

Abstract
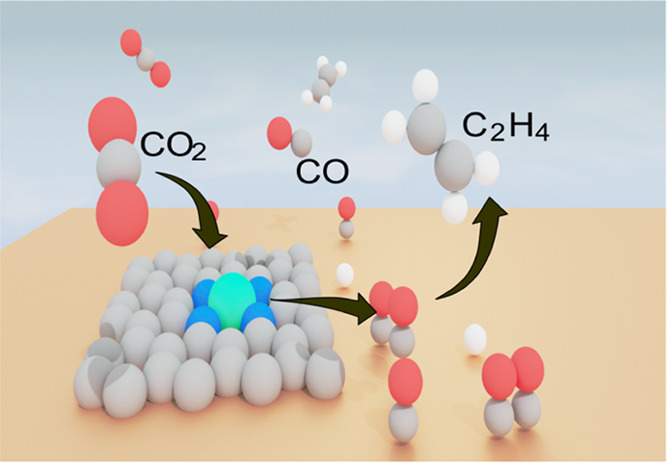
The in-tandem catalyst holds great promise for addressing the limitation of low *CO coverage on Cu-based materials for selective C2H4 generation during CO2 electroreduction. However, the potential mismatch between the CO-formation catalyst and the favorable C–C coupling Cu catalyst represents a bottleneck in these types of electrocatalysts, resulting in low tandem efficiencies. In this study, we propose a robust solution to this problem by introducing a wide-CO generation-potential window nickel single atom catalyst (Ni SAC) supported on a Cu catalyst. The selection of Ni SAC was based on theoretical calculations, and its excellent performance was further confirmed by using in situ IR spectroscopy. The facilitated carbon dimerization in our tandem catalyst led to a ∼370 mA/cm2 partial current density of C2H4, corresponding to a faradic efficiency of ∼62%. This performance remained stable and consistent for at least ∼14 h at a high current density of 500 mA/cm2 in a flow-cell reactor, outperforming most tandem catalysts reported so far.
Introduction
The substantial increase in anthropogenic carbon dioxide (CO2) emissions has had profound and adverse effects on our environment so far. This motivates the development of innovative catalysts that can efficiently capture and reduce atmospheric CO2, leading to a net zero emission goal by 2050. One of the main strategies adopted worldwide is the electroreduction of CO2 (CO2RR), in which the CO2 is converted to a large set of products spanning from hydrocarbons to oxygenates. Ethylene (C2H4) and ethanol are particularly appealing compounds among the products, since the former is utilized as chemical feedstock for plastics and fibers manufacturing,1−3 whereas the latter serves as an effective source of renewable energy.4−8 Transforming CO2 into molecules with more than 1 carbon atom (C2+) involves first the reduction of CO2 into CO, followed by the coupling between activated carbon monoxide (*CO). Metallic copper (Cu) is acknowledged as the most effective catalyst performing the C–C coupling step, owing to moderate *CO adsorption energies, thus enabling the chemical bonding of adjacent *CO molecules.9,10 However, Cu suffers from limitations in the initial step (CO2-to-CO), resulting in poor CO surface coverage and, thus, low CO2-to-C2H4 current density.11,12
To circumvent this challenge, new technologies that combine components with complementary functionalities have been introduced. In such designs, the aim is to leverage the higher CO2-to-CO reduction performance from a cocatalyst, whose function is to deliver *CO for subsequent dimerization reaction toward C2H4 on the Cu surface.13−15 These systems are known as “in-tandem” catalysts. Examples of this advantageous blend are the combination of precious metals (Au, Ag),11,16−18 metal phthalocyanine (M Pc),15,19 or single atom 3d metals,1−3,10,20 employed for the CO-formation step, and metallic Cu that promotes the C–C coupling. Despite the advancements made in CO2-to-C2H4 conversion using in-tandem catalysts, a fundamental issue still hampers the full exploitation of these hybrid catalysts, namely, potential mismatch or dephase. Usually, the potential window in which Cu performs best for the C–C coupling (−1.8 < E < – 1.0 V vs RHE) is not the potential in which the CO-supplier catalyst performs best in CO2-to-CO reduction (−1.0 < E < – 0.5 V vs RHE).21,22 Due to the potential mismatch, the cocatalysts often facilitate the competing hydrogen evolution reaction (HER) at the optimal potential of *CO dimerization reaction on the Cu surface.14
We present an approach to tackle this central concept of in-tandem catalysts, in which we developed a robust CO2 electrocatalyst composed of a nickel single atom catalyst (Ni SAC) dispersed on a Cu matrix. The Ni SAC was selected based on its efficient and broad CO generation-potential window, as well as long stability. Consistent with these theoretical predictions, in situ attenuated total reflection-infrared absorption spectroscopy (ATR-IR) confirmed a larger CO coverage on Cu surfaces when coupled to Ni SAC, thus facilitating the *CO dimerization. As such, the Ni SAC + Cu tandem catalyst exhibits an ∼370 mA/cm2 partial current density of C2H4 with a faradic efficiency of ∼62% and ∼14 h stability at 500 mA/cm2 current density in a flow-cell reactor, suggesting a promising application prospect for industrialization. In situ X-ray absorption near-edge structure (XANES) demonstrated that during this period, Ni atoms remained isolated, contributing to this unprecedented performance.
Results and Discussion
Theoretical Calculations
To identify the most effective cocatalyst for pairing Cu in-tandem catalysts, we first employed density functional theory (DFT) calculations. We screened the relative changes in the activation barrier of *CO–*CO coupling (ΔEreaction) to form *OCCO as a function of both additional *CO and *H coverages (Figures S1–S2). This activation barrier was chosen as a proxy for C2H4 formation since this is the first step in CO dimerization.9,15,19 Cu (111) surface (6 × 6 unit cell) was employed as a model for metallic Cu. The theoretical results revealed that ΔEreaction for *CO–*CO coupling decreases with increasing *CO coverage, as expected, reaching an almost 40% drop when increasing from no additional *CO to 4 *CO molecules (Figure 1a). The opposite trend was observed for *H, since increasing *H coverage impedes the high *CO coverage needed to achieve *CO–*CO coupling (Figure 1b). These simulations clearly indicate that a larger *CO coverage facilitates the C2H4 pathway. Hence, the cocatalyst that can deliver a large amount of CO to Cu surfaces at the potential in which Cu performs best is a suitable candidate for efficient in-tandem catalysis for C2H4 production. In this regard, we used these results to investigate and compare nickel single atom catalyst (Ni SAC), silver nanoparticles (Ag NPs), and nickel phtalocyanine (Ni Pc) as CO producer cocatalysts, all sketched in Figure 1c–e.
Figure 1.

Relative activation barriers (ΔEreaction) for *OCCO formation via the *CO coupling step at (a) different *CO coverage and (b) different *H coverage on the Cu (111) surface. The *CO and *H coverage indicate the number of additional *CO and *H on the Cu (111) surface, respectively, while the label pristine refers to only two adsorbed *CO molecules. Schematic illustration of the CO2RR process for different tandem catalysts at the optimal potential of *CO dimerization reaction, (c) Ag NPs, (d) Ni Pc, and (e) Ni SAC loading on Cu (111) surface, respectively. The colors in the bottom panels represent orange: Cu; light gray: Ag; gray: C; red: O; blue: N; white: H, and green: Ni.
Catalyst Preparation and Characterization
To verify the theoretical results, a wide CO generation-potential window Ni SAC was prepared by using our reported electrostatic self-assembly methods. Cu catalyst (Cu-R, reduction state) was synthesized via one-step wet chemical reduction (Figure S3).23−25 The loadings of metal elements in Ni SAC and Ni Pc are 1.14 and 1.17 wt %, respectively. Then, through sufficient ultrasound, uniform mixing was obtained between Ni SAC and Cu-R to form the tandem catalyst (Ni SAC + Cu-R) (Figure S4). Figure 2 depicts the morphology as well as the electronic structure of the resulting catalyst.
Figure 2.

SEM image (a) and TEM image (b) of the Ni SAC+Cu-R catalyst. (c) AC HAADF-STEM image of the Ni SAC. (d) HRTEM image of Cu-R. (e) EDS mapping image. (f) High-resolution XPS of the Cu 2p spectra of Cu-R. (g) High-resolution XPS of N 1s spectra of Ni SAC and Ni Pc. (h) Ni K-edge XANES spectra of Ni SAC and Ni Pc. (i) k3 weighted Fourier transform spectra from EXAFS.
Scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM) were employed to identify the spatial distribution of the Ni SAC (Figures 2a–b and S5–S7). The presence of Ni SAC was confirmed by observing bright spots on the carbon nanotubes (CNT) when employing aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM), and no clusters were observed (Figure 2c). The metallic nature of the Cu particles as well as the presence of surface Cu2O were confirmed by HRTEM from their respective (111) interplanar spacings (0.239 nm for Cu and 0.203 nm for Cu2O), as illustrated in Figure 2d. The chemical state of the Cu matrix was also investigated by analyzing the 2p signal of its X-ray photoelectron spectroscopy (XPS) spectrum. The Cu catalyst was mostly composed of Cu0 (metallic Cu), although Cu(II) was also observed, suggesting a native oxide formation on its surface (Figure 2f). Elemental analysis via energy-dispersive X-ray spectroscopy (EDS) enabled the mapping of the materials within the catalysts (Figure 2e), which was consistent with the microscopy data.
To evaluate the electronic structure of atomically dispersed Ni atoms in the sample, XPS and X-ray absorption spectra (XAS) were conducted (Figure 2g–i). When the Ni 2p signal was evaluated in XPS, peaks at 855.6 and 873.2 eV were observed, indicating that Ni exhibits an oxidation state via N coordination (Figure S8),26 as also confirmed via the analysis of N 1s in which peaks around 398.8 and 400.0 eV were observed (Figure 2g). These two signals correspond to pyridinic-N and pyrrolic-N, respectively.24 Further information on characterization is shown in Figures S8–S11. The XAS data enabled us to gain information about the chemical state of Ni atoms in the sample. Ni K-edge absorption spectrum of Ni SAC is close to those of Ni Pc and NiO, which leads to the interpretation that Ni atoms remain with a +2 chemical state in Ni SAC (Figure 2h). Fourier transformed extended X-ray absorption fine structure (EXAFS) spectra demonstrate the absence of Ni–Ni bonding in Ni SAC, confirming the atomic dispersion feature of Ni atoms (Figure 2i).
This thorough experimental characterization was also conducted for the other cocatalysts. The morphologies of these cocatalysts are shown in Figures S12–S15.
In Situ Spectroscopy
To investigate the ability of Ni SAC to provide *CO to metallic Cu for CO2 electroreduction, ATR-IR was employed to study CO production from the cocatalysts, as well as the *CO coverage in hybrid systems during CO2RR at different potentials (−1.45 < E< – 0.85 V vs RHE). The scheme of the setup can be found in Figure S16. Panels a–c in Figure 3 show the spectra obtained for Ni SAC, Ni Pc, and Ag NPs within the range of 1200–1700 cm–1. This spectral range contains the most intense absorption peaks from the functional groups of *COOH, which are used as a proxy for CO generation by the cocatalysts. The bands at ∼1300 and ∼1425 cm–1 can be assigned to C–OH and C–O stretching modes from *COOH, respectively.27−29 It is worth noting that the intensity of *COOH stretching modes is higher for Ni SAC (Figure 3a) when compared to both Ni Pc and Ag NPs (Figure 3b,c), indicating a higher CO production rate on Ni SAC at very negative potentials (E < – 1.05 V vs RHE).
Figure 3.

In situ ATR-IR spectra. Spectral region within 1200–1700 cm–1 for Ni SAC (a), Ni Pc (b), and Ag NPs (c), in which CO-derived products are probed, indicating CO generation. Spectral region between 1800–2200 cm–1 for Ni SAC + Cu-R (d), Cu-R (e), and Ni Pc + Cu-R (f), where adsorbed CO was probed for each system. All spectra were acquired while sweeping the potential (−1.45 < E< – 0.85 V vs RHE).
Figure 3d,e displays the ATR-IR spectra within the spectral range of CO vibrational modes (1800–2200 cm–1) for Ni SAC + Cu-R and pristine Cu-R, respectively. The spectra obtained for Ni SAC + Cu-R during CO2RR (Figure 3d) presented a band at ∼2025 cm–1 corresponding to the vibrational excitation of adsorbed CO molecules.30 This band was observed across the entire potential window, although it decreased toward more negative potentials. This behavior suggests fast CO consumption at the expense of *CO dimerization. Unlike the hybrid structure, the *CO coverage at the surface of pristine Cu-R (Figure 3e) was observed only at very negative potentials, evidencing the benefit of incorporating Ni SAC in the catalyst. Notably, poor CO coverage was also observed when Cu-R was paired to either Ni Pc or Ag NPs (Figures 3f and S17), which could be attributed to the preferred *H delivery from the cocatalysts, competing with *CO for the adsorption site.30−32 This hypothesis was confirmed by the lower Stark rate for Ni Pc + Cu-R (24 cm–1/V) and Ag + Cu-R (21 cm–1/V) in comparison with Ni SAC + Cu-R (40 cm–1/V). These results reveal that wide-CO generation-potential-window Ni SAC can effectively provide *CO at the potential of the CO dimerization reaction for Cu-R, confirming our potential-matching tandem strategy.
To understand how the adsorbed *CO overflows from the Ni SAC sites to the Cu-R catalysts for undergoing *CO–*CO coupling, we have designed a comparative experiment using 3 different catalyst configurations (Figure S18). There are three possible configurations for constructing the tandem catalyst. While Ni SAC + Cu-R was prepared through mixing Ni SAC and Cu-R under ultrasonic treatment, the other configurations can be achieved by sequentially depositing each constituent on the electrode (named here as Cu-R|Ni SAC and Ni SAC|Cu-R). These configurations were prepared with the same loadings for both Ni SAC and Cu-R on GDE. The mass transport of CO in Cu-R|Ni SAC and Ni SAC|Cu-R catalysts must take place through desorption and readsorption due to the nearly mutual isolation between Ni SAC and Cu-R. Therefore, these two catalyst configurations show a lower FEC2H4 than that of the Ni SAC + Cu-R catalyst. Based on these results, we believe that the main mass transport mode of CO is the surface transport of adsorbed *CO. This overflow is effective at subnanometric scale, especially for catalysts that have a weak CO adsorption energy.
CO2 Electroreduction Performance
In this section, we present the performance of the Ni SAC + Cu-R on CO2 electroreduction. The tests were conducted in a flow-cell reactor employing a three-electrode configuration and using 1 M KHCO3 solution as the electrolyte (Figure S19–S21). Figure 4a portrays the C2H4 faradic efficiency (FEC2H4) for pristine Cu-R, and the CO faradic efficiency (FECO) for each of the cocatalysts (Ni SAC, Ag NPs, and Ni Pc). Notably, the Cu-R is able to selectively form C2H4 peaks between −1.4 < E < – 1.05 V vs RHE, as pointed out by the greenish background. Within that regime, it is visible that Ni SAC reaches a larger FECO (>90%) in a large potential window in comparison with the other catalysts, being able to enrich the Cu surface with CO better.
Figure 4.

(a) Potential-matching between Cu-R and Ni SAC. (b) Partial current densities of C2H4 at different potentials of catalysts in a flow-cell reactor. (c) FEC2H4 of tandem catalysts. (d) In situ XANES spectra of Ni SAC measured at different potentials. (e) In situ k3 weighted Fourier transform EXAFS spectra of Ni SAC. (f) Stability of Ni SAC+Cu-R at a current density of −500 mA/cm2.
Since the Ni Pc and Ag catalysts show a potential mismatch with the Cu-R catalyst, they mainly play the role of the H-formation catalyst in the tandem catalyst. Based on DFT calculation of the *H coverage (Figure 1b), the competitive adsorption of *H results in the release of CO generated by Cu-R itself. As a result, the CO faradic efficiencies of Ni Pc + Cu-R and Ag + Cu-R are higher than that of the bare Cu-R catalyst (Figure S21). In other words, instead of increasing the *CO coverage, Ni Pc and Ag decrease the *CO coverage on Cu-R surface, which harms the *CO–*CO coupling process. Hence, the faradic efficiency of ethylene on Ni Pc + Cu-R and Ag + Cu-R is lower than that of Cu-R. The reasons for the decreased H2 selectivity of Ni SAC + Cu-R and Ni Pc + Cu-R or Ag + Cu-R observed in Figure S21 are different. For Ni SAC + Cu-R, the *CO delivery from the Ni SAC increases the *CO coverage of Cu-R, which results in an increase in the relative proportion for FEC2H4 and decreases the relative proportion for FEH2. Regarding Ni Pc + Cu-R and Ag + Cu-R, the *H delivery from Ni Pc or Ag accelerates the release of CO generated by Cu-R due to competitive *H adsorption, which results in an increase of the relative proportion for FECO and decreases the relative proportion for FEH2.
To further highlight the role of *CO coverage on the FEC2H4 production, we conducted carbon monoxide electroreduction (CORR) measurements under the same experimental conditions as for CO2RR (Figure S22). As shown in Figure S22, the FEC2H4 of Cu-R during the CORR is much higher than that during the CO2RR, especially at large current densities. This result shows that increasing *CO coverage can improve the generation of C2H4 on Cu-R and that *CO coverage is the main factor affecting FEC2H4 for Cu-R at large current densities.
Figure 4b shows the C2H4 partial current density for Cu-R and all hybrid combinations, whereas the corresponding total current density is shown in Figure S23. The Ni-SAC + Cu-R outperformed the rest of the tandem catalysts in the entire potential window, reaching a C2H4 partial current density >300 mA/cm2 for E < −1.2 vs RHE, which was obtained at the expense of a drop in CO partial current density (Figure S24). The other combinations seemed to have no significant effect on the C2H4 partial current density and presented a similar behavior to pristine Cu-R. The obtained C2H4 partial current density increased from 350 to 600 mA/cm2 when the FE of C2H4 increased from 50 to 60% (Figure 4c). These unprecedented values highlight the successful combination of Ni SAC and Cu-R for CO2 electroreduction (Figures 4c and S25 and Table S1).
To gain insight into the stability of the fabricated Ni SAC + Cu-R, the Ni K-edge XANES spectrum was monitored at different electrochemical potentials. At the open circuit potential, the Ni atoms presented the same features observed before. When sweeping from −0.8 to −1.4 V vs RHE, no significant changes were observed in the spectral response of Ni, implying that the single atoms on Cu-R remain stable and do not aggregate during the CO2 electroreduction (Figure 4d). In situ k3 weighted Fourier transform EXAFS spectra of Ni SAC indicate that the environment near the Ni atoms was not altered under reaction conditions (Figure 4e).
Finally, we tested the stability of the overall catalyst as a function of time. When conducting the CO2 electroreduction at −1.4 V vs RHE, a FEC2H4 of 60% was maintained for the course of 14 h, while a current of −500 mA/cm2 was measured experimentally (Figure 4f). The stability was also confirmed by XRD when the catalyst was evaluated before and after the CO2RR (Figure S26).
Conclusions
To conclude, we developed a binary catalyst aiming to match the potentials for CO-formation on one catalyst and the *CO–*CO coupling potential on Cu for improving the conversion of CO2 to C2H4. In this regard, we first investigated theoretically different cocatalysts, identifying Ni SAC as the one that can maximize the supply of CO in the range of potentials in which Cu-R best performs for the C–C coupling in the selective C2H4 formation. Upon preparation of the Ni SAC + Cu-R catalyst, the larger CO coverage of Cu surfaces was confirmed by in situ ATR-IR studies. This larger CO coverage facilitates the CO dimerization across a wide range of potentials, spanning from −1.05 to −1.4 V vs RHE and resulting in 60% of FEC2H4 while operated at −500 mA/cm2 in a flow-cell reactor. The catalyst demonstrated excellent stability, maintaining its performance for at least 14 h. Our results pave the way for cooperative catalyst design principles for CO2 electroreduction in hybrid catalysts.
Acknowledgments
The authors acknowledge funding and support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy–EXC 2089/1–390776260 and the e-conversion Research Cluster, the Bavarian program Solar Energies Go Hybrid (SolTech), the Center for NanoScience (CeNS), and the European Commission through the ERC Starting Grant CATALIGHT (802989). L.Z. acknowledges the LMU-CSC program for a doctoral fellowship, and A.S. acknowledges the A. von Humboldt Foundation for a postdoctoral fellowship. The authors thank Prof. Beatriz Roldan Cuenya and Dr. Arno Bergmann from FHI Berlin for fruitful discussions while preparing the manuscript. The authors gratefully thank the Natural Science Foundation of China (Grant Numbers: 22002189, 22376222, 52202125, and 52372253), Central South University Research Programme of Advanced Interdisciplinary Studies (Grant No. 2023QYJC012), and Central South University Innovation-Driven Research Programme (Grant No. 2023CXQD042). The authors are grateful for resources from the High Performance Computing Center of Central South University. The authors would like to acknowledge the help from Beamlines BL01C1 in the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan) for various synchrotron-based measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c09632.
Additional experimental details, materials, and methods, including schemes of the experimental setups and computational details; TEM, SEM, XRD, and XPS characterization for all catalysts; additional gas chromatograph measurements and CO2 electroreduction efficiencies for all catalysts (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Yan D.; Mebrahtu C.; Wang S.; Palkovits R. Innovative Electrochemical Strategies for Hydrogen Production: From Electricity Input to Electricity Output. Angew. Chem., Int. Ed. 2023, 62, e202214333 10.1002/anie.202214333. [DOI] [PubMed] [Google Scholar]
- Yin Z.; Yu J.; Xie Z.; Yu S. W.; Zhang L.; Akauola T.; Chen J. G.; Huang W.; Qi L.; Zhang S. Hybrid Catalyst Coupling Single-atom Ni and Nanoscale Cu for Efficient CO2 Electroreduction to Ethylene. J. Am. Chem. Soc. 2022, 144, 20931–20938. 10.1021/jacs.2c09773. [DOI] [PubMed] [Google Scholar]
- Mistry H.; Varela A. S.; Bonifacio C. S.; Zegkinoglou I.; Sinev I.; Choi Y. W.; Kisslinger K.; Stach E. A.; Yang J. C.; Strasser P.; Cuenya B. R. Highly Selective Plasma-Activated Copper Catalysts for Carbon Dioxide Reduction to Ethylene. Nat. Commun. 2016, 7, 12123 10.1038/ncomms12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B.; Liu K.; Li H.; Liu C.; Fu J.; Li H.; Huang J. E.; Ou P.; Alkayyali T.; Cai C.; Duan Y.; Liu H.; An P.; Zhang N.; Li W.; Qiu X.; Jia C.; Hu J.; Chai L.; Lin Z.; Gao Y.; Miyauchi M.; Cortés E.; Maier S. A.; Liu M. Accelerating CO2 Electroreduction to Multi-Carbon Products via Synergistic Electric-thermal Field on Copper Nanoneedles. J. Am. Chem. Soc. 2022, 144, 3039–3049. 10.1021/jacs.1c11253. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Che F.; Liu M.; Zou C.; Liang Z.; De L. P.; Yuan H.; Li J.; Wang Z.; Xie H.; Li H.; Chen P.; Bladt E.; Quintero-Bermudez R.; Sham T. K.; Bals S.; Hofkens J.; Sinton D.; Chen G.; Sargent E. H. Dopant-Induced Electron Localization Drives CO2 Reduction to C2 Hydrocarbons. Nat. Chem. 2018, 10, 974–980. 10.1038/s41557-018-0092-x. [DOI] [PubMed] [Google Scholar]
- Jin J.; Wicks J.; Min Q.; Li J.; Hu Y.; Ma J.; Wang Y.; Jiang Z.; Xu Y.; Lu R.; Si G.; Papangelakis P.; Shakouri M.; Xiao Q.; Ou P.; Wang X.; Chen Z.; Zhang W.; Yu K.; Song J.; Jiang X.; Qiu P.; Lou Y.; Wu D.; Mao Y.; Ozden A.; Wang C.; Xia B.; Hu X.; Dravid V. P.; Yiu Y. M.; Sham T. K.; Wang Z.; Sinton D.; Mai L.; Sargent E. H.; Pang Y. Constrained C2 Adsorbate Orientation Enables CO-to-Acetate Electroreduction. Nature 2023, 617, 724–729. 10.1038/s41586-023-05918-8. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Cai C.; Dai M.; Fu J.; Zhang X.; Li H.; Zhang H.; Chen K.; Lin Y.; Li H.; Hu J.; Miyauchi M.; Liu M. Recent Advances in Strategies for Improving The Performance of CO2 Reduction Reaction on Single Atom Catalysts. Small Sci. 2021, 1, 2000028 10.1002/smsc.202000028. [DOI] [Google Scholar]
- Ezendam S.; Herran M.; Nan L.; Gruber C.; Kang Y.; Gröbmeyer F.; Lin R.; Gargiulo J.; Sousa-Castillo A.; Cortés E. Hybrid Plasmonic Nanomaterials for Hydrogen Generation and Carbon Dioxide Reduction. ACS Energy Lett. 2022, 7, 778–815. 10.1021/acsenergylett.1c02241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan C.; Dattila F.; Rettenmaier C.; Bergmann A.; Kühl S.; García-Muelas R.; López N.; Cuenya B. R. Revealing The CO Coverage-driven C-C coupling mechanism for electroChemical CO2 Reduction on Cu2O Nanocubes via Operando Raman Spectroscopy. ACS Catal. 2021, 11, 7694–7701. 10.1021/acscatal.1c01478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W.; He X.; Wang W.; Xie S.; Zhang Q.; Wang Y. Electrocatalytic Reduction of CO2 and CO to Multi-carbon Compounds Over Cu-based Catalysts. Chem. Soc. Rev. 2021, 50, 12897–12914. 10.1039/D1CS00535A. [DOI] [PubMed] [Google Scholar]
- Chen C.; Li Y.; Yu S.; Louisia S.; Jin J.; Li M.; Ross M. B.; Yang P. Cu-Ag Tandem Catalysts for High-rate CO2 Electrolysis Toward Multi-Carbons. Joule 2020, 4, 1688–1699. 10.1016/j.joule.2020.07.009. [DOI] [Google Scholar]
- Wang P.; Yang H.; Tang C.; Wu Y.; Zheng Y.; Cheng T.; Davey K.; Huang X.; Qiao S. Z. Boosting Electrocatalytic CO2-to-Ethanol Production via Asymmetric C-C coupling. Nat. Commun. 2022, 13, 3754 10.1038/s41467-022-31427-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B.; Li F. Z.; Gu J. Designing Cu-based Tandem Catalysts for CO2 Electroreduction Based on Mass Transport of CO Intermediate. ACS Catal. 2022, 12, 9735–9752. 10.1021/acscatal.2c02579. [DOI] [Google Scholar]
- Zhang B.; Wang L.; Li D.; Li Z.; Bu R.; Lu Y. Tandem Strategy for Electrochemical CO2 Reduction Reaction. Chem. Catal. 2022, 2, 3395–3429. 10.1016/j.checat.2022.10.017. [DOI] [Google Scholar]
- Li F.; Li Y. C.; Wang Z.; Li J.; Nam D. H.; Lum Y.; Luo M.; Wang X.; Ozden A.; Hung S. F.; Chen B.; Wang Y.; Wicks J.; Xu Y.; Li Y.; Gabardo C. M.; Dinh C. T.; Wang Y.; Zhuang T. T.; Sinton D.; Sargent E. H. Cooperative CO2-to-ethanol Conversion via Enriched Intermediates at Molecule-Metal Catalyst Interfaces. Nat. Catal. 2020, 3, 75–82. 10.1038/s41929-019-0383-7. [DOI] [Google Scholar]
- Iyengar P.; Kolb M. J.; Pankhurst J. R.; Calle-Vallejo F.; Buonsanti R. Elucidating the Facet-Dependent Selectivity for CO2 Electroreduction to Ethanol of Cu-Ag Tandem Catalysts. ACS Catal. 2021, 11, 4456–4463. 10.1021/acscatal.1c00420. [DOI] [Google Scholar]
- Huang J.; Mensi M.; Oveisi E.; Mantella V.; Buonsanti R. Structural Sensitivities in Bimetallic Catalysts for Electrochemical CO2 Reduction Revealed by Ag-Cu Nanodimers. J. Am. Chem. Soc. 2019, 141, 2490–2499. 10.1021/jacs.8b12381. [DOI] [PubMed] [Google Scholar]
- Yang R.; Duan J.; Dong P.; Wen Q.; Wu M.; Liu Y.; Liu Y.; Li H.; Zhai T. In situ Halogen-Ion Leaching Regulates Multiple Sites on Tandem Catalysts for Efficient CO2 Electroreduction to C2+ Products. Angew. Chem., Int. Ed. 2022, 61, e202116706 10.1002/anie.202116706. [DOI] [PubMed] [Google Scholar]
- Kong X.; Zhao J.; Ke J.; Wang C.; Li S.; Si R.; Liu B.; Zeng J.; Geng Z. Understanding the Effect of *CO Coverage on C-C Coupling toward CO2 Electroreduction. Nano Lett. 2022, 22, 3801–3808. 10.1021/acs.nanolett.2c00945. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Li P.; Zhao C.; Zhou G.; Zhou F.; Zhang Q.; Su C.; Wu Y. Multicarbons Generation Factory: CuO/Ni Single Atoms Tandem Catalyst for Boosting the Productivity of CO2 Electrocatalysis. Sci. Bull. 2022, 67, 1679–1687. 10.1016/j.scib.2022.07.029. [DOI] [PubMed] [Google Scholar]
- Nam D. H.; De L. P.; Rosas-Hernández A.; Thevenon A.; Li F.; Agapie T.; Peters J. C.; Shekhah O.; Eddaoudi M.; Sargent E. H. Molecular Enhancement of Heterogeneous CO2 Reduction. Nat. Mater. 2020, 19, 266–276. 10.1038/s41563-020-0610-2. [DOI] [PubMed] [Google Scholar]
- Nitopi S.; Bertheussen E.; Scott S. B.; Liu X.; Engstfeld A. K.; Horch S.; Seger B.; Stephens I. E.; Chan K.; Hahn C.; Nørskov J. K.; Jaramillo T. F.; Chorkendorff I. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. 10.1021/acs.chemrev.8b00705. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Liu K.; Hu K.; Cai C.; Li H.; Li H.; Herran M.; Lu Y. R.; Chan T. S.; Ma C.; Fu J.; Zhang S.; Liang Y.; Cortés E.; Liu M. Attenuating Metal-Substrate Conjugation in Atomically Dispersed Nickel Catalysts for Electroreduction of CO2 to CO. Nat. Commun. 2022, 13, 6082 10.1038/s41467-022-33692-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Luo T.; Chen K.; Lin Y.; Fu J.; Liu K.; Cai C.; Wang Q.; Li H.; Li X.; Hu J.; Li H.; Zhu M.; Liu M. Chemical Identification of Catalytically Active Sites on Oxygen-Doped Carbon Nanosheet to Decipher the High Activity for Electro-Synthesis Hydrogen Peroxide. Angew. Chem., Int. Ed. 2021, 60, 16607–16614. 10.1002/anie.202104480. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Liu K.; Fu J.; Cai C.; Li H.; Long Y.; Chen S.; Liu B.; Li H.; Li W.; Qiu X. Atomically Dispersed s-Block Magnesium Sites for Electroreduction of CO2 to CO. Angew. Chem., Int. Ed. 2021, 60, 25241–25245. 10.1002/anie.202109329. [DOI] [PubMed] [Google Scholar]
- Yang H. B.; Hung S. F.; Liu S.; Yuan K.; Miao S.; Zhang L.; Huang X.; Wang H. Y.; Cai W.; Chen R.; Gao J.; Yang X.; Chen W.; Huang Y.; Chen H. M.; Li C. M.; Zhang T.; Liu B. Atomically Dispersed Ni(i) as the Active Site for Electrochemical CO2 Reduction. Nat. Energy 2018, 3, 140–147. 10.1038/s41560-017-0078-8. [DOI] [Google Scholar]
- Cao X.; Zhao L.; Wulan B.; Tan D.; Chen Q.; Ma J.; Zhang J. Atomic Bridging Structure of Nickel-Nitrogen-Carbon for Highly Efficient Electrocatalytic Reduction of CO2. Angew. Chem., Int. Ed. 2022, 134, e202113918 10.1002/anie.202113918. [DOI] [PubMed] [Google Scholar]
- Firet N. J.; Smith W. A. Probing the Reaction Mechanism of CO2 Electroreduction over Ag Films via Operando Infrared Spectroscopy. ACS Catal. 2017, 7, 606–612. 10.1021/acscatal.6b02382. [DOI] [Google Scholar]
- Zhu S.; Li T.; Cai W. B.; Shao M. CO2 Electrochemical Reduction as Probed through Infrared Spectroscopy. ACS Energy Lett. 2019, 4, 682–689. 10.1021/acsenergylett.8b02525. [DOI] [Google Scholar]
- Zhu S.; Jiang B.; Cai W. B.; Shao M. Direct Observation on Reaction Intermediates and the Role of Bicarbonate Anions in CO2 Electrochemical Reduction Reaction on Cu Surfaces. J. Am. Chem. Soc. 2017, 139, 15664–15667. 10.1021/jacs.7b10462. [DOI] [PubMed] [Google Scholar]
- Ding J.; Li F.; Zhang J.; Zhang Q.; Liu Y.; Wang W.; Liu W.; Wang B.; Cai J.; Su X.; Yang H. B.; Yang X.; Huang Y.; Zhai Y.; Liu B. Circumventing CO2 Reduction Scaling Relations Over the Heteronuclear Diatomic Catalytic Pair. J. Am. Chem. Soc. 2023, 145, 11829–11836. 10.1021/jacs.3c03426. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Dai M.; Li H.; Lu Y. R.; Chan T. S.; Ma C.; Liu K.; Fu J.; Liao W.; Chen S.; Pensa E.; Wang Y.; Zhang S.; Sun Y.; Cortés E.; Liu M. Asymmetric Coordination Induces Electron Localization at Ca Sites for Robust CO2 Electroreduction to CO. Adv. Mater. 2023, 35, 2300695 10.1002/adma.202300695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.