

Abstract

The electrochemical transition metal-catalyzed cross-dehydrogenative reaction has emerged as a promising platform to achieve a sustainable and atom-economic organic synthesis that avoids hazardous oxidants and minimizes undesired byproducts and circuitous functional group operations. However, a poor mechanistic understanding still prevents the widespread adoption of this strategy. In this regard, we herein present an electrochemical palladium-catalyzed oxidative coupling strategy to access biaryls in the absence of a stoichiometric chemical oxidant. The robust palladaelectrocatalysis considerably suppresses the occurrence of homocoupling and oxygenation, being compatible even with electron-deficient arenes. Late-stage functionalization and Boscalid precursor synthesis further highlighted the practical importance of our electrolysis. Remarkably, mechanistic studies including the evaluation of the reaction order of each component by variable time normalization analysis (VTNA) and initial rate analysis, H/D exchange experiment, kinetic isotope effect, and stoichiometric organometallic experiments provided strong support for the involvement of transmetalation between two organopalladium complexes in the turnover limiting step. Therefore, matching the concentrations or lifetimes of two distinct organopalladium intermediates is revealed to be a pivot to the success of electrooxidative catalysis. Moreover, the presence of cationic copper(II) seems to contribute to the stabilization of the palladium(0) catalyst instead of playing a role in the oxidation of the catalyst.
Introduction
Biaryl scaffolds represent an important class of structural motifs embedded in non-natural pharmaceuticals, agrochemicals, ligands, and π-conjugated materials.1 Conventional halogen- and organometal-based cross-coupling reactions that access biaryls usually generate superstoichiometric chemical waste through multiple functional group manipulations.2 In sharp contrast, palladium-catalyzed cross-dehydrogenative coupling3 of simple arenes has emerged as a direct and rapid avenue in line with an atom-economic and green synthesis (Figure 1a). Significant progress has been made in palladium-catalyzed double C–H activation for biaryl formation since the seminal works by inter alia Lu, Fagnou, Deboef, and Sanford.4 Nevertheless, challenges such as reduced catalytic efficacy, limited substrate scope, and byproduct formation have raised intriguing questions about the mechanism of such transformation.5
Figure 1.

Novel electrochemical cross-dehydrogenative coupling.
Mechanistically, a commonly accepted rational catalytic cycle involves one palladium(II) center, which undergoes two sequential C–H cleavages via concerted metalation deprotonation (CMD),6 base-assisted internal electrophilic substitution (BIES),7 σ-bond metathesis,8 or electrophilic metalation. However, high-energy barriers have been associated with such elementary steps, casting doubt on the reaction mechanism.9 Alternatively, organometallic reactions built on a Pd(II)/Pd(IV) redox system10 have been explored as a feasible platform for addressing such unfavored energetic limitations. In this context, Sanford, Michael, and Yu, among others, put forward C–H activation at palladium(IV) species to provide distinguished selectivity and functional group tolerance under mild conditions.11 As an alternative, catalysis involving binuclear palladium(III) was reported by Ritter,12 providing new insights into palladium catalysis. Remarkably, mechanistic studies by Echavarren pointed to transmetalation-type reactions between palladium(II) complexes being more facile than a Pd(II)/Pd(IV) redox cycle within the Catellani regime.13 Although the Pd-to-Pd transmetalation was recognized by Davidson and Triggs as early as 1968,14 few reports have provided experimental evidence for such a pathway.15 In 2003, Osakada elegantly illustrated an aryl transmetalation process via an intramolecular ligand exchange (Figure 1b).16 Additionally, Hartwig and Stahl conducted detailed mechanistic studies for the Pd–Pd cooperative modus operandi for the direct arylation of aryl halide and the homocoupling of xylene, respectively.9b,17
Recently, the merger of transition metal-catalyzed C–H activation18 and electro-organic synthesis19 has surfaced as a uniquely effective approach for sustainable molecular synthesis.20 Harnessing the advantages of replacing toxic and undesirable stoichiometric chemical oxidants with electricity, our group has significantly contributed to the progress on electrochemical C–H activation catalyzed by 3d-, 4d-, and 5d-metals.21 Referring to palladaelectrocatalysis,22 we have extended the scope of oxidative coupling to asymmetric catalysis23 and undirected C–H olefination.24 However, to the best of our knowledge, biaryl formation via electrochemical palladium-catalyzed double C–H activation has proven elusive.
Herein, inspired by the elegant multiple C–H activation developed by Shi (Figure 1c),5o we report on a novel electrochemical palladium-catalyzed cross-dehydrogenative transformation for the synthesis of biaryl devoid of stoichiometric chemical oxidant and prefunctionalized fragments (Figure 1d). The electrooxidative conditions exhibit broad applicability, including electron-deficient arenes. Late-stage functionalization as well as Boscalid precursor synthesis has been proved feasible under our electrolysis conditions. Notably, a rare bimetallic mechanism featuring a Pd-to-Pd aryl transfer process as the turnover limiting step was disclosed. Mechanistic studies comprising reaction order studies by VTNA and initial rate analysis, isotope experiments, and stoichiometric organometallic reactions provided strong support for a bimetallic Pd-to-Pd transmetalation mechanism. Moreover, Cu(OTf)2 seems to be crucial for the stabilization of palladium(0) intermediates rather than participating in the oxidation of catalysts.5o,25
Results and Discussion
We initiated our studies for the envisioned electrochemical dual C–H activation using N-acetyltetrahydroquinoline (1a) and o-xylene (2a) as substrates in a divided cell setup (Scheme 1, Entry 1). Using dichloroethane (DCE) as the solvent resulted in a drastic reduction in the yield of product 3 (Entry 2). Similarly, changing the solvent ratio led to a drop in performance, highlighting the H-bonding donor ability of HFIP on stabilizing intermediates (Supplementary Table 7).26 The metallaelectrocatalysis occurred in the absence of Cu(OTf)2 or 2,6-lutidine, whereas when present in catalytic amounts, an improvement in the turnover number and robustness was observed (entries 3 and 4). Control experiments revealed the indispensable role of both the palladium catalyst and the electricity in the electrooxidative double C–H arylation (entries 5 and 6). A divided cell electrolyzer was beneficial to provide good reactivity and chemoselectivity. (Entry 7 and Supplementary Table 4).27 Further optimization demonstrated that adjusting the stoichiometry of reactant 2a had a substantial influence on the isolated yield (Entry 8). In addition, 2,6-bis(trifluoromethyl)pyridine (L2) was found to be an inferior substitute for lutidine (Entry 9). Interestingly, similar reaction efficiency was obtained when replacing Cu(OTf)2 and lutidine with 2,6-di-tert-butyl benzoquinone (L3; Entry 10 and Supplementary Figure 9).28
Scheme 1. Control Experiments for Palladaelectro Cross-Dehydrogenative Arylation.

General reaction conditions: divided cell, anodic chamber: 1a (0.20 mmol), 2a (1.0 mmol), Pd(OAc)2 (10 mol %), 2.6-lutidine (20 mol %), Cu(OTf)2 (10 mol %), nBu4NBF4 (40 mg), HFIP/AcOH (1.0 mL: 2.0 mL), cathodic chamber: 2a (1.0 mmol), nBu4NBF4 (40 mg), HFIP/AcOH (1.0 mL: 2.0 mL), 100 °C, electrolysis (CCE) at 1.0 mA, 18 h, graphite felt (GF) anode (10 mm × 15 mm × 2 mm), Pt plate cathode (10 mm × 15 mm × 0.25 mm), NMR yields using CH2Br2 as an internal standard.
Isolated yield.
80 °C.
Without Cu(OTf)2.
With the optimized reaction conditions in hand, we explored the versatility of electro-oxidation (Scheme 2). We were pleased to find that a wide variety of functional groups involving labile halides and potential Shono-type oxidation alkylated amide motifs were compatible with the robust palladaelectrocatalysis. Acetanilide (1b) and benzanilide (1c) provided both mono- and bis-arylated products 4 and 5, respectively. Anilide derivatives bearing a methyl group on the m-position significantly inhibited the formation of difunctionalized products, thus delivering monoarylated products 6 and 7 with excellent site selectivity. When a wide range of o-functionalities were introduced into the acetanilides, a significant improvement in reactivity was observed when compared to those bearing no o-substituents (8–15). The trifluoromethyl group was also identified as a compatible moiety, thus affording product 12 in moderate yield. Notably, N-methylacetanilide 1n furnished solely monoarylated products 16, possibly due to steric effects. Furthermore, substrates containing different ring-size directing groups like pyrrolidinone (1o), piperidinone (1p), and azepinone (1q) were also converted, affording uniquely monoarylated products 17–19. Next, we explored substrates equipped with alternative directing groups. Gratifyingly, N-methylbenzamide and N,N-dimethylbenzamide were compatible under the reaction conditions (20–21). Unfortunately, carboxylic acid did not mirror the reactivity and was unable to afford the desired product.
Scheme 2. Versatility for Electrochemical Palladium-Catalyzed Double C–H Activation.

Standard reaction conditions. The amount of arenes used is indicated. See SI for more experimental details.
20 mol % Pd(OAc)2, 0.5 mL of TFA as a cosolvent, 90 °C.
As an isomeric mixture.
Triisopropyl(phenoxy)silane as a substrate.
20 mol % Pd(OAc)2 was used.
Thereafter, we examined the scope of directing-group-free arenes 2 in the electrocatalysis (Scheme 2). A set of electronically diverse arenes 2 were compatible with the robust electrochemical conditions, providing products 22–46 in moderate-to-excellent yields. Hence, benzene (2b) and naphthalene (2c) were tested, giving good yields for the respective arylated products (22 and 23). Likewise, toluene (2d) and anisole (2e) were suitable substrates, providing p-arylated products 24 and 25 as major regioisomers. Interestingly, in situ deprotected product 26 was observed when triisopropyl(phenoxy)silane (2f) was subjected to the reaction conditions. Phenylacetate (2i) was also found to be a suitable substrate, furnishing the desired product (29) in good yield. Notably, electronically deficient arenes were compatible under the electrolysis conditions in conjugation with trifluoroacetyl (TFA) as a cosolvent, providing the desired biaryls (30–37) in low-to-excellent yields. Here, TFA was thought to accelerate the C–H activation of electron-poor arenes,5k while for electron-rich arenes, it led to the formation of a homocoupling product as the major product. Unfortunately, bromobenzene was not tolerated under electrocatalysis conditions. Veratrol (2r) and 2,3-dihydrobenzo[b][1,4]dioxine (2t) were identified as amenable substrates; however, we observed that arenes with higher electron densities such as 1,3,5-trimethoxybenzene usually delivered the self-polymerized product. Furthermore, 1,3-disubstituted and asymmetrical 1,2-disubstituted arenes were selectively functionalized, affording products 41–46. We have applied our electrochemical methodology to late-stage diversification of Tamibarotene ester, affording a series of arylated products (47–49) in excellent yields. Furthermore, the precursor (50) for Boscalid was successfully furnished by our electrocatalysis. However, the presence of the methyl group on the N-center was revealed to be necessary for the reactivity to unwind.
Hitherto, our preliminary studies of the electrochemical palladium-catalyzed cross-dehydrogenative arylation left several key questions unanswered. First, the failure of bromobenzene as a starting material and the observation of palladium black jeopardized the proposal of Pd(IV) in our mechanism. Second, low yields for the desired products were usually associated with the homocoupling of simple arenes, which surpassed the cross arylation. Third, the catalytic efficiency was sensitive to the concentration of the palladium catalyst. These questions motivated us to explore the reaction mechanism in detail.
We began our mechanistic interrogation by determining the turnover limiting step through kinetic studies (Figure 2). The kinetic order of the reaction components was determined by using variable time normalization analysis (VTNA) derived from the reaction progress kinetic analysis developed by Blackmond.29Figure 2a shows the kinetic profile of the reaction with two different Pd(OAc)2 concentrations, where two distinct slopes were observed. When the two profiles were replotted as product concentration versus normalized time scale by a first-order factor of catalyst concentration (t [Pd]1), two reaction progress curves failed to overlap (Figure 2b) until a second-order correlation was used (Figure 2c), indicating that the kinetic order for palladium is the second order rather than the first order. The observation of [Pd]2 suggests that two intramolecular or intermolecular palladium nuclei are involved in the turnover limiting step. Next, inferior reaction progress was observed when increasing substrate 1a loading (Figure 2d,e), corresponding to an inverse first order. Therefore, we hypothesized that losing 1 equiv of 1a from a palladium off-cycle species is necessary to activate the catalyst.30 Likewise, an experimental order of one for substrate 2a was obtained by using an analogous procedure (Figure 2f,g). To further corroborate the kinetic data obtained from VTNA, we conducted initial rate analyses using dichlorobenzene (2p) as the substrate (see the SI, Section 7.2). As a result, the kinetic orders obtained from VTNA were supported by the supplementary initial rate analyses, suggesting that the reaction pathway of electron-poor arenes is relevant to the mechanism of the reaction of electron-rich arenes. In addition, initial rate analyses pointed at zero kinetic orders for copper, lutidine, and current (Figure 2h–j). Notably, identical initial rates were obtained even in the absence of these components, supporting the idea that copper, lutidine, and electricity are solely involved in the regeneration of the active catalyst. Based on the above mechanistic findings, we assumed that 2 equiv of palladium, 1 equiv of 1a, and 1 equiv of 2a were involved in the rate-determining step. With this in mind, two possible reaction pathway candidates could be accounted for: a Pd(II)-to-Pd(II) transmetalation mechanism or a previously reported bimetallic Pd(IV) manifold.30 However, the transmetalation pathway seems to be a more plausible pathway over the dimeric palladium catalyst for three reasons: (1) electricity and Cu(OTf)2 were not involved in the oxidation of Pd(II) to Pd(IV), (2) the dimeric catalyst was generally considered as the precatalyst,31 and (3) the observed inverse first order for 1a contradicted the described transformation between the resting-state catalyst and the dimeric palladium complex in the literature.30
Figure 2.

Mechanistic studies; see the Supporting Information for more reaction details. (a–g) VTNA analysis using o-xylene as a substrate. (h–j) Initial rate studies using 1,2-dichlorobenzene as a substrate. (k, l) H/D exchange experiment. Dideuterated dimethoxybenzene could be a possible product. (m, n) Kinetic isotope effect.
To further assess the transmetalation mechanism based on two organopalladium complexes, it is of interest to know if the two C–H activation steps proceed before the transmetalation step or not; thus, we conducted isotope experiments to investigate the nature of the C–H activation step. H/D exchange experiments (Figure 2k,l) illustrated that for both isotopes, the yields exceeded the catalytic amount of Pd(OAc)2, which can be indicative of a reversible metalation for each of the substrates. Additionally, kinetic isotope effect (KIE) studies revealed a secondary KIE or no KIE for substrate 1a and a primary KIE for substrate 2a (Figure 2m,n), indicative of a facile C–H cleavage of 1a, whereas the step for 2a is slow. The observation of deuterated products and KIE are consistent with C–H cleavages occurring during the catalytic cycle but before the turnover limiting step.32 Moreover, the large KIE for 2a implied that the C–H activation of 2a could replace the transmetalation as the rate-determining step when lowering the temperature or reducing the stoichiometry in 2a.
To further validate our finding on cooperative aryl transfer between the two palladium centers, it is necessary to identify the relations between the resting-state catalyst and the active catalyst in the rate-determining step, in particular for the organopalladium complexes with anilide. Therefore, we first conducted an HRMS analysis to detect the possible intermediates under our catalytic conditions. Three intermediates (51–53) could be postulated from the interpretation of the HRMS spectrum (Figure 3a and Supplementary Figures 64 and 65). Next, we synthesized known dimeric complexes 54 and 55 (Figure 3b) with diacetate bridges using noncoordinating dichloromethane as a solvent.5j,5k,33 The easy access to complexes 54 and 55 under mild conditions agrees with the observed KIE value for 1a. Treating dimeric palladium complex 55 with MeCN at room temperature led to the formation of monomeric palladium complex 56 in near-quantitive yield (Figure 3c).34 The stoichiometric organometallic reaction between 55 and 2a afforded 44% of product 8 in 1 h and 55% in 2 h (Figure 3d), implying that organopalladium 55 could presumably be a precursor for the active catalyst. The assumption was substantiated by in situ NMR studies on the reaction of 55 and xylene,35 where an induction period of precatalyst 55 was observed (Figure 3e and Supplementary Figure 83).36 Reaction profiles of complexes 55 and 56 obtained from ex situ GC measurements showed a comparable reaction rate for both intermediates (Figure 3f). When considering the fact that complex 56 was stabilized by the strongly coordinated acetonitrile, monomeric palladium was the more kinetically favored active catalyst. Additionally, DFT calculations were carried out at the B3LYP-D4/6-311+G(2d,p)-SDD+ SMD(AcOH)/B3LYP-D3(BJ)/6-31G(d,p)-LANL2DZ level of theory (see the SI, Section 7.8), revealing the C–H activation of xylene on Pd(OAc)2 to be energetically favorable with an energy barrier of 16.4 kcal mol–1. However, on a dimeric catalyst, the same elementary step proved to be more energetically disfavored with a barrier of 25.2 kcal mol–1. Moreover, cyclic voltammetry (CV) measurements revealed good stability for complex 55 at room temperature in HFIP/AcOH (Figure 3g). No catalytic current was observed at room temperature when adding an excess amount of 2a, repudiating the proposal of second C–H activation at the Pd(III) or Pd(IV) center.22d,37 Heating complex 54 to 90 °C in the solvent mixture used for catalysis induced the occurrence of complex 58 (Figure 3h and Supplementary Figure 66). With these observations as well as our kinetic studies, we proposed that intermediate 52 could be identified as the resting state and monomer 51 was the on-cycle active catalyst.
Figure 3.
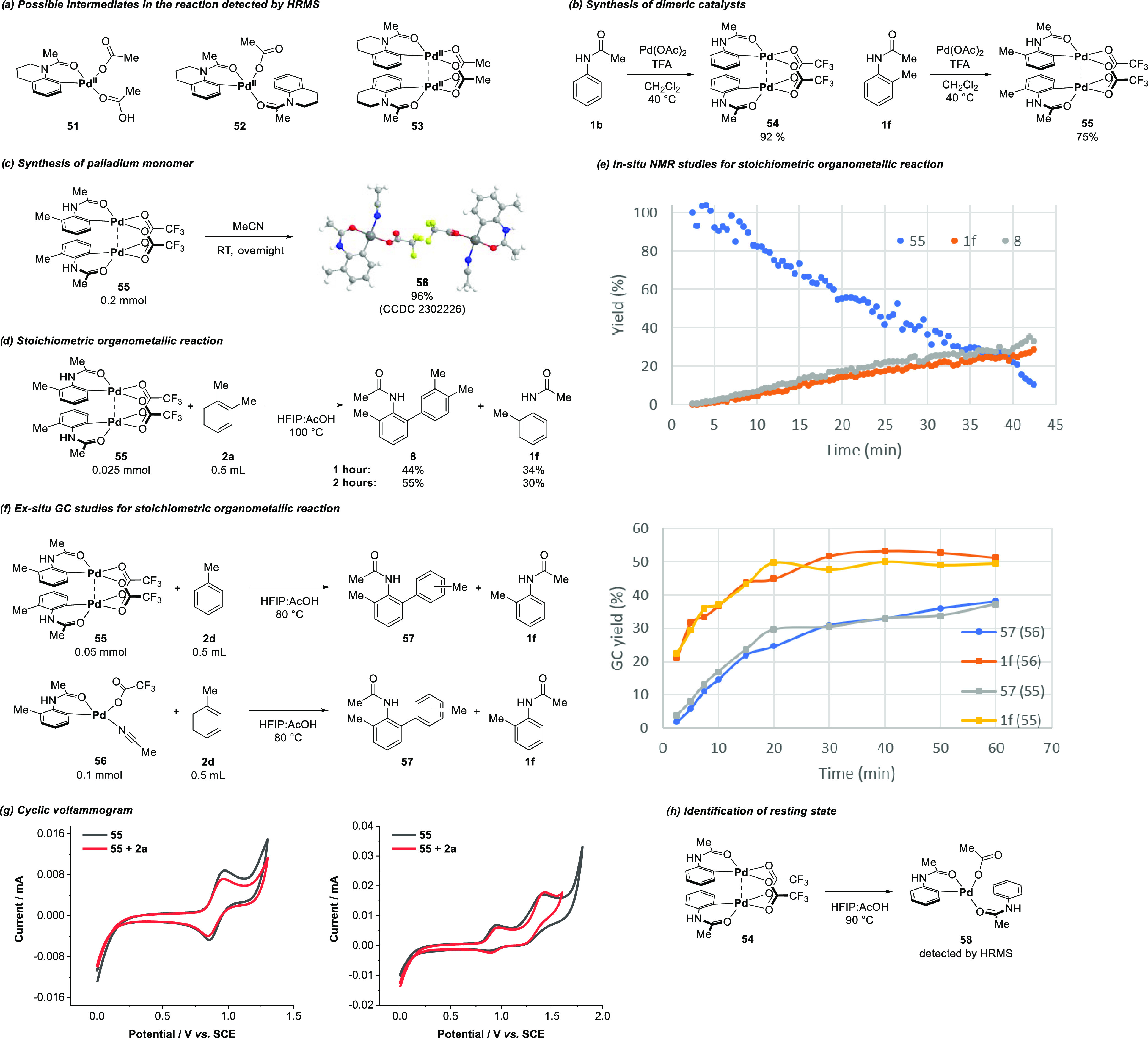
Organopalladium studies. See the SI for more details.
Additionally, we turned our attention to the exact roles of Cu(OTf)2 and 2,6-lutidine in the regeneration of the active catalyst. When increasing the amount of 2,6-lutidine in the presence of 1 equiv of Cu(OTf)2 as a chemical oxidant, deterioration in the yield of product 3 from 88 to 20% was observed (Figure 4a), while the reaction using electricity as the oxidation agent retained its catalytic efficiency. This entailed a diminishing oxidative ability of Cu2+ in the presence of lutidine, which was supported by CV studies (Figure 4b). Moreover, the new oxidative event observed in Figure 4c when palladium and copper were mixed endorsed a heterometallic interaction. Hence, we proposed that copper salt serves as a palladium(0) stabilizer rather than a redox catalyst.38 Further experimentation and electroanalytical studies have been conducted (see the Supporting Information).
Figure 4.
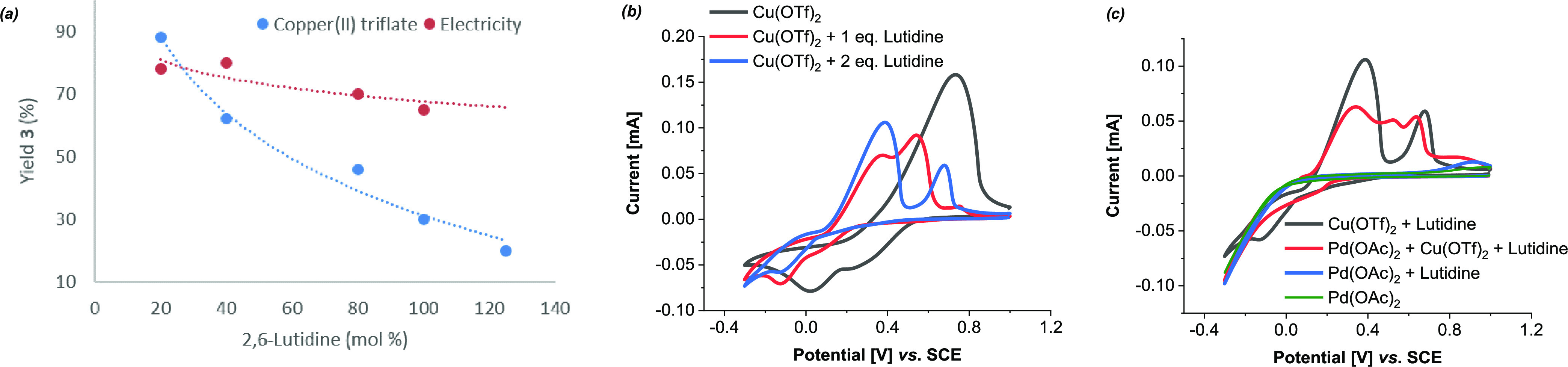
Role of Cu(OTf)2 and lutidine. See the SI for more details. (a) Reaction efficacy dependency on lutidine using Cu(OTf)2 or electricity as the oxidant. (b, c) Cyclic voltammograms. Cu(OTf)2 (5 mM), Pd(OAc)2 (5 mM), lutidine (5 or 10 mM), and 0.1 M nBu4NBF4 in HFIP/AcOH (1:2), 100 mV s–1, room temperature.
Based on our mechanistic studies, a plausible catalytic cycle is presented in Figure 5. C–H activations of 1a and 2a occur concurrently, giving rise to complexes 51 and 59, respectively. Here, dimeric catalyst 53 is considered a precatalyst for monomeric palladacycle 51. Off-cycle species 52 could exist in a different level of concentration depending on the ratio of substrate 1 and Pd(OAc)2. Then, intermolecular transmetalation of 51 to 59 affords 60, followed by reductive elimination, giving desired product 3. The transmetalation between two organopalladium complexes was determined to be the turnover limiting step of the overall reaction. The generated Pd(0) during product formation is then stabilized by Cu complexes, which through anodic oxidation form active Pd(II), thus closing the catalytic cycle.
Figure 5.
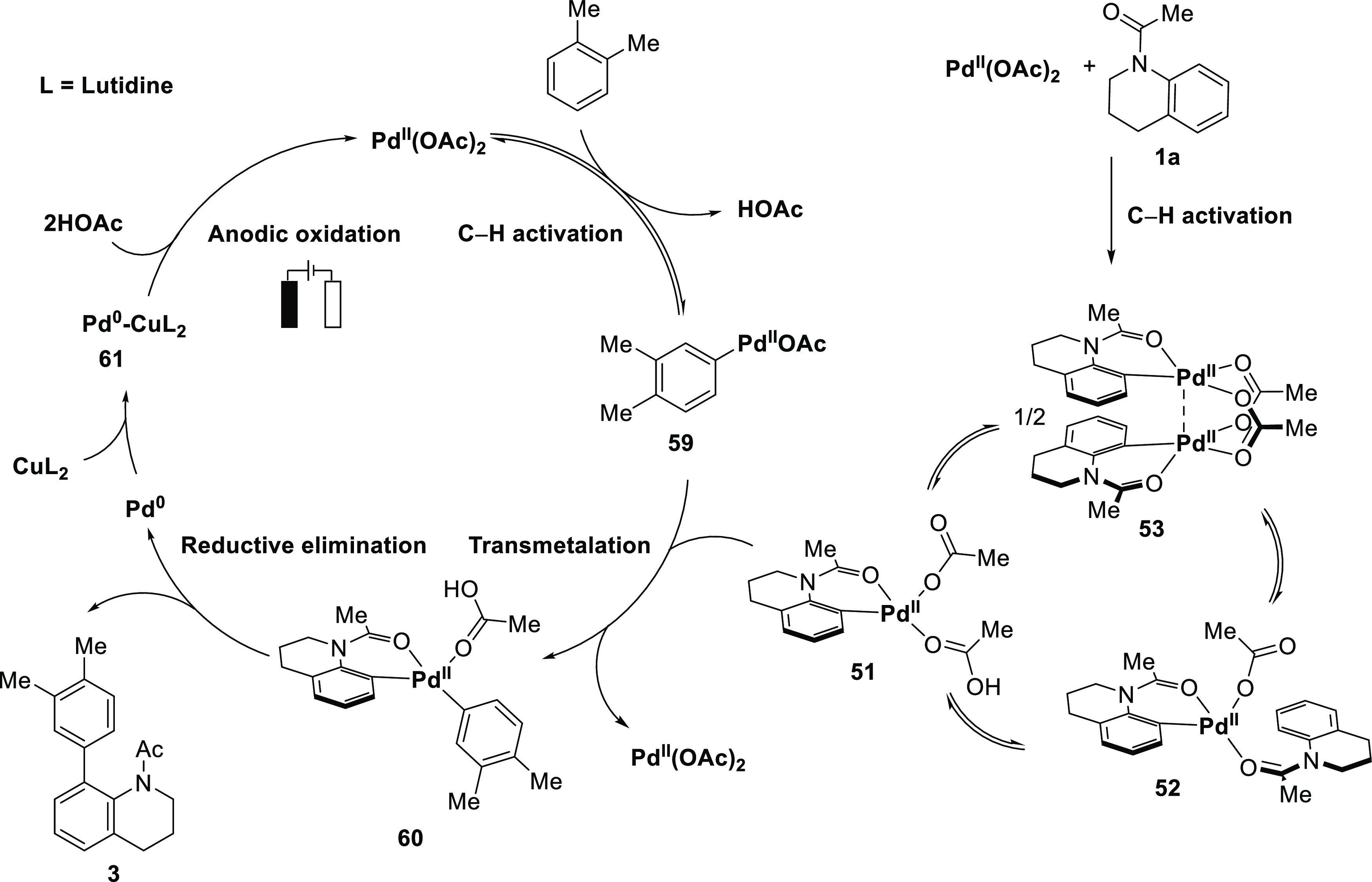
Proposed catalytic cycle.
In summary, we have reported on versatile electrochemical palladium-catalyzed oxidative double C–H arylation without chemical oxidants. The robust electrolysis condition exhibited extraordinary reactivity; thus, a variety of arenes including electron-deficient arenes were compatible. Late-stage functionalization highlighted the synthetic value of our methodology. In addition, detailed mechanistic studies were conducted, thus supporting a bimetallic mechanism involving a transmetalation process as the rate-determining step.
Acknowledgments
Generous support by the ERC Advanced Grant (101021358), the DFG (Gottfried Wilhelm Leibniz award and SPP 2363), and the CSC (scholarship to Z.L.) is gratefully acknowledged. The authors thank our colleagues Svenja and Tristan for their careful and meticulous proofreading of the manuscript. The authors thank Dr. Christopher Golz (Göttingen University) for assistance with the X-ray diffraction analysis. The authors thank Dr. Michael John and Christiane Siebert (Göttingen University) for assistance with the VT-NMR analysis.
Glossary
Abbreviations
- CMD
concerted metalation deprotonation
- DCE
dichloroethane
- HFIP
hexafluorisopropanol
- VTNA
variable time normalization analysis
- KIE
kinetic isotope effect
- CV
cyclic voltammogram
- HRMS
high-resolution mass spectrometry
- NMR
nuclear magnetic resonance
- CDA
cross-dehydrogenative arylation
- CDC
cross-dehydrogenative coupling
- DFT
density functional theory
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c08479.
Experimental procedures and compound characterization data including 1H and 13C NMR spectra and kinetic analyses (PDF)
ERC Advanced Grant (101021358); DFG (Gottfried Wilhelm Leibniz award); CSC (scholarship to Z. L.).
The authors declare no competing financial interest.
Supplementary Material
References
- a Ghasemabadi P. G.; Yao T.; Bodwell G. J. Cyclophanes containing large polycyclic aromatic hydrocarbons. Chem. Soc. Rev. 2015, 44, 6494–6518. 10.1039/C5CS00274E. [DOI] [PubMed] [Google Scholar]; b Surry D. S.; Buchwald S. L. Biaryl Phosphane Ligands in Palladium-Catalyzed Amination. Angew. Chem., Int. Ed. 2008, 47, 6338–6361. 10.1002/anie.200800497. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Horton D. A.; Bourne G. T.; Smythe M. L. The Combinatorial Synthesis of Bicyclic Privileged Structures or Privileged Substructures. Chem. Rev. 2003, 103, 893–930. 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- a De Meijere A.; Bräse S.; Oestreich M.. Metal Catalyzed Cross-Coupling Reactions and More; John Wiley & Sons, 2013. [Google Scholar]; b Miyaura N.; Buchwald S. L.. Cross-Coupling Reactions: A Practical Guide; Springer, 2002. [Google Scholar]; c Stanforth S. P. Catalytic cross-coupling reactions in biaryl synthesis. Tetrahedron 1998, 54, 263–303. 10.1016/S0040-4020(97)10233-2. [DOI] [Google Scholar]
- Selected references for CDC:; a Li C.-J. Cross-Dehydrogenative Coupling (CDC): Exploring C–C Bond Formations beyond Functional Group Transformations. Acc. Chem. Res. 2009, 42, 335–344. 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]; b Zhang H.-B.; Liu L.; Chen Y.-J.; Wang D.; Li C.-J. On Water”-Promoted Direct Coupling of Indoles with 1,4-Benzoquinones without Catalyst. Eur. J. Org. Chem. 2006, 2006, 869–873. 10.1002/ejoc.200500863. [DOI] [Google Scholar]; c Li Z.; Bohle D. S.; Li C.-J. Cu-catalyzed cross-dehydrogenative coupling: A versatile strategy for C–C bond formations via the oxidative activation of sp3 C–H bonds. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 8928–8933. 10.1073/pnas.0601687103. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Li Z.; Li C.-J. CuBr-Catalyzed Direct Indolation of Tetrahydroisoquinolines via Cross-Dehydrogenative Coupling between sp3 C–H and sp2 C–H Bonds. J. Am. Chem. Soc. 2005, 127, 6968–6969. 10.1021/ja0516054. [DOI] [PubMed] [Google Scholar]; e Masui K.; Ikegami H.; Mori A. Palladium-Catalyzed C–H Homocoupling of Thiophenes: Facile Construction of Bithiophene Structure. J. Am. Chem. Soc. 2004, 126, 5074–5075. 10.1021/ja031855p. [DOI] [PubMed] [Google Scholar]; f Jia C.; Kitamura T.; Fujiwara Y. Catalytic functionalization of arenes and alkanes via C–H bond activation. Acc. Chem. Res. 2001, 34, 633–639. 10.1021/ar000209h. [DOI] [PubMed] [Google Scholar]; g Jia C.; Lu W.; Kitamura T.; Fujiwara Y. Highly efficient Pd-catalyzed coupling of arenes with olefins in the presence of tert-butyl hydroperoxide as oxidant. Org. Lett. 1999, 1, 2097–2100. 10.1021/ol991148u. [DOI] [Google Scholar]; h Shiotani A.; Itatani H. Dibenzofurans by Intramolecular Ring Closure Reactions. Angew. Chem., Int. Ed. 1974, 13, 471–472. 10.1002/anie.197404711. [DOI] [Google Scholar]; i Fujiwara Y.; Moritani I.; Danno S.; Asano R.; Teranishi S. Aromatic substitution of olefins. VI. Arylation of olefins with palladium (II) acetate. J. Am. Chem. Soc. 1969, 91, 7166–7169. 10.1021/ja01053a047. [DOI] [PubMed] [Google Scholar]; j Moritanl I.; Fujiwara Y. Aromatic substitution of styrene-palladium chloride complex. Tetrahedron Lett. 1967, 8, 1119–1122. 10.1016/S0040-4039(00)90648-8. [DOI] [Google Scholar]
- a Pereira K. C.; Porter A. L.; Potavathri S.; LeBris A. P.; DeBoef B. Insight into the palladium-catalyzed oxidative arylation of benzofuran: heteropoly acid oxidants evoke a Pd (II)/Pd (IV) mechanism. Tetrahedron 2013, 69, 4429–4435. 10.1016/j.tet.2013.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lyons T. W.; Hull K. L.; Sanford M. S. Controlling Site Selectivity in Pd-Catalyzed Oxidative Cross-Coupling Reactions. J. Am. Chem. Soc. 2011, 133, 4455–4464. 10.1021/ja1097918. [DOI] [PubMed] [Google Scholar]; c Potavathri S.; Pereira K. C.; Gorelsky S. I.; Pike A.; LeBris A. P.; DeBoef B. Regioselective Oxidative Arylation of Indoles Bearing N-Alkyl Protecting Groups: Dual C–H Functionalization via a Concerted Metalation–Deprotonation Mechanism. J. Am. Chem. Soc. 2010, 132, 14676–14681. 10.1021/ja107159b. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hull K. L.; Sanford M. S. Mechanism of Benzoquinone-Promoted Palladium-Catalyzed Oxidative Cross-Coupling Reactions. J. Am. Chem. Soc. 2009, 131, 9651–9653. 10.1021/ja901952h. [DOI] [PubMed] [Google Scholar]; e Potavathri S.; Dumas A. S.; Dwight T. A.; Naumiec G. R.; Hammann J. M.; DeBoef B. Oxidant-controlled regioselectivity in the oxidative arylation of N-acetylindoles. Tetrahedron Lett. 2008, 49, 4050–4053. 10.1016/j.tetlet.2008.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Liégault B.; Fagnou K. Palladium-Catalyzed Intramolecular Coupling of Arenes and Unactivated Alkanes in Air. Organometallics 2008, 27, 4841–4843. 10.1021/om800780f. [DOI] [Google Scholar]; g Liégault B.; Lee D.; Huestis M. P.; Stuart D. R.; Fagnou K. Intramolecular Pd(II)-Catalyzed Oxidative Biaryl Synthesis Under Air: Reaction Development and Scope. J. Org. Chem. 2008, 73, 5022–5028. 10.1021/jo800596m. [DOI] [PubMed] [Google Scholar]; h Hull K. L.; Sanford M. S. Catalytic and Highly Regioselective Cross-Coupling of Aromatic C–H Substrates. J. Am. Chem. Soc. 2007, 129, 11904–11905. 10.1021/ja074395z. [DOI] [PubMed] [Google Scholar]; i Stuart D. R.; Fagnou K. The Catalytic Cross-Coupling of Unactivated Arenes. Science 2007, 316, 1172–1175. 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]; j Dwight T. A.; Rue N. R.; Charyk D.; Josselyn R.; DeBoef B. C–C bond formation via double C–H functionalization: aerobic oxidative coupling as a method for synthesizing heterocoupled biaryls. Org. Lett. 2007, 9, 3137–3139. 10.1021/ol071308z. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Stuart D. R.; Villemure E.; Fagnou K. Elements of Regiocontrol in Palladium-Catalyzed Oxidative Arene Cross-Coupling. J. Am. Chem. Soc. 2007, 129, 12072–12073. 10.1021/ja0745862. [DOI] [PubMed] [Google Scholar]; l Rong Y.; Li R.; Lu W. Palladium(II)-Catalyzed Coupling of p-Xylene via Regioselective C–H Activation in TFA. Organometallics 2007, 26, 4376–4378. 10.1021/om700418h. [DOI] [Google Scholar]; m Li R.; Jiang L.; Lu W. Intermolecular Cross-Coupling of Simple Arenes via C–H Activation by Tuning Concentrations of Arenes and TFA. Organometallics 2006, 25, 5973–5975. 10.1021/om060889d. [DOI] [Google Scholar]
- a von Münchow T.; Dana S.; Xu Y.; Yuan B.; Ackermann L. Enantioselective electrochemical cobalt-catalyzed aryl C–H activation reactions. Science 2023, 379, 1036–1042. 10.1126/science.adg2866. [DOI] [PubMed] [Google Scholar]; b Frey J.; Hou X.; Ackermann L. Atropoenantioselective palladaelectro-catalyzed anilide C–H olefinations viable with natural sunlight as sustainable power source. Chem. Sci. 2022, 13, 2729–2734. 10.1039/D1SC06135F. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rogge T.; Ackermann L. Arene-Free Ruthenium(II/IV)-Catalyzed Bifurcated Arylation for Oxidative C–H/C–H Functionalizations. Angew. Chem., Int. Ed. 2019, 58, 15640–15645. 10.1002/anie.201909457. [DOI] [PubMed] [Google Scholar]; d Sun D.; Li B.; Lan J.; Huang Q.; You J. Chelation-assisted Pd-catalysed ortho-selective oxidative C–H/C–H cross-coupling of aromatic carboxylic acids with arenes and intramolecular Friedel–Crafts acylation: one-pot formation of fluorenones. Chem. Commun. 2016, 52, 3635–3638. 10.1039/C6CC00103C. [DOI] [PubMed] [Google Scholar]; e Bechtoldt A.; Tirler C.; Raghuvanshi K.; Warratz S.; Kornhaaß C.; Ackermann L. Ruthenium Oxidase Catalysis for Site-Selective C–H Alkenylations with Ambient O2 as the Sole Oxidant. Angew. Chem., Int. Ed. 2016, 55, 264–267. 10.1002/anie.201507801. [DOI] [PubMed] [Google Scholar]; f Liu B.; Huang Y.; Lan J.; Song F.; You J. Pd-catalyzed oxidative C–H/C–H cross-coupling of pyridines with heteroarenes. Chem. Sci. 2013, 4, 2163–2167. 10.1039/c3sc50348h. [DOI] [Google Scholar]; g Graczyk K.; Ma W.; Ackermann L. Oxidative Alkenylation of Aromatic Esters by Ruthenium-Catalyzed Twofold C–H Bond Cleavages. Org. Lett. 2012, 14, 4110–4113. 10.1021/ol301759v. [DOI] [PubMed] [Google Scholar]; h Wang Z.; Li K.; Zhao D.; Lan J.; You J. Palladium-Catalyzed Oxidative C–H/C–H Cross-Coupling of Indoles and Pyrroles with Heteroarenes. Angew. Chem., Int. Ed. 2011, 50, 5365–5369. 10.1002/anie.201101416. [DOI] [PubMed] [Google Scholar]; i Li H.; Liu J.; Sun C.-L.; Li B.-J.; Shi Z.-J. Palladium-Catalyzed Cross-Coupling of Polyfluoroarenes with Simple Arenes. Org. Lett. 2011, 13, 276–279. 10.1021/ol102688e. [DOI] [PubMed] [Google Scholar]; j Yeung C. S.; Zhao X.; Borduas N.; Dong V. M. Pd-catalyzed ortho-arylation of phenylacetamides, benzamides, and anilides with simple arenes using sodium persulfate. Chem. Sci. 2010, 1, 331–336. 10.1039/c0sc00231c. [DOI] [Google Scholar]; k Zhao X.; Yeung C. S.; Dong V. M. Palladium-Catalyzed Ortho-Arylation of O-Phenylcarbamates with Simple Arenes and Sodium Persulfate. J. Am. Chem. Soc. 2010, 132, 5837–5844. 10.1021/ja100783c. [DOI] [PubMed] [Google Scholar]; l Xi P.; Yang F.; Qin S.; Zhao D.; Lan J.; Gao G.; Hu C.; You J. Palladium(II)-Catalyzed Oxidative C–H/C–H Cross-Coupling of Heteroarenes. J. Am. Chem. Soc. 2010, 132, 1822–1824. 10.1021/ja909807f. [DOI] [PubMed] [Google Scholar]; m Cho S. H.; Hwang S. J.; Chang S. Palladium-Catalyzed C–H Functionalization of Pyridine N-Oxides: Highly Selective Alkenylation and Direct Arylation with Unactivated Arenes. J. Am. Chem. Soc. 2008, 130, 9254–9256. 10.1021/ja8026295. [DOI] [PubMed] [Google Scholar]; n Brasche G.; García-Fortanet J.; Buchwald S. L. Twofold C–H Functionalization: Palladium-Catalyzed Ortho Arylation of Anilides. Org. Lett. 2008, 10, 2207–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Li B. J.; Tian S. L.; Fang Z.; Shi Z. J. Multiple C–H activations to construct biologically active molecules in a process completely free of organohalogen and organometallic components. Angew. Chem., Int. Ed. 2008, 47, 1115–1118. 10.1002/anie.200704092. [DOI] [PubMed] [Google Scholar]; p Xia J.-B.; You S.-L. Carbon–Carbon Bond Formation through Double sp2 C–H Activations: Synthesis of Ferrocenyl Oxazoline Derivatives. Organometallics 2007, 26, 4869–4871. 10.1021/om700806e. [DOI] [Google Scholar]
- a Lafrance M.; Fagnou K. Palladium-Catalyzed Benzene Arylation: Incorporation of Catalytic Pivalic Acid as a Proton Shuttle and a Key Element in Catalyst Design. J. Am. Chem. Soc. 2006, 128, 16496–16497. 10.1021/ja067144j. [DOI] [PubMed] [Google Scholar]; b Davies D. L.; Donald S. M. A.; Macgregor S. A. Computational Study of the Mechanism of Cyclometalation by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13754–13755. 10.1021/ja052047w. [DOI] [PubMed] [Google Scholar]
- a Ma W.; Mei R.; Tenti G.; Ackermann L. Ruthenium(II)-Catalyzed Oxidative C–H Alkenylations of Sulfonic Acids, Sulfonyl Chlorides and Sulfonamides. Chem. - Eur. J. 2014, 20, 15248–15251. [DOI] [PubMed] [Google Scholar]; b Ackermann L.; Vicente R.; Althammer A. Assisted Ruthenium-Catalyzed C–H Bond Activation: Carboxylic Acids as Cocatalysts for Generally Applicable Direct Arylations in Apolar Solvents. Org. Lett. 2008, 10, 2299–2302. 10.1021/ol800773x. [DOI] [PubMed] [Google Scholar]
- a Watson P. L.; Parshall G. W. Organolanthanides in catalysis. Acc. Chem. Res. 1985, 18, 51–56. 10.1021/ar00110a004. [DOI] [Google Scholar]; b Tremont S. J.; Rahman H. U. Ortho-alkylation of acetanilides using alkyl halides and palladium acetate. J. Am. Chem. Soc. 1984, 106, 5759–5760. 10.1021/ja00331a073. [DOI] [Google Scholar]; c Watson P. L. Methane exchange reactions of lanthanide and early-transition-metal methyl complexes. J. Am. Chem. Soc. 1983, 105, 6491–6493. 10.1021/ja00359a023. [DOI] [Google Scholar]
- a Wang L.; Carrow B. P. Oligothiophene Synthesis by a General C–H Activation Mechanism: Electrophilic Concerted Metalation–Deprotonation (eCMD). ACS Catal. 2019, 9, 6821–6836. 10.1021/acscatal.9b01195. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tan Y.; Barrios-Landeros F.; Hartwig J. F. Mechanistic studies on direct arylation of pyridine N-oxide: evidence for cooperative catalysis between two distinct palladium centers. J. Am. Chem. Soc. 2012, 134, 3683–3686. 10.1021/ja2122156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sehnal P.; Taylor R. J. K.; Fairlamb I. J. S. Emergence of Palladium(IV) Chemistry in Synthesis and Catalysis. Chem. Rev. 2010, 110, 824–889. 10.1021/cr9003242. [DOI] [PubMed] [Google Scholar]; b Lyons T. W.; Sanford M. S. Palladium-catalyzed ligand-directed C–H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Amatore C.; Catellani M.; Deledda S.; Jutand A.; Motti E. Rates of the Oxidative Addition of Benzyl Halides to a Metallacyclic Palladium(II) Complex and of the Reductive Elimination from a Benzyl-Palladium(IV) Complex. Organometallics 2008, 27, 4549–4554. 10.1021/om800015x. [DOI] [Google Scholar]; d Whitfield S. R.; Sanford M. S. Reactivity of Pd(II) Complexes with Electrophilic Chlorinating Reagents: Isolation of Pd(IV) Products and Observation of C–Cl Bond-Forming Reductive Elimination. J. Am. Chem. Soc. 2007, 129, 15142–15143. 10.1021/ja077866q. [DOI] [PubMed] [Google Scholar]; e Canty A. J. The + IV Oxidation State in Organopalladium Chemistry. Recent Advances and Potential Intermediates in Organic Synthesis and Catalysis. Platinum Met. Rev. 1993, 37, 2–7. [Google Scholar]; f Byers P. K.; Canty A. J.; Skelton B. W.; White A. H. The oxidative addition of lodomethane to [PdMe2(bpy)] and the X-ray structure of the organopalladium(IV) product fac-[PdMe3(bpy)l](bpy = 2,2′-bipyridyl). J. Chem. Soc., Chem. Commun. 1986, 0, 1722–1724. 10.1039/C39860001722. [DOI] [Google Scholar]; g Catellani M.; Chiusoli G. P. Palladium-catalyzed synthesis of 1,2,3,4,4a,12b-hexahydro-1,4-methanotriphenylenes. J. Organomet. Chem. 1985, 286, c13–c16. 10.1016/0022-328X(85)87247-8. [DOI] [Google Scholar]; h Milstein D.; Stille J. Mechanism of reductive elimination. Reaction of alkylpalladium (II) complexes with tetraorganotin, organolithium, and Grignard reagents. Evidence for palladium (IV) intermediacy. J. Am. Chem. Soc. 1979, 101, 4981–4991. 10.1021/ja00511a031. [DOI] [Google Scholar]
- Pd(IV) in C–H activation:; a Maleckis A.; Kampf J. W.; Sanford M. S. A Detailed Study of Acetate-Assisted C–H Activation at Palladium(IV) Centers. J. Am. Chem. Soc. 2013, 135, 6618–6625. 10.1021/ja401557m. [DOI] [PubMed] [Google Scholar]; b Hickman A. J.; Sanford M. S. High-valent organometallic copper and palladium in catalysis. Nature 2012, 484, 177–185. 10.1038/nature11008. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hickman A. J.; Sanford M. S. Catalyst Control of Site Selectivity in the PdII/IV-Catalyzed Direct Arylation of Naphthalene. ACS Catal. 2011, 1, 170–174. 10.1021/cs1001543. [DOI] [Google Scholar]; d Wang X.; Leow D.; Yu J.-Q. Pd(II)-Catalyzed para-Selective C–H Arylation of Monosubstituted Arenes. J. Am. Chem. Soc. 2011, 133, 13864–13867. 10.1021/ja206572w. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Sibbald P. A.; Rosewall C. F.; Swartz R. D.; Michael F. E. Mechanism of N-Fluorobenzenesulfonimide Promoted Diamination and Carboamination Reactions: Divergent Reactivity of a Pd(IV) Species. J. Am. Chem. Soc. 2009, 131, 15945–15951. 10.1021/ja906915w. [DOI] [PubMed] [Google Scholar]; f Rosewall C. F.; Sibbald P. A.; Liskin D. V.; Michael F. E. Palladium-Catalyzed Carboamination of Alkenes Promoted by N-Fluorobenzenesulfonimide via C–H Activation of Arenes. J. Am. Chem. Soc. 2009, 131, 9488–9489. 10.1021/ja9031659. [DOI] [PubMed] [Google Scholar]
- a Powers D. C.; Ritter T. Bimetallic Redox Synergy in Oxidative Palladium Catalysis. Acc. Chem. Res. 2012, 45, 840–850. 10.1021/ar2001974. [DOI] [PubMed] [Google Scholar]; b Powers D. C.; Xiao D. Y.; Geibel M. A. L.; Ritter T. On the Mechanism of Palladium-Catalyzed Aromatic C–H Oxidation. J. Am. Chem. Soc. 2010, 132, 14530–14536. 10.1021/ja1054274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Livendahl M.; Echavarren A. M. Palladium-Catalyzed Arylation Reactions: A Mechanistic Perspective. Isr. J. Chem. 2010, 50, 630–651. 10.1002/ijch.201000040. [DOI] [Google Scholar]; b Cárdenas D. J.; Martín-Matute B.; Echavarren A. M. Aryl Transfer between Pd(II) Centers or Pd(IV) Intermediates in Pd-Catalyzed Domino Reactions. J. Am. Chem. Soc. 2006, 128, 5033–5040. 10.1021/ja056661j. [DOI] [PubMed] [Google Scholar]
- Davidson J. M.; Triggs C. Reaction of metal ion complexes with hydrocarbons. Part I. ‘Palladation’ and some other new electrophilic substitution reactions. The preparation of palladium(I). J. Chem. Soc. A 1968, 0, 1324–1330. 10.1039/J19680001324. [DOI] [Google Scholar]
- Reports that provide evidence for Pd-to-Pd transmetalation:; a Casado A. L.; Casares J. A.; Espinet P. An aryl exchange reaction with full retention of configuration of the complexes: Mechanism of the aryl exchange between [PdR2L2] complexes in chloroform (R = Pentahalophenyl, L = Thioether). Organometallics 1997, 16, 5730–5736. 10.1021/om970721f. [DOI] [Google Scholar]; b Ozawa F.; Fujimori M.; Yamamoto T.; Yamamoto A. Mechanism of the reaction of trans-bis(diethylphenylphosphine) di-m-tolylpalladium(II) with methyl iodide affording m-xylene. Evidence for a reductive elimination process involving the intermolecular exchange of organic groups. Organometallics 1986, 5, 2144–2149. 10.1021/om00141a036. [DOI] [Google Scholar]
- Suzaki Y.; Osakada K. Cyclization of Dinuclear Aryl- and Aroylpalladium Complexes with the Metal Centers Tethered by an Oligo(ethylene oxide) Chain. Intramolecular Transmetalation of the Cationic Dinuclear Arylpalladium Complexes. Organometallics 2003, 22, 2193–2195. 10.1021/om030035i. [DOI] [Google Scholar]
- Wang D.; Izawa Y.; Stahl S. S. Pd-catalyzed aerobic oxidative coupling of arenes: evidence for transmetalation between two Pd(II)-aryl intermediates. J. Am. Chem. Soc. 2014, 136, 9914–9917. 10.1021/ja505405u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C–H activation:; a Guillemard L.; Kaplaneris N.; Ackermann L.; Johansson M. J. Late-stage C–H functionalization offers new opportunities in drug discovery. Nat. Rev. Chem. 2021, 5, 522–545. 10.1038/s41570-021-00300-6. [DOI] [PubMed] [Google Scholar]; b Rogge T.; Kaplaneris N.; Chatani N.; Kim J.; Chang S.; Punji B.; Schafer L. L.; Musaev D. G.; Wencel-Delord J.; Roberts C. A. C–H activation. Nat. Rev. Methods Primers 2021, 1, 43. [Google Scholar]; c Rej S.; Ano Y.; Chatani N. Bidentate directing groups: an efficient tool in C–H bond functionalization chemistry for the expedient construction of C–C bonds. Chem. Rev. 2020, 120, 1788–1887. 10.1021/acs.chemrev.9b00495. [DOI] [PubMed] [Google Scholar]; d He J.; Wasa M.; Chan K. S.; Shao Q.; Yu J.-Q. Palladium-catalyzed transformations of alkyl C–H bonds. Chem. Rev. 2017, 117, 8754–8786. 10.1021/acs.chemrev.6b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Gensch T.; Hopkinson M.; Glorius F.; Wencel-Delord J. Mild metal-catalyzed C–H activation: examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. 10.1039/C6CS00075D. [DOI] [PubMed] [Google Scholar]; f Daugulis O.; Roane J.; Tran L. D. Bidentate, monoanionic auxiliary-directed functionalization of carbon–hydrogen bonds. Acc. Chem. Res. 2015, 48, 1053–1064. 10.1021/ar5004626. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Ackermann L.; Vicente R.; Kapdi A. R. Transition-metal-catalyzed direct arylation of (hetero) arenes by C–H bond cleavage. Angew. Chem., Int. Ed. 2009, 48, 9792–9826. 10.1002/anie.200902996. [DOI] [PubMed] [Google Scholar]; h Bergman R. G. C–H activation. Nature 2007, 446, 391–393. 10.1038/446391a. [DOI] [PubMed] [Google Scholar]
- Reviews on organic electrosynthesis:; a Malapit C. A.; Prater M. B.; Cabrera-Pardo J. R.; Li M.; Pham T. D.; McFadden T. P.; Blank S.; Minteer S. D. Advances on the Merger of Electrochemistry and Transition Metal Catalysis for Organic Synthesis. Chem. Rev. 2022, 122, 3180–3218. 10.1021/acs.chemrev.1c00614. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Novaes L. F. T.; Liu J.; Shen Y.; Lu L.; Meinhardt J. M.; Lin S. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 2021, 50, 7941–8002. 10.1039/D1CS00223F. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ma C.; Fang P.; Liu D.; Jiao K.-J.; Gao P.-S.; Qiu H.; Mei T.-S. Transition metal-catalyzed organic reactions in undivided electrochemical cells. Chem. Sci. 2021, 12, 12866–12873. 10.1039/D1SC04011A. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Chen Z.; Villani E.; Inagi S. Recent progress in bipolar electropolymerization methods toward one-dimensional conducting polymer structures. Curr. Opin Electrochem 2021, 28, 100702 10.1016/j.coelec.2021.100702. [DOI] [Google Scholar]; e Zhu C.; Ang N. W.; Meyer T. H.; Qiu Y.; Ackermann L. Organic electrochemistry: molecular syntheses with potential. ACS Cent. Sci. 2021, 7, 415–431. 10.1021/acscentsci.0c01532. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Liu J.; Lu L.; Wood D.; Lin S. New redox strategies in organic synthesis by means of electrochemistry and photochemistry. ACS Cent. Sci. 2020, 6, 1317–1340. 10.1021/acscentsci.0c00549. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Siu J. C.; Fu N.; Lin S. Catalyzing electrosynthesis: a homogeneous electrocatalytic approach to reaction discovery. Acc. Chem. Res. 2020, 53, 547–560. 10.1021/acs.accounts.9b00529. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Xiong P.; Xu H.-C. Chemistry with electrochemically generated N-centered radicals. Acc. Chem. Res. 2019, 52, 3339–3350. 10.1021/acs.accounts.9b00472. [DOI] [PubMed] [Google Scholar]; i Yamamoto K.; Kuriyama M.; Onomura O. Anodic oxidation for the stereoselective synthesis of heterocycles. Acc. Chem. Res. 2020, 53, 105–120. 10.1021/acs.accounts.9b00513. [DOI] [PubMed] [Google Scholar]; j Kärkäs M. D. Electrochemical strategies for C–H functionalization and C–N bond formation. Chem. Soc. Rev. 2018, 47, 5786–5865. 10.1039/C7CS00619E. [DOI] [PubMed] [Google Scholar]; k Moeller K. D. Using physical organic chemistry to shape the course of electrochemical reactions. Chem. Rev. 2018, 118, 4817–4833. 10.1021/acs.chemrev.7b00656. [DOI] [PubMed] [Google Scholar]; l Tang S.; Liu Y.; Lei A. Electrochemical oxidative cross-coupling with hydrogen evolution: a green and sustainable way for bond formation. Chem. 2018, 4, 27–45. 10.1016/j.chempr.2017.10.001. [DOI] [Google Scholar]; m Yan M.; Kawamata Y.; Baran P. S. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 2017, 117, 13230–13319. 10.1021/acs.chemrev.7b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Francke R.; Little R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 2014, 43, 2492–2521. 10.1039/c3cs60464k. [DOI] [PubMed] [Google Scholar]
- Reviews on electrocatalytic C–H activation:; a Gandeepan P.; Finger L. H.; Meyer T. H.; Ackermann L. 3d metallaelectrocatalysis for resource economical syntheses. Chem. Soc. Rev. 2020, 49, 4254–4272. 10.1039/D0CS00149J. [DOI] [PubMed] [Google Scholar]; b Jiao K.-J.; Xing Y.-K.; Yang Q.-L.; Qiu H.; Mei T.-S. Site-selective C–H functionalization via synergistic use of electrochemistry and transition metal catalysis. Acc. Chem. Res. 2020, 53, 300–310. 10.1021/acs.accounts.9b00603. [DOI] [PubMed] [Google Scholar]; c Meyer T. H.; Choi I.; Tian C.; Ackermann L. Powering the future: how can electrochemistry make a difference in organic synthesis?. Chem 2020, 6, 2484–2496. 10.1016/j.chempr.2020.08.025. [DOI] [Google Scholar]; d Meyer T. H.; Finger L. H.; Gandeepan P.; Ackermann L. Resource economy by metallaelectrocatalysis: merging electrochemistry and CH activation. Trends Chem. 2019, 1, 63–76. 10.1016/j.trechm.2019.01.011. [DOI] [Google Scholar]; e Ackermann L. Metalla-electrocatalyzed C–H activation by earth-abundant 3d metals and beyond. Acc. Chem. Res. 2020, 53, 84–104. 10.1021/acs.accounts.9b00510. [DOI] [PubMed] [Google Scholar]; f Sauermann N.; Meyer T. H.; Qiu Y.; Ackermann L. Electrocatalytic C–H Activation. ACS Catal. 2018, 8, 7086–7103. 10.1021/acscatal.8b01682. [DOI] [Google Scholar]; g Ma C.; Fang P.; Mei T.-S. Recent advances in C–H functionalization using electrochemical transition metal catalysis. ACS Catal. 2018, 8, 7179–7189. 10.1021/acscatal.8b01697. [DOI] [Google Scholar]
- Electrochemical transition metal catalyzed C–H activation:; a Choi I.; Messinis A. M.; Hou X.; Ackermann L. A Strategy for Site- and Chemoselective C–H Alkenylation through Osmaelectrooxidative Catalysis. Angew. Chem., Int. Ed. 2021, 60, 27005–27012. 10.1002/anie.202110616. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Massignan L.; Zhu C.; Hou X.; Oliveira J. C. A.; Salamé A.; Ackermann L. Manganaelectro-Catalyzed Azine C–H Arylations and C–H Alkylations by Assistance of Weakly Coordinating Amides. ACS Catal. 2021, 11, 11639–11649. 10.1021/acscatal.1c02516. [DOI] [Google Scholar]; c Zhu C.; Stangier M.; Oliveira J. C. A.; Massignan L.; Ackermann L. Iron-Electrocatalyzed C–H Arylations: Mechanistic Insights into Oxidation-Induced Reductive Elimination for Ferraelectrocatalysis. Chem. - Eur. J. 2019, 25, 16382–16389. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tian C.; Dhawa U.; Scheremetjew A.; Ackermann L. Cupraelectro-Catalyzed Alkyne Annulation: Evidence for Distinct C–H Alkynylation and Decarboxylative C–H/C–C Manifolds. ACS Catal. 2019, 9, 7690–7696. 10.1021/acscatal.9b02348. [DOI] [Google Scholar]; e Qiu Y.; Kong W.-J.; Struwe J.; Sauermann N.; Rogge T.; Scheremetjew A.; Ackermann L. Electrooxidative Rhodium-Catalyzed C–H/C–H Activation: Electricity as Oxidant for Cross-Dehydrogenative Alkenylation. Angew. Chem., Int. Ed. 2018, 57, 5828–5832. 10.1002/anie.201803342. [DOI] [PubMed] [Google Scholar]; f Qiu Y.; Tian C.; Massignan L.; Rogge T.; Ackermann L. Electrooxidative Ruthenium-Catalyzed C–H/O–H Annulation by Weak O-Coordination. Angew. Chem., Int. Ed. 2018, 57, 5818–5822. 10.1002/anie.201802748. [DOI] [PubMed] [Google Scholar]; g Qiu Y.; Stangier M.; Meyer T. H.; Oliveira J. C. A.; Ackermann L. Iridium-Catalyzed Electrooxidative C–H Activation by Chemoselective Redox-Catalyst Cooperation. Angew. Chem., Int. Ed. 2018, 57, 14179–14183. 10.1002/anie.201809611. [DOI] [PubMed] [Google Scholar]; h Zhang S.-K.; Samanta R. C.; Sauermann N.; Ackermann L. Nickel-Catalyzed Electrooxidative C–H Amination: Support for Nickel(IV). Chem. - Eur. J. 2018, 24, 19166–19170. [DOI] [PubMed] [Google Scholar]; i Sauermann N.; Meyer T. H.; Tian C.; Ackermann L. Electrochemical Cobalt-Catalyzed C–H Oxygenation at Room Temperature. J. Am. Chem. Soc. 2017, 139, 18452–18455. 10.1021/jacs.7b11025. [DOI] [PubMed] [Google Scholar]
- Electrochemical palladium catalyzed C–H activation:; a Yang Q.-L.; Li C.-Z.; Zhang L.-W.; Li Y.-Y.; Tong X.; Wu X.-Y.; Mei T.-S. Palladium-catalyzed electrochemical C–H alkylation of arenes. Organometallics 2019, 38, 1208–1212. 10.1021/acs.organomet.8b00550. [DOI] [Google Scholar]; b Yang Q.-L.; Li Y.-Q.; Ma C.; Fang P.; Zhang X.-J.; Mei T.-S. Palladium-Catalyzed C(sp3)–H Oxygenation via Electrochemical Oxidation. J. Am. Chem. Soc. 2017, 139, 3293–3298. 10.1021/jacs.7b01232. [DOI] [PubMed] [Google Scholar]; c Konishi M.; Tsuchida K.; Sano K.; Kochi T.; Kakiuchi F. Palladium-catalyzed ortho-selective C–H chlorination of benzamide derivatives under anodic oxidation conditions. J. Org. Chem. 2017, 82, 8716–8724. 10.1021/acs.joc.7b01137. [DOI] [PubMed] [Google Scholar]; d Ma C.; Zhao C.-Q.; Li Y.-Q.; Zhang L.-P.; Xu X.-T.; Zhang K.; Mei T.-S. Palladium-catalyzed C–H activation/C–C cross-coupling reactions via electrochemistry. Chem. Commun. 2017, 53, 12189–12192. 10.1039/C7CC07429H. [DOI] [PubMed] [Google Scholar]; e Dudkina Y. B.; Mikhaylov D. Y.; Gryaznova T. V.; Tufatullin A. I.; Kataeva O. N.; Vicic D. A.; Budnikova Y. H. Electrochemical ortho functionalization of 2-phenylpyridine with perfluorocarboxylic acids catalyzed by palladium in higher oxidation states. Organometallics 2013, 32, 4785–4792. 10.1021/om400492g. [DOI] [Google Scholar]; f Kakiuchi F.; Kochi T.; Mutsutani H.; Kobayashi N.; Urano S.; Sato M.; Nishiyama S.; Tanabe T. Palladium-catalyzed aromatic C–H halogenation with hydrogen halides by means of electrochemical oxidation. J. Am. Chem. Soc. 2009, 131, 11310–11311. 10.1021/ja9049228. [DOI] [PubMed] [Google Scholar]; g Amatore C.; Cammoun C.; Jutand A. Electrochemical Recycling of Benzoquinone in the Pd/Benzoquinone-Catalyzed Heck-Type Reactions from Arenes. Adv. Synth. Catal. 2007, 349, 292–296. 10.1002/adsc.200600389. [DOI] [Google Scholar]
- Dhawa U.; Tian C.; Wdowik T.; Oliveira J. C. A.; Hao J.; Ackermann L. Enantioselective Pallada-Electrocatalyzed C–H Activation by Transient Directing Groups: Expedient Access to Helicenes. Angew. Chem., Int. Ed. 2020, 59, 13451–13457. 10.1002/anie.202003826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z.; Dhawa U.; Hou X.; Surke M.; Yuan B.; Li S.-W.; Liou Y.-C.; Johansson M. J.; Xu L.-C.; Chao C.-H.; et al. Electrocatalyzed direct arene alkenylations without directing groups for selective late-stage drug diversification. Nat. Commun. 2023, 14, 4224 10.1038/s41467-023-39747-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonogashira K. Development of Pd–Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem. 2002, 653, 46–49. 10.1016/S0022-328X(02)01158-0. [DOI] [Google Scholar]
- a Motiwala H. F.; Armaly A. M.; Cacioppo J. G.; Coombs T. C.; Koehn K. R. K.; Norwood V. M.; Aube J. HFIP in Organic Synthesis. Chem. Rev. 2022, 122, 12544–12747. 10.1021/acs.chemrev.1c00749. [DOI] [PubMed] [Google Scholar]; b Berkessel A.; Adrio J. A. Dramatic Acceleration of Olefin Epoxidation in Fluorinated Alcohols: Activation of Hydrogen Peroxide by Multiple H-Bond Networks. J. Am. Chem. Soc. 2006, 128, 13412–13420. 10.1021/ja0620181. [DOI] [PubMed] [Google Scholar]; c Berkessel A.; Adrio J. A.; Hüttenhain D.; Neudörfl J. M. Unveiling the “Booster Effect” of Fluorinated Alcohol Solvents: Aggregation-Induced Conformational Changes and Cooperatively Enhanced H-Bonding. J. Am. Chem. Soc. 2006, 128, 8421–8426. 10.1021/ja0545463. [DOI] [PubMed] [Google Scholar]
- Sun G.-Q.; Yu P.; Zhang W.; Zhang W.; Wang Y.; Liao L.-L.; Zhang Z.; Li L.; Lu Z.; Yu D.-G.; Lin S. Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations. Nature 2023, 615, 67–72. 10.1038/s41586-022-05667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kong W.-J.; Reil M.; Feng L.; Li M.-B.; Jan E. B. Aerobic Heterogeneous Palladium-Catalyzed Oxidative Allenic C–H Arylation: Benzoquinone as a Direct Redox Mediator between O2 and Pd. CCS Chem. 2021, 3, 1127–1137. 10.31635/ccschem.021.202100816. [DOI] [Google Scholar]; b Salazar C. A.; Flesch K. N.; Haines B. E.; Zhou P. S.; Musaev D. G.; Stahl S. S. Tailored quinones support high-turnover Pd catalysts for oxidative C–H arylation with O2. Science 2020, 370, 1454–1460. 10.1126/science.abd1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Choudhary S.; Cannas D. M.; Wheatley M.; Larrosa I. A manganese(I)tricarbonyl-catalyst for near room temperature alkene and alkyne hydroarylation. Chem. Sci. 2022, 13, 13225–13230. 10.1039/D2SC04295A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Burés J. A Simple Graphical Method to Determine the Order in Catalyst. Angew. Chem., Int. Ed. 2016, 55, 2028–2031. 10.1002/anie.201508983. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Burés J. Variable Time Normalization Analysis: General Graphical Elucidation of Reaction Orders from Concentration Profiles. Angew. Chem., Int. Ed. 2016, 55, 16084–16087. 10.1002/anie.201609757. [DOI] [PubMed] [Google Scholar]; d Blackmond D. G. Kinetic Profiling of Catalytic Organic Reactions as a Mechanistic Tool. J. Am. Chem. Soc. 2015, 137, 10852–10866. 10.1021/jacs.5b05841. [DOI] [PubMed] [Google Scholar]; e Baxter R. D.; Sale D.; Engle K. M.; Yu J. Q.; Blackmond D. G. Mechanistic rationalization of unusual kinetics in Pd-catalyzed C–H olefination. J. Am. Chem. Soc. 2012, 134, 4600–4606. 10.1021/ja207634t. [DOI] [PubMed] [Google Scholar]
- Deprez N. R.; Sanford M. S. Synthetic and Mechanistic Studies of Pd-Catalyzed C–H Arylation with Diaryliodonium Salts: Evidence for a Bimetallic High Oxidation State Pd Intermediate. J. Am. Chem. Soc. 2009, 131, 11234–11241. 10.1021/ja904116k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rosner T.; Le Bars J.; Pfaltz A.; Blackmond D. G. Kinetic studies of Heck coupling reactions using palladacycle catalysts: experimental and kinetic modeling of the role of dimer species. J. Am. Chem. Soc. 2001, 123, 1848–1855. 10.1021/ja003191e. [DOI] [PubMed] [Google Scholar]; b Rosner T.; Pfaltz A.; Blackmond D. G. Observation of unusual kinetics in Heck reactions of aryl halides: the role of non-steady-state catalyst concentration. J. Am. Chem. Soc. 2001, 123, 4621–4622. 10.1021/ja005872f. [DOI] [PubMed] [Google Scholar]
- a Altus K. M.; Love J. A. The continuum of carbon–hydrogen (C–H) activation mechanisms and terminology. Commun. Chem. 2021, 4, 173. 10.1038/s42004-021-00611-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bauer M.; Cadge J.; Davies D.; Durand D. J.; Eisenstein O.; Ess D.; Fey N.; Gallarati S.; George M.; Hamilton A.; et al. Computational and theoretical approaches for mechanistic understanding: general discussion. Faraday Discuss. 2019, 220, 464–488. 10.1039/C9FD90073J. [DOI] [PubMed] [Google Scholar]; c Simmons E. M.; Hartwig J. F. On the Interpretation of Deuterium Kinetic Isotope Effects in C–H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem., Int. Ed. 2012, 51, 3066–3072. 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- Váňa J.; Terencio T.; Petrović V.; Tischler O.; Novák Z.; Roithová J. Palladium-Catalyzed C–H Activation: Mass Spectrometric Approach to Reaction Kinetics in Solution. Organometallics 2017, 36, 2072–2080. 10.1021/acs.organomet.6b00960. [DOI] [Google Scholar]
- Rauf W.; Thompson A. L.; Brown J. M. Anilide activation of adjacent C–H bonds in the palladium-catalysed Fujiwara–Moritani reaction. Dalton Trans. 2010, 39, 10414–10421. 10.1039/c0dt00378f. [DOI] [PubMed] [Google Scholar]
- Ben-Tal Y.; Boaler P. J.; Dale H. J. A.; Dooley R. E.; Fohn N. A.; Gao Y.; García-Domínguez A.; Grant K. M.; Hall A. M. R.; Hayes H. L. D.; et al. Mechanistic analysis by NMR spectroscopy: A users guide. Prog. Nucl. Magn. Reson. Spectrosc. 2022, 129, 28–106. 10.1016/j.pnmrs.2022.01.001. [DOI] [PubMed] [Google Scholar]
- Bowring M. A.; Bergman R. G.; Tilley T. D. Pt-Catalyzed C–C Activation Induced by C–H Activation. J. Am. Chem. Soc. 2013, 135, 13121–13128. 10.1021/ja406260j. [DOI] [PubMed] [Google Scholar]
- a Yang Q.-L.; Li C.-Z.; Zhang L.-W.; Li Y.-Y.; Tong X.; Wu X.-Y.; Mei T.-S. Palladium-Catalyzed Electrochemical C–H Alkylation of Arenes. Organometallics 2019, 38, 1208–1212. 10.1021/acs.organomet.8b00550. [DOI] [Google Scholar]; b Li Y.-Q.; Yang Q.-L.; Fang P.; Mei T.-S.; Zhang D. Palladium-Catalyzed C(sp2)–H Acetoxylation via Electrochemical Oxidation. Org. Lett. 2017, 19, 2905–2908. 10.1021/acs.orglett.7b01138. [DOI] [PubMed] [Google Scholar]
- a Oeschger R. J.; Bissig R.; Chen P. Model Compounds for Intermediates and Transition States in Sonogashira and Negishi Coupling: d(8)-d(10) Bonds in Large Heterobimetallic Complexes Are Weaker than Computational Chemistry Predicts. J. Am. Chem. Soc. 2022, 144, 10330–10343. 10.1021/jacs.2c01641. [DOI] [PubMed] [Google Scholar]; b Berry J. F.; Lu C. C. Metal-Metal Bonds: From Fundamentals to Applications. Inorg. Chem. 2017, 56, 7577–7581. 10.1021/acs.inorgchem.7b01330. [DOI] [PubMed] [Google Scholar]; c Oeschger R. J.; Chen P. Structure and Gas-Phase Thermochemistry of a Pd/Cu Complex: Studies on a Model for Transmetalation Transition States. J. Am. Chem. Soc. 2017, 139, 1069–1072. 10.1021/jacs.6b12152. [DOI] [PubMed] [Google Scholar]; d Paenurk E.; Gershoni-Poranne R.; Chen P. Trends in Metallophilic Bonding in Pd–Zn and Pd–Cu Complexes. Organometallics 2017, 36, 4854–4863. 10.1021/acs.organomet.7b00748. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.