

Abstract
Impaired glucose tolerance (IGT) and β-cell dysfunction in insulin resistance associated with obesity lead to type 2 diabetes (T2D). Glucose-stimulated insulin secretion (GSIS) from β-cells occurs via a canonical pathway that involves glucose metabolism, ATP generation, inactivation of KATP channels, plasma membrane depolarization, and increases in cytosolic concentrations of [Ca2+]c. However, optimal insulin secretion requires amplification of GSIS by increases in cyclic adenosine monophosphate (cAMP) signaling. The cAMP effectors protein kinase A (PKA) and exchange factor activated by cyclic-AMP (Epac) regulate membrane depolarization, gene expression, and trafficking and fusion of insulin granules to the plasma membrane for amplifying GSIS. The widely recognized lipid signaling generated within β-cells by the β-isoform of Ca2+-independent phospholipase A2 enzyme (iPLA2β) participates in cAMP-stimulated insulin secretion (cSIS). Recent work has identified the role of a G-protein coupled receptor (GPCR) activated signaling by the complement 1q like-3 (C1ql3) secreted protein in inhibiting cSIS. In the IGT state, cSIS is attenuated, and the β-cell function is reduced. Interestingly, while β-cell-specific deletion of iPLA2β reduces cAMP-mediated amplification of GSIS, the loss of iPLA2β in macrophages (MØ) confers protection against the development of glucose intolerance associated with diet-induced obesity (DIO). In this article, we discuss canonical (glucose and cAMP) and novel noncanonical (iPLA2β and C1ql3) pathways and how they may affect β-cell (dys)function in the context of impaired glucose intolerance associated with obesity and T2D. In conclusion, we provide a perspective that in IGT states, targeting noncanonical pathways along with canonical pathways could be a more comprehensive approach for restoring β-cell function in T2D.
Introduction
Glucose is the major in vivo regulator of insulin secretion from the pancreatic islet β-cells. Elevations in circulating levels of glucose prompt increased transport of glucose into the β-cells, where it enters the glycolytic pathway. Second messengers derived during the metabolism of glucose signal through multiple pathways to trigger the exocytosis of insulin from β-cells to affect a return to a euglycemic state. With obesity and/or type 2 diabetes (T2D), there is a progressive defect in β-cell function to the extent that insulin release is insufficient to control blood glucose levels optimally. A primary defect in these individuals is the inability of the β-cells to respond to glucose.
Molecular mechanisms contributing to glucose-stimulated insulin secretion (GSIS) from β-cells have been described in the context of well-characterized canonical pathways. These are initiated by the transport of glucose into the β-cells, followed by the metabolism of glucose, leading to adenosine 5′-triphosphate (ATP) generation, inactivation of the KATP channels, and increases in [Ca2+]c, triggering insulin exocytosis. It has long been recognized that to achieve optimal insulin secretion, cyclic adenosine monophosphate (cAMP) generation and cAMP-mediated activation of downstream pathways that transduce signals to the plasma membrane and nucleus levels are integral to the amplification of GSIS. As such, cAMP might be regarded as a nexus through which various factors can modulate insulin secretion, ipso facto, β-cell function.
Ongoing studies of β-cell function during the healthy and diseased states continue to reveal additional pathways or modulators of GSIS. These may be considered to encompass noncanonical pathways that have important roles in the overall secretory function of β-cells. It has long been recognized that lipid signaling generated within the β-cells through activation of the β-isoform of Ca2+-independent phospholipase A2 enzyme (iPLA2β) participates in GSIS. More recent work has provided evidence for a reciprocal impact of a β-cell secreted protein, complement 1q like-3 (C1ql3), on β-cell function. Intriguingly, both iPLA2β and C1ql3 signaling intersect with the cAMP amplification pathway. In this article, we discuss canonical and novel noncanonical pathways involving iPLA2β and C1ql3 and suggest how they may converge to affect β-cell (dys)function and modulate glucose intolerance in obesity and T2D.
Canonical Mechanism for Insulin Secretion
Biphasic GSIS
In response to elevations in circulating glucose concentrations, pancreatic islet β-cells secrete insulin. Glucose enters the cells via the plasma membrane transporter Glut2 in rodents or Glut1 in humans. It is rapidly metabolized by oxidative glycolysis to increase the ratio of ATP to adenosine diphosphate (ADP). Elevations in the intracellular ATP levels promote the inhibition of ATP-sensitive potassium channels (KATP), leading to depolarization of the plasma membrane. Consequently, voltage-dependent Ca2+ channels (VDCC) open to permit Ca2+ influx, facilitating the formation of the soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) protein complex-dependent fusion of insulin granules to the plasma membrane. This triggering of the exocytotic process releases a short but robust burst of insulin for the first 10 min of glucose stimulation (16, 158, 395). This is referred to as the early phase of the biphasic glucose response. During this early phase, the fusion of insulin granules primarily occurs from the readily releasable pool (RRP) of predocked granules located within 100 to 200nm of the plasma membrane (59, 289, 372). Subsequently, a sustained second phase of insulin secretion persists for several hours with relatively reduced insulin secretion rates. During this sustained phase, insulin granules from the storage pool present in the cytoplasm distal to the plasma membrane are recruited and undergo plasma membrane fusion (372). These granules, referred to as newcomers, undergo fusion mainly via two modes of action: “short dock” or “no dock.” The short-dock newcomer insulin granules traffic to the plasma membrane and undergo docking before fusion, whereas no-dock newcomer insulin granules are “fast-release” and do not require docking before undergoing fusion. In addition to the predocked granules, the fusion of newcomer granules contributes partly to the insulin released in the early phase (238, 251, 311). The burst of insulin released in the early phase is critical for priming the skeletal muscle for sustained, long-term insulin-dependent glucose clearance. During the development of impaired glucose tolerance (IGT), typically associated with insulin resistance due to obesity, a reduction of early-phase insulin secretion occurs. In T2D, insulin secretion from both early and sustained phases is severely reduced. Overall, reduced GSIS is associated with IGT (18, 148, 186, 245, 250, 261, 311, 372).
Triggering and amplifying pathways of GSIS
Glucose metabolism generates signals for triggering and amplifying pathways in insulin secretion (Figure 1). The triggering pathway involves increases in [Ca2+]c, which directly contribute to the exocytosis of insulin granules (151, 153, 341). Multiple factors or mechanisms can contribute to the increases in β-cell [Ca2+]c (34, 39, 87, 95, 268, 290, 295, 387). However, the best elucidated mechanism for the influx of Ca2+ is the opening of the VDCC in response to depolarization of the plasma membrane potential achieved by the closure of KATP channels. The KATP channels are complex structures comprising pore-forming Kir6.2 and regulatory sulfonylurea receptor 1 (SUR1) subunits (198, 223). Nutrient signaling effectors such as ATP generated during glycolysis or nonnutrient insulin secretagogues such as sulfonylureas (SUs) bind to the SUR1 or Kir6.2, causing the closure of KATP channels, which leads to membrane depolarization, increases in Ca2+ influx, and triggering of insulin secretion.
Figure 1. Triggering and amplifying pathways of glucose in increasing insulin secretion (GSIS) from β-cells.
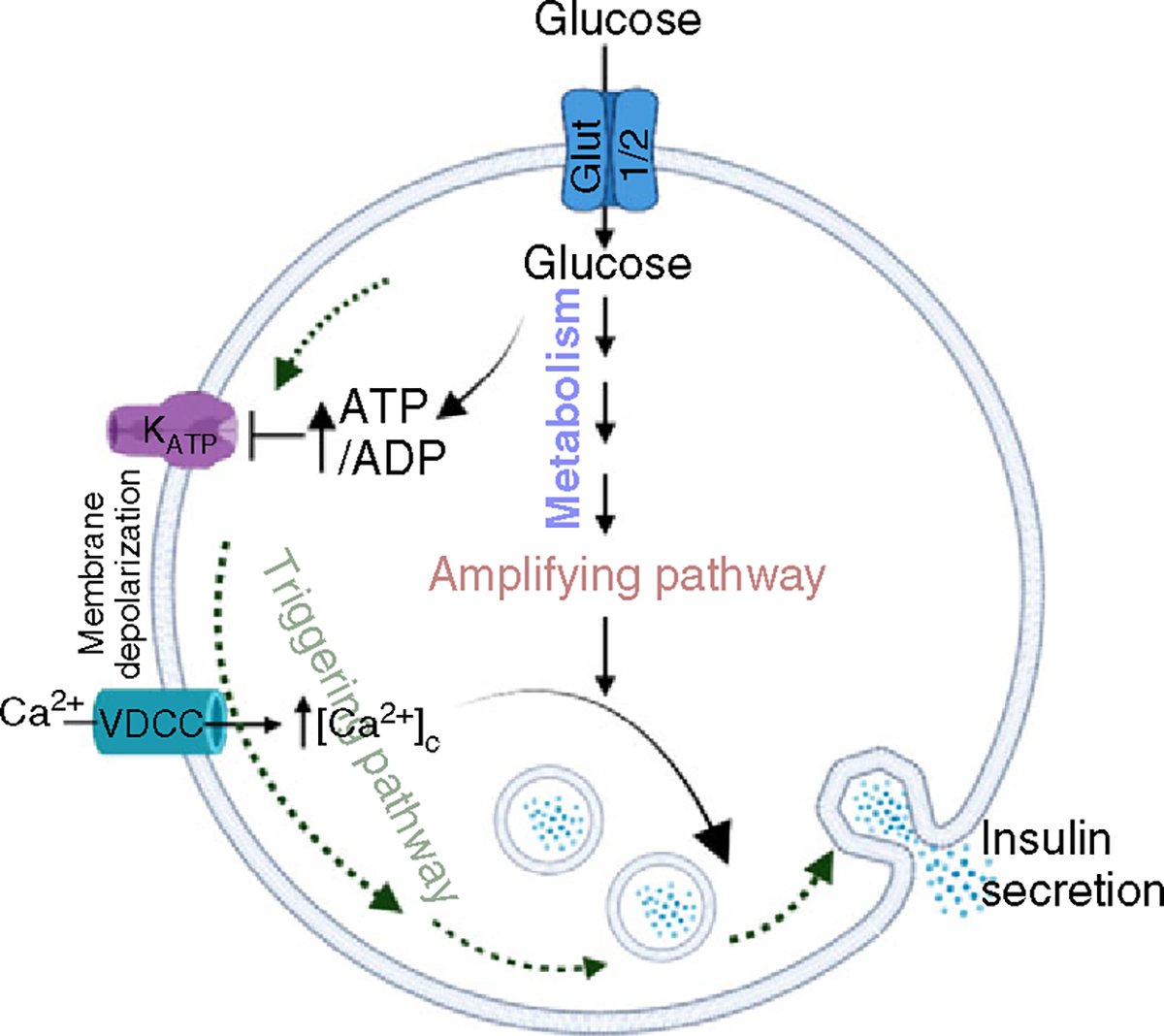
Glucose enters the β-cells via the Glut2 in rodents and Glut1 in humans transporters and undergoes metabolism by the glycolytic pathway. The increase in the ATP/ADP ratio causes inhibition of the KATP channels. This causes the depolarization of the plasma membrane, causing the influx of Ca2+ through VDCC. The subsequent increase in [Ca2+]c facilitates the fusion of insulin granules to the plasma membrane. This step is called the triggering pathway. The metabolism of glucose leads to generation of metabolites or second messengers that amplify the ability of [Ca2+]c to increase insulin secretion. This is called the amplifying pathway of GSIS.
While there is ample evidence for the role of KATP in GSIS, the ability of glucose to amplify insulin secretion was demonstrated using a pharmacological bypass of the KATP channels when [Ca2+]c was clamped at an elevated level. This was achieved by treating islets with diazoxide in combination with KCl. Diazoxide binds directly to SUR1 to keep the KATP channels open, while KCl affects the depolarization of the plasma membrane. In this experimental paradigm, even when KATP channels were held open by diazoxide, KCl treatment caused plasma membrane depolarization, leading to increased [Ca2+]c and insulin secretion. Adding glucose in the presence of KCl and diazoxide resulted in higher increases in insulin secretion with minimal or no further increases in [Ca2+]c compared to the KCl and diazoxide treatment (91, 92, 116). Another study (91, 92, 116) showed that treating islets with high concentrations of SUs (e.g., tolbutamide), which bind to and elicit the closure of KATP channels, caused the clamping of β-cell [Ca2+]c at elevated levels, leading to increases in insulin secretion. The addition of glucose caused further increases in insulin secretion without affecting [Ca2+]c levels. These studies demonstrated the presence of an amplifying pathway in GSIS that is independent of KATP channels and does not require further increases in [Ca2+]c, suggesting that the amplifying pathway essentially increases the efficacy of [Ca2+]c in insulin secretion. Henquin et al. (121) evaluated the physiological significance of the amplifying pathway by assessing insulin secretion in response to glucose where [Ca2+]c is allowed to fluctuate freely. Under these conditions, a sigmoidal relationship was observed between the [glucose]-dependent curves, where the insulin secretion-response curve was shifted to the right of [Ca2+]c. This implies that signals, in addition to [Ca2+]c, are also generated by glucose that contribute to insulin secretion (114, 121). Complementing these studies, islets from mice with deletion of SUR1 or Kir6.2 (inactivation of KATP channel) also exhibited plasma membrane depolarization and increases in [Ca2+]c and insulin secretion (231, 241, 279). Furthermore, plasma membrane depolarization-mediated increases in [Ca2+]c were observed upon the deletion of KATP channels, implicating a role of transducers other than the KATP channels in increasing [Ca2+]c and insulin secretion. The amplifying pathway contributes approximately 50% to GSIS (118) during the early and sustained phases of insulin secretion. While its physiological importance in mouse and human islets (337) is accepted, the underlying molecular mechanisms are not completely elucidated.
cAMP signaling in stimulus-coupled insulin secretion
In addition to an increase in [Ca2+]c, which is required for triggering insulin secretion, cAMP functions as a key second messenger to amplify GSIS. Increased β-cell production of cAMP facilitates the exocytosis of insulin in both the early and sustained phases of secretion (12, 257, 341) and contributes to whole-body glucose homeostasis (29, 75, 98, 126, 138, 173, 176, 177, 310, 311, 347). The major effects of cAMP in promoting insulin release are attributed to plasma membrane depolarization, [Ca2+]c signaling, recruitment of insulin granules to the plasma membrane, and activation of the exocytosis machinery (12) (Figure 2).
Figure 2. Cyclic AMP signaling amplifies GSIS in β-cells.
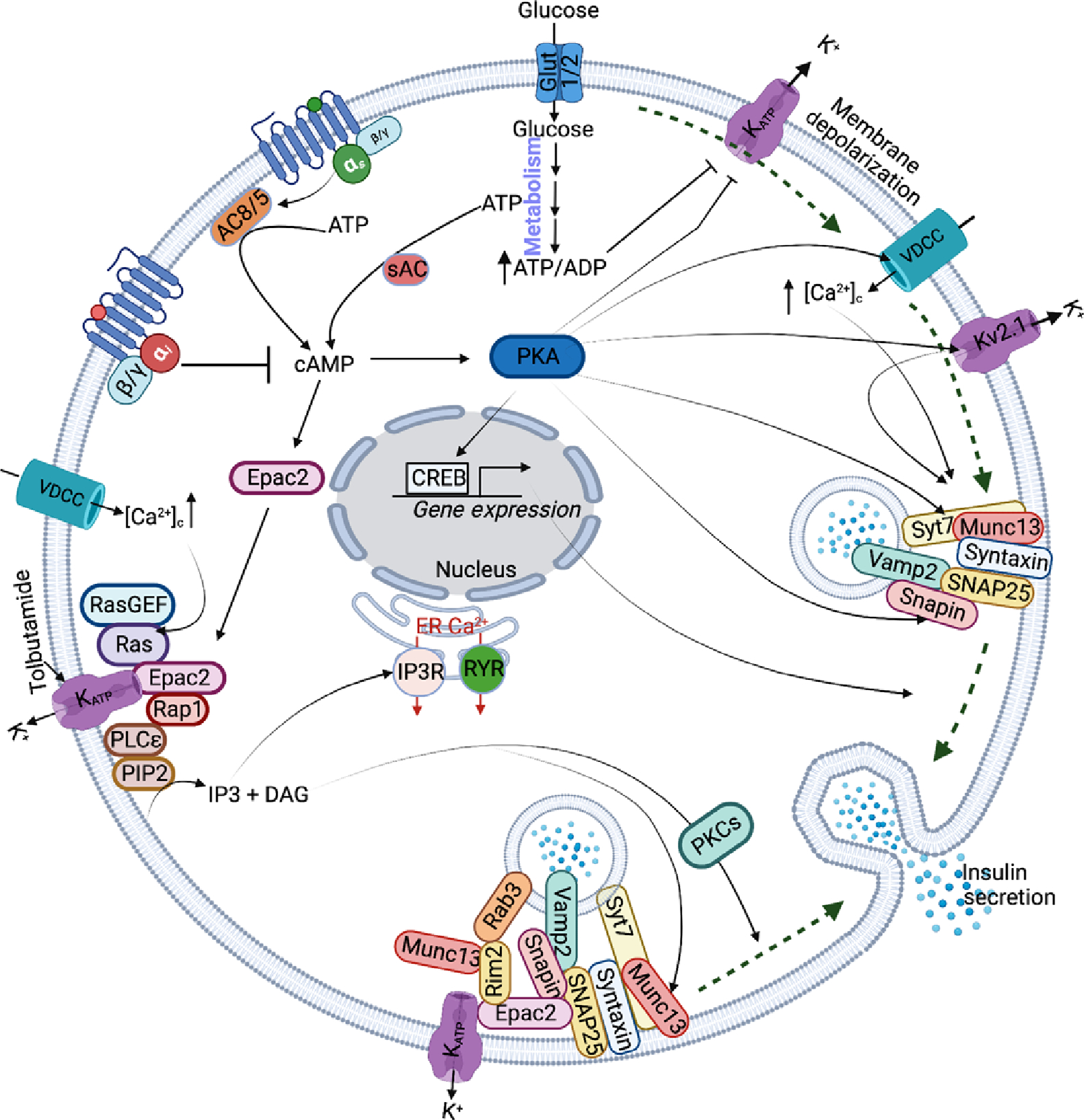
The green dotted line shows the triggering pathway of glucose in increasing insulin secretion. The activated Gs-coupled GPCR signaling increases cAMP levels thereby activating its downstream effectors Epac2 and PKA. Subsequently, PKA increases insulin secretion by modulating the function of VDCC, Kv2.1, KATP, Snapin, and Syt7, and nuclear translocation and activation of CREB. Epac2 binds KATP channels, causing activation of IP3 and DAG. The IP3 increases Ca2+ release from the ER, and DAG binds and activates the Munc13–1 priming factor and PKC to increase insulin secretion by facilitating the fusion of insulin granules to the plasma membrane.
The Gαs coupled G-protein coupled receptor (GPCR) agonists such as glucagon, glucose-dependent insulinotropic peptide (GIP), glucagon-like peptide-1 (GLP1), pituitary adenylyl cyclase (AC) activating polypeptide (PACAP), and adrenocorticotropic hormone (ACTH) increase GSIS by increasing cAMP via activation of ACs, which convert ATP to cAMP. Increased cAMP signaling accounts for approximately 60% to 70% of total insulin output from β-cells in response to oral glucose stimulation (75, 176). In contrast, reduced cAMP signaling and inhibition of insulin secretion by somatostatin, epinephrine (130), neuropeptide Y (NPY), and EP3 occur through the activation of Gi-coupled coupled receptors (GPCRs) (65, 259). Relatively less is known about GPCRs that inhibit cAMP production and GSIS. Kimple and Gannon recently assessed the role of cAMP-inhibitory (i.e., Gi-coupled) prostaglandin E2 (PGE2)-EP3 receptor (EP3) signaling, and they reported that PGE2 production and secretion, and EP3 expression are up-regulated in islets isolated from T2D mice and humans (174, 300). Consequentially, GSIS was mitigated, and the maximal potentiating effect of GLP1R ligands on the process was blunted. The EP3 receptor is among a small group of inhibitory GPCRs whose role in β-cells has been well-characterized (36, 37, 47, 174–176, 243, 293, 297, 298, 300, 352, 382). Additionally, evidence of increased cAMP levels in response to glucose was obtained based on studies where Ca2+ was clamped (75) or by using a fluorescence resonance energy transfer (FRET)-based cAMP indicator (172). Moreover, a requirement of cAMP in insulin secretion was demonstrated using a membrane-permeable antagonist of cAMP, para-acetoxybenzyl (pAB) ester prodrug (304). The mechanisms by which exposure of islets to high glucose concentrations promotes increases in cAMP levels and how increases in cAMP signaling via activation of soluble and membrane-bound ACs function in amplifying insulin secretion require further clarification.
The Gαs protein is critical for optimal β-cell function, and the ablation of Gαs in mice leads to impaired β-cell proliferation, reduced insulin gene expression, and glucose intolerance (389). It is known that Gαs activates ACs and potentiates both phases of insulin secretion (267). The ACs are localized in the membrane (AC1–AC9) and cytosol (referred to as soluble AC), AC10 (62, 88, 102, 179, 347). Most AC isoforms are expressed in pancreatic islets and insulinoma cells (102, 192), and their function is affected by [Ca2+] (50, 62, 365), providing a link between cAMP and Ca2+ signaling. The Ca2+-activated (AC1 (179), AC3 (308), AC6 (192), AC8 (62, 69)) and Ca2+-insensitive AC9 (256) have regulatory functions in rodent mouse β-cells (347). In human β-cells, AC5 was reported to regulate GSIS (124, 192, 397). The soluble AC is activated by HCO3− (derived from CO2 generated during glycolysis) and Ca2+ and acts as a potential sensor of ATP generated during glycolysis (128). Its activation has been reported to increase cAMP oscillations and is implicated in increasing insulin secretion in response to high glucose (75). Overall, activated ACs increase cAMP levels, which causes the amplification of insulin secretion. Another class of enzymes, the phosphodiesterases (PDEs), decrease intracellular (cAMP) by hydrolyzing cAMP to 5′ AMP, leading to a reduction in insulin secretion (reviewed in (88, 171, 346)).
Protein kinase A (PKA) is an effector of cAMP
The PKA is a well-characterized serine/threonine kinase and is comprised of two regulatory (RI and RII) and catalytic subunits each. The binding of cAMP to binding sites in the R subunits of PKA leads to conformational changes in the R subunits, causing the release of the catalytic subunits to confer kinase activity. Agonists of PKA amplify GSIS and tolbutamide-stimulated insulin secretion by augmenting the actions of [Ca2+]c on insulin exocytosis (120). The alpha (α) isoform of the type I R-subunit of PKA is abundantly expressed in mouse and human islets (136, 260, 319). Ablation of RIα from β-cells increased GSIS and glucose tolerance (136, 330), demonstrating its inhibitory function. Consistently, mutations identified in RIα caused increases in insulin release from human β-cells (330), illustrating its relevance in human β-cell function. Moreover, expressing a constitutively active catalytic subunit of PKA in mouse β-cells promotes increases in both phases of GSIS (157). Intriguingly, these genetic alterations of PKA that lead to increases in insulin secretion do not induce hypoglycemia, highlighting the importance of cAMP/PKA signaling and suggesting that it is a viable target for developing therapeutic drugs to improve insulin secretion in obesity and T2D (136).
The PKA-dependent phosphorylation of VDCC (12, 165), KATP (201), and Kv (191) channels affect stimulus-coupled insulin secretion. Voltage-dependent K+ currents are modulated by delayed rectifying K+ (Kv) channels that repolarize the action potential in the plasma membrane (39). The Kv2.1 and Kv2.2 are major channel types in both mouse and human β-cells, and their inactivation increases insulin secretion (143, 200). PKA promotes the inhibitory effects of cAMP on the Kv channels (191, 206). This prolongs plasma membrane depolarization, increasing [Ca2+]c and insulin secretion. In addition to Kv channels, PKA has been implicated in inhibiting the KATP channels in an ADP-dependent manner in response to high glucose (201); the cellular ADP levels are low in β-cells in response to high glucose. Under this condition, PKA binds to the Kir6.2 subunit at serine-372 or SUR1 subunit at serine-1448 to inhibit KATP channels (25), potentially aiding in plasma membrane depolarization and consequently increasing insulin secretion. PKA-induced phosphorylation of VDCC, primarily of Cav1.3, and the mobilization of internal Ca2+ stores by Ca2+-induced Ca2+ release (CICR) (74, 162) are also implicated in insulin secretion (204). While increases in PKA activity are proportional to increases in cAMP levels (246), cAMP-elevating agents have no effect in increasing β-cell [Ca2+]c in the absence of stimulatory concentrations of glucose. These observations suggest that the potentiating effects of cAMP/PKA require triggering levels of [Ca2+]c, which facilitate the PKA-mediated amplification of insulin secretion.
The PKA is also implicated in increasing the sensitivity of [Ca2+]c for insulin exocytosis by acting more distally to regulate β-cell [Ca2+]c and by sensitizing the effects of Ca2+ on the insulin secretory machinery (325, 369). This is achieved by increasing RRP of insulin granules and the trafficking of insulin granules to the plasma membrane (123). Several proteins have been identified as targets of PKA; however, only a few in the secretory machinery, such as synaptotagmin-7 and snapin, upon phosphorylation, have been found to directly regulate the fusion of insulin granules to the plasma membrane during insulin exocytosis. Synaptotagmin-7 is an insulin granule protein that acts as a Ca2+ sensor and functions to accelerate the formation of the SNARE complexes (386). PKA-dependent phosphorylation of snapin (330) increases its interaction with the SNARE complex proteins synaptosomal-associated protein (SNAP25) and vesicle-associated membrane protein 2 (Vamp2), and of synaptotagmin-7 at serine 103 (386) to increase SNARE complex-mediated fusion of insulin granules during insulin exocytosis (330).
Exchange protein activated by cyclic-AMP (Epac2) is an effector of cAMP
The cAMP guanine nucleotide exchange factor (GEF), belonging to the receptor-associated protein (Rap) family of small GTPases (99), is the main Epac isoform that regulates insulin secretion (126, 167, 192, 247, 254). Epac2 contains a regulatory region with two cAMP binding domains and a catalytic region. The binding of cAMP to the regulatory region frees the catalytic region for Epac2 function (281). The Epac2 selective analog 8-pCPT-2′O-Me-cAMP was reported to increase the early and sustained phases of insulin secretion from both mouse (163, 167) and human islets (51). Antisense oligonucleotide-mediated knockdown of Epac2 decreased incretin-mediated potentiation of insulin secretion by 50%, attenuating both phases of insulin secretion from mouse islets (167). Though the Epac2 analog does not activate PKA or PKA substrates (CREB, cAMP-response element binding protein, and kemptide), PKA inhibitors blocked the ability of the Epac2 analog to potentiate GSIS, suggesting that PKA has a permissive role in insulin secretion (51). Epac2 has a PKA-independent role in potentiating the effects of cAMP on insulin secretion (78, 283), and mice lacking Epac2 exhibit reduced potentiation of GSIS in response to cAMP (316). Furthermore, Shibasaki et al. showed that the cAMP potentiation of the early phase insulin secretion was through Epac2 (316). Elevation of cAMP facilitates the localization of Epac2 to the plasma membrane near the insulin granules (11). Moreover, Epac2 clusters are present at the docking site for the insulin granules, facilitating priming and exocytosis (11). Increases in the early phase of insulin secretion are achieved by Epac2-mediated increase in the abundance of RRP (187, 316) and the recruitment of insulin granules from RRP fusion to the plasma membrane (101).
A plausible mechanism suggested for the potentiation of insulin secretion by Epac2 implicates modulation of the fusion of insulin granules to the plasma membrane by the small GTPase Rap1, which Epac2 activates in a cAMP-dependent manner (187). Subsequently, Rap1 activates phospholipase C-epsilon (PLCε) (328), which contains a Rap1-associated domain, allowing activation by cAMP-Epac2-Rap1. The PLCε hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to generate inositol 1,4,5-trisphosphate (IP3) and the lipid metabolite diacylglycerol (DAG) (9). As a second messenger, DAG binds and activates protein kinase C (317) and insulin granule priming factor Munc13–1 (26) to facilitate insulin secretion. Consistently, knockout of PLCε disrupts the ability of the Epac2 analog to potentiate GSIS (76), further supporting the role of cAMP-Epac2-Rap1-PLCε in regulating insulin secretion.
Overall, PKCs potentiate GSIS by regulating cortical actin rearrangement and phosphorylation of exocytotic proteins such as synaptotagmin, SNAP25, and Munc18 (key for regulating syntaxin-mediated plasma membrane fusion of insulin granules) (183, 351). Munc13–1, in turn, binds and activates the insulin granule fusion complex assembly to facilitate insulin secretion. Thus, PLCε links cAMP signaling to DAG-mediated potentiation of GSIS via PKC and Munc13–1. Additionally, Rab-interacting molecule 2α (Rim2α) has been implicated in mediating the effects of Epac2 on insulin secretion, as evidenced by an impairment in insulin secretion with Rim2α deletion (392). Rim2α has been reported to interact with an insulin granule-associated Rab3 GTPase (76) to regulate the docking and priming of insulin granules by interacting with Munc13–1 and the SNARE-core complex protein syntaxin to facilitate insulin exocytosis (72, 139). Epac2 interacts with Rim2α in a cAMP-dependent and PKA-independent manner (76), and this interaction is required for the potentiation of incretins on insulin secretion (167). The distinct roles of Rap1-PLCε and Rim2α-Munc13–1 signaling pathways in cAMP-Epac2 regulated potentiation of insulin secretion remain to be elucidated.
K+-channels as targets of cAMP signaling
Studies by several groups have also noted a dependence of cSIS on K+-channel activity. The SUs are known to bind to the SUR1 subunit of the KATP channels, causing membrane depolarization, the influx of Ca2+, increases in [Ca2+]c, and subsequent increases in insulin secretion. SUs such as tolbutamide have been reported to bind and activate Epac2 (122, 394). However, while the interaction between Epac2 and SUs (280, 354) could not be confirmed, the ability of tolbutamide to increase insulin secretion was attenuated in Epac2-null mice (394). These findings support the role of Epac2 in mediating the insulin-promoting effects of SUs via the KATP channels (145). Epac2 was found to interact with the SUR1 subunit of KATP channels (254) and functions by activating the Ras-like small G protein Rap in a cAMP-dependent manner. In β-cells, upon stimulation by SUs, Epac2 activates Rap1 to facilitate its interaction with PLCε. As expected, the ability of Epac2-specific analogs to potentiate GSIS and increase β-cell [Ca2+]c was found to be disrupted in mice lacking plasma membrane-associated PLCε (76, 200). These outcomes suggest that Rap1-regulated PLCε links Epac2 activation by SUs to Ca2+-dependent exocytosis of insulin (76, 200). In addition to the KATP channels, activation of Epac2 inhibits voltage-dependent potassium (Kv) channels, thus prolonging the duration of the action potential and causing a delay in membrane repolarization, leading to increases in Ca2+ influx and insulin secretion (396). However, the molecular mechanism by which Epac2 modulates the activity of Kv channels to regulate insulin exocytosis remains to be elucidated. Molecular docking stimulation and site-directed mutagenesis analyses have identified that SUs and cAMP cooperatively activate Epac2 by binding to cyclic nucleotide-binding domains (340). Mice lacking Epac2 had a severe reduction in insulin secretion in response to the combined treatment of incretin, GLP1, and SUs (339), suggesting that Epac2 is a critical node that links incretin hormone signaling and SUs in β-cells to increases in insulin secretion, thus connecting the triggering and amplifying pathways to insulin exocytosis.
cAMP signaling in β-cell adaptation
The development of insulin resistance typically associated with obesity triggers a compensatory β-cell adaptive response, which involves altered insulin secretory capacity and increased β-cell mass. Alterations in these attributes evoke responses in the β-cells that culminate in the increased net output of insulin into the plasma to maintain euglycemia during an insulin-resistant state. When β-cells are unable to compensate completely (i.e., fail) to meet the increased demand for insulin due to genetics and/or environmental factors in the face of insulin resistance, glucose intolerance increases and leads to the development of an IGT state, which initially results in mild hyperglycemia and then to elevated hyperglycemia leading to T2D development.
The compensatory β-cell response to insulin resistance ensures that sufficient insulin is released from the pancreas into the plasma to maintain euglycemia. Moreover, complete compensation involves increased plasma insulin levels with normal glycemia, whereas, in partial compensation, plasma insulin levels are elevated with mild hyperglycemia. In these compensating phases, the increases in plasma insulin levels are not particularly associated with increases in the secretory capacity of β-cells but are attributed to increases in the β-cell mass (291). Details on the role of β-cell mass during compensation are thoroughly discussed elsewhere (52, 141, 203, 234, 375).
Multiple studies have shown that early β-cell dysfunction occurs with the onset of insulin resistance in the pathogenesis of T2D. As insulin resistance continues to increase, progressive β-cell dysfunction contributes to hyperglycemia in T2D (81, 86, 147, 233, 331, 364, 379). This has been shown in populations such as Japanese Americans and Asian Americans (331, 364), who are generally not obese, and in Pima Indians (379), where insulin secretion defects occur early in T2D progression. Furthermore, groups with increased risk of T2D, such as normoglycemic first-degree relatives of individuals with T2D (54, 263), women with polycystic ovary syndrome (73), or with a history of gestational diabetes (373, 388) were shown to have early onset of β-cell dysfunction. In 1987, Nesher et al. showed that individuals with T2D have reduced pancreatic responsiveness without differences in peripheral insulin sensitivity compared to obese nondiabetic individuals (242). DeFronzo’s group assessed β-cell function in relation to peripheral insulin sensitivity in lean, obese normal glucose tolerant, obese IGT, and obese T2D individuals. They reported that obese individuals with normal glucose tolerance have significantly reduced β-cell function compared to healthy individuals (4, 5, 60, 85). They further demonstrated that IGT reduced β-cell function by nearly 70%. These studies show that by the time a diagnosis of T2D is made, patients have significantly reduced β-cell function. Prentki and Khan corroborated these observations in studies performed using high-fat diet-induced obese (DIO) mice (135, 261). Their work showed that the DIO mice exhibited hyperinsulinemia, impaired glucose tolerance, with reduced ex vivo islets insulin secretion in response to glucose compared to mice on the standard chow diet (135, 261). However, another study reported increases in GSIS in islets isolated from DIO mice compared to controls (96). It is essential to determine the causes for the reductions in early and progressive β-cell functions with IGT during the compensation phase to delineate the mechanisms contributing to T2D. This will facilitate the development of more targeted and effective therapeutics for T2D.
GLP1 incretin contributes to β-cell adaptation by increasing cAMP signaling to facilitate β-cell proliferation and differentiation while decreasing apoptosis (149, 166). Studies in rodents reveal that activation of the cAMP downstream effector PKA causes translocation of the transcription factor CREB and transducer of regulated CREB activity-2 (TORC2) into the nucleus and induction of genes such as MafA1, Nkx6.1, Pdx1, recognized to impact β-cell mass and function (8, 21, 30, 366, 368). Exogenous treatment of GLP1 or GLP1 agonists in mice increases insulin secretion and glucose clearance (315), and genetic ablation of GLP1R or pharmacological blockage of GLP1R attenuates insulin secretion and glucose clearance (292, 305). The GLP1R signaling is known to be dysfunctional in T2D (53, 70, 180, 282, 353). Moreover, the sensitivity of GLP1 for β-cells is reduced in obese and T2D, compared to healthy individuals (20), and is attributed to reduced cAMP signaling. The β-cell cAMP levels are reduced in hyperglycemia and T2D, and islets from individuals with IGT or T2D have reduced early and sustained phases of GSIS and a blunted response to GLP1 (282, 353). Increasing cAMP signaling improves overall β-cell function in obesity and T2D and enhances the ability of GLP1 to improve β-cell dysfunction in T2D (348). Also, injecting GLP1 increased β-cell mass and insulin secretion in DIO mice treated with streptozotocin (135). These studies suggest that GLP1/cAMP signaling has beneficial effects in improving β-cell function and β-cell mass. However, the underlying molecular mechanisms that reduce GLP1/cAMP signaling in T2D require clarification.
The reduction in the early phase of insulin secretion indicates β-cell dysfunction is observed in insulin-resistant prediabetic individuals wherein the cellular insulin content is still not altered (78). Treatment with GLP1 receptor agonists can completely restore this secretory defect in prediabetes, implicating that GLP1 potentially functions by improving glycolysis-generated ATP-mediated closure of the KATP channels (78). This is supported by the findings that GLP1 cannot potentiate insulin secretion in mice lacking SUR1 and Kir6.2 subunits of the KATP channels (230, 239, 319). Conversely, neonatal diabetes patients harboring a gain-of-function mutation in the SUR1 have attenuated GSIS due to overactive KATP channels (38), whereas treatment with GLP1R agonists potentiated GSIS in these patients. The Epac2 and PKA are effectors of GLP1/cAMP signaling that inhibit KATP channels; Epac2 by enhancing the ability of ATP to inhibit the Kir6.2 (164), and PKA by reducing the stimulatory actions of Mg2+-ADP at SUR1 (201). Thus, it is possible that GLP1/cAMP, via its effectors Epac2 and PKA, directly inhibits KATP channels. However, the molecular mechanism by which an increase in GLP1R signaling modulates KATP channel activity is not completely elucidated. In addition to the KATP channels, GLP1 signaling also inhibits the delayed rectifier voltage-dependent K+ current (Kv) (217), prolonging the duration of the action potential, and thereby increasing the influx of Ca2+ by VDCC and enhancing insulin secretion.
The role of Epac2 in regulating insulin secretion from β-cells has been demonstrated in insulin resistance and obesity (127). Ablation of Epac2 in DIO mice was reported to increase body weight and reduce glucose intolerance and insulin secretion compared to mice on the control diet (137, 329). Furthermore, the glucose-lowering effects of SUs and GLP1 agonist liraglutide treatment were attenuated in Epac2−/− DIO mice. This suggests a role for Epac2 in potentiating insulin secretion in response to the combination treatment of SU and liraglutide in T2D (339). In addition, it was found that the disruption of PKA inhibitor B (PKIB) gene (a potent inhibitor of PKA catalytic activity) improved glucose sensitivity and GSIS in DIO mice, suggesting that PKA potentiates insulin secretion (30). These studies show that cAMP/Epac2 and cAMP/PKA are beneficial in potentiating insulin secretion in obesity and T2D. However, the mechanisms by which Epac2 and PKA mediate the effects of GLP1/cAMP signaling on the RRP, priming of insulin granules, Ca2+ sensitivity, and insulin exocytosis, and how these attributes are altered in obesity and T2D remain to be elucidated.
Noncanonical Mechanisms for cSIS
Contribution of lipid signaling to insulin secretion
It is well-recognized that lipids, in particular eicosanoids and sphingolipids, participate in inflammatory processes. Here, we discuss their identified roles in β-cell secretory function and how modulation of their signaling can impact GSIS, in the context of obesity and T2D.
Fatty acids
GSIS is potentiated by nonesterified fatty acids (NEFAs), with the potency correlating directly with chain length and desaturation of the fatty acid (220, 252, 265, 333). The NEFAs can promote insulin secretion at basal glucose concentrations (129) and are particularly critical for insulin secretion in healthy nonobese individuals following prolonged fasting (67). During periods of short starvation, unsaturated fatty acids can stimulate, whereas saturated short/medium chain fatty acids decrease insulin secretion (332). It has been suggested that this may be related to the modulation of intercellular signaling that influences higher oxygen consumption rate (48), greater activation of FFAR1/GPR40 signaling (48), or increased triglyceride esterification (77). Consistently, GSIS is enhanced by diets enriched in monounsaturated fatty acids (MUFAs) or PUFAs and diminished by diets enriched in saturated fatty acids (262). Studies in the BRIN BD11 insulinoma cell line suggest that arachidonic acid (AA) stimulates β-cell proliferation and insulin secretion and that these effects are mitigated by hyperglycemia or the saturated fatty acid palmitate, as in T2D (66).
In addition to the ω-6 AA (C20:4 double bonds), other polyunsaturated fatty acids (PUFAs) hydrolyzed from the sn-2 position of membrane glycerophospholipids include the long-chain ω-3 polyunsaturated eicosapentaenoic acid (EPA, C20:5) and docosahexaenoic acid (DHA, C22:6). Through islet lipid enrichment studies, EPA and DHA have been recognized to provide signaling to improve β-cell secretion under T2D, obesity, or insulin resistance states. In an early study, EPA enrichment was found to prevent lipotoxicity-induced activation of the uncoupling protein-2 (154), which can recover ATP production and preserve insulin secretion (168). Analogously, a diet enriched with both EPA and DHA reduced palmitate-mediated decreases in GSIS and apoptosis by reducing palmitate-induced increases in β-cell reactive oxygen species (207). Concomitantly, a high-fat diet (HFD) enriched in EPA mitigated insulin resistance in a DIO mouse model (205). In an elegant study, supplementing the diet of BTBR Leptinob/ob mice with EPA was reported to decrease the production of PGE2 from AA and increase the production of the EPA metabolite PGE3, which is 10-fold less potent in reducing GSIS when compared to PGE2 (244, 284). Moreover, EPA also decreased the expression of the PGE2 receptor, EP3. Fewer studies have been performed with DHA alone. Nevertheless, DHA supplementation has been reported to reduce insulin resistance in obese humans (2). Moreover, EPA and DHA index correlated positively with increased insulin sensitivity in obese men (10). Interestingly, overweight obese and diabetic women, but not men, had lower plasma levels of EPA and DHA (3), implying a sex-difference association of these ω-3 fatty acids with T2D. The protective effects of these ω-3 fatty acids in obesity and T2D were reasoned to be related to reductions in AA levels that occur with EPA and/or DHA supplementation. Furthermore, EPA and DHA potentially function to alter membrane structure, fluidity, and signal transduction, which are beneficial for β-cell function (13, 156). Also, EPA and DHA are thought to signal through the GPCR (GPR)120 receptor (249) to induce GLP-1 secretion from enteroendocrine L-cells to promote insulin secretion (27). In islets, GPR120 expression has been identified in β-cells, and lipid agonists of GPR120 were found to promote insulin secretion through inhibition of somatostatin secretion, and GPR120 signaling was found to be impaired in the db/db mouse (71). An ω-3 metabolite of EPA, 5-hydroxyicosapentaenoic acid (5-HEPE), has been reported to enhance GSIS and cAMP generation (181) by signaling through the GPR119 receptor, which is predominantly expressed in β-cells (253).
Eicosanoids
Metabolism of AA in β-cells generates oxidized bioactive lipids (Figure 3) that have been implicated in insulin secretion. Forty years ago, studies identified an inhibitory effect of PGE2 on GSIS (229, 287, 306). This was later attributed to PGE2 signaling via the EP3 receptor to inhibit c-Jun N-terminal kinase (JNK1) activation of AKT serine/threonine kinase 1 (AKT) phosphorylation leading to downstream dephosphorylation of the transcription factor forkhead box protein O1 (FOXO1), facilitating its entry into the nucleus to dysregulate insulin synthesis and GSIS (226, 227). Alternatively, reduced GSIS has also been associated with EP3 receptor signalling-mediated decreases in cAMP production (47, 174), and inhibiting EP3 receptor signaling improves insulin secretion from islets of T2D donors (174). Reports along these lines have led to further exploration of targeting EP3 receptors to counter β-cell dysfunction and the progression of T2D (243). Similarly, PGD2 has been reported to reduce GSIS by signaling through the PGD2 receptor GPR44/DP2, highly expressed in human β-cells (202), to inhibit cAMP production (1). In T2D, GPR44/DP2 expression is increased in islets (115), and while in vitro inhibition of this receptor increased GSIS from human islets, short-term in vivo inhibition was without affect (326). Additionally, cytokines contribute to β-cell dysfunction in T2D (68, 327, 378). In particular, interleukin-1β (IL-1β) is elevated in islets from T2D donors and induces cyclooxygenase 2 (COX2), leading to increased production of PGE2 and EP3 receptor expression, with an overall effect of amplifying PGE2-EP3 signaling in the β-cells (56, 255, 350) and reducing GSIS. Consistently intervening with IL-1β receptor signaling improves GSIS (188, 219). Further, PGE2 generated through the elevated COX2 expression in human islets from T2D donors decreases the expression of genes involved in β-cell function, such as Pdx1, Nkx6.1, and MafA1 (371). This raises the possibility of dysregulated metabolism of AA to PGE2 in T2D islets. Further, PGE1, which is derived from dihomo-γ-linolenic acid (DGLA) and signals through EP2 receptor, has been reported to decrease in vitro and in vivo GSIS (229, 285). A potential mechanism for this is the PGE1-mediated activation of the G-protein Gz to inhibit β-cell endocytosis, which is an integral component of the exocytosis-endocytosis process that needs to be maintained in equilibrium to preserve β-cell volume for optimal β-cell function (400). In contrast to the above-discussed PGs, prostacyclin (prostaglandin I2, PGI2) through PGI2 receptor (IP)-GPCR signaling induces adenyl cyclase to increase the production of cAMP, which directly activates Epac2 to induce GSIS (105). The potential of PGI2 analogs in mitigating glucose intolerance and insulin resistance associated with T2D has also been identified (296). Collectively, these studies suggest that several different PGs may serve as therapeutic targets to improve β-cell function in T2D.
Figure 3. Contribution of β-cell-derived lipids to insulin secretion.
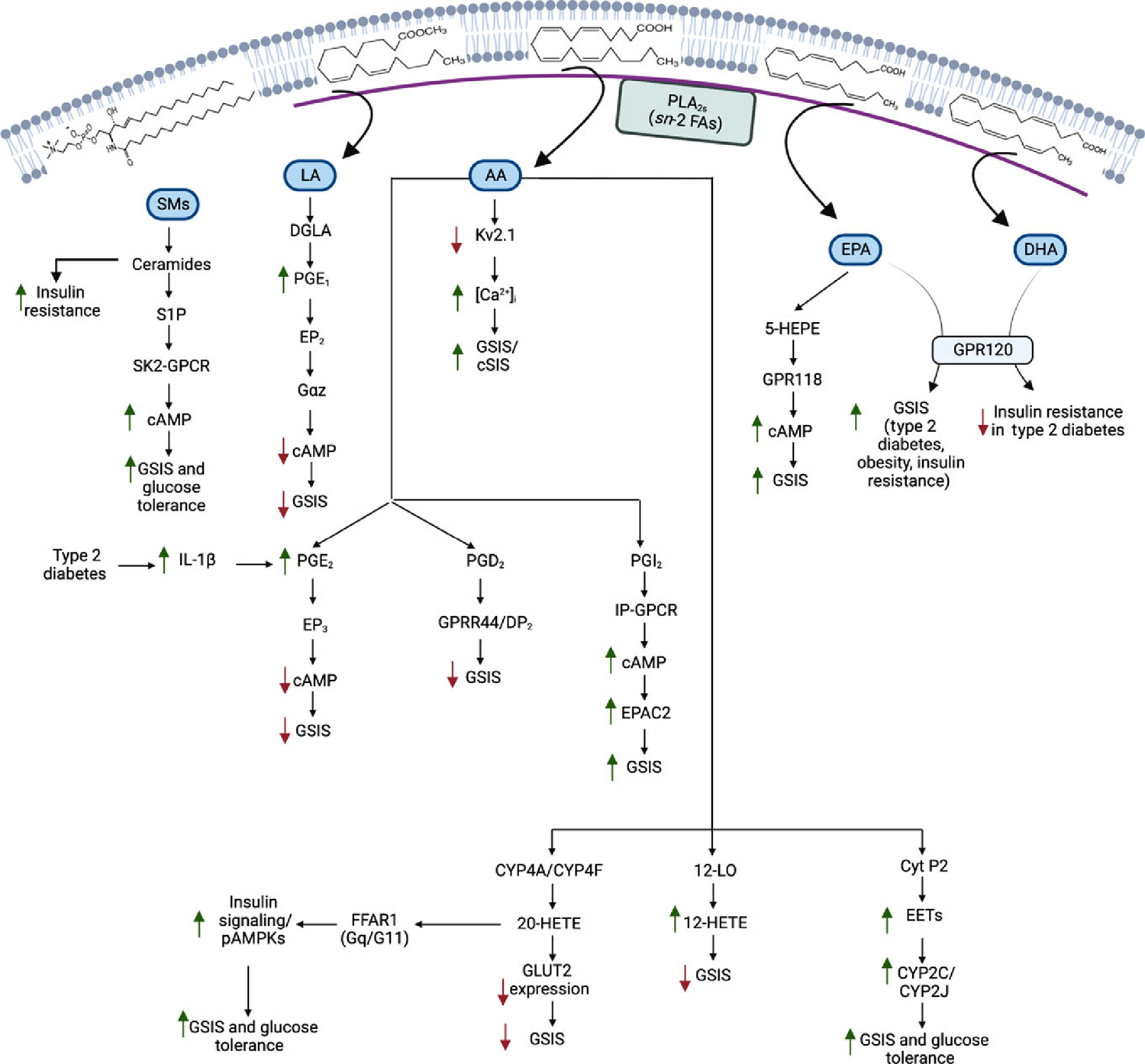
Elevations in circulating glucose concentrations induce phospholipases A2 (PLA2s)-catalyzed hydrolysis of the sn-2 fatty acid substituent from β-cell membranes. The released fatty acids can then be metabolized by oxidases to generate bioactive lipid mediators (e.g., PGs, prostaglandins; HETEs, hydroxyeicosatetraenoic acids; and EETs, epoxyeicosatrienoic acids). Further, membrane-associated sphingomyelins can be hydrolyzed by sphingomyelinases to ceramides, which can be converted to sphingosine-1-phosphate (S1P). These lipids via intracrine, autocrine, paracrine, and endocrine mechanisms are recognized modulators of insulin secretion.
HETEs
Other metabolites of AA, such as hydroxyeicosatetraenoic acids (HETE), have also been shown to have profound effects on GSIS. These include 12-HETE and 20-HETE generated by lipoxygenases (LOX) and cytochromes P450 (CYP4A & CYP4F), respectively. Islets from T2D donors are reported to have increased expression of 12-LOX (140). Consistently, the deletion of 12-LOX in a rodent model of T2D is associated with reductions in 12-HETE, preservation of GSIS (345), and inhibition of insulin resistance associated with DIO (248, 307). Similarly, increases in 20-HETE appear to decrease GSIS, and rescue of GSIS is observed with the inhibition of 20-HETE-induced decrease in Glut2 (393). Moreover, the production of epoxyeicosatrienoic acids (EETs) (209), which promote GSIS (83), by CYP2C & CYP2J, also modulates the effects of 20-HETE on GSIS. In contrast to these studies, 20-HETE has been suggested to provide a positive feedback regulation of GSIS by signaling through the Gq/G11-coupled free fatty acid receptor 1 (FFAR1) expressed highly in β-cells (142, 185, 286, 309) to facilitate insulin exocytosis. Interestingly, though the basal levels of 20-HETE in islets from multiple rodent models of T2D and human T2D donors were increased, under conditions of GSIS, the levels of 20-HETE were reported to be decreased, and the addition of selective FFAR1 agonists failed to rescue GSIS in the T2D islets (356). This suggested impairment in the positive regulatory loop on GSIS mediated by 20-HETE in T2D. Human islet FFRA1 mRNA levels were consistently found to correlate positively with the insulinogenic index (349).
EETs
The CYP2-catalyzed metabolism of AA leads to the generation of EETs, which are rapidly metabolized to dihydroxyeicosatrienoic acids (DHETs) by soluble epoxide hydrolase (sEH). Among the EETs, 5,6-EET has been reported to induce insulin secretion (83). However, all isomers of EETs in the plasma of humans correlated positively with insulin sensitivity (90). In a model of KATP-independent insulin secretion, imidazoline-induced GSIS was reported to occur through AA-derived EET generation (314). Further, because of their rapid degradation, the potential beneficial effects of EETs have mostly been gleaned from examining the effects of inhibiting or reducing the expression of sEH. Luo et al. were the first to demonstrate an improvement of in vivo insulin secretion and a reduction in blood glucose levels of streptozotocin diabetic mice with inhibition or deletion of sEH, and an increase in GSIS with sEH inhibition (209). Similar manipulation of sEH in a DIO model also reduced plasma glucose levels and mitigated insulin resistance development (210). In this regard, the expression of CYP2 isoforms was decreased, sEH expression increased with obesity in the Zucker rat model (399), and the knockout of CYP2C44 decreased glucose tolerance and insulin sensitivity (90). Consistently, CYP2J gene therapy resulted in increases in EET levels and reversal of insulin resistance in T2D rodent models (db/db and fructose-treated mice) (390). Further, deletion of sEH in db/db mice markedly reduced blood glucose levels (131). The effects of EETs in ameliorating T2D are likely related to the prevention of decreases in insulin receptor signaling and phosphorylation of AMP-activated protein kinases (AMPKs) in a variety of peripheral tissues (390). The expression of sEH has been found to increase in islets from mice and macaque maintained on HFD and from T2D donors (182). In the same study, sEH inhibition, genetic reduction, and EET treatment were demonstrated to potentiate GSIS and reduce HFD-induced β-cell death. At the β-cell level, the positive impact of sEH inhibition on GSIS appears to be due to the potentiation of KATP-independent amplification of insulin secretion involving cAMP signaling (117). Consistently, sEH inhibition or genetic reduction increased glucose-stimulated ATP and cAMP levels in the islets (182). Given these observations, modulating sEH activity is under consideration as therapy for many disorders, including obesity and diabetes (107).
Sphingolipids (SLs)
Another major class of lipids implicated in modulating GSIS and T2D is the SLs (6, 336). Among them, ceramides induce insulin resistance (32, 338) and decrease insulin gene transcription (103) and are reported to be elevated in T2D (6, 108). In contrast, decreasing ceramide synthesis promotes insulin sensitivity (125, 398) and β-cell survival (193, 195). Moreover, elevations in dihydroceramides have been reported in the plasma of subjects that eventually developed T2D (380). Intriguingly, inhibiting de novo production of hypothalamic ceramides in the obese Zucker T2D model reduced glucose intolerance and enhanced GSIS (45). In contrast, the ceramide metabolite sphingosine-1-phosphate (S1P), which has the opposite effects of ceramides (190, 318), promotes GSIS by activating sphingosine kinase 2 (SK2) GPCR (46), the predominant SKR expressed in β-cells (225). Other ceramide-derived SLs have also been suggested to affect GSIS positively or negatively (35).
Phospholipases A2-derived lipids and β-cell function
Phospholipases A2 (PLA2s)
The family of phospholipases A2 (PLA2s) catalyzes the hydrolysis of the sn-2 substituent from the glycerol backbone of membrane glycerophospholipids to yield a free fatty acid and a lysophospholipid (96). The major constituents of the PLA2 family are (s)ecretory, (c)ytosolic, and Ca2+-(i)ndependent PLA2s, with the iPLA2s being the most recent family of PLA2s described and characterized (41, 64). Ca2+ at mM and μM concentrations are essential for the activity of sPLA2s and the membrane association of cPLA2, respectively. In contrast, the iPLA2s mobilize and manifest activity in the absence of Ca2+ (270). Among the iPLA2s, the cytosolic iPLA2, designated Group VIA iPLA2 or iPLA2β, was discovered in 1997 (22, 213, 344) and was soon followed by the description of the membrane-associated iPLA2γ (Group VIB iPLA2) in 2002 (221). Other iPLA2s (iPLA2δ, iPLA2ε, iPLA2ζ, and iPLA2η) (42, 146, 301, 367) have since been described but remain to be characterized in detail. Several members of the sPLA2 family have been demonstrated to negatively (group X (322) and V (321)) or positively (IB (276)) influence GSIS, whereas cPLA2 overexpression can promote insulin exocytosis (155), but sustained cPLA2 activation reduces β-cell function and GSIS (152, 232).
iPLA2β and GSIS
Given recent studies concerning mechanisms that promote β-cell decompensation during the development of obesity and T2D, this article focuses on the very first functional role ascribed to iPLA2β, relating to its involvement in insulin secretion. Before its formal description in 1997, work done by the Turk and Gross groups identified a phospholipase A2 activity in islet β-cells, but not in non-β-cells, that was manifested in the absence of Ca2+ and was stimulatable by ATP (100, 273), leading to its initial designation as ASCI-PLA2 (ATP-stimulatable Ca2+-independent phospholipase A2) (357). Subsequently, its predominant expression in the β-cells of the islets was demonstrated at the mRNA (213) and protein (194) levels. Cloning of iPLA2β from hamster, mouse, and rat identified protein homologs that are 85 kDa (752 amino acids) (22, 213, 344) with a serine lipase consensus sequence (GTSGT), preceded by eight N-terminal ankyrin repeats (213, 344). In human lymphocyte lines and in testis, iPLA2β was described as containing a 54-amino acid insert interrupting the eighth ankyrin repeat (189), and analyses in human pancreatic islets identified cDNA species that encoded two distinct 85 kDa (VIA-1) and 88 kDa (VIA-2) human iPLA2β isoforms (215). Subsequent characterization of iPLA2β by several groups identified other features in the protein that included: GXGXXG nucleotide-binding motif (214, 221), N-terminal caspase-3 cleavage site (DVTD) (19), putative bipartite nuclear localization sequence (KREFGEHTKMTDVKKPK) (214), and C-terminal 1–9-14 calmodulin-binding motif (IRKGQGNKVKKLSI) and a calmodulin-binding peptide (AWSEMVGIQYFR) (146, 214, 359). Studies in numerous laboratories over the past three decades have identified iPLA2β as a ubiquitous enzyme that has roles in phospholipid remodeling, cell proliferation, apoptosis, and signaling.
Earlier works revealed that fuel secretagogue-induced hydrolysis of β-cell membrane phospholipids promotes accumulations of AA and its metabolites in β-cells (28, 362). The islets are particularly enriched in AA-containing glycerophospholipids, with 31% residing in glycerophosphoethanolamine (GPE), 53% in glycerophosphocholine, 10% in glycerophosphoinositol, and 3% in glycerophosphoerine (271, 272). The fraction of AA in islet GPE is greater than that in liver, heart, or brain (275) and nearly half of this is contained in plasmenylethanolamine species, as in the brain (275). Further, islet subcellular organelles (mixed membranes, secretory granules, plasma membranes, endoplasmic reticulum) are enriched in AA-containing plasmenylethanolamine molecular species (275), and this pool is the major source of AAs that are hydrolyzed during GSIS (271) and subsequent accumulations in AA correlated with temporal increases in GSIS (273, 274, 278).
Studies by the Turk group revealed that AA at concentrations that accumulate within the β-cell with high glucose stimulation promotes increases in [Ca2+]c levels (228, 383, 384). Subsequent collaborative efforts with the Phillipson group led to delineation of a molecular mechanism whereby the AA inactivates the β-cell-delayed rectifier potassium channel, Kv2.1, causing a prolongation of glucose-induced action potentials and elevation in [Ca2+]c. These observations suggest that arachidonic acid also plays a key second messenger role in GSIS (23, 144).
Prior to the 1990s, the hydrolysis of membrane phospholipids in β-cells was understood to occur via phospholipases that are expressed in β-cells and activated by increases in [Ca2+]c. However, if the hydrolysis of AA induced by glucose were dependent on the Ca2+ influx, it would not align temporally with the pathway through which it affects Ca2+ influx. A solution to this conundrum was provided by studies demonstrating that glucose-induced release of AA occurred, in part, in the absence of Ca2+ or in the presence of Ca2+-channel blockers (358, 383), suggesting the expression of a Ca2+-independent PLA2 activity. In fact, in the early 1990s, the Gross group reported expression of a myocardial ASCI-PLA2 (110–113). Subsequently, analogous studies not only identified a similar iPLA2 in β-cells but determined that its activation led to AA accumulation in β-cells and that this correlated with GSIS (100, 273). This iPLA2 was subsequently designated iPLA2β, based on the presence of the GXSXG lipase consensus motif and a GXGXXG nucleotide-binding motif that was sequentially first identified in the potato enzyme patatin (14).
The recognition of an iPLA2β activity in the islets prompted a detailed characterization of its role in GSIS, especially in light of the recognition that T2D is characterized by a select defect in GSIS (266). Studies in insulinoma cells and islets (rodent and human) utilizing multiple approaches to modify iPLA2β activity or expression strongly supported the following: (i) GSIS correlates with the iPLA2β-dependent accumulation of AA in the β-cells and the release of PGE2 from β-cells (268, 273, 274, 278), (ii) iPLA2β promotes glucose, but not AA-induced increase in [Cai2+] (273) and (iii) iPLA2β activation reduces depolarization-induced Kv2.1 currents, thus promoting Ca2+ flux and amplifying GSIS (23, 144). These observations brought to light a positive impact of iPLA2β on promoting insulin secretion from β-cells.
These observations suggest that dysfunction of iPLA2β in β-cells is critical to the development of β-cell decompensation associated with T2D. Indeed, nonglucose secretagogues, but not glucose, have been reported to increase PLA2 activity and insulin secretion from islets isolated from mice fed a HFD. This raises the possibility that PLA2 activation is a mechanism that participates in β-cell compensation associated with insulin resistance (324). In support of this possibility, a recent study demonstrated that mice with a conditional knockout in β-cell iPLA2β have elevated fasting glucose levels and exhibit glucose intolerance that is exacerbated when maintained on a HFD (360). Deficiency in iPLA2β was also associated with mitigating obesity in a variety of models (49, 63), reductions in plasma glucose and insulin levels, and improvement in glucose tolerance (63).
Role of complement1q-like 3 (C1ql3) in insulin secretion
Secreted proteins are critical metabolic regulators that play significant roles in cell-cell communication between different cell types. These proteins relay the metabolic status of the origin cell to impart metabolic changes in the target cells in an autocrine, paracrine, or endocrine manner. The roles of islet endocrine hormones such as glucagon, insulin, somatostatin (SST), pancreatic polypeptide (PPY), and ghrelin secreted by alpha (α), beta (β), delta (δ), PP, and epsilon (ε) cells, respectively, have been well-characterized in whole-body glucose homeostasis. Additionally, significant crosstalk occurs within islets between distinct islet cells and a few notable autocrine and paracrine regulators have been identified, such as SST, urocortin-3, islet amyloid polypeptide (IAPP), and glucagon. The SST secreted from δ-cells act on α- and β-cells to inhibit glucagon and insulin secretion, respectively (40, 79, 134). A late β-cell maturity marker, urocortin-3, secreted from β-cells, decreases SST secretion from δ-cells (132). Moreover, glucagon has an intra-islet role in increasing insulin secretion from β-cells. Islet autocrine and paracrine regulators have been previously reviewed (44, 84, 119, 134, 288, 355).
Eleven percent of the human and mouse genome transcripts encode for secreted proteins, and approximately 3000 secreted proteins have been annotated, but the function for most of them remains undescribed (363). To this end, Koltes et al. used an unbiased approach of the weighted gene expression network analysis to identify secreted protein regulators of islet function (184). Using lean and obese islet transcriptomics data, they identified that islets express 850 transcripts encoded for secreted proteins whose expression significantly changes with obesity. This suggested that several islet-derived secreted proteins potentially manifest roles in response to obesity. Islets by mass are only 1% of the total pancreas weight. So, it is conceivable that several of the islet-derived secreted proteins are involved in crosstalk between distinct islet cells in an autocrine and/or paracrine manner in obesity. They further identified 44 hub genes with high connectivity attributes encoding secreted proteins to affect islet function in obesity. Of those, the complement 1q like 3 (C1ql3) secreted protein was reported as a top hub gene, illustrating the utility of unbiased network analysis in identifying secreted protein regulators affecting islet function in obesity. The complement 1q (C1q)/tumor necrosis factor (Tnf)-related proteins (CTRP) are secreted proteins (e.g., adiponectin) that are evolutionarily conserved from zebrafish to humans (385). They have been implicated in lipid and carbohydrate metabolism, immunity, inflammation, and synapse homeostasis (33, 94, 312, 323). C1ql3, also called CTRP13, is a soluble secreted protein whose primary structure resembles proteins encoded by highly homologous genes: C1ql-1, -2, and -4. It is expressed in adipose tissue and the brain of lean mice. Obesity due to a HFD or homozygosity for the LeptinOb mutation was reported to increase the expression of C1ql3 in the brain and islets (184), whereas caloric restriction decreases its expression (43). Serum C1ql3 levels are increased in LeptinOb mice (43). Wei et al. have reported that C1ql3 regulates glucose metabolism (374), and epidemiological studies have shown an association between serum C1ql3 levels and T2D and metabolic disorders in humans (7, 80, 82, 313).
C1ql3 is abundantly expressed in islets, and its expression is increased in islets from db/db, DIO, and LeptinOb (>30-fold) mice (104). Localization of C1ql3 is in insulin-positive β-cells, but not in α- or δ-cells, identifying β-cells as the source of the secreted C1ql3 protein. The molecular mechanism of obesity-induced transcriptional regulation of C1ql3 and how C1ql3 is secreted from β-cells is unknown. A recent report by Gupta et al. (104) demonstrated that C1ql3 inhibits high glucose (16.7 mM)-stimulated insulin secretion without affecting insulin secretion in response to low (2.8 mM) or sub-maximal (11.1 mM) glucose from mouse islets. Remarkably, C1ql3 almost completely blocked insulin secretion in response to stimulation with exendin-4 (a stable analog of GLP1) and cAMP from mouse and human islets. In addition, C1ql3 also decreased cAMP levels in response to high glucose. Such an inhibitory impact of C1ql3 on insulin secretion was confirmed using mouse and human C1ql3 recombinant proteins, C1ql3-conditioned media, knockdown approaches in clonal β-cells, and mouse and human islets. Since high glucose treatment is known to increase cAMP levels in β-cells by activating soluble AC (124, 192, 397), the inhibitory effect of C1ql3 on insulin secretion in response to high glucose could be due to the attenuation of cAMP signaling. Remaining to be elucidated are the molecular mechanisms and the physiological significance of β-cell C1ql3 in regulating islet function to affect whole-body glucose homeostasis in lean vs. obesity states (Figure 4).
Figure 4. C1ql3 regulates cAMP signaling and amplifies GSIS in β-cells.
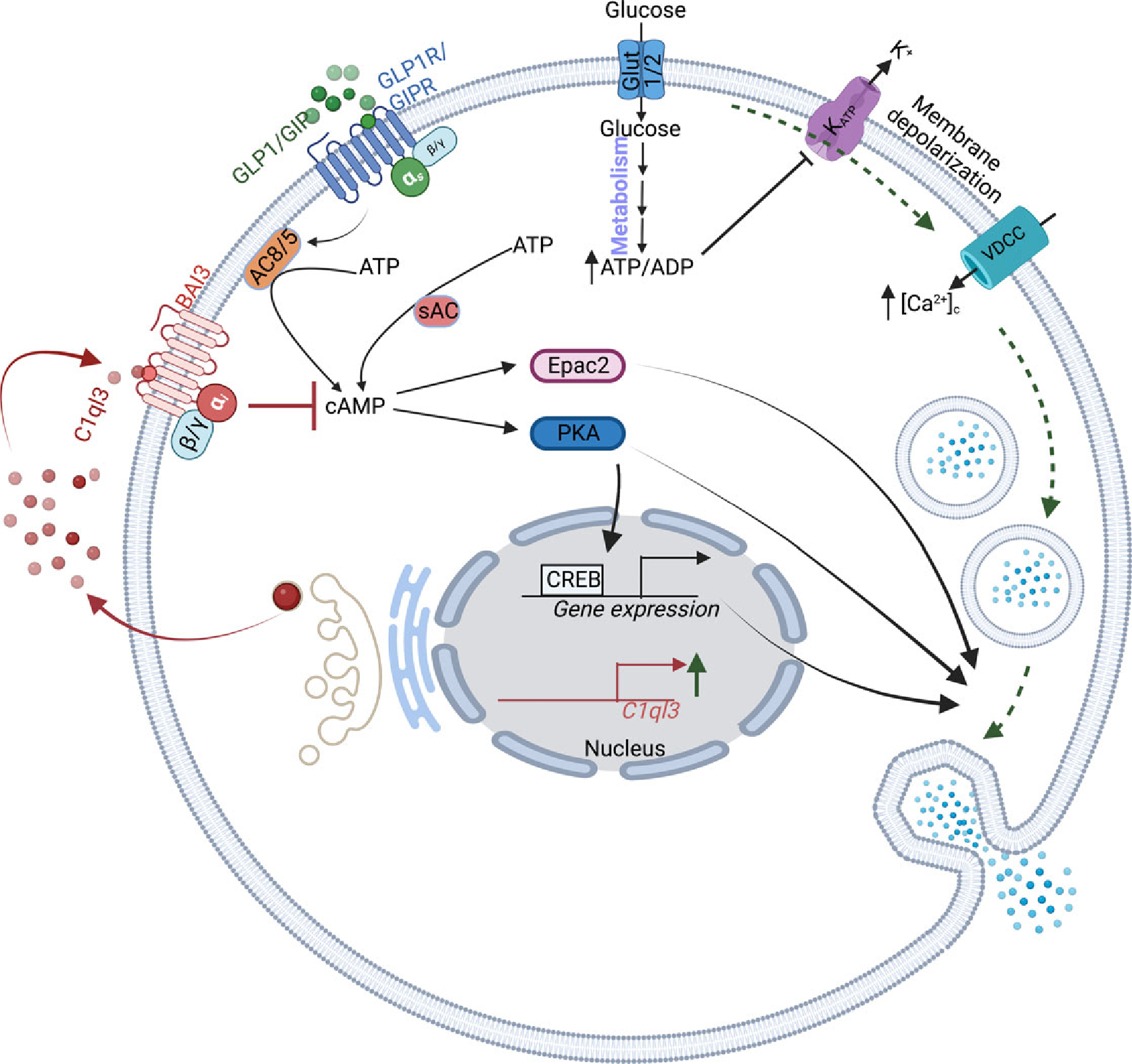
Upon secretion from β-cells, C1ql3 signals in an autocrine manner by binding to its receptor BAI3 to regulate insulin secretion by inhibiting cAMP signaling.
The brain-specific angiogenesis inhibitor 3 (BAI3) has been identified as a receptor for C1ql3 (224). The BAI3 is a member of the BAI subfamily of adhesion GPCRs. These proteins are involved in synaptogenesis, angiogenesis, and tumor suppression (57, 235, 320). Structurally, BAI3 is comprised of an extracellular N-terminal CUB domain, four thrombospondin type 1 repeats, a hormonebinding domain, the GPCR auto-proteolysis-inducing domain (GAIN) with an auto-proteolysis site (GPS), the characteristic 7-transmembrane repeat of a GPCR, an intracellular α-helical RKR motif, and PDZ domain-binding motif (57). BAI3 is expressed primarily in the brain and has been implicated in psychiatric disorders such as schizophrenia, bipolar disorder, and addiction (169, 197). It is also expressed in islets and muscles, and mutations in BAI3 have been linked to T2D in human GWAS studies (109, 150). The BAI3 receptor is coupled to Gi and has a role in early-onset venous thromboembolism risk (15), stimulation of testosterone secretion (342), and myoblast fusion (106). Further, BAI3 signaling is required in the synaptic organization, acquisition of social transmission of food preference (370), and mediation of cell-cell adhesion (334). In β-cells, siRNA-mediated knockdown of BAI3 increased insulin secretion from INS1(832/13) cells and is implicated in mediating the inhibitory effects of C1ql3 on insulin secretion (104). These studies suggest a role for this poorly characterized BAI3 signaling in insulin secretion affecting β-cell function. The molecular mechanism by which C1ql3/BAI3 regulates islet function remains to be elucidated.
Perspective: Potential Interplay Between iPLA2β and C1ql3 During T2D Development
Nearly 35 million people in the USA have T2D, and obesity is a major risk factor for T2D (196, 303). A key feature of T2D development is that during the initial phase of obesity-induced metabolic stress, β-cells compensate for increasing insulin secretion to achieve glucose homeostasis. However, chronic obesity manifests insulin resistance, prompting further attempts of β-cell compensation until the β-cells cannot overcome the metabolic demands and begin to fail (133). The concept of β-cell compensation progressing to β-cell decompensation is entrenched in the T2D field. However, mechanisms that contribute to this eventuality are still not completely understood.
There is strong evidence in the literature that defects in insulin granule exocytosis machinery, metabolism, and electrical activity contribute to β-cell dysfunction in IGT associated with obesity and T2D (81, 89). The early phase insulin secretion loss is the initial event during the development of β-cell dysfunction in IGT (81). This is attributed primarily to defects in insulin exocytosis, as cellular insulin content is not significantly altered during this state. Treatment with GLP1 receptor agonists that increase cAMP signaling can restore early phase insulin secretion deficits (315), highlighting the role of downstream cAMP effectors Epac2 and PKA in improving glucose responsivity of the β-cells to secrete insulin. The overall mechanism of how PKA and Epac2 mediate GLP1/cAMP effects on improving insulin secretion in IGT is not completely understood. It is possible that upon activation, PKA and Epac2 increase the ability of glucose to inactivate KATP and Kv2.1 channels, delaying membrane repolarization, thus increasing the duration of the action potential and enhancing the rise in [Cac]2+ (58, 191, 199, 218, 335, 391). These events promote the fusion of insulin granules to the plasma membrane for insulin exocytosis. Epac2 binds to and inhibits KATP channels (145, 254), whereas PKA phosphorylates KATP (25) and Kv (191, 206) channels. In addition to regulating plasma membrane electrical activity, Epac2 and PKA can directly affect the secretory machinery (325, 369) of the insulin granules by facilitating the RRP pool, docking, priming, and fusion of insulin granules to the plasma membrane (386). Furthermore, cAMP/PKA regulates β-cell gene expression through CREB to increase insulin secretion (8, 21, 30, 366, 368). The beneficial effects of GLP1/cAMP signaling in improving β-cell function are well established. However, the precise mechanism by which PKA and Epac2 function in improving β-cell dysfunction in IGT remain to be determined (Figure 5).
Figure 5. IGT and T2D can affect multiple aspects of the cAMP signaling to decrease insulin secretion.
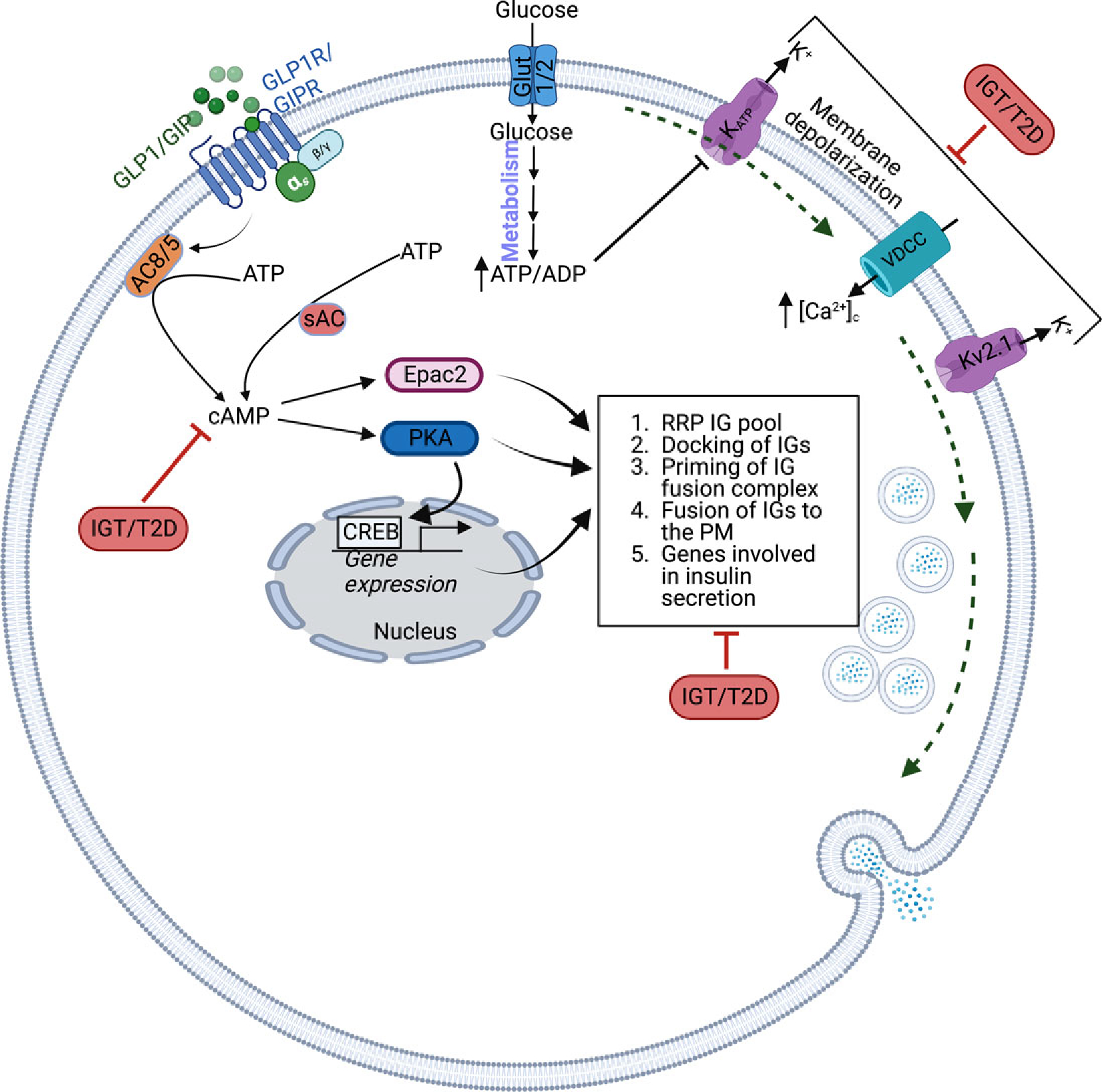
The IGT/T2D states can potentially reduce cAMP levels, size of the RRP insulin granule (IG) pool, docking of the IGs to the plasma membrane, priming of the IGs to the plasma membrane, the fusion of the IGs to the plasma membrane, and PKA-CREB mediated β-cell gene expression. Additionally, IGT/TD can also decrease the ability of glucose to cause plasma membrane depolarization-regulated insulin secretion.
When the clinical diagnosis of T2D is made, the β-cell function is already reduced by 70% (4, 5, 60, 85). Moreover, normal and impaired glucose-tolerant obese individuals also have significantly decreased β-cell function (4, 5, 60, 85). The mechanisms that cause this reduction in β-cell function are not yet completely understood. Stable GLP1 analogs (e.g., exenatide, liraglutide, semaglutide) are currently wellaccepted T2D therapeutics with many advantages over older β-cell secretagogues that function in a glucose-independent manner (e.g., SUs, glinides) (264). These GLP1 agonists have been the most successful approaches in T2D treatment, and their effect in improving β-cell function is via the GPCR-mediated activation of cAMP signaling. Even though stable GLP1 analogs function in a physiologically relevant manner, they remain ineffective or insufficiently effective in 20% to 40% of T2D individuals in improving β-cell function (55, 161, 178). Thus, a complete understanding of how β-cell cAMP signaling is regulated in the healthy, obese, and T2D states is essential for developing interventions for improving β-cell function in IGT for treating individuals with T2D. The canonical approach involves elucidating the downstream molecular mechanism by which cAMP regulates β-cell function. An additional approach involves identifying and characterizing signaling pathways that either directly activates, antagonize, or synergize with the cAMP signaling to modulate β-cell function. Elucidating these cell-signaling pathways that modulate cAMP signaling would be key to improving our understanding of β-cell adaption that can also be targeted to increase the efficacy of GLP1 analogs that are used for the treatment of T2D. In this regard, we have identified C1ql3 and iPLA2β as manifesting opposing effects on the cAMP pathway in the β-cells.
C1ql3 negatively impacts insulin secretion
C1ql3 is secreted from islet β-cells and has its maximal inhibitory effect on insulin secretion in response to stimulation by exendin-4 (GLP1 agonist) and cAMP (104, 184), implicating it as an autocrine regulator of insulin secretion. Moreover, C1ql3 was found to not affect insulin secretion in response to submaximal or basal glucose concentrations, mitochondrial metabolites (isocitrate, α-ketoglutarate, malate, and glutamate), amino acids (leucine, glutamine, alanine, arginine), or fatty acids (monoacylglycerols, DAGs, palmitate) (104, 184). The expression of C1ql3 and BAI3 is increased by greater than 30-fold and 4-fold, respectively, in islets obtained from euglycemic obese mice, during conditions when β-cells are undergoing functional adaption to obesity or IGT associated with insulin resistance. Thus, it is plausible that the activation of C1ql3/BAI3 signaling in β-cells during obesity contributes to regulating cAMP signaling mediated β-cell adaptation during IGT/T2D state. Elucidating the precise molecular mechanism of how C1ql3/BAI3 regulates cAMP-signaling in β-cells within a healthy pancreas, as well as under pathogenic conditions such as obesity and T2D, will elucidate whether C1ql3 contributes to β-cell dysfunction associated with IGT in obesity and T2D. It is broadly accepted that β-cell mass is increased to adapt to obesity-associated insulin resistance (52, 141, 203, 234, 375). However, the relationship between proliferation and functional maturity of β-cells is poorly understood. Several studies have suggested that proliferating β-cells are functionally immature (31, 222, 269, 302). Thus, it is possible that C1ql3 signaling also has a role in reducing the responsivity of β-cells to glucose/cAMP for optimal insulin secretion in the proliferating β-cell population.
iPLA2β positively impacts insulin secretion
The ubiquitous Ca2+-independent PLA2 (iPLA2β) (22, 146, 221, 344) hydrolyzes the sn-2 substituent of membrane glycerophospholipids to yield a free fatty acid and a 2-lysophospholipid (237, 361). The iPLA2β participates in cellular lipid metabolism, signaling, energy homeostasis (170, 237, 381), and amplification of cSIS (212, 216, 271, 274, 277, 278). Supporting a role for iPLA2β in insulin secretion are the findings that islets from global iPLA2β-null mice exhibit impaired GSIS (23, 24) in the presence of forskolin, which activates AC to generate cAMP. These mice exhibit normal glucose tolerance in the unstressed state (24) but develop more severe glucose intolerance while exhibiting better insulin sensitivity than wild-type littermates when subjected to DIO (24). This discordance of the effects of DIO on insulin secretion and sensitivity in the global iPLA2β-null mice suggests that iPLA2β plays distinct roles in the molecular mechanisms underlying insulin secretion and insulin action and in the impact of DIO on these processes. Arachidonic acid is a major sn-2 substituent in β-cell membranes, and its iPLA2β-mediated hydrolysis correlates with stimulated insulin secretion (24, 100, 212, 216, 271, 274, 277, 278). Mechanistically, arachidonic acid has been reported to inactivate the inward rectifier Kv2.1 channel to prolong β-cell membrane polarization and enhance Ca2+ influx (23, 144). This is recapitulated in β-cells overexpressing iPLA2β and mitigated in iPLA2β-null β-cells (23, 144) treated with glucose and carbachol, which signals through acetylcholine receptors to increase cAMP generation in the β-cells.
Linking C1ql3 and cell-specific iPLA2β to cAMP signaling
The ability of cAMP signaling to improve β-cell function is reduced in IGT (obese and T2D) states (282, 353). It has been reported that glucose tolerance and cSIS in mice with select deficiency in β-cell-iPLA2β (iPLA2ββKO) are impaired (360) and are exacerbated in DIO mice. Conversely, cSIS insulin secretion and glucose tolerance improvement occur in mice overexpressing iPLA2β (iPLA2ββTG) in β-cells. These observations support a role for β-cell-iPLA2β in maintaining glucose tolerance and improving β-cell function. Understudied but critical contributors to islet inflammation in T2D are the macrophages (MØ), and MØ and precursor monocytes express iPLA2β (97, 236, 258, 343). Activation of iPLA2β favors polarization of MØ to an inflammatory M1 phenotype (17, 240), which triggers events that contribute to diet-induced insulin resistance in diabetes and obesity (159, 160, 208, 294, 376, 377). We find that mice with select iPLA2β deficiency in MØ (iPLA2βMØKO) have improved β-cell function, glucose tolerance, and insulin sensitivity (360). Thus, increased MØ-iPLA2β signaling contributes to the detrimental effects of MØ on β-cell dysfunction in IGT/T2D, countering the beneficial effects of β-cell iPLA2β signaling in improving cSIS. The expression of C1ql3 and BAI3 is increased in islets during IGT, implicating that enhanced C1ql3/BAI3 signaling in β-cells could potentially be exacerbating the reduction of cAMP signaling, thereby contributing to β-cell dysfunction in IGT/T2D. This could be achieved by C1ql3/BAI3 signaling to decrease the ability of iPLA2β to increase cSIS (Figure 6).
Figure 6. Proposed role of the noncanonical signaling pathways affecting the β-cell secretory function during IGT and T2D.
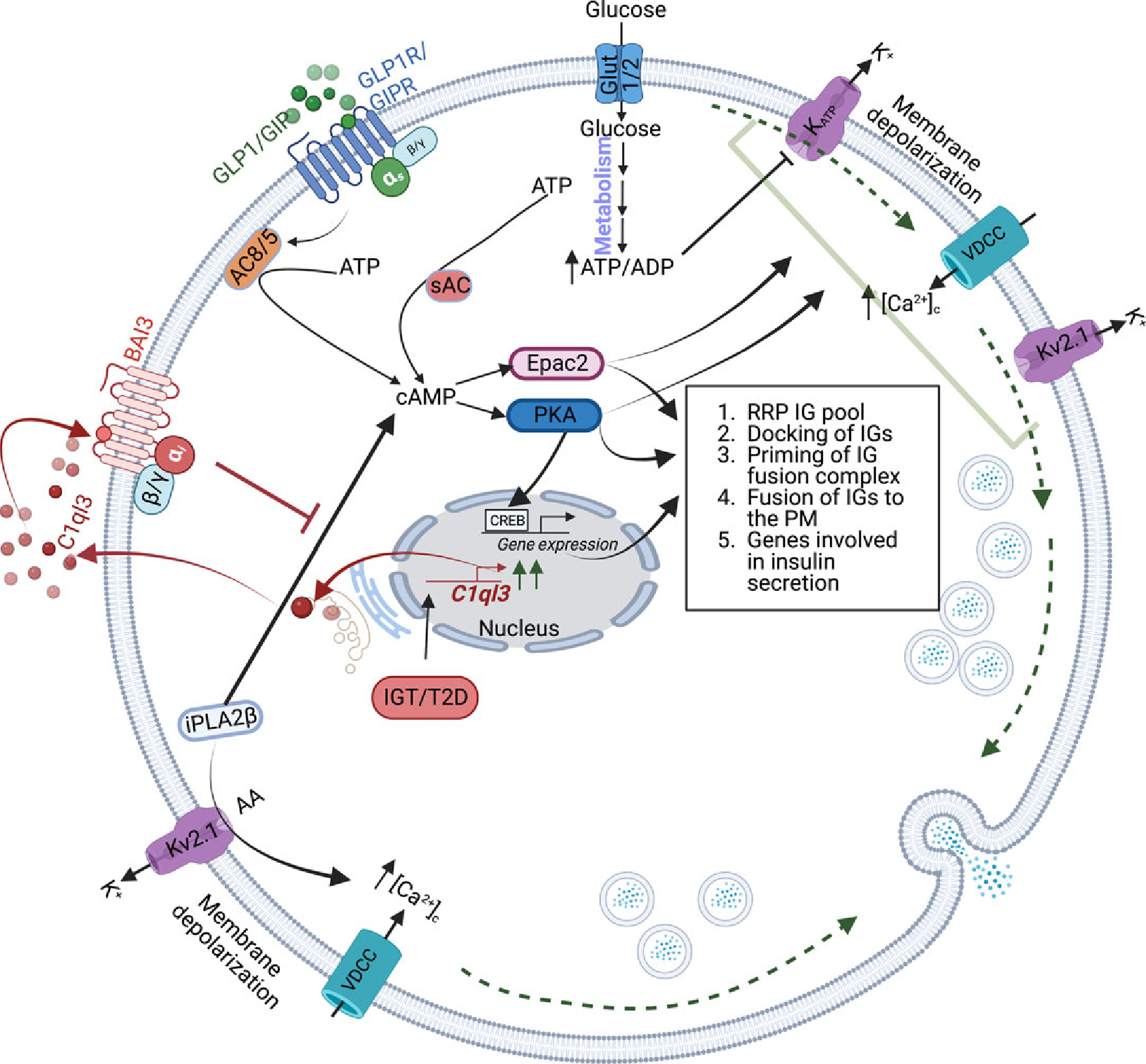
During IGT/T2D, mRNA abundance and the secretion of C1ql3 are increased from the β-cells. The C1ql3 binds to and activates its receptor BAI3 to decrease cAMP signaling, affecting the RRP of IGs, docking of IGs, the priming of IGs, fusion of IGs, and PKA-CREB-regulated expression of genes. Another signaling event mediated by iPLA2β causes an increase in cSIS. The interplay between C1ql3 and iPLA2β in IGT/TD could be important in modulating cAMP signaling, affecting insulin secretion from β-cells during IGT/T2D.
GSIS is reduced from pancreatic β-cells, in the context of impaired glucose tolerance (IGT) and T2D (18, 148, 250, 261, 311, 372). Furthermore, amplification of GSIS in response to the incretins is also blunted. Decades of work established that cAMP is essential for promoting insulin secretion and maintaining whole-body glucose homeostasis (176, 299, 310, 347). β-cell cAMP levels and signaling are reduced in islets from individuals with IGT or T2D (282, 353). The Gs-coupled receptor signaling, including for GLP1, in β-cells, is very well characterized, however, little is known about factors that function to regulate cAMP signaling. Targeting C1ql3 and iPLA2β that function reciprocally to increase GLP1/cAMP signaling is an innovative strategy to improve β-cell function in obesity/T2D. Excitingly, it could also provide a solution for improving the effectiveness of GLP1 drugs that are used for T2D treatment. An intriguing path is to resolve the previously unrecognized crosstalk between two well-established pathways involving iPLA2β and GLP1 signaling, and how C1ql3 modulates their roles to affect β-cell (dys)function. We posit that the study of a link between iPLA2β and C1ql3 could elucidate the initial signaling events that contribute to β-cell dysfunction in the IGT state associated with obesity before the clinical diagnosis of T2D (61, 93, 211, 233). Table 1 lists the abbreviations used throughout the article.
Table 1.
Abbreviation List
| AA | arachidonic acid |
| AC | adenylate cyclase |
| ADP | adenosine diphosphate |
| AKT | Ak strain transforming serine/threonine kinase 1 |
| AMK | AMP-activated protein kinases |
| ATP | adenosine 5′-triphosphate |
| BAI3 | brain-specific angiogenesis inhibitor 3 |
| Ca2+ | calcium ion |
| [Ca2+]c | cytosolic Ca2+ concentration |
| cAMP | cyclic adenosine monophosphate |
| CICR | Ca2+-induced calcium release |
| C1ql3 | complement 1q-like-3 |
| COX | cyclooxygenase |
| cPLA2 | cytosolic phospholipase A2 |
| cSIS | cAMP-stimulated insulin secretion |
| CTRP13 | C1q/TNF-related protein 13 |
| CUB | complement C1r/C1s, Uegf, Bmp1 |
| CREB | cAMP-response element binding protein |
| CYP | cytochrome P450 |
| DAG | diacylglyceride |
| DGLA | dihomo-γ-linolenic acid |
| DHA | docosahexaenoic acid |
| DHET | dihydroxyeicosatrienoic acids |
| DIO | diet-induced obesity |
| DP2 | prostaglandin D2 receptor |
| EET | epoxyeicosatrienoic acids |
| EP | PGE2 receptor |
| EPA | eicosapentaenoic acid |
| Epac | exchange protein activated by cyclic-AMP |
| FFAR1 | free fatty acid receptor 1 |
| FOXO1 | forkhead box protein O1 |
| FRET | fluorescence resonance energy transfer |
| GAIN | G-protein-coupled receptor (GPCR) autoproteolysis-inducing domain |
| GEF | guanine nucleotide exchange factor |
| GRP | G protein-coupled receptor |
| Gs | Gs protein |
| Gi | Gi protein |
| Gα | Gs alpha subunit |
| GIP | glucose-dependent insulinotropic polypeptide |
| GLP1 | glucagon-like peptide-1 |
| GPCR | G-protein coupled receptor |
| GPE | glycerophosphatidylethanolamine |
| GPR120 | G-protein coupled receptor 120 |
| GPR119 | G-protein coupled receptor 119 |
| GPR44 | G-protein coupled receptor 44 |
| GPS | glycerophosphatidylserine |
| GSIS | glucose-stimulated insulin secretion |
| GWAS | genome-wide association studies |
| HEPE | hydroxyeicosapentaenoic acid |
| HETE | hydroxyeicosatetraenoic acid |
| HFD | high-fat diet |
| IAPP | islet amyloid polypeptide |
| IG | insulin granule |
| IGT | impaired glucose tolerance |
| IP3 | inositol trisphosphate |
| iPLA2 | Ca2+-independent phospholipase A2 |
| iPLA2β | β-isoform of Ca2+-independent phospholipase A2 |
| iPLA2ββKO | select-deficiency of iPLA2β in β-cells |
| iPLA2ββTG | select overexpression of iPLA2β in β-cells |
| JNK1 | c-Jun N-terminal kinase |
| K+ | potassium ion |
| KATP | potassium-sensitive ATP-channel |
| Kv2.1 | delayed rectifier K+-channel |
| LOX | lipo-oxygenase |
| MØ | macrophages |
| M1 | inflammatory MØ phenotype |
| MAFA1 | MAF BZIP Transcription Factor A |
| MUFA | mono-unsaturated fatty acid |
| Munc | mammalian uncoordinated-18 |
| NEFA | nonesterified fatty acid |
| NKX6.2 | NK6 Homeobox 2 |
| PDE | phosphodiesterase |
| PDXl | pancreatic and duodenal homeobox l |
| PDZ | modular protein-protein interacting domain |
| PKA | protein kinase A |
| PGE2 | prostaglandin E2 |
| PGI2 | prostaglandin I2 |
| PKC | protein kinase C |
| PLA2 | phospholipase A2 |
| PLCε | phospholipase C-epsilon |
| PUFAs | polyunsaturated fatty acids |
| P450 | cytochrome P450 |
| PGD2 | prostaglandin D2 |
| PUFA | polyunsaturated fatty acid |
| Rab | Ras superfamily of small G proteins |
| Rap | receptor-associated protein |
| RIα | R-subunit |
| RIM | Rab-interacting molecules |
| RRP | readily releasable pool |
| sEH | soluble epoxide hydrolase |
| siRNA | small interfering RNA |
| SL | sphingolipid |
| SMase | sphingomyelinase |
| SNAP | synaptosomal-associated protein |
| SNARE | soluble N-ethylmaleimide-sensitive factor attachment protein receptor |
| S1P | sphingosine-1-phosphate |
| sPLA2 | secretory phospholipase A2 |
| SU | sulfonylurea |
| SUR1 | sulfonylurea receptor 1 |
| T2D | type 2 diabetes |
| TF | transcription factor |
| Tnf | tumor necrosis factor |
| TORC2 | transducer of regulated CREB activity-2 |
| Vamp2 | vesicle-associated membrane protein 2 |
| VDCC | voltage-dependent Ca2+-channels |
Didactic Synopsis.
Major teaching points
β-Cell dysfunction is observed during impaired glucose tolerance states associated with obesity and type 2 diabetes (T2D).
Insulin secretion from β-cells occurs through well-understood canonical and less-understood noncanonical pathways.
The canonical pathways include: (i) triggering and amplifying pathways that regulate glucose-stimulated insulin secretion (GSIS) and (ii) the amplification of GSIS by activating cAMP signaling via PKA/Epac proteins.
A requisite for optimal stimulus-coupled insulin secretion is the activation of cAMP signaling in β-cells.
Noncanonical pathways include signaling by C1ql3 secreted protein and iPLA2β-derived lipids generated within the β-cells.
C1ql3 and iPLA2β signaling intersect at the cAMP level to produce opposing effects on insulin secretion.
It is speculated that the dysregulation of C1ql3 and iPLA2β proteins contributes to the β-cell dysfunction associated with obesity and T2D.
Detailed studies of these noncanonical pathways will improve our understanding of β-cell secretory function/capacity and offer new therapeutic avenues to counter impaired β-cell function in obesity and T2D.
Acknowledgements
This work was supported by NIH/NIDDK (R01DK126444-01 and R01DK110292), NIH/HIRN (U01DK127786), UAB Department of Cell, Developmental, and Integrative Biology, and UAB Comprehensive Diabetes Center to SR; and SB laboratory has been supported by UAB Department of Medicine, UAB Pilot awards (5P30DK079626-10, 5P30DK079626-10), NIDDK (R01DK120684-01, 1R21DK129968-01, 4R00DK095975-03), and American Diabetes Association (Grant #1-18-PDF-103). The authors would like to thank Dr. Mary Wohltmann, Mr. Alan Bohrer, Dr. Zhongmin (Alex) Ma, Dr. Shunzhong Bao, Dr. Haowei Song, Dr. Douglas Stickle, Dr. Zhepeng Wang, Dr. Willian Nowatzke, Ms. Min Tan, Dr. Chun Jin, Ms. Sheng Zhang, Dr. Xiaoyong Lei, Dr. Rajesh Gupta, Dr. James Koltes, Dr. Itika Arora for their years of dedicated work and contributions to the areas addressed in this article.
References
- 1.Abadpour S, Tyrberg B, Schive SW, Huldt CW, Gennemark P, Ryberg E, Ryden-Bergsten T, Smith DM, Korsgren O, Skrtic S, Scholz H, Winzell MS. Inhibition of the prostaglandin D2-GPR44/DP2 axis improves human islet survival and function. Diabetologia 63: 1355–1367, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbott KA, Burrows TL, Acharya S, Thota RN, Garg ML. DHA-enriched fish oil reduces insulin resistance in overweight and obese adults. Prostaglandins Leukot Essent Fatty Acids 159: 102154, 2020. [DOI] [PubMed] [Google Scholar]
- 3.Abbott KA, Burrows TL, Thota RN, Alex A, Acharya S, Attia J, McEvoy M, Garg ML. Association between plasma phospholipid omega-3 polyunsaturated fatty acids and type 2 diabetes is sex dependent: The Hunter Community Study. Clin Nutr 39: 1059–1066, 2020. [DOI] [PubMed] [Google Scholar]
- 4.Abdul-Ghani MA, Jenkinson CP, Richardson DK, Tripathy D, DeFronzo RA. Insulin secretion and action in subjects with impaired fasting glucose and impaired glucose tolerance: Results from the Veterans Administration Genetic Epidemiology Study. Diabetes 55: 1430–1435, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Abdul-Ghani MA, Tripathy D, DeFronzo RA. Contributions of beta-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care 29: 1130–1139, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Adams JM 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 53: 25–31, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Afrookhteh A, Emamgholipour S, Alipoor B, Moradi N, Meshkani R, Nasli-Esfahani E, Rahimipour A, Shanaki M. The circulating levels of complement-C1q/TNF-related protein 13 (CTRP13) in patients with type 2 diabetes and its association with insulin resistance. Clin Lab 63: 327–333, 2017. [DOI] [PubMed] [Google Scholar]
- 8.Aigha II, Abdelalim EM. NKX6.1 transcription factor: A crucial regulator of pancreatic beta cell development, identity, and proliferation. Stem Cell Res Ther 11: 459, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akita Y Protein kinase C-epsilon (PKC-epsilon): Its unique structure and function. J Biochem 132: 847–852, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Albert BB, Derraik JG, Brennan CM, Biggs JB, Smith GC, Garg ML, Cameron-Smith D, Hofman PL, Cutfield WS. Higher omega-3 index is associated with increased insulin sensitivity and more favourable metabolic profile in middle-aged overweight men. Sci Rep 4: 6697, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alenkvist I, Gandasi NR, Barg S, Tengholm A. Recruitment of Epac2A to insulin granule docking sites regulates priming for exocytosis. Diabetes 66: 2610–2622, 2017. [DOI] [PubMed] [Google Scholar]
- 12.Ammala C, Ashcroft FM, Rorsman P. Calcium-independent potentiation of insulin release by cyclic AMP in single beta-cells. Nature 363: 356–358, 1993. [DOI] [PubMed] [Google Scholar]
- 13.Anderson BM, Ma DW. Are all n-3 polyunsaturated fatty acids created equal? Lipids Health Dis 8: 33, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andrews DL, Beames B, Summers MD, Park WD. Characterization of the lipid acyl hydrolase activity of the major potato (Solanum tuberosum) tuber protein, patatin, by cloning and abundant expression in a baculovirus vector. Biochem J 252: 199–206, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antoni G, Morange PE, Luo Y, Saut N, Burgos G, Heath S, Germain M, Biron-Andreani C, Schved JF, Pernod G, Galan P, Zelenika D, Alessi MC, Drouet L, Visvikis-Siest S, Wells PS, Lathrop M, Emmerich J, Tregouet DA, Gagnon F. A multi-stage multi-design strategy provides strong evidence that the BAI3 locus is associated with early-onset venous thromboembolism. J Thromb Haemost 8: 2671–2679, 2010. [DOI] [PubMed] [Google Scholar]
- 16.Araki K, Araki A, Honda D, Izumoto T, Hashizume A, Hijikata Y, Yamada S, Iguchi Y, Hara A, Ikumi K, Kawai K, Ishigaki S, Nakamichi Y, Tsunekawa S, Seino Y, Yamamoto A, Takayama Y, Hidaka S, Tominaga M, Ohara-Imaizumi M, Suzuki A, Ishiguro H, Enomoto A, Yoshida M, Arima H, Muramatsu SI, Sobue G, Katsuno M. TDP-43 regulates early-phase insulin secretion via CaV1.2-mediated exocytosis in islets. J Clin Invest 129: 3578–3593, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashley JW, Hancock WD, Nelson AJ, Bone RN, Tse HM, Wohltmann M, Turk J, Ramanadham S. Polarization of macrophages toward M2 phenotype is favored by reduction in iPLA2beta (group VIA phospholipase A2). J Biol Chem 291: 23268–23281, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aslamy A, Thurmond DC. Exocytosis proteins as novel targets for diabetes prevention and/or remediation? Am J Physiol Regul Integr Comp Physiol 312: R739–R752, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atsumi G, Murakami M, Kojima K, Hadano A, Tajima M, Kudo I. Distinct roles of two intracellular phospholipase A2s in fatty acid release in the cell death pathway. Proteolytic fragment of type IVA cytosolic phospholipase A2alpha inhibits stimulus-induced arachidonate release, whereas that of type VI Ca2+-independent phospholipase A2 augments spontaneous fatty acid release. J Biol Chem 275: 18248–18258, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Aulinger BA, Vahl TP, Wilson-Perez HE, Prigeon RL, D’Alessio DA. beta-Cell Sensitivity to GLP-1 in healthy humans is variable and proportional to insulin sensitivity. J Clin Endocrinol Metab 100: 2489–2496, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bae JS, Kim TH, Kim MY, Park JM, Ahn YH. Transcriptional regulation of glucose sensors in pancreatic beta-cells and liver: An update. Sensors 10: 5031–5053, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balboa MA, Balsinde J, Jones SS, Dennis EA. Identity between the Ca2+-independent phospholipase A2 enzymes from P388D1 macrophages and Chinese hamster ovary cells. J Biol Chem 272: 8576–8580, 1997. [DOI] [PubMed] [Google Scholar]
- 23.Bao S, Jacobson DA, Wohltmann M, Bohrer A, Jin W, Philipson LH, Turk J. Glucose homeostasis, insulin secretion, and islet phospholipids in mice that overexpress iPLA2beta in pancreatic beta-cells and in iPLA2beta-null mice. Am J Physiol Endocrinol Metab 294: E217–E229, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bao S, Song H, Wohltmann M, Ramanadham S, Jin W, Bohrer A, Turk J. Insulin secretory responses and phospholipid composition of pancreatic islets from mice that do not express Group VIA phospholipase A2 and effects of metabolic stress on glucose homeostasis. J Biol Chem 281: 20958–20973, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beguin P, Nagashima K, Nishimura M, Gonoi T, Seino S. PKA-mediated phosphorylation of the human KATP channel: Separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J 18: 4722–4732, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Sudhof TC, Rettig J, Brose N. Munc13–1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron 21: 123–136, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Bhaswant M, Poudyal H, Brown L. Mechanisms of enhanced insulin secretion and sensitivity with n-3 unsaturated fatty acids. J Nutr Biochem 26: 571–584, 2015. [DOI] [PubMed] [Google Scholar]
- 28.Biden TJ, Peter-Riesch B, Schlegel W, Wollheim CB. Ca2+-mediated generation of inositol 1,4,5-triphosphate and inositol 1,3,4,5-tetrakisphosphate in pancreatic islets. Studies with K+, glucose, and carbamylcholine. J Biol Chem 262: 3567–3571, 1987. [PubMed] [Google Scholar]
- 29.Black MA, Heick HM, Begin-Heick N. Abnormal regulation of cAMP accumulation in pancreatic islets of obese mice. Am J Phys 255: E833–E838, 1988. [DOI] [PubMed] [Google Scholar]
- 30.Blanchet E, Van de Velde S, Matsumura S, Hao E, LeLay J, Kaestner K, Montminy M. Feedback inhibition of CREB signaling promotes beta cell dysfunction in insulin resistance. Cell Rep 10: 1149–1157, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blum B, Hrvatin S, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol 30: 261–264, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boden G Ceramide: A contributor to insulin resistance or an innocent bystander? Diabetologia 51: 1095–1096, 2008. [DOI] [PubMed] [Google Scholar]
- 33.Bolliger MF, Martinelli DC, Sudhof TC. The cell-adhesion G protein-coupled receptor BAI3 is a high-affinity receptor for C1q-like proteins. Proc Natl Acad Sci U S A 108: 2534–2539, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borge PD, Moibi J, Greene SR, Trucco M, Young RA, Gao Z, Wolf BA. Insulin receptor signaling and sarco/endoplasmic reticulum calcium ATPase in beta-cells. Diabetes 51 (Suppl 3): S427–S433, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Boslem E, Meikle PJ, Biden TJ. Roles of ceramide and sphingolipids in pancreatic beta-cell function and dysfunction. Islets 4: 177–187, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bosma KJ, Andrei SR, Katz LS, Smith AA, Dunn JC, Ricciardi VF, Ramirez MA, Baumel-Alterzon S, Pace WA, Carroll DT, Overway EM, Wolf EM, Kimple ME, Sheng Q, Scott DK, Breyer RM, Gannon M. Pharmacological blockade of the EP3 prostaglandin E2 receptor in the setting of type 2 diabetes enhances beta-cell proliferation and identity and relieves oxidative damage. Mol Metab 54: 101347, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bosma KJ, Kaiser CE, Kimple ME, Gannon M. Effects of arachidonic acid and its metabolites on functional beta-cell mass. Metabolites 12: 342, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bourron O, Chebbi F, Halbron M, Saint-Martin C, Bellanne-Chantelot C, Abed A, Charbit B, Magnan C, Lacorte JM, Hartemann A. Incretin effect of glucagon-like peptide 1 receptor agonist is preserved in presence of ABCC8/SUR1 mutation in beta-cell. Diabetes Care 35: e76, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR, Rorsman P. Voltage-gated ion channels in human pancreatic beta-cells: Electrophysiological characterization and role in insulin secretion. Diabetes 57: 1618–1628, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Braun M, Ramracheya R, Rorsman P. Autocrine regulation of insulin secretion. Diabetes Obes Metab 14 (Suppl 3): 143–151, 2012. [DOI] [PubMed] [Google Scholar]
- 41.Burke JE, Dennis EA. Phospholipase A2 biochemistry. Cardiovasc Drugs Ther 23: 49–59, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res 50 (Suppl): S237–S242, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Byerly MS, Swanson R, Wei Z, Seldin MM, McCulloh PS, Wong GW. A central role for C1q/TNF-related protein 13 (CTRP13) in modulating food intake and body weight. PLoS One 8: e62862, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paracrine Caicedo A. and autocrine interactions in the human islet: More than meets the eye. Semin Cell Dev Biol 24: 11–21, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campana M, Bellini L, Rouch C, Rachdi L, Coant N, Butin N, Bandet CL, Philippe E, Meneyrol K, Kassis N, Dairou J, Hajduch E, Colsch B, Magnan C, Le Stunff H. Inhibition of central de novo ceramide synthesis restores insulin signaling in hypothalamus and enhances beta-cell function of obese Zucker rats. Mol Metab 8: 23–36, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cantrell Stanford J, Morris AJ, Sunkara M, Popa GJ, Larson KL, Ozcan S. Sphingosine 1-phosphate (S1P) regulates glucosestimulated insulin secretion in pancreatic beta cells. J Biol Chem 287: 13457–13464, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carboneau BA, Breyer RM, Gannon M. Regulation of pancreatic beta-cell function and mass dynamics by prostaglandin signaling. J Cell Commun Signal 11: 105–116, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cen J, Sargsyan E, Bergsten P. Fatty acids stimulate insulin secretion from human pancreatic islets at fasting glucose concentrations via mitochondria-dependent and -independent mechanisms. Nutr Metab 13: 59, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chamulitrat W, Jansakun C, Li H, Liebisch G. Rescue of hepatic phospholipid remodeling Defectin iPLA2beta-null mice attenuates obese but not non-obese fatty liver. Biomolecules 10, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Charles MA, Lawecki J, Pictet R, Grodsky GM. Insulin secretion. Interrelationships of glucose, cyclic adenosine 3:5-monophosphate, and calcium. J Biol Chem 250: 6134–6140, 1975. [PubMed] [Google Scholar]
- 51.Chepurny OG, Kelley GG, Dzhura I, Leech CA, Roe MW, Dzhura E, Li X, Schwede F, Genieser HG, Holz GG. PKA-dependent potentiation of glucose-stimulated insulin secretion by Epac activator 8-pCPT-2’-O-Me-cAMP-AM in human islets of Langerhans. Am J Physiol Endocrinol Metab 298: E622–E633, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Christensen AA, Gannon M. The beta-cell in type 2 diabetes. Curr Diab Rep 19: 81, 2019. [DOI] [PubMed] [Google Scholar]
- 53.Chueire VB, Muscelli E. Effect of free fatty acids on insulin secretion, insulin sensitivity and incretin effect—A narrative review. Arch Endocrinol Metab 65: 24–31, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cnop M, Vidal J, Hull RL, Utzschneider KM, Carr DB, Schraw T, Scherer PE, Boyko EJ, Fujimoto WY, Kahn SE. Progressive loss of beta-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes. Diabetes Care 30: 677–682, 2007. [DOI] [PubMed] [Google Scholar]
- 55.Collet-Gaudillat C, Petit-Aubert G, Desforges-Bullet V, Beressi JP. Exenatide efficacy in unselected patients: comparison with clinical trials. J Diabetes Mellitus 2: 118–121, 2012. [Google Scholar]
- 56.Corbett JA, Wang JL, Misko TP, Zhao W, Hickey WF, McDaniel ML. Nitric oxide mediates IL-1 beta-induced islet dysfunction and destruction: prevention by dexamethasone. Autoimmunity 15: 145–153, 1993. [DOI] [PubMed] [Google Scholar]
- 57.Cork SM, Van Meir EG. Emerging roles for the BAI1 protein family in the regulation of phagocytosis, synaptogenesis, neurovasculature, and tumor development. J Mol Med 89: 743–752, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dai XQ, Manning Fox JE, Chikvashvili D, Casimir M, Plummer G, Hajmrle C, Spigelman AF, Kin T, Singer-Lahat D, Kang Y, Shapiro AM, Gaisano HY, Lotan I, Macdonald PE. The voltage-dependent potassium channel subunit Kv2.1 regulates insulin secretion from rodent and human islets independently of its electrical function. Diabetologia 55: 1709–1720, 2012. [DOI] [PubMed] [Google Scholar]
- 59.Daniel S, Noda M, Straub SG, Sharp GW. Identification of the docked granule pool responsible for the first phase of glucose-stimulated insulin secretion. Diabetes 48: 1686–1690, 1999. [DOI] [PubMed] [Google Scholar]
- 60.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 58: 773–795, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32 (Suppl 2): S157–S163, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Delmeire D, Flamez D, Hinke SA, Cali JJ, Pipeleers D, Schuit F. Type VIII adenylyl cyclase in rat beta cells: Coincidence signal detector/generator for glucose and GLP-1. Diabetologia 46: 1383–1393, 2003. [DOI] [PubMed] [Google Scholar]
- 63.Deng X, Wang J, Jiao L, Utaipan T, Tuma-Kellner S, Schmitz G, Liebisch G, Stremmel W, Chamulitrat W. iPLA2beta deficiency attenuates obesity and hepatic steatosis in ob/ob mice through hepatic fatty-acyl phospholipid remodeling. Biochim Biophys Acta 449–461: 2016, 1861. [DOI] [PubMed] [Google Scholar]
- 64.Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev 111: 6130–6185, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dezaki K, Damdindorj B, Sone H, Dyachok O, Tengholm A, Gylfe E, Kurashina T, Yoshida M, Kakei M, Yada T. Ghrelin attenuates cAMP-PKA signaling to evoke insulinostatic cascade in islet beta-cells. Diabetes 60: 2315–2324, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dixon G, Nolan J, McClenaghan NH, Flatt PR, Newsholme P. Arachidonic acid, palmitic acid and glucose are important for the modulation of clonal pancreatic beta-cell insulin secretion, growth and functional integrity. Clin Sci 106: 191–199, 2004. [DOI] [PubMed] [Google Scholar]
- 67.Dobbins RL, Chester MW, Daniels MB, McGarry JD, Stein DT. Circulating fatty acids are essential for efficient glucose-stimulated insulin secretion after prolonged fasting in humans. Diabetes 47: 1613–1618, 1998. [DOI] [PubMed] [Google Scholar]
- 68.Donath MY. Targeting inflammation in the treatment of type 2 diabetes: Time to start. Nat Rev Drug Discov 13: 465–476, 2014. [DOI] [PubMed] [Google Scholar]
- 69.Dou H, Wang C, Wu X, Yao L, Zhang X, Teng S, Xu H, Liu B, Wu Q, Zhang Q, Hu M, Wang Y, Wang L, Wu Y, Shang S, Kang X, Zheng L, Zhang J, Raoux M, Lang J, Li Q, Su J, Yu X, Chen L, Zhou Z. Calcium influx activates adenylyl cyclase 8 for sustained insulin secretion in rat pancreatic beta cells. Diabetologia 58: 324–333, 2015. [DOI] [PubMed] [Google Scholar]
- 70.Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab 27: 740–756, 2018. [DOI] [PubMed] [Google Scholar]
- 71.Du YQ, Sha XY, Cheng J, Wang J, Lin JY, An WT, Pan W, Zhang LJ, Tao XN, Xu YF, Jia YL, Yang Z, Xiao P, Liu M, Sun JP, Yu X. Endogenous lipid-GPR120 signaling modulates pancreatic islet homeostasis to different extents. Diabetes 71: 1454–1471, 2022. [DOI] [PubMed] [Google Scholar]
- 72.Dulubova I, Lou X, Lu J, Huryeva I, Alam A, Schneggenburger R, Sudhof TC, Rizo J. A Munc13/RIM/Rab3 tripartite complex: From priming to plasticity? EMBO J 24: 2839–2850, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dunaif A, Finegood DT. Beta-cell dysfunction independent of obesity and glucose intolerance in the polycystic ovary syndrome. J Clin Endocrinol Metab 81: 942–947, 1996. [DOI] [PubMed] [Google Scholar]
- 74.Dyachok O, Gylfe E. Ca2+-induced Ca2+ release via inositol 1,4,5-trisphosphate receptors is amplified by protein kinase A and triggers exocytosis in pancreatic beta-cells. J Biol Chem 279: 45455–45461, 2004. [DOI] [PubMed] [Google Scholar]
- 75.Dyachok O, Idevall-Hagren O, Sagetorp J, Tian G, Wuttke A, Arrieumerlou C, Akusjarvi G, Gylfe E, Tengholm A. Glucose-induced cyclic AMP oscillations regulate pulsatile insulin secretion. Cell Metab 8: 26–37, 2008. [DOI] [PubMed] [Google Scholar]
- 76.Dzhura I, Chepurny OG, Leech CA, Roe MW, Dzhura E, Xu X, Lu Y, Schwede F, Genieser HG, Smrcka AV, Holz GG. Phospholipase C-epsilon links Epac2 activation to the potentiation of glucosestimulated insulin secretion from mouse islets of Langerhans. Islets 3: 121–128, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.El-Azzouny M, Evans CR, Treutelaar MK, Kennedy RT, Burant CF. Increased glucose metabolism and glycerolipid formation by fatty acids and GPR40 receptor signaling underlies the fatty acid potentiation of insulin secretion. J Biol Chem 289: 13575–13588, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eliasson L, Ma X, Renstrom E, Barg S, Berggren PO, Galvanovskis J, Gromada J, Jing X, Lundquist I, Salehi A, Sewing S, Rorsman P. SUR1 regulates PKA-independent cAMP-induced granule priming in mouse pancreatic B-cells. J Gen Physiol 121: 181–197, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.English A, Irwin N. Nonclassical islet peptides: Pancreatic and extrapancreatic actions. Clin Med Insights Endocrinol Diabetes 12: 1179551419888871, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Erbas IM, Paketci A, Turan S, Sisman AR, Demir K, Bober E, Abaci A. Low complement C1q/TNF-related protein-13 levels are associated with childhood obesity but not binge eating disorder. J Clin Res Pediatr Endocrinol 14: 179–187, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Esser N, Utzschneider KM, Kahn SE. Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia 63: 2007–2021, 2020. [DOI] [PubMed] [Google Scholar]
- 82.Fadaei R, Moradi N, Baratchian M, Aghajani H, Malek M, Fazaeli AA, Fallah S. Association of C1q/TNF-related protein-3 (CTRP3) and CTRP13 serum levels with coronary artery disease in subjects with and without type 2 diabetes mellitus. PLoS One 11: e0168773, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Falck JR, Manna S, Moltz J, Chacos N, Capdevila J. Epoxyeicosatrienoic acids stimulate glucagon and insulin release from isolated rat pancreatic islets. Biochem Biophys Res Commun 114: 743–749, 1983. [DOI] [PubMed] [Google Scholar]
- 84.Fava GE, Dong EW, Wu H. Intra-islet glucagon-like peptide 1. J Diabetes Complicat 30: 1651–1658, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA. beta-Cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: A new analysis. J Clin Endocrinol Metab 90: 493–500, 2005. [DOI] [PubMed] [Google Scholar]
- 86.Festa A, Williams K, D’Agostino R Jr, Wagenknecht LE, Haffner SM. The natural course of beta-cell function in nondiabetic and diabetic individuals: The insulin resistance Atherosclerosis Study. Diabetes 55: 1114–1120, 2006. [DOI] [PubMed] [Google Scholar]
- 87.Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci Signal 3: ra70, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Furman B, Ong WK, Pyne NJ. Cyclic AMP signaling in pancreatic islets. Adv Exp Med Biol 654: 281–304, 2010. [DOI] [PubMed] [Google Scholar]
- 89.Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, Ostolaza H, Martin C. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci 21, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gangadhariah MH, Dieckmann BW, Lantier L, Kang L, Wasserman DH, Chiusa M, Caskey CF, Dickerson J, Luo P, Gamboa JL, Capdevila JH, Imig JD, Yu C, Pozzi A, Luther JM. Cytochrome P450 epoxygenase-derived epoxyeicosatrienoic acids contribute to insulin sensitivity in mice and in humans. Diabetologia 60: 1066–1075, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gembal M, Detimary P, Gilon P, Gao ZY, Henquin JC. Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J Clin Invest 91: 871–880, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gembal M, Gilon P, Henquin JC. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J Clin Invest 89: 1288–1295, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gerich JE. Is reduced first-phase insulin release the earliest detectable abnormality in individuals destined to develop type 2 diabetes? Diabetes 51 (Suppl 1): S117–S121, 2002. [DOI] [PubMed] [Google Scholar]
- 94.Ghai R, Waters P, Roumenina LT, Gadjeva M, Kojouharova MS, Reid KB, Sim RB, Kishore U. C1q and its growing family. Immunobiology 212: 253–266, 2007. [DOI] [PubMed] [Google Scholar]
- 95.Gibson TB, Lawrence MC, Gibson CJ, Vanderbilt CA, McGlynn K, Arnette D, Chen W, Collins J, Naziruddin B, Levy MF, Ehrlich BE, Cobb MH. Inhibition of glucose-stimulated activation of extracellular signal-regulated protein kinases 1 and 2 by epinephrine in pancreatic beta-cells. Diabetes 55: 1066–1073, 2006. [DOI] [PubMed] [Google Scholar]
- 96.Gijon MA, Leslie CC. Phospholipases A2. Semin Cell Dev Biol 8: 297–303, 1997. [DOI] [PubMed] [Google Scholar]
- 97.Gil-de-Gomez L, Astudillo AM, Guijas C, Magrioti V, Kokotos G, Balboa MA, Balsinde J. Cytosolic group IVA and calcium-independent group VIA phospholipase A2s act on distinct phospholipid pools in zymosan-stimulated mouse peritoneal macrophages. J Immunol 192: 752–762, 2014. [DOI] [PubMed] [Google Scholar]
- 98.Gillis KD, Misler S. Enhancers of cytosolic cAMP augment depolarization-induced exocytosis from pancreatic B-cells: Evidence for effects distal to Ca2+ entry. Pflugers Arch 424: 195–197, 1993. [DOI] [PubMed] [Google Scholar]
- 99.Gloerich M, Bos JL. Epac: Defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol 50: 355–375, 2010. [DOI] [PubMed] [Google Scholar]
- 100.Gross RW, Ramanadham S, Kruszka KK, Han X, Turk J. Rat and human pancreatic islet cells contain a calcium ion independent phospholipase A2 activity selective for hydrolysis of arachidonate which is stimulated by adenosine triphosphate and is specifically localized to islet beta-cells. Biochemistry 32: 327–336, 1993. [DOI] [PubMed] [Google Scholar]
- 101.Gucek A, Gandasi NR, Omar-Hmeadi M, Bakke M, Doskeland SO, Tengholm A, Barg S. Fusion pore regulation by cAMP/Epac2 controls cargo release during insulin exocytosis. elife 8: e41711, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Guenifi A, Portela-Gomes GM, Grimelius L, Efendic S, Abdel-Halim SM. Adenylyl cyclase isoform expression in non-diabetic and diabetic Goto-Kakizaki (GK) rat pancreas. Evidence for distinct overexpression of type-8 adenylyl cyclase in diabetic GK rat islets. Histochem Cell Biol 113: 81–89, 2000. [DOI] [PubMed] [Google Scholar]
- 103.Guo J, Qian Y, Xi X, Hu X, Zhu J, Han X. Blockage of ceramide metabolism exacerbates palmitate inhibition of pro-insulin gene expression in pancreatic beta-cells. Mol Cell Biochem 338: 283–290, 2010. [DOI] [PubMed] [Google Scholar]
- 104.Gupta R, Nguyen DC, Schaid MD, Lei X, Balamurugan AN, Wong GW, Kim JA, Koltes JE, Kimple ME, Bhatnagar S. Complement 1q-like-3 protein inhibits insulin secretion from pancreatic β-cells via the cell adhesion G protein-coupled receptor BAI3. J Biol Chem 293: 18086–18098, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gurgul-Convey E, Hanzelka K, Lenzen S. Mechanism of prostacyclin-induced potentiation of glucose-induced insulin secretion. Endocrinology 153: 2612–2622, 2012. [DOI] [PubMed] [Google Scholar]
- 106.Hamoud N, Tran V, Aimi T, Kakegawa W, Lahaie S, Thibault MP, Pelletier A, Wong GW, Kim IS, Kania A, Yuzaki M, Bouvier M, Côté JF. Spatiotemporal regulation of the GPCR activity of BAI3 by C1qL4 and Stabilin-2 controls myoblast fusion. Nat Commun 9: 4470, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Harris TR, Hammock BD. Soluble epoxide hydrolase: Gene structure, expression and deletion. Gene 526: 61–74, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA, Kirwan JP. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 58: 337–343, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hayes MG, Pluzhnikov A, Miyake K, Sun Y, Ng MC, Roe CA, Below JE, Nicolae RI, Konkashbaev A, Bell GI, Cox NJ, Hanis CL. Identification of type 2 diabetes genes in Mexican Americans through genome-wide association studies. Diabetes 56: 3033–3044, 2007. [DOI] [PubMed] [Google Scholar]
- 110.Hazen SL, Ford DA, Gross RW. Activation of a membrane-associated phospholipase A2 during rabbit myocardial ischemia which is highly selective for plasmalogen substrate. J Biol Chem 266: 5629–5633, 1991. [PubMed] [Google Scholar]
- 111.Hazen SL, Gross RW. ATP-dependent regulation of rabbit myocardial cytosolic calcium-independent phospholipase A2. J Biol Chem 266: 14526–14534, 1991. [PubMed] [Google Scholar]
- 112.Hazen SL, Stuppy RJ, Gross RW. Purification and characterization of canine myocardial cytosolic phospholipase A2. A calcium-independent phospholipase with absolute f1–2 regiospecificity for diradyl glycerophospholipids. J Biol Chem 265: 10622–10630, 1990. [PubMed] [Google Scholar]
- 113.Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2. Mechanism-based discrimination between calcium-dependent and -independent phospholipases A2. J Biol Chem 266: 7227–7232, 1991. [PubMed] [Google Scholar]
- 114.Heart E, Corkey RF, Wikstrom JD, Shirihai OS, Corkey BE. Glucose-dependent increase in mitochondrial membrane potential, but not cytoplasmic calcium, correlates with insulin secretion in single islet cells. Am J Physiol Endocrinol Metab 290: E143–E148, 2006. [DOI] [PubMed] [Google Scholar]
- 115.Hellstrom-Lindahl E, Danielsson A, Ponten F, Czernichow P, Korsgren O, Johansson L, Eriksson O. GPR44 is a pancreatic protein restricted to the human beta cell. Acta Diabetol 53: 413–421, 2016. [DOI] [PubMed] [Google Scholar]
- 116.Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49: 1751–1760, 2000. [DOI] [PubMed] [Google Scholar]
- 117.Henquin JC. Pathways in beta-cell stimulus-secretion coupling as targets for therapeutic insulin secretagogues. Diabetes 53 (Suppl 3): S48–S58, 2004. [DOI] [PubMed] [Google Scholar]
- 118.Henquin JC. Regulation of insulin secretion: A matter of phase control and amplitude modulation. Diabetologia 52: 739–751, 2009. [DOI] [PubMed] [Google Scholar]
- 119.Henquin JC. Paracrine and autocrine control of insulin secretion in human islets: Evidence and pending questions. Am J Physiol Endocrinol Metab 320: E78–E86, 2021. [DOI] [PubMed] [Google Scholar]
- 120.Henquin JC, Nenquin M. Activators of PKA and Epac distinctly influence insulin secretion and cytosolic Ca2+ in female mouse islets stimulated by glucose and tolbutamide. Endocrinology 155: 3274–3287, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Henquin JC, Nenquin M, Stiernet P, Ahren B. In vivo and in vitro glucose-induced biphasic insulin secretion in the mouse: Pattern and role of cytoplasmic Ca2+ and amplification signals in beta-cells. Diabetes 55: 441–451, 2006. [DOI] [PubMed] [Google Scholar]
- 122.Herbst KJ, Coltharp C, Amzel LM, Zhang J. Direct activation of Epac by sulfonylurea is isoform selective. Chem Biol 18: 243–251, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hisatomi M, Hidaka H, Niki I. Ca2+/calmodulin and cyclic 3,5’ adenosine monophosphate control movement of secretory granules through protein phosphorylation/dephosphorylation in the pancreatic beta-cell. Endocrinology 137: 4644–4649, 1996. [DOI] [PubMed] [Google Scholar]
- 124.Hodson DJ, Mitchell RK, Marselli L, Pullen TJ, Gimeno Brias S, Semplici F, Everett KL, Cooper DM, Bugliani M, Marchetti P, Lavallard V, Bosco D, Piemonti L, Johnson PR, Hughes SJ, Li D, Li WH, Shapiro AM, Rutter GA. ADCY5 couples glucose to insulin secretion in human islets. Diabetes 63: 3009–3021, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5: 167–179, 2007. [DOI] [PubMed] [Google Scholar]
- 126.Holz GG. Epac: A new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes 53: 5–13, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Holz GG, Chepurny OG, Leech CA. Epac2A makes a new impact in beta-cell biology. Diabetes 62: 2665–2666, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Holz GG, Leech CA, Chepurny OG. New insights concerning the molecular basis for defective glucoregulation in soluble adenylyl cyclase knockout mice. Biochim Biophys Acta 2593–2600: 2014, 1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hosokawa H, Corkey BE, Leahy JL. Beta-cell hypersensitivity to glucose following 24-h exposure of rat islets to fatty acids. Diabetologia 40: 392–397, 1997. [DOI] [PubMed] [Google Scholar]
- 130.Hsu WH, Xiang HD, Rajan AS, Boyd AE 3rd. Activation of alpha 2-adrenergic receptors decreases Ca2+ influx to inhibit insulin secretion in a hamster beta-cell line: An action mediated by a guanosine triphosphate-binding protein. Endocrinology 128: 958–964, 1991. [DOI] [PubMed] [Google Scholar]
- 131.Huang H, Weng J, Wang MH. EETs/sEH in diabetes and obesity-induced cardiovascular diseases. Prostaglandins Other Lipid Mediat 125: 80–89, 2016. [DOI] [PubMed] [Google Scholar]
- 132.Huang JL, Lee S, Hoek P, van der Meulen T, Van R, Huising MO. Genetic deletion of Urocortin 3 does not prevent functional maturation of beta cells. J Endocrinol 246: 69–78, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hudish LI, Reusch JE, Sussel L. beta Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J Clin Invest 129: 4001–4008, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Huising MO. Paracrine regulation of insulin secretion. Diabetologia 63: 2057–2063, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hull RL, Kodama K, Utzschneider KM, Carr DB, Prigeon RL, Kahn SE. Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: Evidence for specificity of impaired beta cell adaptation. Diabetologia 48: 1350–1358, 2005. [DOI] [PubMed] [Google Scholar]
- 136.Hussain MA, Stratakis C, Kirschner L. Prkar1a in the regulation of insulin secretion. Horm Metab Res 44: 759–765, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hwang M, Go Y, Park JH, Shin SK, Song SE, Oh BC, Im SS, Hwang I, Jeon YH, Lee IK, Seino S, Song DK. Epac2a-null mice exhibit obesity-prone nature more susceptible to leptin resistance. Int J Obes 41: 279–288, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Idevall-Hagren O, Barg S, Gylfe E, Tengholm A. cAMP mediators of pulsatile insulin secretion from glucose-stimulated single beta-cells. J Biol Chem 285: 23007–23018, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Iezzi M, Regazzi R, Wollheim CB. The Rab3-interacting molecule RIM is expressed in pancreatic beta-cells and is implicated in insulin exocytosis. FEBS Lett 474: 66–70, 2000. [DOI] [PubMed] [Google Scholar]
- 140.Imai Y, Dobrian AD, Weaver JR, Butcher MJ, Cole BK, Galkina EV, Morris MA, Taylor-Fishwick DA, Nadler JL. Interaction between cytokines and inflammatory cells in islet dysfunction, insulin resistance and vascular disease. Diabetes Obes Metab 15 (Suppl 3): 117–129, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Inaishi J, Saisho Y. Beta-cell mass in obesity and type 2 diabetes, and its relation to pancreas fat: A mini-review. Nutrients 12, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422: 173–176, 2003. [DOI] [PubMed] [Google Scholar]
- 143.Jacobson DA, Shyng SL. Ion channels of the islets in type 2 diabetes. J Mol Biol 432: 1326–1346, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jacobson DA, Weber CR, Bao S, Turk J, Philipson LH. Modulation of the pancreatic islet beta-cell-delayed rectifier potassium channel Kv2.1 by the polyunsaturated fatty acid arachidonate. J Biol Chem 282: 7442–7449, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jarrard RE, Wang Y, Salyer AE, Pratt EP, Soderling IM, Guerra ML, Lange AM, Broderick HJ, Hockerman GH. Potentiation of sulfonylurea action by an EPAC-selective cAMP analog in INS-1 cells: Comparison of tolbutamide and gliclazide and a potential role for EPAC activation of a 2-APB-sensitive Ca2+ influx. Mol Pharmacol 83: 191–205, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem 279: 48968–48975, 2004. [DOI] [PubMed] [Google Scholar]
- 147.Jensen CC, Cnop M, Hull RL, Fujimoto WY, Kahn SE, American Diabetes Association GSG. Beta-cell function is a major contributor to oral glucose tolerance in high-risk relatives of four ethnic groups in the U.S. Diabetes 51: 2170–2178, 2002. [DOI] [PubMed] [Google Scholar]
- 148.Jewell JL, Oh E, Thurmond DC. Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. Am J Physiol Regul Integr Comp Physiol 298: R517–R531, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M, Montminy M. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev 17: 1575–1580, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Johnson AD, O’Donnell CJ. An open access database of genome-wide association results. BMC Med Genet 10: 6, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jonas JC, Li G, Palmer M, Weller U, Wollheim CB. Dynamics of Ca2+ and guanosine 5’-[gamma-thio]triphosphate action on insulin secretion from alpha-toxin-permeabilized HIT-T15 cells. Biochem J 301 (Pt 2): 523–529, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jones PM, Burns CJ, Belin VD, Roderigo-Milne HM, Persaud SJ. The role of cytosolic phospholipase A2 in insulin secretion. Diabetes 53 (Suppl 1): S172–S178, 2004. [DOI] [PubMed] [Google Scholar]
- 153.Jones PM, Stutchfield J, Howell SL. Effects of Ca2+ and a phorbol ester on insulin secretion from islets of Langerhans permeabilised by high-voltage discharge. FEBS Lett 191: 102–106, 1985. [DOI] [PubMed] [Google Scholar]
- 154.Joseph JW, Koshkin V, Saleh MC, Sivitz WI, Zhang CY, Lowell BB, Chan CB, Wheeler MB. Free fatty acid-induced beta-cell defects are dependent on uncoupling protein 2 expression. J Biol Chem 279: 51049–51056, 2004. [DOI] [PubMed] [Google Scholar]
- 155.Juhl K, Hoy M, Olsen HL, Bokvist K, Efanov AM, Hoffmann EK, Gromada J. cPLA2alpha-evoked formation of arachidonic acid and lysophospholipids is required for exocytosis in mouse pancreatic beta-cells. Am J Physiol Endocrinol Metab 285: E73–E81, 2003. [DOI] [PubMed] [Google Scholar]
- 156.Jump DB. The biochemistry of n-3 polyunsaturated fatty acids. J Biol Chem 277: 8755–8758, 2002. [DOI] [PubMed] [Google Scholar]
- 157.Kaihara KA, Dickson LM, Jacobson DA, Tamarina N, Roe MW, Philipson LH, Wicksteed B. Beta-Cell-specific protein kinase A activation enhances the efficiency of glucose control by increasing acute-phase insulin secretion. Diabetes 62: 1527–1536, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kalwat MA, Cobb MH. Mechanisms of the amplifying pathway of insulin secretion in the beta cell. Pharmacol Ther 179: 17–30, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, Ohtsuka-Kowatari N, Kumagai K, Sakamoto K, Kobayashi M, Yamauchi T, Ueki K, Oishi Y, Nishimura S, Manabe I, Hashimoto H, Ohnishi Y, Ogata H, Tokuyama K, Tsunoda M, Ide T, Murakami K, Nagai R, Kadowaki T. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem 281: 26602–26614, 2006. [DOI] [PubMed] [Google Scholar]
- 160.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116: 1494–1505, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kaneto H, Kimura T, Shimoda M, Obata A, Sanada J, Fushimi Y, Nakanishi S, Mune T, Kaku K. Favorable effects of GLP-1 receptor agonist against pancreatic beta-cell glucose toxicity and the development of arteriosclerosis: "The earlier, the better" in therapy with incretin-based medicine. Int J Mol Sci 22, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kang G, Chepurny OG, Rindler MJ, Collis L, Chepurny Z, Li WH, Harbeck M, Roe MW, Holz GG. A cAMP and Ca2+ coincidence detector in support of Ca2+-induced Ca2+ release in mouse pancreatic beta cells. J Physiol 566: 173–188, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kang G, Joseph JW, Chepurny OG, Monaco M, Wheeler MB, Bos JL, Schwede F, Genieser HG, Holz GG. Epac-selective cAMP analog 8-pCPT-2’-O-Me-cAMP as a stimulus for Ca2+-induced Ca2+ release and exocytosis in pancreatic beta-cells. J Biol Chem 278: 8279–8285, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kang G, Leech CA, Chepurny OG, Coetzee WA, Holz GG. Role of the cAMP sensor Epac as a determinant of KATP channel ATP sensitivity in human pancreatic beta-cells and rat INS-1 cells. J Physiol 586: 1307–1319, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kanno T, Suga S, Wu J, Kimura M, Wakui M. Intracellular cAMP potentiates voltage-dependent activation of L-type Ca2+ channels in rat islet beta-cells. Pflugers Arch 435: 578–580, 1998. [DOI] [PubMed] [Google Scholar]
- 166.Karaca M, Magnan C, Kargar C. Functional pancreatic beta-cell mass: Involvement in type 2 diabetes and therapeutic intervention. Diabetes Metab 35: 77–84, 2009. [DOI] [PubMed] [Google Scholar]
- 167.Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S. Critical role of cAMP-GEFII—Rim2 complex in incretin-potentiated insulin secretion. J Biol Chem 276: 46046–46053, 2001. [DOI] [PubMed] [Google Scholar]
- 168.Kato T, Shimano H, Yamamoto T, Ishikawa M, Kumadaki S, Matsuzaka T, Nakagawa Y, Yahagi N, Nakakuki M, Hasty AH, Takeuchi Y, Kobayashi K, Takahashi A, Yatoh S, Suzuki H, Sone H, Yamada N. Palmitate impairs and eicosapentaenoate restores insulin secretion through regulation of SREBP-1c in pancreatic islets. Diabetes 57: 2382–2392, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kee HJ, Ahn KY, Choi KC, Won Song J, Heo T, Jung S, Kim JK, Bae CS, Kim KK. Expression of brain-specific angiogenesis inhibitor 3 (BAI3) in normal brain and implications for BAI3 in ischemia-induced brain angiogenesis and malignant glioma. FEBS Lett 569: 307–316, 2004. [DOI] [PubMed] [Google Scholar]
- 170.Kienesberger PC, Oberer M, Lass A, Zechner R. Mammalian patatin domain containing proteins: a family with diverse lipolytic activities involved in multiple biological functions. J Lipid Res 50 (Suppl): S63–S68, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kilanowska A, Ziolkowska A. Role of phosphodiesterase in the biology and pathology of diabetes. Int J Mol Sci 21, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kim JW, Roberts CD, Berg SA, Caicedo A, Roper SD, Chaudhari N. Imaging cyclic AMP changes in pancreatic islets of transgenic reporter mice. PLoS One 3: e2127, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kimple ME, Joseph JW, Bailey CL, Fueger PT, Hendry IA, Newgard CB, Casey PJ. Gαz negatively regulates insulin secretion and glucose clearance. J Biol Chem 283: 4560–4567, 2008. [DOI] [PubMed] [Google Scholar]
- 174.Kimple ME, Keller MP, Rabaglia MR, Pasker RL, Neuman JC, Truchan NA, Brar HK, Attie AD. Prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes 62: 1904–1912, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kimple ME, Moss JB, Brar HK, Rosa TC, Truchan NA, Pasker RL, Newgard CB, Casey PJ. Deletion of GalphaZ protein protects against diet-induced glucose intolerance via expansion of beta-cell mass. J Biol Chem 287: 20344–20355, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kimple ME, Neuman JC, Linnemann AK, Casey PJ. Inhibitory G proteins and their receptors: emerging therapeutic targets for obesity and diabetes. Exp Mol Med 46: e102, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kimple ME, Nixon AB, Kelly P, Bailey CL, Young KH, Fields TA, Casey PJ. A role for G(z) in pancreatic islet beta-cell biology. J Biol Chem 280: 31708–31713, 2005. [DOI] [PubMed] [Google Scholar]
- 178.Kimura T, Kaneto H, Shimoda M, Hirukawa H, Okauchi S, Kohara K, Hamamoto S, Tawaramoto K, Hashiramoto M, Kaku K. Protective effects of pioglitazone and/or liraglutide on pancreatic beta-cells in db/db mice: comparison of their effects between in an early and advanced stage of diabetes. Mol Cell Endocrinol 400: 78–89, 2015. [DOI] [PubMed] [Google Scholar]
- 179.Kitaguchi T, Oya M, Wada Y, Tsuboi T, Miyawaki A. Extracellular calcium influx activates adenylate cyclase 1 and potentiates insulin secretion in MIN6 cells. Biochem J 450: 365–373, 2013. [DOI] [PubMed] [Google Scholar]
- 180.Klen J, Dolzan V. Glucagon-like peptide-1 receptor agonists in the management of type 2 diabetes mellitus and obesity: the impact of pharmacological properties and genetic factors. Int J Mol Sci 23, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kogure R, Toyama K, Hiyamuta S, Kojima I, Takeda S. 5-Hydroxyeicosapentaenoic acid is an endogenous GPR119 agonist and enhances glucose-dependent insulin secretion. Biochem Biophys Res Commun 416: 58–63, 2011. [DOI] [PubMed] [Google Scholar]
- 182.Koike S, Hsu MF, Bettaieb A, Chu B, Matsumoto N, Morisseau C, Havel PJ, Huising MO, Hammock BD, Haj FG. Genetic deficiency or pharmacological inhibition of soluble epoxide hydrolase ameliorates high fat diet-induced pancreatic beta-cell dysfunction and loss. Free Radic Biol Med 172: 48–57, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kolczynska K, Loza-Valdes A, Hawro I, Sumara G. Diacylglycerol-evoked activation of PKC and PKD isoforms in regulation of glucose and lipid metabolism: a review. Lipids Health Dis 19: 113, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Koltes JE, Arora I, Gupta R, Nguyen DC, Schaid M, Kim JA, Kimple ME, Bhatnagar S. A gene expression network analysis of the pancreatic islets from lean and obese mice identifies complement 1q like-3 secreted protein as a regulator of beta-cell function. Sci Rep 9: 10119, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kotarsky K, Nilsson NE, Flodgren E, Owman C, Olde B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem Biophys Res Commun 301: 406–410, 2003. [DOI] [PubMed] [Google Scholar]
- 186.Kwan EP, Gaisano HY. Rescuing the subprime meltdown in insulin exocytosis in diabetes. Ann N Y Acad Sci 1152: 154–164, 2009. [DOI] [PubMed] [Google Scholar]
- 187.Kwan EP, Gao X, Leung YM, Gaisano HY. Activation of exchange protein directly activated by cyclic adenosine monophosphate and protein kinase A regulate common and distinct steps in promoting plasma membrane exocytic and granule-to-granule fusions in rat islet beta cells. Pancreas 35: e45–e54, 2007. [DOI] [PubMed] [Google Scholar]
- 188.Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356: 1517–1526, 2007. [DOI] [PubMed] [Google Scholar]
- 189.Larsson PK, Claesson HE, Kennedy BP. Multiple splice variants of the human calcium-independent phospholipase A2 and their effect on enzyme activity. J Biol Chem 273: 207–214, 1998. [DOI] [PubMed] [Google Scholar]
- 190.Laychock SG, Sessanna SM, Lin MH, Mastrandrea LD. Sphingosine 1-phosphate affects cytokine-induced apoptosis in rat pancreatic islet beta-cells. Endocrinology 147: 4705–4712, 2006. [DOI] [PubMed] [Google Scholar]
- 191.Lee CH, Chu CS, Tsai HJ, Ke LY, Lee HC, Yeh JL, Chen CH, Wu BN. Xanthine-derived KMUP-1 reverses glucotoxicity-activated Kv channels through the cAMP/PKA signaling pathway in rat pancreatic beta cells. Chem Biol Interact 279: 171–176, 2018. [DOI] [PubMed] [Google Scholar]
- 192.Leech CA, Castonguay MA, Habener JF. Expression of adenylyl cyclase subtypes in pancreatic beta-cells. Biochem Biophys Res Commun 254: 703–706, 1999. [DOI] [PubMed] [Google Scholar]
- 193.Lei X, Zhang S, Bohrer A, Bao S, Song H, Ramanadham S. The group VIA calcium-independent phospholipase A2 participates in ER stress-induced INS-1 insulinoma cell apoptosis by promoting ceramide generation via hydrolysis of sphingomyelins by neutral sphingomyelinase. Biochemistry 46: 10170–10185, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lei X, Zhang S, Bohrer A, Barbour SE, Ramanadham S. Role of calcium-independent phospholipase A2beta in human pancreatic islet beta-cell apoptosis. Am J Physiol Endocrinol Metab 303: E1386–E1395, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lei X, Zhang S, Bohrer A, Ramanadham S. Calcium-independent phospholipase A2 (iPLA2 beta)-mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. J Biol Chem 283: 34819–34832, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Leitner DR, Fruhbeck G, Yumuk V, Schindler K, Micic D, Woodward E, Toplak H. Obesity and type 2 diabetes: Two diseases with a need for combined treatment strategies—EASO can lead the way. Obes Facts 10: 483–492, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Leung CCY, Wong YH. Role of G protein-coupled receptors in the regulation of structural plasticity and cognitive function. Molecules 22, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Li N, Wu JX, Ding D, Cheng J, Gao N, Chen L. Structure of a pancreatic ATP-sensitive potassium channel. Cell 168: 101–110 e110, 2017. [DOI] [PubMed] [Google Scholar]
- 199.Li Q, Bartley AF, Dobrunz LE. Endogenously released neuropeptide Y suppresses hippocampal short-term facilitation and is impaired by stress-induced anxiety. J Neurosci 37: 23–37, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Li XN, Herrington J, Petrov A, Ge L, Eiermann G, Xiong Y, Jensen MV, Hohmeier HE, Newgard CB, Garcia ML, Wagner M, Zhang BB, Thornberry NA, Howard AD, Kaczorowski GJ, Zhou YP. The role of voltage-gated potassium channels Kv2.1 and Kv2.2 in the regulation of insulin and somatostatin release from pancreatic islets. J Pharmacol Exp Ther 344: 407–416, 2013. [DOI] [PubMed] [Google Scholar]
- 201.Light PE, Manning Fox JE, Riedel MJ, Wheeler MB. Glucagon-like peptide-1 inhibits pancreatic ATP-sensitive potassium channels via a protein kinase A- and ADP-dependent mechanism. Mol Endocrinol 16: 2135–2144, 2002. [DOI] [PubMed] [Google Scholar]
- 202.Lindskog C, Korsgren O, Ponten F, Eriksson JW, Johansson L, Danielsson A. novel pancreatic beta cell-specific proteins: antibody-based proteomics for identification of new biomarker candidates. J Proteome 75: 2611–2620, 2012. [DOI] [PubMed] [Google Scholar]
- 203.Linnemann AK, Baan M, Davis DB. Pancreatic beta-cell proliferation in obesity. Adv Nutr 5: 278–288, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Liu G, Jacobo SM, Hilliard N, Hockerman GH. Differential modulation of Cav1.2 and Cav1.3-mediated glucose-stimulated insulin secretion by cAMP in INS-1 cells: distinct roles for exchange protein directly activated by cAMP 2 (Epac2) and protein kinase A. J Pharmacol Exp Ther 318: 152–160, 2006. [DOI] [PubMed] [Google Scholar]
- 205.Liu X, Xue Y, Liu C, Lou Q, Wang J, Yanagita T, Xue C, Wang Y. Eicosapentaenoic acid-enriched phospholipid ameliorates insulin resistance and lipid metabolism in diet-induced-obese mice. Lipids Health Dis 12: 109, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Liu Y, Zhong X, Ding Y, Ren L, Bai T, Liu M, Liu Z, Guo Y, Guo Q, Zhang Y, Yang J, Zhang Y . Inhibition of voltage-dependent potassium channels mediates cAMP-potentiated insulin secretion in rat pancreatic beta cells. Islets 9: 11–18, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lucena CF, Roma LP, Graciano MF, Veras K, Simoes D, Curi R, Carpinelli AR. Omega-3 supplementation improves pancreatic islet redox status: in vivo and in vitro studies. Pancreas 44: 287–295, 2015. [DOI] [PubMed] [Google Scholar]
- 208.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117: 175–184, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Luo P, Chang HH, Zhou Y, Zhang S, Hwang SH, Morisseau C, Wang CY, Inscho EW, Hammock BD, Wang MH. Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J Pharmacol Exp Ther 334: 430–438, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Luria A, Bettaieb A, Xi Y, Shieh GJ, Liu HC, Inoue H, Tsai HJ, Imig JD, Haj FG, Hammock BD. Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc Natl Acad Sci U S A 108: 9038–9043, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lyssenko V, Almgren P, Anevski D, Perfekt R, Lahti K, Nissen M, Isomaa B, Forsen B, Homstrom N, Saloranta C, Taskinen MR, Groop L, Tuomi T, Botnia study g. Predictors of and longitudinal changes in insulin sensitivity and secretion preceding onset of type 2 diabetes. Diabetes 54: 166–174, 2005. [DOI] [PubMed] [Google Scholar]
- 212.Ma Z, Bohrer A, Wohltmann M, Ramanadham S, Hsu FF, Turk J. Studies of phospholipid metabolism, proliferation, and secretion of stably transfected insulinoma cells that overexpress group VIA phospholipase A2. Lipids 36: 689–700, 2001. [DOI] [PubMed] [Google Scholar]
- 213.Ma Z, Ramanadham S, Kempe K, Chi XS, Ladenson J, Turk J. Pancreatic islets express a Ca2+-independent phospholipase A2 enzyme that contains a repeated structural motif homologous to the integral membrane protein binding domain of ankyrin. J Biol Chem 272: 11118–11127, 1997. [PubMed] [Google Scholar]
- 214.Ma Z, Turk J. The molecular biology of the group VIA Ca2+-independent phospholipase A2. Prog Nucleic Acid Res Mol Biol 67: 1–33, 2001. [DOI] [PubMed] [Google Scholar]
- 215.Ma Z, Wang X, Nowatzke W, Ramanadham S, Turk J. Human pancreatic islets express mRNA species encoding two distinct catalytically active isoforms of group VI phospholipase A2 (iPLA2) that arise from an exon-skipping mechanism of alternative splicing of the transcript from the iPLA2 gene on chromosome 22q13.1. J Biol Chem 274: 9607–9616, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ma Z, Zhang S, Turk J, Ramanadham S. Stimulation of insulin secretion and associated nuclear accumulation of iPLA2beta in INS-1 insulinoma cells. Am J Physiol Endocrinol Metab 282: E820–E833, 2002. [DOI] [PubMed] [Google Scholar]
- 217.MacDonald PE, Wang X, Xia F, El-kholy W, Targonsky ED, Tsushima RG, Wheeler MB. Antagonism of rat beta-cell voltage-dependent K+ currents by exendin 4 requires dual activation of the cAMP/protein kinase A and phosphatidylinositol 3-kinase signaling pathways. J Biol Chem 278: 52446–52453, 2003. [DOI] [PubMed] [Google Scholar]
- 218.MacDonald PE, Wheeler MB. Voltage-dependent K+ channels in pancreatic beta cells: Role, regulation and potential as therapeutic targets. Diabetologia 46: 1046–1062, 2003. [DOI] [PubMed] [Google Scholar]
- 219.Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110: 851–860, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 50: 69–76, 2001. [DOI] [PubMed] [Google Scholar]
- 221.Mancuso DJ, Jenkins CM, Gross RW. The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A2. J Biol Chem 275: 9937–9945, 2000. [DOI] [PubMed] [Google Scholar]
- 222.Martens GA, Motte E, Kramer G, Stange G, Gaarn LW, Hellemans K, Nielsen JH, Aerts JM, Ling Z, Pipeleers D. Functional characteristics of neonatal rat beta cells with distinct markers. J Mol Endocrinol 52: 11–28, 2014. [DOI] [PubMed] [Google Scholar]
- 223.Martin GM, Yoshioka C, Rex EA, Fay JF, Xie Q, Whorton MR, Chen JZ, Shyng SL. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. elife 6: e24149, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Martinelli DC, Chew KS, Rohlmann A, Lum MY, Ressl S, Hattar S, Brunger AT, Missler M, Sudhof TC. Expression of C1ql3 in discrete neuronal populations controls efferent synapse numbers and diverse behaviors. Neuron 91: 1034–1051, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Mastrandrea LD, Sessanna SM, Laychock SG. Sphingosine kinase activity and sphingosine-1 phosphate production in rat pancreatic islets and INS-1 cells: Response to cytokines. Diabetes 54: 1429–1436, 2005. [DOI] [PubMed] [Google Scholar]
- 226.Meng Z, Lv J, Luo Y, Lin Y, Zhu Y, Nie J, Yang T, Sun Y, Han X. Forkhead box O1/pancreatic and duodenal homeobox 1 intracellular translocation is regulated by c-Jun N-terminal kinase and involved in prostaglandin E2-induced pancreatic beta-cell dysfunction. Endocrinology 150: 5284–5293, 2009. [DOI] [PubMed] [Google Scholar]
- 227.Meng ZX, Sun JX, Ling JJ, Lv JH, Zhu DY, Chen Q, Sun YJ, Han X. Prostaglandin E2 regulates Foxo activity via the Akt pathway: implications for pancreatic islet beta cell dysfunction. Diabetologia 49: 2959–2968, 2006. [DOI] [PubMed] [Google Scholar]
- 228.Metz SA, Draznin B, Sussman KE, Leitner JW. Unmasking of arachidonate-induced insulin release by removal of extracellular calcium. Arachidonic acid mobilizes cellular calcium in rat islets of Langerhans. Biochem Biophys Res Commun 142: 251–258, 1987. [DOI] [PubMed] [Google Scholar]
- 229.Metz SA, Robertson RP, Fujimoto WY. Inhibition of prostaglandin E synthesis augments glucose-induced insulin secretion is cultured pancreas. Diabetes 30: 551–557, 1981. [DOI] [PubMed] [Google Scholar]
- 230.Miki T, Minami K, Shinozaki H, Matsumura K, Saraya A, Ikeda H, Yamada Y, Holst JJ, Seino S. Distinct effects of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 on insulin secretion and gut motility. Diabetes 54: 1056–1063, 2005. [DOI] [PubMed] [Google Scholar]
- 231.Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci U S A 95: 10402–10406, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Milne HM, Burns CJ, Squires PE, Evans ND, Pickup J, Jones PM, Persaud SJ. Uncoupling of nutrient metabolism from insulin secretion by overexpression of cytosolic phospholipase A2. Diabetes 54: 116–124, 2005. [DOI] [PubMed] [Google Scholar]
- 233.Mitrakou A, Kelley D, Mokan M, Veneman T, Pangburn T, Reilly J, Gerich J. Role of reduced suppression of glucose production and diminished early insulin release in impaired glucose tolerance. N Engl J Med 326: 22–29, 1992. [DOI] [PubMed] [Google Scholar]
- 234.Mondal P, Prasad A, Girdhar K. Interventions to improve beta-cell mass and function. Ann Endocrinol 78: 469–477, 2017. [DOI] [PubMed] [Google Scholar]
- 235.Moon SY, Shin SA, Oh YS, Park HH, Lee CS. Understanding the role of the BAI subfamily of adhesion G protein-coupled receptors (GPCRs) in pathological and physiological conditions. Genes 9: 597, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Moran JM, Buller RM, McHowat J, Turk J, Wohltmann M, Gross RW, Corbett JA. Genetic and pharmacologic evidence that calcium-independent phospholipase A2beta regulates virus-induced inducible nitric-oxide synthase expression by macrophages. J Biol Chem 280: 28162–28168, 2005. [DOI] [PubMed] [Google Scholar]
- 237.Mouchlis VD, Dennis EA. Phospholipase A2 catalysis and lipid mediator lipidomics. Biochim Biophys Acta Mol Cell Biol Lipids 1864: 766–771, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Nagamatsu S, Ohara-Imaizumi M, Nakamichi Y, Kikuta T, Nishiwaki C. Imaging docking and fusion of insulin granules induced by antidiabetes agents: sulfonylurea and glinide drugs preferentially mediate the fusion of newcomer, but not previously docked, insulin granules. Diabetes 55: 2819–2825, 2006. [DOI] [PubMed] [Google Scholar]
- 239.Nakazaki M, Crane A, Hu M, Seghers V, Ullrich S, Aguilar-Bryan L, Bryan J. cAMP-activated protein kinase-independent potentiation of insulin secretion by cAMP is impaired in SUR1 null islets. Diabetes 51: 3440–3449, 2002. [DOI] [PubMed] [Google Scholar]
- 240.Nelson AJ, Stephenson DJ, Cardona CL, Lei X, Almutairi A, White TD, Tusing YG, Park MA, Barbour SE, Chalfant CE, Ramanadham S. Macrophage polarization is linked to Ca2+-independent phospholipase A2beta-derived lipids and cross-cell signaling in mice. J Lipid Res 61: 143–158, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Nenquin M, Szollosi A, Aguilar-Bryan L, Bryan J, Henquin JC. Both triggering and amplifying pathways contribute to fuel-induced insulin secretion in the absence of sulfonylurea receptor-1 in pancreatic beta-cells. J Biol Chem 279: 32316–32324, 2004. [DOI] [PubMed] [Google Scholar]
- 242.Nesher R, Della Casa L, Litvin Y, Sinai J, Del Rio G, Pevsner B, Wax Y, Cerasi E. Insulin deficiency and insulin resistance in type 2 (non-insulin-dependent) diabetes: quantitative contributions of pancreatic and peripheral responses to glucose homeostasis. Eur J Clin Investig 17: 266–274, 1987. [DOI] [PubMed] [Google Scholar]
- 243.Neuman JC, Kimple ME. The EP3 receptor: Exploring a new target for type 2 diabetes therapeutics. J Endocrinol Diabetes Obes 1, 2013. [PMC free article] [PubMed] [Google Scholar]
- 244.Neuman JC, Schaid MD, Brill AL, Fenske RJ, Kibbe CR, Fontaine DA, Sdao SM, Brar HK, Connors KM, Wienkes HN, Eliceiri KW, Merrins MJ, Davis DB, Kimple ME. Enriching islet phospholipids with eicosapentaenoic acid reduces prostaglandin E2 signaling and enhances diabetic beta-cell function. Diabetes 66: 1572–1585, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Nevins AK, Thurmond DC. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am J Physiol Cell Physiol 285: C698–C710, 2003. [DOI] [PubMed] [Google Scholar]
- 246.Ni Q, Ganesan A, Aye-Han NN, Gao X, Allen MD, Levchenko A, Zhang J. Signaling diversity of PKA achieved via a Ca2+-cAMP-PKA oscillatory circuit. Nat Chem Biol 7: 34–40, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Niimura M, Miki T, Shibasaki T, Fujimoto W, Iwanaga T, Seino S. Critical role of the N-terminal cyclic AMP-binding domain of Epac2 in its subcellular localization and function. J Cell Physiol 219: 652–658, 2009. [DOI] [PubMed] [Google Scholar]
- 248.Nunemaker CS, Chen M, Pei H, Kimble SD, Keller SR, Carter JD, Yang Z, Smith KM, Wu R, Bevard MH, Garmey JC, Nadler JL. 12-Lipoxygenase-knockout mice are resistant to inflammatory effects of obesity induced by Western diet. Am J Physiol Endocrinol Metab 295: E1065–E1075, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142: 687–698, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Oh E, Thurmond DC. Munc18c depletion selectively impairs the sustained phase of insulin release. Diabetes 58: 1165–1174, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Ohara-Imaizumi M, Fujiwara T, Nakamichi Y, Okamura T, Akimoto Y, Kawai J, Matsushima S, Kawakami H, Watanabe T, Akagawa K, Nagamatsu S. Imaging analysis reveals mechanistic differences between first- and second-phase insulin exocytosis. J Cell Biol 177: 695–705, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Opara EC, Garfinkel M, Hubbard VS, Burch WM, Akwari OE. Effect of fatty acids on insulin release: Role of chain length and degree of unsaturation. Am J Phys 266: E635–E639, 1994. [DOI] [PubMed] [Google Scholar]
- 253.Overton HA, Fyfe MC, Reynet C. GPR119, a novel G protein-coupled receptor target for the treatment of type 2 diabetes and obesity. Br J Pharmacol 153 (Suppl 1): S76–S81, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol 2: 805–811, 2000. [DOI] [PubMed] [Google Scholar]
- 255.Parazzoli S, Harmon JS, Vallerie SN, Zhang T, Zhou H, Robertson RP. cyclooxygenase-2, not microsomal prostaglandin E synthase-1, is the mechanism for interleukin-1beta-induced prostaglandin E2 production and inhibition of insulin secretion in pancreatic islets. J Biol Chem 287: 32246–32253, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Parker JC, VanVolkenburg MA, Ketchum RJ, Brayman KL, Andrews KM. Cyclic AMP phosphodiesterases of human and rat islets of Langerhans: Contributions of types III and IV to the modulation of insulin secretion. Biochem Biophys Res Commun 217: 916–923, 1995. [DOI] [PubMed] [Google Scholar]
- 257.Pedersen MG, Tagliavini A, Henquin JC. Calcium signaling and secretory granule pool dynamics underlie biphasic insulin secretion and its amplification by glucose: Experiments and modeling. Am J Physiol Endocrinol Metab 316: E475–E486, 2019. [DOI] [PubMed] [Google Scholar]
- 258.Perez R, Balboa MA, Balsinde J. Involvement of group VIA calcium-independent phospholipase A2 in macrophage engulfment of hydrogen peroxide-treated U937 cells. J Immunol 176: 2555–2561, 2006. [DOI] [PubMed] [Google Scholar]
- 259.Peschke E, Muhlbauer E. New evidence for a role of melatonin in glucose regulation. Best Pract Res Clin Endocrinol Metab 24: 829–841, 2010. [DOI] [PubMed] [Google Scholar]
- 260.Petyuk VA, Qian WJ, Hinault C, Gritsenko MA, Singhal M, Monroe ME, Camp DG 2nd, Kulkarni RN, Smith RD. Characterization of the mouse pancreatic islet proteome and comparative analysis with other mouse tissues. J Proteome Res 7: 3114–3126, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Peyot ML, Pepin E, Lamontagne J, Latour MG, Zarrouki B, Lussier R, Pineda M, Jetton TL, Madiraju SR, Joly E, Prentki M. Beta-cell failure in diet-induced obese mice stratified according to body weight gain: secretory dysfunction and altered islet lipid metabolism without steatosis or reduced beta-cell mass. Diabetes 59: 2178–2187, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Picinato MC, Curi R, Machado UF, Carpinelli AR. Soybean- and olive-oils-enriched diets increase insulin secretion to glucose stimulus in isolated pancreatic rat islets. Physiol Behav 65: 289–294, 1998. [DOI] [PubMed] [Google Scholar]
- 263.Pimenta W, Korytkowski M, Mitrakou A, Jenssen T, Yki-Jarvinen H, Evron W, Dailey G, Gerich J. Pancreatic beta-cell dysfunction as the primary genetic lesion in NIDDM. Evidence from studies in normal glucose-tolerant individuals with a first-degree NIDDM relative. JAMA 273: 1855–1861, 1995. [PubMed] [Google Scholar]
- 264.Pinkney J, Fox T, Ranganath L. Selecting GLP-1 agonists in the management of type 2 diabetes: differential pharmacology and therapeutic benefits of liraglutide and exenatide. Ther Clin Risk Manag 6: 401–411, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Poitout V Fatty acids and insulin secretion: From FFAR and near? Diabetes 67: 1932–1934, 2018. [DOI] [PubMed] [Google Scholar]
- 266.Porte D Jr. Banting lecture. Beta-cells in type II diabetes mellitus. Diabetes 40: 166, 1991–180, 1990. [DOI] [PubMed] [Google Scholar]
- 267.Prentki M, Matschinsky FM. Ca2+, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiol Rev 67: 1185–1248, 1987. [DOI] [PubMed] [Google Scholar]
- 268.Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuelinduced insulin secretion. Cell Metab 18: 162–185, 2013. [DOI] [PubMed] [Google Scholar]
- 269.Puri S, Roy N, Russ HA, Leonhardt L, French EK, Roy R, Bengtsson H, Scott DK, Stewart AF, Hebrok M. Replication confers beta-cell immaturity. Nat Commun 9: 485, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Quach ND, Arnold RD, Cummings BS. Secretory phospholipase A2 enzymes as pharmacological targets for treatment of disease. Biochem Pharmacol 90: 338–348, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Ramanadham S, Bohrer A, Gross RW, Turk J. Mass spectrometric characterization of arachidonate-containing plasmalogens in human pancreatic islets and in rat islet beta-cells and subcellular membranes. Biochemistry 32: 13499–13509, 1993. [DOI] [PubMed] [Google Scholar]
- 272.Ramanadham S, Bohrer A, Mueller M, Jett P, Gross RW, Turk J. Mass spectrometric identification and quantitation of arachidonate-containing phospholipids in pancreatic islets: prominence of plasmenylethanolamine molecular species. Biochemistry 32: 5339–5351, 1993. [DOI] [PubMed] [Google Scholar]
- 273.Ramanadham S, Gross RW, Han X, Turk J. Inhibition of arachidonate release by secretagogue-stimulated pancreatic islets suppresses both insulin secretion and the rise in beta-cell cytosolic calcium ion concentration. Biochemistry 32: 337–346, 1993. [DOI] [PubMed] [Google Scholar]
- 274.Ramanadham S, Hsu FF, Bohrer A, Ma Z, Turk J. Studies of the role of group VI phospholipase A2 in fatty acid incorporation, phospholipid remodeling, lysophosphatidylcholine generation, and secretagogue-induced arachidonic acid release in pancreatic islets and insulinoma cells. J Biol Chem 274: 13915–13927, 1999. [DOI] [PubMed] [Google Scholar]
- 275.Ramanadham S, Hsu FF, Bohrer A, Nowatzke W, Ma Z, Turk J. Electrospray ionization mass spectrometric analyses of phospholipids from rat and human pancreatic islets and subcellular membranes: Comparison to other tissues and implications for membrane fusion in insulin exocytosis. Biochemistry 37: 4553–4567, 1998. [DOI] [PubMed] [Google Scholar]
- 276.Ramanadham S, Ma Z, Arita H, Zhang S, Turk J. Type IB secretory phospholipase A2 is contained in insulin secretory granules of pancreatic islet beta-cells and is co-secreted with insulin from glucose-stimulated islets. Biochim Biophys Acta 1390: 301–312, 1998. [DOI] [PubMed] [Google Scholar]
- 277.Ramanadham S, Song H, Hsu FF, Zhang S, Crankshaw M, Grant GA, Newgard CB, Bao S, Ma Z, Turk J. Pancreatic islets and insulinoma cells express a novel isoform of group VIA phospholipase A2 (iPLA2 beta) that participates in glucose-stimulated insulin secretion and is not produced by alternate splicing of the iPLA2 beta transcript. Biochemistry 42: 13929–13940, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ramanadham S, Wolf MJ, Jett PA, Gross RW, Turk J. Characterization of an ATP-stimulatable Ca2+-independent phospholipase A2 from clonal insulin-secreting HIT cells and rat pancreatic islets: a possible molecular component of the beta-cell fuel sensor. Biochemistry 33: 7442–7452, 1994. [DOI] [PubMed] [Google Scholar]
- 279.Ravier MA, Nenquin M, Miki T, Seino S, Henquin JC. Glucose controls cytosolic Ca2+ and insulin secretion in mouse islets lacking adenosine triphosphate-sensitive K+ channels owing to a knockout of the poreforming subunit Kir6.2. Endocrinology 150: 33–45, 2009. [DOI] [PubMed] [Google Scholar]
- 280.Rehmann H Epac2: A sulfonylurea receptor? Biochem Soc Trans 40: 6–10, 2012. [DOI] [PubMed] [Google Scholar]
- 281.Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its autoinhibited state. Nature 439: 625–628, 2006. [DOI] [PubMed] [Google Scholar]
- 282.Reimann F, Gribble FM. G protein-coupled receptors as new therapeutic targets for type 2 diabetes. Diabetologia 59: 229–233, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Renstrom E, Eliasson L, Rorsman P. Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic B-cells. J Physiol 502 (Pt 1): 105–118, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Robertson RP. The COX-2/PGE2/EP3/Gi/o/cAMP/GSIS pathway in the islet: The beat goes on. Diabetes 66: 1464–1466, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Robertson RP, Gavareski DJ, Porte D Jr, Bierman EL. Inhibition of in vivo insulin secretion by prostaglandin E1. J Clin Invest 54: 310–315, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 53 (Suppl 1): S119–S124, 2004. [DOI] [PubMed] [Google Scholar]
- 287.Robertson RP, Tsai P, Little SA, Zhang HJ, Walseth TF. Receptor-mediated adenylate cyclase-coupled mechanism for PGE2 inhibition of insulin secretion in HIT cells. Diabetes 36: 1047–1053, 1987. [DOI] [PubMed] [Google Scholar]
- 288.Rodriguez-Diaz R, Tamayo A, Hara M, Caicedo A. The local paracrine actions of the pancreatic alpha-cell. Diabetes 69: 550–558, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Rorsman P, Renstrom E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 46: 1029–1045, 2003. [DOI] [PubMed] [Google Scholar]
- 290.Sabatini PV, Speckmann T, Lynn FC. Friend and foe: Beta-cell Ca2+ signaling and the development of diabetes. Mol Metab 21: 1–12, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. Beta-cell mass and turnover in humans: Effects of obesity and aging. Diabetes Care 36: 111–117, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Salehi M, Vahl TP, D’Alessio DA. Regulation of islet hormone release and gastric emptying by endogenous glucagon-like peptide 1 after glucose ingestion. J Clin Endocrinol Metab 93: 4909–4916, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Sandhu HK, Neuman JC, Schaid MD, Davis SE, Connors KM, Challa R, Guthery E, Fenske RJ, Patibandla C, Breyer RM, Kimple ME. Rat prostaglandin EP3 receptor is highly promiscuous and is the sole prostanoid receptor family member that regulates INS-1 (832/13) cell glucose-stimulated insulin secretion. Pharmacol Res Perspect 9: e00736, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci U S A 100: 7265–7270, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Sasaki S, Nakagaki I, Kondo H, Hori S. Involvement of the ryanodine-sensitive Ca2+ store in GLP-1-induced Ca2+ oscillations in insulin-secreting HIT cells. Pflugers Arch 445: 342–351, 2002. [DOI] [PubMed] [Google Scholar]
- 296.Sato N, Kaneko M, Tamura M, Kurumatani H. The prostacyclin analog beraprost sodium ameliorates characteristics of metabolic syndrome in obese Zucker (fatty) rats. Diabetes 59: 1092–1100, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Schaid MD, Green CL, Peter DC, Gallagher SJ, Guthery E, Carbajal KA, Harrington JM, Kelly GM, Reuter A, Wehner ML, Brill AL, Neuman JC, Lamming DW, Kimple ME. Agonist-independent Galphaz activity negatively regulates beta-cell compensation in a diet-induced obesity model of type 2 diabetes. J Biol Chem 296: 100056, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Schaid MD, Harrington JM, Kelly GM, Sdao SM, Merrins MJ, Kimple ME. EP3 signaling is decoupled from regulation of glucose-stimulated insulin secretion in β-cells compensating for obesity and insulin resistance. BioRxiv, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Schaid MD, Wisinski JA, Kimple ME. The EP3 Receptor/Gz signaling axis as a therapeutic target for diabetes and cardiovascular disease. AAPS J 19: 1276–1283, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Schaid MD, Zhu Y, Richardson NE, Patibandla C, Ong IM, Fenske RJ, Neuman JC, Guthery E, Reuter A, Sandhu HK, Fuller MH, Cox ED, Davis DB, Layden BT, Brasier AR, Lamming DW, Ge Y, Kimple ME. Systemic metabolic alterations correlate with islet-level prostaglandin E2 production and signaling mechanisms that predict beta-cell dysfunction in a mouse model of type 2 diabetes. Metabolites 11: 58, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta 1761: 1246–1259, 2006. [DOI] [PubMed] [Google Scholar]
- 302.Scharfmann R, Pechberty S, Hazhouz Y, von Bulow M, Bricout-Neveu E, Grenier-Godard M, Guez F, Rachdi L, Lohmann M, Czernichow P, Ravassard P. Development of a conditionally immortalized human pancreatic beta cell line. J Clin Invest 124: 2087–2098, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Schnurr TM, Jakupovic H, Carrasquilla GD, Angquist L, Grarup N, Sorensen TIA, Tjonneland A, Overvad K, Pedersen O, Hansen T, Kilpelainen TO. Obesity, unfavourable lifestyle and genetic risk of type 2 diabetes: A case-cohort study. Diabetologia 63: 1324–1332, 2020. [DOI] [PubMed] [Google Scholar]
- 304.Schwede F, Chepurny OG, Kaufholz M, Bertinetti D, Leech CA, Cabrera O, Zhu Y, Mei F, Cheng X, Manning Fox JE, MacDonald PE, Genieser HG, Herberg FW, Holz GG. Rp-cAMPS prodrugs reveal the cAMP dependence of first-phase glucose-stimulated insulin secretion. Mol Endocrinol 29: 988–1005, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Scrocchi LA, Brown TJ, MaClusky N, Brubaker PL, Auerbach AB, Joyner AL, Drucker DJ. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat Med 2: 1254–1258, 1996. [DOI] [PubMed] [Google Scholar]
- 306.Seaquist ER, Walseth TF, Nelson DM, Robertson RP. Pertussis toxinsensitive G protein mediation of PGE2 inhibition of cAMP metabolism and phasic glucose-induced insulin secretion in HIT cells. Diabetes 38: 1439–1445, 1989. [DOI] [PubMed] [Google Scholar]
- 307.Sears DD, Miles PD, Chapman J, Ofrecio JM, Almazan F, Thapar D, Miller YI. 12/15-lipoxygenase is required for the early onset of high fat diet-induced adipose tissue inflammation and insulin resistance in mice. PLoS One 4: e7250, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Seed Ahmed M, Kovoor A, Nordman S, Abu Seman N, Gu T, Efendic S, Brismar K, Ostenson CG, Gu HF. Increased expression of adenylyl cyclase 3 in pancreatic islets and central nervous system of diabetic Goto-Kakizaki rats: A possible regulatory role in glucose homeostasis. Islets 4: 343–348, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Segerstolpe A, Palasantza A, Eliasson P, Andersson EM, Andreasson AC, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, Smith DM, Kasper M, Ammala C, Sandberg R. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 24: 593–607, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev 85: 1303–1342, 2005. [DOI] [PubMed] [Google Scholar]
- 311.Seino S, Shibasaki T, Minami K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J Clin Invest 121: 2118–2125, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Seldin MM, Tan SY, Wong GW. Metabolic function of the CTRP family of hormones. Rev Endocr Metab Disord 15: 111–123, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Shanaki M, Fadaei R, Moradi N, Emamgholipour S, Poustchi H. The Circulating CTRP13 in type 2 diabetes and non-alcoholic fatty liver patients. PLoS One 11: e0168082, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Sharoyko VV, Zaitseva II, Leibiger B, Efendic S, Berggren PO, Zaitsev SV. Arachidonic acid signaling is involved in the mechanism of imidazoline-induced KATP channel-independent stimulation of insulin secretion. Cell Mol Life Sci 64: 2985–2993, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Shen CA, Fagan S, Fischman AJ, Carter EE, Chai JK, Lu XM, Yu YM, Tompkins RG. Effects of glucagon-like peptide 1 on glycemia control and its metabolic consequence after severe thermal injury—studies in an animal model. Surgery 149: 635–644, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J, Seino S. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci U S A 104: 19333–19338, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Shigeto M, Cha CY, Rorsman P, Kaku K. A role of PLC/PKC-dependent pathway in GLP-1-stimulated insulin secretion. J Mol Med 95: 361–368, 2017. [DOI] [PubMed] [Google Scholar]
- 318.Shimizu H, Okajima F, Kimura T, Ohtani K, Tsuchiya T, Takahashi H, Kuwabara A, Tomura H, Sato K, Mori M. Sphingosine 1-phosphate stimulates insulin secretion in HIT-T 15 cells and mouse islets. Endocr J 47: 261–269, 2000. [DOI] [PubMed] [Google Scholar]
- 319.Shiota C, Larsson O, Shelton KD, Shiota M, Efanov AM, Hoy M, Lindner J, Kooptiwut S, Juntti-Berggren L, Gromada J, Berggren PO, Magnuson MA. Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem 277: 37176–37183, 2002. [DOI] [PubMed] [Google Scholar]
- 320.Shiratsuchi T, Nishimori H, Ichise H, Nakamura Y, Tokino T. Cloning and characterization of BAI2 and BAI3, novel genes homologous to brain-specific angiogenesis inhibitor 1 (BAI1). Cytogenet Cell Genet 79: 103–108, 1997. [DOI] [PubMed] [Google Scholar]
- 321.Shridas P, Noffsinger VP, Trumbauer AC, Webb NR. The dual role of group V secretory phospholipase A2 in pancreatic beta-cells. Endocrine 58: 47–58, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Shridas P, Zahoor L, Forrest KJ, Layne JD, Webb NR. Group X secretory phospholipase A2 regulates insulin secretion through a cyclooxygenase-2-dependent mechanism. J Biol Chem 289: 27410–27417, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Sigoillot SM, Iyer K, Binda F, Gonzalez-Calvo I, Talleur M, Vodjdani G, Isope P, Selimi F. The secreted protein C1QL1 and Its Receptor BAI3 control the synaptic connectivity of excitatory inputs converging on cerebellar purkinje cells. Cell Rep 10: 820–832, 2015. [DOI] [PubMed] [Google Scholar]
- 324.Simonsson E, Karlsson S, Ahren B. Islet phospholipase A2 activation is potentiated in insulin resistant mice. Biochem Biophys Res Commun 272: 539–543, 2000. [DOI] [PubMed] [Google Scholar]
- 325.Skelin M, Rupnik M. cAMP increases the sensitivity of exocytosis to Ca2+ primarily through protein kinase A in mouse pancreatic beta cells. Cell Calcium 49: 89–99, 2011. [DOI] [PubMed] [Google Scholar]
- 326.Skrtic S, Tyrberg B, Broberg M, Ericsson H, Schnecke V, Kjaer M, Hompesch M, Andersson EM, Ryberg E, Aivazidis A, Wennberg Huldt C, Lofgren L, Morrow L, Parkinson J, Ryden-Bergsten T, Watkins E, Sorhede WM. Exploring the insulin secretory properties of the PGD2-GPR44/DP2 axis in vitro and in a randomized phase-1 trial of type 2 diabetes patients. PLoS One 13: e0208998, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Soleimanpour SA, Stoffers DA. The pancreatic beta cell and type 1 diabetes: Innocent bystander or active participant? Trends Endocrinol Metab 24: 324–331, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Song C, Satoh T, Edamatsu H, Wu D, Tadano M, Gao X, Kataoka T. Differential roles of Ras and Rap1 in growth factor-dependent activation of phospholipase C epsilon. Oncogene 21: 8105–8113, 2002. [DOI] [PubMed] [Google Scholar]
- 329.Song WJ, Mondal P, Li Y, Lee SE, Hussain MA. Pancreatic beta-cell response to increased metabolic demand and to pharmacologic secretagogues requires EPAC2A. Diabetes 62: 2796–2807, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Song WJ, Seshadri M, Ashraf U, Mdluli T, Mondal P, Keil M, Azevedo M, Kirschner LS, Stratakis CA, Hussain MA. Snapin mediates incretin action and augments glucose-dependent insulin secretion. Cell Metab 13: 308–319, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Staimez LR, Weber MB, Ranjani H, Ali MK, Echouffo-Tcheugui JB, Phillips LS, Mohan V, Narayan KM. Evidence of reduced beta-cell function in Asian Indians with mild dysglycemia. Diabetes Care 36: 2772–2778, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Stein DT, Esser V, Stevenson BE, Lane KE, Whiteside JH, Daniels MB, Chen S, McGarry JD. Essentiality of circulating fatty acids for glucose-stimulated insulin secretion in the fasted rat. J Clin Invest 97: 2728–2735, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Stein DT, Stevenson BE, Chester MW, Basit M, Daniels MB, Turley SD, McGarry JD. The insulinotropic potency of fatty acids is influenced profoundly by their chain length and degree of saturation. J Clin Invest 100: 398–403, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Sticco MJ, Peña Palomino PA, Lukacsovich D, Thompson BL, Földy C, Ressl S, Martinelli DC. C1QL3 promotes cell-cell adhesion by mediating complex formation between ADGRB3/BAI3 and neuronal pentraxins. FASEB J 35: e21194, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Stozer A, Paradiz Leitgeb E, Pohorec V, Dolensek J, Krizancic Bombek L, Gosak M, Skelin KM. The role of cAMP in beta cell stimulus-secretion and intercellular coupling. Cell 10: 1658, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Straczkowski M, Kowalska I, Baranowski M, Nikolajuk A, Otziomek E, Zabielski P, Adamska A, Blachnio A, Gorski J, Gorska M. Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia 50: 2366–2373, 2007. [DOI] [PubMed] [Google Scholar]
- 337.Straub SG, James RF, Dunne MJ, Sharp GW. Glucose activates both KATP channel-dependent and KATP channel-independent signaling pathways in human islets. Diabetes 47: 758–763, 1998. [DOI] [PubMed] [Google Scholar]
- 338.Summers SA. Sphingolipids and insulin resistance: The five Ws. Curr Opin Lipidol 21: 128–135, 2010. [DOI] [PubMed] [Google Scholar]
- 339.Takahashi H, Shibasaki T, Park JH, Hidaka S, Takahashi T, Ono A, Song DK, Seino S. Role of Epac2A/Rap1 signaling in interplay between incretin and sulfonylurea in insulin secretion. Diabetes 64: 1262–1272, 2015. [DOI] [PubMed] [Google Scholar]
- 340.Takahashi T, Shibasaki T, Takahashi H, Sugawara K, Ono A, Inoue N, Furuya T, Seino S. Antidiabetic sulfonylureas and cAMP cooperatively activate Epac2A. Sci Signal 6: ra94, 2013. [DOI] [PubMed] [Google Scholar]
- 341.Tamagawa T, Niki H, Niki A. Insulin release independent of a rise in cytosolic free Ca2+ by forskolin and phorbol ester. FEBSLett 183: 430–432, 1985. [DOI] [PubMed] [Google Scholar]
- 342.Tan A, Ke S, Chen Y, Chen L, Lu X, Ding F, Yang L, Tang Y, Yu Y. Expression patterns of C1ql4 and its cell-adhesion GPCR Bai3 in the murine testis and functional roles in steroidogenesis. FASEB J 33: 4893–4906, 2019. [DOI] [PubMed] [Google Scholar]
- 343.Tan C, Day R, Bao S, Turk J, Zhao QD. Group VIA phospholipase A2 mediates enhanced macrophage migration in diabetes mellitus by increasing expression of nicotinamide adenine dinucleotide phosphate oxidase 4. Arterioscler Thromb Vasc Biol 34: 768–778, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J Biol Chem 272: 8567–8575, 1997. [DOI] [PubMed] [Google Scholar]
- 345.Taylor-Fishwick DA, Weaver J, Glenn L, Kuhn N, Rai G, Jadhav A, Simeonov A, Dudda A, Schmoll D, Holman TR, Maloney DJ, Nadler JL. Selective inhibition of 12-lipoxygenase protects islets and beta cells from inflammatory cytokine-mediated beta cell dysfunction. Diabetologia 58: 549–557, 2015. [DOI] [PubMed] [Google Scholar]
- 346.Tengholm A Cyclic AMP dynamics in the pancreatic beta-cell. Ups J Med Sci 117: 355–369, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Tengholm A, Gylfe E. cAMP signalling in insulin and glucagon secretion. Diabetes Obes Metab 19 (Suppl 1): 42–53, 2017. [DOI] [PubMed] [Google Scholar]
- 348.Tomas A, Jones B, Leech C. New insights into beta-cell GLP-1 receptor and cAMP signaling. J Mol Biol 432: 1347–1366, 2020. [DOI] [PubMed] [Google Scholar]
- 349.Tomita T, Masuzaki H, Iwakura H, Fujikura J, Noguchi M, Tanaka T, Ebihara K, Kawamura J, Komoto I, Kawaguchi Y, Fujimoto K, Doi R, Shimada Y, Hosoda K, Imamura M, Nakao K. Expression of the gene for a membrane-bound fatty acid receptor in the pancreas and islet cell tumours in humans: Evidence for GPR40 expression in pancreatic beta cells and implications for insulin secretion. Diabetologia 49: 962–968, 2006. [DOI] [PubMed] [Google Scholar]
- 350.Tran PO, Gleason CE, Robertson RP. Inhibition of interleukin-1beta-induced COX-2 and EP3 gene expression by sodium salicylate enhances pancreatic islet beta-cell function. Diabetes 51: 1772–1778, 2002. [DOI] [PubMed] [Google Scholar]
- 351.Trexler AJ, Taraska JW. Regulation of insulin exocytosis by calcium-dependent protein kinase C in beta cells. Cell Calcium 67: 1–10, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Truchan NA, Sandhu HK, Fenske RJ, Buchanan R, Moeller J, Reuter A, Harrington JM, Kimple ME. Differential effects of prostaglandin E2 production and signaling through the prostaglandin EP3 receptor on human beta-cell compensation. BioRxiv, 2019. [Google Scholar]
- 353.Trujillo JM, Nuffer W. GLP-1 receptor agonists for type 2 diabetes mellitus: Recent developments and emerging agents. Pharmacotherapy 34: 1174–1186, 2014. [DOI] [PubMed] [Google Scholar]
- 354.Tsalkova T, Gribenko AV, Cheng X. Exchange protein directly activated by cyclic AMP isoform 2 is not a direct target of sulfonylurea drugs. Assay Drug Dev Technol 9: 88–91, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Tse LH, Wong YH. GPCRs in autocrine and paracrine regulations. Front Endocrinol 10: 428, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Tunaru S, Bonnavion R, Brandenburger I, Preussner J, Thomas D, Scholich K, Offermanns S. 20-HETE promotes glucose-stimulated insulin secretion in an autocrine manner through FFAR1. Nat Commun 9: 177, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Turk J, Gross RW, Ramanadham S. Amplification of insulin secretion by lipid messengers. Diabetes 42: 367–374, 1993. [DOI] [PubMed] [Google Scholar]
- 358.Turk J, Mueller M, Bohrer A, Ramanadham S. Arachidonic acid metabolism in isolated pancreatic islets. VI. Carbohydrate insulin secretagogues must be metabolized to induce eicosanoid release. Biochim Biophys Acta 1125: 280–291, 1992. [DOI] [PubMed] [Google Scholar]
- 359.Turk J, Ramanadham S. The expression and function of a group VIA Ca2+-independent phospholipase A2 (iPLA2beta) in beta-cells. Can J Physiol Pharmacol 82: 824–832, 2004. [DOI] [PubMed] [Google Scholar]
- 360.Turk J, Song H, Wohltmann M, Frankfater C, Lei X, Ramanadham S. Metabolic effects of selective deletion of group VIA phospholipase A2 from macrophages or pancreatic islet beta-cells. Biomolecules 10, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Turk J, White TD, Nelson AJ, Lei X, Ramanadham S. iPLA2beta and its role in male fertility, neurological disorders, metabolic disorders, and inflammation. Biochim Biophys Acta Mol Cell Biol Lipids 846–860: 2019, 1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Turk J, Wolf BA, McDaniel ML. The role of phospholipid-derived mediators including arachidonic acid, its metabolites, and inositoltrisphosphate and of intracellular Ca2+ in glucose-induced insulin secretion by pancreatic islets. Prog Lipid Res 26: 125–181, 1987. [DOI] [PubMed] [Google Scholar]
- 363.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. Proteomics. Tissue-based map of the human proteome. Science 347: 1260419, 2015. [DOI] [PubMed] [Google Scholar]
- 364.Utzschneider KM, Prigeon RL, Faulenbach MV, Tong J, Carr DB, Boyko EJ, Leonetti DL, McNeely MJ, Fujimoto WY, Kahn SE. Oral disposition index predicts the development of future diabetes above and beyond fasting and 2-h glucose levels. Diabetes Care 32: 335–341, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Valverde I, Vandermeers A, Anjaneyulu R, Malaisse WJ. Calmodulin activation of adenylate cyclase in pancreatic islets. Science 206: 225–227, 1979. [DOI] [PubMed] [Google Scholar]
- 366.Van de Velde S, Wiater E, Tran M, Hwang Y, Cole PA, Montminy M. CREB promotes beta cell gene expression by targeting its coactivators to tissue-specific enhancers. Mol Cell Biol 39, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.van Tienhoven M, Atkins J, Li Y, Glynn P. Human neuropathy target esterase catalyzes hydrolysis of membrane lipids. J Biol Chem 277: 20942–20948, 2002. [DOI] [PubMed] [Google Scholar]
- 368.Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem 276: 13505–13508, 2001. [DOI] [PubMed] [Google Scholar]
- 369.Wan QF, Dong Y, Yang H, Lou X, Ding J, Xu T. Protein kinase activation increases insulin secretion by sensitizing the secretory machinery to Ca2+. J Gen Physiol 124: 653–662, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Wang CY, Liu Z, Ng YH, Südhof TC. A synaptic circuit required for acquisition but not recall of social transmission of food preference. Neuron 107: 144–157.e144, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Wang G, Liang R, Liu T, Wang L, Zou J, Liu N, Liu Y, Cai X, Liu Y, Ding X, Zhang B, Wang Z, Wang S, Shen Z. Opposing effects of IL-1beta/COX-2/PGE2 pathway loop on islets in type 2 diabetes mellitus. Endocr J 66: 691–699, 2019. [DOI] [PubMed] [Google Scholar]
- 372.Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis—roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci 122: 893–903, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Ward WK, Johnston CL, Beard JC, Benedetti TJ, Halter JB, Porte D Jr. Insulin resistance and impaired insulin secretion in subjects with histories of gestational diabetes mellitus. Diabetes 34: 861–869, 1985. [DOI] [PubMed] [Google Scholar]
- 374.Wei Z, Peterson JM, Wong GW. Metabolic regulation by C1q/TNF-related protein-13 (CTRP13): Activation OF AMP-activated protein kinase and suppression of fatty acid-induced JNK signaling. J Biol Chem 286: 15652–15665, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Weir GC, Gaglia J, Bonner-Weir S. Inadequate beta-cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol 8: 249–256, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116: 115–124, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Westwell-Roper C, Ehses JA. Is there a role for the adaptive immune system in pancreatic beta cell failure in type 2 diabetes? Diabetologia 57: 447–450, 2014. [DOI] [PubMed] [Google Scholar]
- 379.Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest 104: 787–794, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Wigger L, Cruciani-Guglielmacci C, Nicolas A, Denom J, Fernandez N, Fumeron F, Marques-Vidal P, Ktorza A, Kramer W, Schulte A, Le Stunff H, Liechti R, Xenarios I, Vollenweider P, Waeber G, Uphues I, Roussel R, Magnan C, Ibberson M, Thorens B. Plasma dihydroceramides are diabetes susceptibility biomarker candidates in mice and humans. Cell Rep 18: 2269–2279, 2017. [DOI] [PubMed] [Google Scholar]
- 381.Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. Characterization of the human patatin-like phospholipase family. J Lipid Res 47: 1940–1949, 2006. [DOI] [PubMed] [Google Scholar]
- 382.Wisinski JA, Reuter A, Peter DC, Schaid MD, Fenske RJ, Kimple ME. Prostaglandin EP3 receptor signaling is required to prevent insulin hypersecretion and metabolic dysfunction in a non-obese mouse model of insulin resistance. Am J Physiol Endocrinol Metab 321: E479–E489, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Wolf BA, Pasquale SM, Turk J. Free fatty acid accumulation in secretagogue-stimulated pancreatic islets and effects of arachidonate on depolarization-induced insulin secretion. Biochemistry 30: 6372–6379, 1991. [DOI] [PubMed] [Google Scholar]
- 384.Wolf BA, Turk J, Sherman WR, McDaniel ML. Intracellular Ca2+ mobilization by arachidonic acid. Comparison with myo-inositol 1,4,5-trisphosphate in isolated pancreatic islets. J Biol Chem 261: 3501–3511, 1986. [PubMed] [Google Scholar]
- 385.Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, Revett T, Gimeno R, Lodish HF. Molecular, biochemical and functional characterizations of C1q/TNF family members: adipose-tissue-selective expression patterns, regulation by PPAR-gamma agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem J 416: 161–177, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Wu B,Wei S,Petersen N,Ali Y,Wang X,Bacaj T, Rorsman P,Hong W, Sudhof TC, Han W. Synaptotagmin-7 phosphorylation mediates GLP-1-dependent potentiation of insulin secretion from beta-cells. Proc Natl Acad Sci U S A 112: 9996–10001, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Wu Y, Shyng SL, Chen PC. Concerted trafficking regulation of Kv2.1 and KATP channels by leptin in pancreatic beta-cells. J Biol Chem 290: 29676–29690, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Xiang AH, Takayanagi M, Black MH, Trigo E, Lawrence JM, Watanabe RM, Buchanan TA. Longitudinal changes in insulin sensitivity and beta cell function between women with and without a history of gestational diabetes mellitus. Diabetologia 56: 2753–2760, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Xie T, Chen M, Weinstein LS. Pancreas-specific Gsalpha deficiency has divergent effects on pancreatic alpha- and beta-cell proliferation. J Endocrinol 206: 261–269, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Xu X, Zhao CX, Wang L, Tu L, Fang X, Zheng C, Edin ML, Zeldin DC, Wang DW. Increased CYP2J3 expression reduces insulin resistance in fructose-treated rats and db/db mice. Diabetes 59: 997–1005, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Yan L, Figueroa DJ, Austin CP, Liu Y, Bugianesi RM, Slaughter RS, Kaczorowski GJ, Kohler MG. Expression of voltage-gated potassium channels in human and rhesus pancreatic islets. Diabetes 53: 597–607, 2004. [DOI] [PubMed] [Google Scholar]
- 392.Yasuda T, Shibasaki T, Minami K, Takahashi H, Mizoguchi A, Uriu Y, Numata T, Mori Y, Miyazaki J, Miki T, Seino S. Rim2alpha determines docking and priming states in insulin granule exocytosis. Cell Metab 12: 117–129, 2010. [DOI] [PubMed] [Google Scholar]
- 393.Zhang B, Lai G, Wu J, Sun R, Xu R, Yang X, Qi Y, Zhao Y. 20-HETE attenuates the response of glucose-stimulated insulin secretion through the AKT/GSK-3beta/Glut2 pathway. Endocrine 54: 371–382, 2016. [DOI] [PubMed] [Google Scholar]
- 394.Zhang CL, Katoh M, Shibasaki T, Minami K, Sunaga Y, Takahashi H, Yokoi N, Iwasaki M, Miki T, Seino S. The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science 325: 607–610, 2009. [DOI] [PubMed] [Google Scholar]
- 395.Zhang E, Mohammed Al-Amily I, Mohammed S, Luan C, Asplund O, Ahmed M, Ye Y, Ben-Hail D, Soni A, Vishnu N, Bompada P, De Marinis Y, Groop L, Shoshan-Barmatz V, Renstrom E, Wollheim CB, Salehi A. Preserving insulin secretion in diabetes by inhibiting VDAC1 overexpression and surface translocation in beta cells. Cell Metab 29: 64–77 e66, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Zhang Y, Guo Q, Li X, Gao J, Liu Y, Yang J, Li Q. P2Y purinergic receptor-regulated insulin secretion is mediated by a cAMP/Epac/Kv channel pathway. Biochem Biophys Res Commun 460: 850–856, 2015. [DOI] [PubMed] [Google Scholar]
- 397.Zhang Y, Han C, Zhu W, Yang G, Peng X, Mehta S, Zhang J, Chen L, Liu Y. Glucagon potentiates insulin secretion via beta-cell GCGR at physiological concentrations of glucose. Cell 10: 2495, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Zhao H, Przybylska M, Wu IH, Zhang J, Siegel C, Komarnitsky S, Yew NS, Cheng SH. Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes 56: 1210–1218, 2007. [DOI] [PubMed] [Google Scholar]
- 399.Zhao X, Dey A, Romanko OP, Stepp DW, Wang MH, Zhou Y, Jin L, Pollock JS, Webb RC, Imig JD. Decreased epoxygenase and increased epoxide hydrolase expression in the mesenteric artery of obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 288: R188–R196, 2005. [DOI] [PubMed] [Google Scholar]
- 400.Zhao Y, Fang Q, Straub SG, Lindau M, Sharp GW. Prostaglandin E1 inhibits endocytosis in the beta-cell endocytosis. J Endocrinol 229: 287–294, 2016. [DOI] [PubMed] [Google Scholar]