containing 0.1% HCOOH (50:50 to 100:0) to yield the known bioactive compound 46 (52.2 mg).
Borohydride Reduction of 46.
To a flask, 9.0 mg of 46 (0.016 mmol) in 4 mL MeOH was added. While stirring, 7.6 mg of sodium borohydride (0.20 mmol) dissolved in 1 mL MeOH was added slowly over a 5 min period. The reaction mixture was stirred at room temperature for 45 min. The reaction was quenched by adding 500 μL of 1M HCl to the flask and stirred for 10 min. Solvent was removed from the reaction mixture under vacuum to yield compound 47 (8.8 mg, 96% yield). 1H NMR (500 MHz, CDCl3): 14.44 (s, 1H), 13.94 (s, 1H), 11.91 (s, 1H), 11.44 (s, 1H), 7.04 (s, 1H), 6.70 (s, 1H), 6.47 (m, 2H), 6.08 (s, 1H), 5.04 (s, 1H), 4.74 (dd, J = 5.5, 2.0 Hz, 1H), 4.26 (dd, J = 4.0, 2.0 Hz, 1H), 3.68 (s, 3H), 3.05 (d, J = 18.0 Hz, 1H), 2.81 (m, 1H), 2.58 (d, J = 18.0 Hz, 1H), 2.40 (s, 3H), 2.37 (m, 1H), 2.12 (m, 1H), 1.91 (m, 1H). 13C NMR (125 MHz, CDCl3): 188.5, 186.7, 184.6, 179.4, 171.1, 161.4, 158.5, 155.9, 148.3, 147.6, 141.8, 134.9, 131.8, 117.5, 117.2×2, 111.7, 111.3, 106.5, 104.6, 100.1, 83.8, 71.9, 66.8, 53.4, 43.6, 37.6, 30.5, 24.4, 23.0, 22.4.
Purification of Tetramic Acid Derivatives 48–56.
5 fungal isolates coded Miller-1 Cz-3, Miller-26 SEA-3, KY6863 TV8–3, Column L5 SEA-1, CA9310 TV8–3, were identified as Fusarium sp. isolate B, Fusarium sp. isolate C, Penicillium sp., Alternaria sp., Phoma sp. P34E5, respectively, based on their ribosomal internal transcribed spacer (ITS) sequence data (GenBank accession numbers MK401898, MK401899, MK401895, MK401896, and MK401897, respectively). These five fungal isolates were fermented on a solid-state medium composed of Cheerios breakfast cereal supplemented with 0.3% sucrose solution for 4 weeks at 25 °C. The fungal isolate designated Column L5 SEA-1 was inoculated into 16 L of PDB media (10 g/L dried mashed potato, 5 g/L glucose) supplemented with 2 g/L NaNO3 and grown for 9 days with shaking.
The fungal culture designated Miller-1 Cz-3 (Fusarium sp. isolate B) was homogenized and partitioned 3× with equal volumes of EtOAc. The resulting EtOAc soluble material (25.0 g) was subjected to silica gel VLC with elution steps of 1:1 hexanes/DCM, DCM, 10:1 DCM/MeOH, and MeOH, yielding 4 fractions. The 10:1 DCM/MeOH fraction was subjected to HP20SS VLC and eluted with a step gradient of MeOH in H2O (30%, 50%, 70%, 90%, and 100%), yielding 5 fractions. The 90% MeOH fraction from HP20SS VLC was further fractionated by preparative C18 HPLC (250 mm × 21.2 mm, 5 μm) with a MeCN-H2O gradient containing 0.1% HCOOH (70:30 to 100:0) to yield compounds 48 (56 mg) and 49 (18.9 mg).
The fungal culture designated Miller-26 SEA-3 (Fusarium sp. isolate C) was homogenized and partitioned 3× with equal volumes of EtOAc. The resulting EtOAc soluble material (14.0 g) was subjected to silica gel VLC with elution steps of 1:1 hexanes/DCM, DCM, 10:1 DCM/MeOH, and MeOH, yielding 4 fractions. The 10:1 DCM/MeOH fraction was subjected to HP20SS VLC and eluted with a step gradient of MeOH in H2O (30%, 50%, 70%, 90%, and 100%), yielding 5 fractions. The 70% MeOH fraction from HP20SS VLC was further fractionated by isocratic preparative C18 HPLC (250 mm × 21.2 mm, 5 μm) with MeOH-H2O (75:25) to yield compound 50 (50 mg).
The fungal culture designated KY6863 TV8–3 (Penicillium sp.) was homogenized and partitioned 3× with equal volumes of EtOAc. The resulting EtOAc soluble material (31.0 g) was subjected to silica gel VLC with elution steps of 1:1 hexanes/DCM, DCM, 10:1 DCM/MeOH, and MeOH, yielding 4 fractions. The 10:1 DCM/MeOH fraction was subjected to HP20SS VLC and eluted with a step gradient of MeOH in H2O (30%, 50%, 70%, 90%, and 100%), yielding 5 fractions. The 90% MeOH fraction from HP20SS VLC was further fractionated by preparative C18 HPLC (250 mm × 21.2 mm, 5 μm) with a MeOH-H2O gradient containing 0.1% HCOOH (85:15 to 100:0) to yield compounds 51 (50 mg) and 52 (15 mg). 5’-epipyrrolocin A (52): pale yellow plates; [α]20D +93 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 238 (2.37), 265 (0.42), 295 (0.77); CD (MeOH) λmax (Δε) 234 (8.1), 291 (6.1); 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 456.2747 [M - H]− (calcd for 456.2755).
Fungal culture broth prepared from the fungus designated Column L5 SEA-1 (Alternaria sp.) was partitioned 3× with equal volumes of EtOAc to provide 3.0 g of EtOAc-soluble residue. The organic residue was subjected to HP20SS VLC and eluted with a step gradient of MeOH in H2O (30%, 50%, 70%, 90%, and 100%), yielding 5 fractions. The 90% MeOH fraction was further fractionated by preparative C18 HPLC (250 mm × 21.2 mm, 5 μm) with a MeOH-H2O gradient containing 0.1% HCOOH (90:10 to 100:0) yielding 6 fractions (Fr.1–6). Bioactive compounds in fractions 2 and 4 were further purified by isocratic semi-preparative C18 HPLC (250 mm × 10 mm, 5 μm) with MeCN-H2O containing 0.1% HCOOH (80:20) to yield compounds 53 (67 mg) and 54 (7.5 mg). 5’-epibeauversetin (54): pale yellow plate; [α]20D +409 (c 0.17, MeOH); UV (MeOH) λmax (log ε) 236 (4.39), 287 (4.11); CD (MeOH) λmax (Δε) 232 (8.2), 285 (9.8); 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 398.2337, [M - H]− (calcd for C24H32NO4, 398.2337).
The fungal culture designated CA9310 TV8–3 (Phoma sp. P34E5) was homogenized and partitioned 3× with equal volumes of EtOAc. The resulting EtOAc soluble material (38.0 g) was subjected to silica gel VLC with elution steps of 1:1 hexanes/DCM, DCM, 10:1 DCM/MeOH, and MeOH, yielding 4 fractions. The 10:1 DCM/MeOH fraction was subjected to HP20SS VLC and eluted with a step gradient of MeOH in H2O (30%, 50%, 70%, 90%, and 100%) yielding 5 fractions. The 70% MeOH fraction from HP20SS VLC was further fractionated by isocratic preparative C18 HPLC (250 mm × 21.2 mm, 5 μm) with MeOH-H2O containing 0.1% HCOOH (85:15) to yield compounds 55 (140 mg) and 56 (4 mg).
Preparation of MTPA Esters.
Compound 52 (0.5 mg) was dissolved in pyridine-d5 and equal portions transferred to two NMR tubes.47 The (R)-α-methoxy-α-trifluoromethyl-phenylacetyl chloride [(R)-MTPA-Cl] (5 μL) was added into one sample under an N2 gas stream. After mixing, it was incubated at 40 °C in a H2O bath and monitored every 2 h by 1H NMR. The same method was used to treat the second sample using (S)-α-methoxy-α-trifluoromethyl-phenylacetyl chloride [(S)-MTPA-Cl] as the reagent to yield the (R)-Mosher ester derivative. (S)-Mosher ester derivative: 1H NMR (500 MHz, pyridine-d5): 6.38 (1H, m, H-14), 6.07 (1H, m, H-15), 5.90 (1H, m, H-16), 5.69 (1H, m, H-13), 3.87 (1H, m, H-12), 2.37 (1H, m, H-17), 2.10 (1H, m, H-17), 1.20 (3H, d, J = 6.5 Hz, CH3-19). (R)-Mosher ester derivative: 1H NMR (500 MHz, pyridine-d5) 6.39 (1H, m, H-14), 6.07 (1H, m, H-15), 5.92 (1H, m, H-16), 5.72 (1H, m, H-13), 3.94 (1H, m, H-12), 2.37 (1H, m, H-17), 2.10 (1H, m, H-17), 1.19 (3H, d, J = 6.5 Hz, CH3-19).
Cleavage of the Tetramic Acid Ring and Marfey’s Reaction.
Samples of compounds (20 mg) were dissolved in MeOH (0.6 mL) followed by addition of 1 M NaOH (75 μL) and a NaOCl solution (available chlorine 10−15%, 0.35 mL) as per prior reports.42–43 The mixture was stirred at room temperature for 8 h and then it was quenched by adding 1 M aqueous Na2SO2 (3 mL) and neutralized by the addition of 1 M HCl. After removal of the solvent, the residue was diluted with H2O, and the resulting mixture was partitioned against EtOAc. The organic layer was retained and the solvent removed in vacuo. The residue was dissolved in 6 M HCl (500 μL) and held overnight at 110 °C. The solvent was removed from the hydrolysate under a stream of N2 and the residue treated with 1 M NaHCO3 (40 μL) and 1% FDAA/acetone (200 μL) at 70 °C for 1 h. The reactants were neutralized with 1 M HCl (40 μL) and diluted with MeCN (400 μL) prior to LC-ESIMS analysis. FDAA derivatives of L-Ser/N-Me-L-Ser and D-Ser/N-Me-D-Ser standards were prepared in a similar manner. Aqueous solutions of serine standards (50 mM, 50 μL) were reacted, neutralized, and diluted following the same procedure.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases (R33AI119713). The LC-ESIMS instrument used for this project was provided in part by a Challenge Grant from the Office of the Vice President for Research, University of Oklahoma, Norman Campus and an award through the Shimadzu Equipment Grant Program. We appreciate the contributions of soil samples from citizen scientists J. Miller, L. Miller, and W. Tierney to the University of Oklahoma Citizen Science Soil Collection Program, which served as the sources for several of the fungi reported in this study.
ABBREVIATIONS
- SI
selectivity index
- PAINS
pan-assay interference compounds
- VLC
vacuum liquid chromatography
- ITS
internal transcribed spacer
- MTPA
α-methoxy-α-trifluoromethyl-phenylacetyl
- FDAA
1-Fluoro-2–4-Dinitrophenyl-5-L-Alanine Amide
- ATCC
the American Type Culture Collection
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.9b00156.
Bioactive compounds from the pure compound screening in T. vaginalis; Structures and therapeutic indices for quinones and related structures; Comparison of antitrichomonad activity of xanthoquinodin A1 (46) and 47 under anaerobic conditions; LC-ESIMS analysis of FDAA derivatives of beauversetin (53), equisetin (48), and 5’-epipyrrolocin A (52) derived hydrolysates; Effects of xanthoquinodin A1 (46) and beauversetin (53) on the growth of T. vaginalis under test conditions involving different oxygen levels; 1D, 2D NMR spectra and HRESIMS data for compounds 47, 52, and 54
REFERENCES
- (1).Sutton M, Sternberg M, Koumans EH, McQuillan G, Berman S, Markowitz L (2007) The prevalence of Trichomonas vaginalis infection among reproductive-age women in the United States, 2001–2004. Clin. Infect. Dis. 45, 1319–1326. DOI: 10.1086/522532. [DOI] [PubMed] [Google Scholar]
- (2).Kissinger P (2015) Trichomonas vaginalis: a review of epidemiologic, clinical and treatment issues. BMC Infect. Dis. 15, 307. DOI: 10.1186/s12879-015-1055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gabriel G, Robertson E, Thin RN (1982) Single dose treatment of trichomoniasis. J. Int. Med. Res. 10, 129–130. DOI: 10.1177/030006058201000212. [DOI] [PubMed] [Google Scholar]
- (4).Aubert JM, Sesta HJ (1982) Treatment of vaginal trichomoniasis. Single, 2-gram dose of metronidazole as compared with a seven-day course. J. Reprod. Med. 27, 743–745. [PubMed] [Google Scholar]
- (5).Howe K, Kissinger PJ (2017) Single-dose compared with multidose metronidazole for the treatment of trichomoniasis in women: a meta-analysis. Sex. Transm. Dis. 44, 29–34. DOI: 10.1097/olq.0000000000000537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bendesky A, Menéndez D, Ostrosky-Wegman P (2002) Is metronidazole carcinogenic? Mutat. Res. 511, 133–144. DOI: 10.1016/S1383-5742(02)00007-8. [DOI] [PubMed] [Google Scholar]
- (7).Dobiás L, Černá M, Rössner P, Šrám R (1994) Genotoxicity and carciogenicity of metronidazole. Mutat. Res. 317, 177–194. DOI: 10.1016/0165-1110(94)90001-9. [DOI] [PubMed] [Google Scholar]
- (8).2015 sexually transmitted diseases treatment guidelines: trichomoniasis. https://www.cdc.gov/std/tg2015/trichomoniasis.htm (accessed June 28, 2018).
- (9).Kirkcaldy RD, Augostini P, Asbel LE, Bernstein KT, Kerani RP, Mettenbrink CJ, Pathela P, Schwebke JR, Secor WE, Workowski KA, Davis D, Braxton J, Weinstock HS (2012) Trichomonas vaginalis antimicrobial drug resistance in 6 U.S. cities, STD Surveillance Network, 2009–2010. Emerging Infect. Dis. 18, 939–943. DOI: 10.3201/eid1806.111590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schwebke JR, Barrientes FJ (2006) Prevalence of Trichomonas vaginalis isolates with resistance to metronidazole and tinidazole. Antimicrob. Agents Chemother. 50, 4209–4210. DOI: 10.1128/aac.00814-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Scopel M, dos Santos O, Frasson A, Abraham W, Tasca T, Henriques A, Macedo A (2013) Anti-Trichomonas vaginalis activity of marine-associated fungi from the South Brazilian Coast. Exp. Parasitol. 133, 211–216. DOI: 10.1016/j.exppara.2012.11.006. [DOI] [PubMed] [Google Scholar]
- (12).Duarte M, Seixas A, de Carvalho M, Tasca T, Macedo A (2016) Amaurocine: anti-Trichomonas vaginalis protein produced by the basidiomycete Amauroderma camerarium. Exp. Parasitol. 161, 6–11. DOI: 10.1016/j.exppara.2015.12.012. [DOI] [PubMed] [Google Scholar]
- (13).Desrivot J, Waikedre J, Cabalion P, Herrenknecht C, Bories C, Hocquemiller R, Fournet A (2007) Antiparasitic activity of some New Caledonian medicinal plants. J. Ethnopharmacol. 112, 7–12. DOI: 10.1016/j.jep.2007.01.026. [DOI] [PubMed] [Google Scholar]
- (14).Calzada F, Yepez-Mulia L, Tapia-Contreras A (2007) Effect of Mexican medicinal plant used to treat trichomoniasis on Trichomonas vaginalis trophozoites. J. Ethnopharmacol. 113, 248–251. DOI: 10.1016/j.jep.2007.06.001. [DOI] [PubMed] [Google Scholar]
- (15).Lara-Díaz V, Gaytán-Ramos A, Dávalos-Balderas A, Santos-Guzmán J, Mata-Cárdenas B, Vargas-Villarreal J, Barbosa-Quintana A, Sanson M, López-Reyes A, Moreno-Cuevas J (2009) Microbiological and toxicological effects of Perla black bean (Phaseolus vulgaris L.) extracts: in vitro and in vivo studies. Basic Clin. Pharmacol. Toxicol. 104, 81–86. DOI: 10.1111/j.1742-7843.2008.00330.x. [DOI] [PubMed] [Google Scholar]
- (16).Moon T, Wilkinson J, Cavanagh H (2006) Antiparasitic activity of two Lavandula essential oils against Giardia duodenalis, Trichomonas vaginalis, and Hexamita inflata. Parasitol. Res. 99, 722–728. DOI: 10.1007/s00436-006-0234-8. [DOI] [PubMed] [Google Scholar]
- (17).Kaneda Y, Tanaka T, Saw T (1990) Effects of berberine, a plant alkaloid, on the growth of anaerobic protozoa in axenic culture. Tokai J. Exp. Clin. Med. 15, 417–423. [PubMed] [Google Scholar]
- (18).Wu J, Zhang M, Ding D, Tan T, Yan B (1995) Effect of Cladonia alpestris on Trichomonas vaginalis in vitro. Chinese Journal of Parasitology and Parasitic Diseases 13, 126–129. [PubMed] [Google Scholar]
- (19).Wang H (1993) Antitrichomonal action of emodin in mice. J. Ethnopharmacol. 40, 111–116. DOI: 10.1016/0378-8741(93)90055-A. [DOI] [PubMed] [Google Scholar]
- (20).Bhagwat P, Gokhale B, Sane H, Thirumalachar M (1964) Assessment of antitrichomonal activity of hamycin. Indian J. Med. Res. 52, 36–37. [PubMed] [Google Scholar]
- (21).He W, VanPuyvelde L, Maes L, Bosselaers J, DeKimpe N (2003) Antitrichomonas in vitro activity of Cussonia holstii Engl. Nat. Prod. Res. 17, 127–133. DOI: 10.1080/1478641031000103713. [DOI] [PubMed] [Google Scholar]
- (22).Loyola L, Bórquez J, Morales G, Araya J, González J, Neira I, Sagua H, San-Martín A (2001) Diterpenoids from Azorella yareta and their trichomonicidal activities. Phytochemistry 56, 177–180. DOI: 10.1016/S0031-9422(00)00380-0. [DOI] [PubMed] [Google Scholar]
- (23).Kayser O, Kiderlen A, Croft S (2003) Natural products as antiparasitic drugs. Parasitol. Res. 90, S55–S62. DOI: 10.1007/s00436-002-0768-3. [DOI] [PubMed] [Google Scholar]
- (24).Newman DJ, Cragg GM (2016) Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661. DOI: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- (25).Campos Aldrete M, Salgado-Zamora H, Luna J, Meléndez E, Meráz-Ríos M (2005) A high-throughput colorimetric and fluorometric microassay for the evaluation of nitroimidazole derivatives anti-Trichomonas activity. Toxicol. in Vitro 19, 1045–1050. DOI: 10.1016/j.tiv.2005.04.007. [DOI] [PubMed] [Google Scholar]
- (26).Duarte M, Giordani R, De Carli G, Zuanazzi J, Macedo A, Tasca T (2009) A quantitative resazurin assay to determinate the viability of Trichomonas vaginalis and the cytotoxicity of organic solvents and surfactant agents. Exp. Parasitol. 123, 195–198. DOI: 10.1016/j.exppara.2009.07.002. [DOI] [PubMed] [Google Scholar]
- (27).Chen JL, Steele TWJ, Stuckey DC (2015) Modeling and application of a rapid fluorescence-based assay for biotoxicity in anaerobic digestion. Environ. Sci. Technol. 49, 13463–13471. DOI: 10.1021/acs.est.5b03050. [DOI] [PubMed] [Google Scholar]
- (28).Forestier C, Späth G, Prina E, Dasari S (2015) Simultaneous multi-parametric analysis of Leishmania and of its hosting mammal cells: a high content imaging-based method enabling sound drug discovery process. Microb. Pathog. 88, 103–108. DOI: 10.1016/j.micpath.2014.10.012. [DOI] [PubMed] [Google Scholar]
- (29).Ruelius H, Gauche A (1950) Fusarubin, a naphthoquinone coloring matter from Fusaria. Justus Liebigs Ann. Chem. 569, 38–59. [Google Scholar]
- (30).Arnstein H, Cook A (1947) Production of antibiotics by fungi. Part III. Javanicin. An antibacterial pigment from Fusarium javanicum. J. Chem. Soc. 1947, 1021–1028. [DOI] [PubMed] [Google Scholar]
- (31).Arsenault G (1968) Fungal metabolites. III. Quinones from Fusarium solani D2 purple and structure of (+)-solaniol. Tetrahedron 24, 4745–4749. DOI: 10.1016/S0040-4020(01)98671-5. [DOI] [Google Scholar]
- (32).Kurobane IZ,N; Fukuda A (1985) New metabolites of Fusarium martii related to dihydrofusarubin. J. Antibiot. 39, 205–214. DOI: 10.7164/antibiotics.39.205. [DOI] [PubMed] [Google Scholar]
- (33).Hashimoto J, Motohashi K, Sakamoto K, Hashimoto S, Yamanouchi M, Tanaka H, Takahashi T, Takagi M, Shin-ya K (2009) Screening and evaluation of new inhibitors of hepatic glucose production. J. Antibiot. 62, 625–629. DOI: 10.1038/ja.2009.93. [DOI] [PubMed] [Google Scholar]
- (34).Hughes W, Gray V, Gutteridge W, Latter V, Pudney M (1990) Efficacy of a hydroxynaphthoquinone, 566C80, in experimental Pneumocystis carinii pneumonitis. Antimicrob. Agents Chemother. 34, 225–228. DOI: 10.1128/AAC.34.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Baggish A, Hill D (2002) Antiparasitic agent atovaquone. Antimicrob. Agents Chemother. 46, 1163–1173. DOI: 10.1128/AAC.46.5.1163-1173.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Baell JB (2016) Feeling nature’s PAINS: Natural products, natural product drugs, and pan assay interference compounds (PAINS). J. Nat. Prod. 79, 616–28. DOI: 10.1021/acs.jnatprod.5b00947. [DOI] [PubMed] [Google Scholar]
- (37).Tabata N, Tomoda H, Matsuzaki K, Omura S (1993) Structure and biosynthesis of xanthoquinodins, anticoccidial antibiotics. J. Am. Chem. Soc. 115, 8558–8564. DOI: 10.1021/ja00072a006. [DOI] [Google Scholar]
- (38).Sobel J (1999) Is there a protective role for vaginal flora? Curr. Infect. Dis. Rep. 1, 379–383. [DOI] [PubMed] [Google Scholar]
- (39).Ma B, Forney L, Ravel J (2012) The vaginal microbiome: rethinking health and diseases. Annu. Rev. Microbiol. 66, 371–389. DOI: 10.1146/annurev-micro-092611-150157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hunter N, Dingsdag S (2017) Metronidazole: an update on metabolism, structure-cytotoxicity and resistance mechanisms. J Antimicrob Chemoth 73, 265–279. DOI: 10.1039/jac/dkx351. [DOI] [PubMed] [Google Scholar]
- (41).Philips N, Goodwin J, Fraiman A, Cole R, Lynn D (1989) Characterization of the Fusarium toxin equisetin- the use of phenylboronates in structure assignment. J. Am. Chem. Soc 111, 8223–8231. [Google Scholar]
- (42).Jadulco RC, Koch M, Kakule TB, Schmidt EW, Orendt A, He H, Janso JE, Carter GT, Larson EC, Pond C, Matainaho TK, Barrows LR (2014) Isolation of pyrrolocins A-C: cis- and trans-decalin tetramic acid antibiotics from an endophytic fungal-derived pathway. J Nat Prod 77, 2537–2544. DOI: 10.1021/np500617u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Minowa NK,Y; Harimaya K; Mikawa T (1998) A degradation study of vermisporin and determination of its absolute configuration. Heterocycles 48, 1639–1642. [Google Scholar]
- (44).Singh SB, Zink DL, Goetz MA, W. DA, Polishook JD, Hazuda DJ (1998) Equisetin and a novel opposite stereochemical homolog phomasetin, two fungal metabolites as inhibitors of HIV-1 integrase. Tetrahedron Lett. 39, 2243–2246. DOI: 10.1016/S0040-4039(98)00269-X. [DOI] [Google Scholar]
- (45).Marfori E, Kajiyama S, Fukusaki E, Kobayashi A (2002) Trichosetin, a novel tetramic acid antibiotic produced in dual culture of Trichoderma harzianum and Catharanthus roseus Callus. Z. Naturforsch., C: Biosci. 57, 465–470. DOI: 10.1515/znc-2002-5-611. [DOI] [PubMed] [Google Scholar]
- (46).Neumann K, Kehraus S, Gütschow M, König G (2009) Cytotoxic and HLE-inhibitory tetramic acid derivatives from marine-derived fungi. Nat. Prod. Commun. 4, 347–354. [PubMed] [Google Scholar]
- (47).Seco JM, Quiñoá E, Riguera R (2001) A practical guide for the assignment of the absolute configuration of alcohols, amines and carboxylic acids by NMR. Tetrahedron: Asymmetry 12, 2915–2925. DOI: 10.1016/S0957-4166(01)00508-0. [DOI] [Google Scholar]
- (48).Yamada S, Hongo C, Yoshioka R, Chibata I (1983) Method for the racemization of optically active amino acids. J. Org. Chem 48, 843–846. DOI: 10.1021/jo00154a019. [DOI] [Google Scholar]
- (49).Fujii K, Ikai Y, Oka H, Suzuki M, Harada K (1997) A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide- combination of Marfey’s method with mass spectrometry and Its practical application. Anal. Chem. 69, 5146–5151. DOI: 10.1021/ac970289b. [DOI] [Google Scholar]
- (50).Ratnaweera P, de Silva E, Williams D, Andersen R (2015) Antimicrobial activities of endophytic fungi obtained from the arid zone invasive plant Opuntia dillenii and the isolation of equisetin, from endophytic Fusarium sp. BMC Complement. Altern. Med. 15, 220. DOI: 10.1186/s12906-015-0722-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Putri S, Kinoshita H, Ihara F, Igarashi Y, Nihira T (2010) Ophiosetin, a new tetramic acid derivative from the mycopathogenic fungus Elaphocordyceps ophioglossoides. J. Antibiot. 63, 195–198. DOI: 10.1038/ja.2010.8. [DOI] [PubMed] [Google Scholar]
- (52).Baell J, Nissink J (2018) Seven year itch: pan-assay interference compounds (PAINS) in 2017-utility and limitations. ACS Chem. Biol. 13, 36–44. DOI: 10.1021/acschembio.7b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Davids B, Gillin F, Methods for Giardia culture, cryopreservation, encystation, and excystation in vitro. In Giardia A Model Organism, Luján H; Svärd S, Eds. SpringerWienNewYork: Wien, Austria, 2011; pp 381–394. [Google Scholar]
- (54).Paget T, Lloyd D (1990) Trichomonas vaginalis requires traces of oxygen and high concentrations of carbon dioxide for optimal growth. Mol. Biochem. Parasitol. 41, 65–72. DOI: 10.1016/0166-6851(90)90097-6. [DOI] [PubMed] [Google Scholar]
- (55).Du L, Risinger A, King J, Powell D, Cichewicz R (2014) A potent HDAC inhibitor, 1-alaninechlamydocin, from a Tolypocladium sp. induces G2/M cell cycle arrest and apoptosis in MIA PaCa-2 cells. J. Nat. Prod. 77, 1753–1757. DOI: 10.1021/np500387h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hansen M, Nielsen S, Berg K (1989) Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods 119, 203–210. DOI: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- (57).Fieser L (1928) The tautomerism of hydroxy quinones. J. Am. Chem. Soc. 50, 439–465. DOI: 10.1021/ja01389a033. [DOI] [Google Scholar]
- (58).Sreelatha T, Kandhasamy S, Dinesh R, Shruthy S, Shweta S, Mukesh D, Karunagaran D, Balaji R, Mathivanan N, Perumal PT (2014) Synthesis and SAR study of novel anticancer and antimicrobial naphthoquinone amide derivatives. Bioorg. Med. Chem. Lett. 24, 3647–3651. DOI: 10.1016/j.bmcl.2014.04.080. [DOI] [PubMed] [Google Scholar]
- (59).Burns C, Gill M, Saubern S (1991) Pigments of fungi. XXI Synthesis of (±)-6-demethoxyaustrocortirubin. Aust. J. Chem. 44, 1427–1445. DOI: 10.1071/CH9911427. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.