
Fig 1.
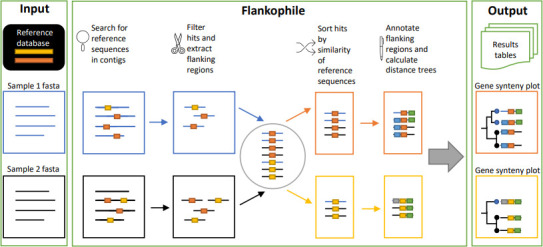
Diagram overview of the Flankophile pipeline. As input data, the pipeline expects DNA contigs, e.g., from assembled genomes or metagenomes. Any collection of user-supplied sequences can be used as a reference database. Both the input data and reference database should be (multi-)FASTA files. Flankophile searches the input sequences for matches to the reference database. Hits with flanking regions of the required length for flank analysis are selected, and their flanking region sequences are extracted. Hits that matched to similar reference sequences are clustered into groups. Genetic features in the flanking regions are annotated, and three distance matrices are calculated on the sequences in each group—one based on the flanking region, one based on the target region, and one based on a combination. Distance trees are made from the distance matrices using hierarchical clustering and plotted along with annotation arrows, gene variant information, and metadata. Output includes plots in PDF format and results tables.