

Abstract
DICER1 syndrome is a rare genetic disorder predisposing young patients to multiple types of cancer. A 17-year-old woman with a history of mixed Sertoli-Leydig cell tumor and juvenile granulosa cell tumor of the left ovary at age 14 presented with a pelvic mass. She underwent fertility preservation cytoreductive surgery and the pathology showed high-grade sarcoma with rhabdomyosarcomatous differentiation. After the surgery, patient received one cycle of chemotherapy but her disease continued to progress. She therefore underwent total hysterectomy, right salpingo-oophorectomy and hyperthermic intraperitoneal chemotherapy followed by consolidation chemotherapy. Magnetic resonance imaging revealed no evidence of the disease before and after the completion of her chemotherapy. Genetic testing confirmed the DICER1 pathogenic variant. However, she presented again with a recurrence of the disease 6 months later and ultimately died of the disease 11 months after the surgery. Our case demonstrates the challenging management of this rare disease in a young patient and the need for new and effective treatments.
Keywords: DICER1, Sertoli-Leydig cell tumor, Juvenile granulosa cell tumor, High-grade sarcoma, Rhabdomyosarcoma
Highlights
-
•
DICER1 syndrome is a rare inherited disorder with an autosomal dominant pattern.
-
•
Young patients with ovarian neoplasm need genetic counseling and testing.
-
•
Embryonal rhabdomyosarcoma (ERMS) appears to be associated with DICER1 mutations.
-
•
The management of DICER1 syndrome is challenging and new treatment options need to be explored.
1. Introduction
DICER1 syndrome is a rare inherited disorder with an autosomal dominant pattern with a heterozygous DICER1 germline mutation that affects mainly young adults [[1], [2], [3], [4]]. The two-hit hypothesis applies to the DICER1 mutation which acts as a tumor suppressor gene. This syndrome is characterized by pleuropulmonary blastoma, pulmonary cysts, thyroid gland tumors, multinodular goiter, cystic nephroma and ovarian tumors such as Sertoli–Leydig cell tumor (SLCT) and rhabdomyosarcomas [5,6]. Additional features include macrocephaly, structural kidney and dental abnormalities. In the general population, the estimated prevalence of pathogenic DICER1 variant is about 1:10,600 and there are approximately 30,000 Americans with a DICER1 mutation [7].
Ovarian sex cord-stromal tumors have been associated with DICER1 mutation and are often diagnosed before the age of 40 [1,3,4]. A systemic review showed that 57% of patients with SLCT had a DICER1 germline mutation [8]. Ovarian SLCTs are responsible for an increase in testosterone levels, which can act a tumor marker. Patients often present with abdominal distention, pelvic pain, pelvic mass, virilization, hirsutism, menstrual irregularities, amenorrhea, voice change and acne [1,4].
DICER1 mutations have also been associated with embryonal rhabdomyosarcoma (ERMS), affecting most commonly the uterine cervix [9]. Other results have linked it to the ovaries and fallopian tubes [10]. ERMS is a malignant tumor of mesenchymal origin and is often present in childhood. It primarily affects the orbit, nasopharynx, vagina, and bladder. It may manifest before, during or after the presence of SLCT in patients with DICER1 [2].
The management of DICER1 syndrome depends on the extent of the disease spread and the presence of other DICER1-related conditions [1]. This case highlights the presence of DICER1 mutation affecting primarily the reproductive system in a young patient, the challenging management, and the need for further treatment strategies to improve survival.
2. Case Presentation
A 17-year-old woman, G0P0, presented with abdominal and pelvic pain associated with nausea and vomiting for one week. Abdominal physical examination was significant for peritoneal signs. Magnetic resonance imaging (MRI) showed a 10-cm pelvic mass. A computed tomography (CT) scan of the chest, abdomen, and pelvis showed complex bilateral pelvic masses with mixed soft tissue and fluid attenuation, without fat or calcification. The right pelvic mass measured 13 × 6 × 12 cm. The left pelvic mass measured 13 × 7 × 8 cm (Fig. 1). Pre-operative AFP, hCG, CA19–9, and CEA were within normal levels. CA 125 was 61 U/mL. Due to the concern of internal bleeding related to tumor, the patient was scheduled for immediate surgery. She underwent diagnostic laparoscopy, exploratory laparotomy, radical cytoreductive surgery with removal of pelvic mass, appendectomy, total omentectomy, and pelvic peritonectomy. The right ovary and uterus were preserved. Pathology was consistent with high-grade sarcoma with rhabdomyosarcomatous differentiation (Fig. 2A). Germline testing identified a pathogenic variant, DICER1 c.3517dup (p.Thr1173Asnfs*4).
Fig. 1.
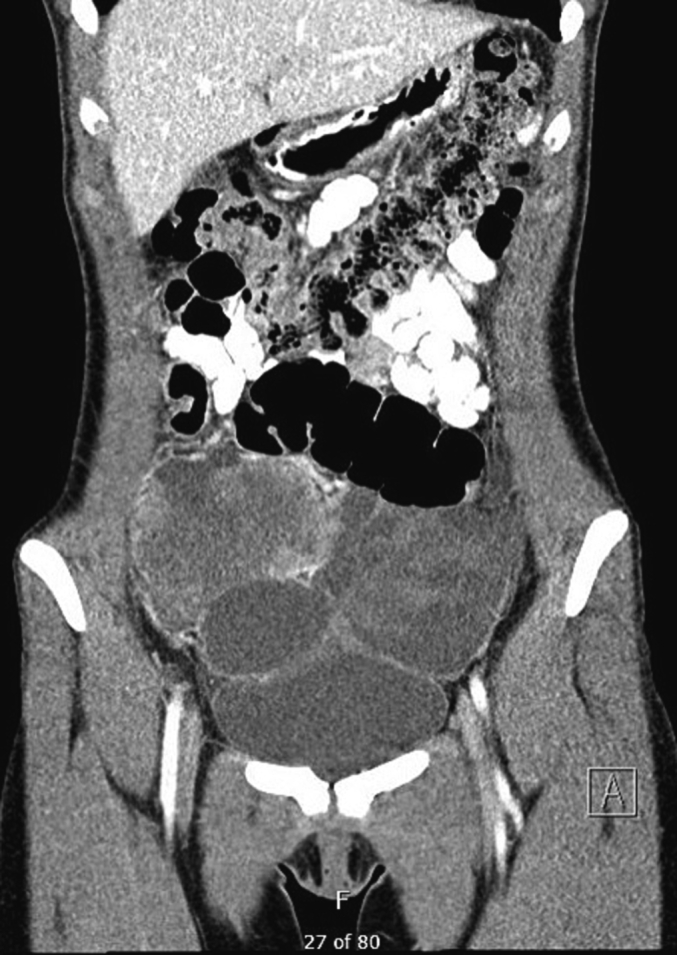
CT scan showed bilateral pelvic masses.
Fig. 2.

Histopathology of nodules.
A: The rhabdomyoblastic features are evident by the strong expression for desmin in the tumor cells in one of the peritoneal nodules (immunoperoxidase, 200×).
B: In areas, the nodules showed frank interlacing fascicles of spindle cells supportive of a spindle variant of rhabdomyosarcoma (Hematoxylin and eosin, 200×).
The patient initiated therapy with VAC/VI (vincristine, dactinomycin, cyclophosphamide (VAC) alternating with vincristine and irinotecan (VI)) per Children's Oncology Group (COG) protocol ARST0431. A month later, due to progression of disease and tumor rupture, she underwent second cytoreductive surgery R0, total hysterectomy, right salpingo-oophorectomy, small bowel resection with side-to-side anastomosis, pelvic peritonectomy and hyperthermic intraperitoneal chemotherapy (HIPEC) with cisplatin. The post-operative course was uncomplicated and pathology was consistent with ERMS (Fig. 2B).
Three weeks later, a positron emission tomography (PET)-CT scan and MRI revealed no residual disease. She received consolidative therapy with a total of 6 cycles of VAC alternating with etoposide, ifosfamide and cisplatin (VIP), with the thought of targeting the SLCT by incorporating a platinum-based regimen [11]. Four months later, the patient presented with pelvic pain with evidence of disease recurrence. Third-line chemotherapy with gemcitabine and nab-paclitaxel was attempted for high-grade ERMS but there was no response after two cycles. Unfortunately, the patient progressed and was offered hospice care and died of the disease 11 months from the initial surgery.
The patient's medical history includes left ovarian cyst at age 14. Due to left ovarian cyst, patient underwent laparoscopic left ovarian cystectomy. Pathology at that time showed sex cord stromal tumor with features of SLCT of intermediate differentiation and juvenile granulosa cell tumor. Based on pathologic findings, the patient underwent laparoscopic left salpingo-oophorectomy, bilateral pelvic lymph node dissection, omentum biopsy, right ovarian cystectomy, and peritoneal biopsy. Pathology failed to reveal any disease and the patient did not receive any adjuvant treatment at that time.
Her family history was significant for her mother having thyroid nodules and her maternal grandfather having a nephrectomy at 3 years of age, her maternal grandmother having an unspecified abdominal cancer at age 50, her maternal grandmother's brother having an unspecified brain tumor, and her paternal grandmother having esophageal cancer.
3. Discussion
We present a challenging case of a young patient with rhabdomyosarcomatous conversion of a prior sex cord stromal tumor with an extensive surgical history.
Very little is known about the management of SLCT and ERMS in the setting of DICER1 syndrome. SLCT is the most common ovarian tumor associated with DICER1 syndrome; it accounts for less than 2% of all ovarian neoplasms [3,7,11,12]. In the presence of rhabdomyosarcoma elements, previous studies have suggested a higher rate of recurrent disease [13].
We reviewed the literature for SLCT and ERMS related to DICER1 syndrome, including their management, which is summarized in Table 1. All the cases listed used imaging of abdomen and pelvis every 6 months to help monitor DICER1 syndrome recurrence. The first patient had a thyroid examination with imaging since she had multinodular goiter (MNG) and a family history of thyroid disorders [14]. The second patient developed a radiological focal nodular liver hyperplasia, underwent partial left nephrectomy due to the development of a cystic nephroma, and had MNG [9]. One case report did not specify the chemotherapeutic regimen used for a 14-year-old girl [10]. The rest of the case reports highlight the pharmacological regimen used without specifying their effectiveness, although all patients seemed to be in remission and alive at the time of publication [6,9,11,13]. Interestingly, in the first case an intensified Ewing sarcoma regimen was used in conjunction with other chemotherapy medication as per a COG study, but without specific details being given [14].
Table 1.
Cases of DICER1 syndrome related Sertoli Leydig cell tumor (SLCT) and embryonal rhabdomyosarcoma (ERMS) as per current literature search.
| Author, year | Case | Mutations | Gynecological Surgery | Pathology | Medical management |
|---|---|---|---|---|---|
| Cowan, M. et al. (2018) [14] | 17-year-old female with hemorrhage and intermenstrual bleeding from a cervical and adnexal mass |
DICER1 VUS1 in: BCOR, BRCA2, MLH1, PALB2 |
|
|
Ewing sarcoma regimen Vincristine Doxorubicin Cyclophosphamide Ifosfamide Etoposide |
| De Kock, L. et al. (2015) [9] | 6-year-old female with abdominal distension, rigidity and difficulty passing urine due to an ovarian mass | DICER1 | Right salpingo-oophorectomy | oERMS3 | Antinomycin D Vincristine Cyclophosphamide Etoposide Ifosfamide Radiation (abdomen) |
| Koo, J. & et al. (2020) [11] | 4 females with mean age of 15.3 years with most commonly abdominal pain/distension | DICER1 |
|
|
8 cycles of: Etoposide Ifosfamide Cisplatin Vincristine Dactinomycin Cyclophosphamide |
| McCluggage, W.G. et al. (2020) [10] | 14-year-old female with abdominal pain, urinary incontinence, and fluid leakage per vagina | DICER1 | Resection of 3 masses in the lower abdomen, cul-de-sac pelvis and right fallopian tube (salpingectomy) | ERMS | Abdominopelvic radiation Multiagent chemotherapy without specification |
| Plastini, T. et al. (2017) [13] |
|
DICER1 |
|
|
|
| Schultz, K. et al. (2016) [6] | 10-year-old female with a history of dysuria and pelvic pressure due to left ovarian and right ovarian (4 years later) mass | DICER1 |
|
SLCT on left ovary with weak staining for desmin | Vincristine Actinomycin D Cyclophosphamide 6 cycles of: Cisplatin Etoposide Bleomycin |
Variants of unknown significance.
Cervical embryonal rhabdomyosarcoma.
Ovarian embryonal rhabdomyosarcoma.
There is no standard chemotherapy regimen for patients with advanced or recurrent SLCT and rhabdomyosarcoma after surgery. A regimen that has been used with some response includes 4 cycles of VIP alternating with VAC; however, this did not provide long-term benefit to our patient [11].There is an agreement that VAC is the backbone for the treatment of rhabdomyosarcoma. Another regimen that has been described for adult patients includes doxorubicin, ifosfamide, and vincristine [13]. The feasibility and efficacy of HIPEC in pediatric sarcoma has also been described and there is an ongoing clinical trial [15].
In our case, the immunohistochemical findings showing muscle differentiation may present an argument that incipient immunophenotypical rhabdomyosarcoma cells were present in the sex cord tumor that did not show typical morphology of heterologous elements of rhabdomyosarcoma. Perhaps, routine staining for muscle markers in sex cord tumors in patients with DICER1 syndrome may predict future and frank manifestation of rhabdomyosarcoma. Also, their presence may prompt establishing therapy for rhabdomyosarcoma from the beginning of the disease manifestation. Whereas ERMS has been found in the setting of DICER1, there are no reports in the literature of spindle cell rhabdomyosarcoma linking it to DICER1 syndrome.
Future studies are warranted to explore new and effective treatment plans in patients with DICER1 syndrome related to SLCT and ERMS to help increase their response rate and improve their overall life expectancy.
4. Conclusion
We present an extremely rare and challenging case of a young patient with SLCT and pelvic ERMS with a diagnosis of a DICER1 syndrome, managed with extensive surgeries, multiple rounds of chemotherapy, and HIPEC. This case highlights the importance of further exploring new and effective treatment options for patients with DICER1 syndrome affecting the reproductive system.
Acknowledgments
Contributors
Nora Shero contributed to conception of the case report, drafting the manuscript, undertaking the literature review and revising the article critically for important intellectual content.
Aditi Dhir contributed to patient care, conception of the case report, acquiring and interpreting the data, and revising the article critically for important intellectual content.
Pablo Bejarano contributed to patient care, conception of the case report, acquiring and interpreting the data, and revising the article critically for important intellectual content.
Sara Rhode contributed to patient care, conception of the case report, and revising the article critically for important intellectual content.
Joel Cardenas Goicocechea contributed to patient care, conception of the case report, acquiring and interpreting the data, drafting the manuscript, undertaking the literature review and revising the article critically for important intellectual content.
All authors approved the final submitted manuscript.
Funding
No funding from an external source supported the publication of this case report.
Patient consent
Informed consent was obtained, from both the patient and her legal representative, for the publication of this case report and associated images in accordance with institutional policy.
Provenance and peer review
This article was not commissioned and was peer reviewed.
Acknowledgments
Conflict of interest statement
The authors declare that they have no conflict of interest regarding the publication of this case report.
Contributor Information
Nora Shero, Email: n.shero@mua.edu.
Aditi Dhir, Email: aditi.dhir@med.miami.edu.
Pablo Bejarano, Email: bejarap@ccf.org.
Sara Rhode, Email: rhodes@ccf.org.
Joel Cardenas Goicocechea, Email: Cardenj3@ccf.org.
References
- 1.Schultz K.A.P., Stewart D.R., Kamihara J., Bauer A.J., Merideth M.A., Stratton P., et al. In: DICER1 tumor predisposition. Adam M.P., Mirzaa G.M., Pagon R.A., Wallace S.E., LJH Bean, Gripp K.W., Amemiya A., editors. GeneReviews((R)); Seattle (WA): 2020. [PubMed] [Google Scholar]
- 2.Schultz K.A.P., Williams G.M., Kamihara J., Stewart D.R., Harris A.K., Bauer A.J., et al. DICER1 and associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin. Cancer Res. 2018;24(10):2251–2261. doi: 10.1158/1078-0432.CCR-17-3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stewart C.J., Charles A., Foulkes W.D. Gynecologic manifestations of the DICER1 syndrome. Surg. Pathol. Clin. 2016;9(2):227–241. doi: 10.1016/j.path.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Robertson J.C., Jorcyk C.L., Oxford J.T. DICER1 syndrome: DICER1 mutations in rare cancers. Cancers (Basel) 2018;10(5) doi: 10.3390/cancers10050143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watanabe T., Soeda S., Endo Y., Okabe C., Sato T., Kamo N., et al. Rare hereditary gynecological cancer syndromes. Int. J. Mol. Sci. 2022;23(3) doi: 10.3390/ijms23031563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schultz K.A., Harris A., Messinger Y., Sencer S., Baldinger S., Dehner L.P., Hill D.A. Ovarian tumors related to intronic mutations in DICER1: a report from the international ovarian and testicular stromal tumor registry. Familial Cancer. 2016;15(1):105–110. doi: 10.1007/s10689-015-9831-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez I.A., Stewart D.R., Schultz K.A.P., Field A.P., Hill D.A., Dehner L.P. DICER1 tumor predisposition syndrome: an evolving story initiated with the pleuropulmonary blastoma. Mod. Pathol. 2022;35(1):4–22. doi: 10.1038/s41379-021-00905-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai S., Zhao W., Nie X., Abbas A., Fu L., Bihi S., et al. Multimorbidity and genetic characteristics of DICER1 syndrome based on systematic review. J. Pediatr. Hematol. Oncol. 2017;39(5):355–361. doi: 10.1097/MPH.0000000000000715. [DOI] [PubMed] [Google Scholar]
- 9.de Kock L., Druker H., Weber E., Hamel N., Traubici J., Malkin D., et al. Ovarian embryonal rhabdomyosarcoma is a rare manifestation of the DICER1 syndrome. Hum. Pathol. 2015;46(6):917–922. doi: 10.1016/j.humpath.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 10.McCluggage W.G., Apellaniz-Ruiz M., Chong A.L., Hanley K.Z., Velazquez Vega J.E., McVeigh T.P., Foulkes W.D. Embryonal rhabdomyosarcoma of the ovary and fallopian tube: rare neoplasms associated with germline and somatic DICER1 mutations. Am. J. Surg. Pathol. 2020;44(6):738–747. doi: 10.1097/PAS.0000000000001442. [DOI] [PubMed] [Google Scholar]
- 11.Koo J., Garrington T.P., Kerr K., Treece A.L., Cost C.R. Pediatric ovarian Sertoli-Leydig cell tumors with heterologous rhabdomyosarcoma elements: clinical case series and review of the literature. Pediatr. Blood Cancer. 2020;67(10) doi: 10.1002/pbc.28621. [DOI] [PubMed] [Google Scholar]
- 12.de Kock L., Terzic T., McCluggage W.G., Stewart C.J.R., Shaw P., Foulkes W.D., Clarke B.A. DICER1 mutations are consistently present in moderately and poorly differentiated Sertoli-Leydig cell tumors. Am. J. Surg. Pathol. 2017;41(9):1178–1187. doi: 10.1097/PAS.0000000000000895. [DOI] [PubMed] [Google Scholar]
- 13.Plastini T., Staddon A. Sertoli-Leydig cell tumor with concurrent rhabdomyosarcoma: three case reports and a review of the literature. Case Rep. Med. 2017;2017:4587296. doi: 10.1155/2017/4587296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cowan M., Suntum T., Olivas A.D., Perpich M., Applebaum M.A., Lastra R.R., Yamada S.D. Second primary rhabdomyosarcoma of the uterine cervix presenting with synchronous ovarian Sertoli-Leydig cell tumor: an illustrative case of DICER1 syndrome. Gynecol. Oncol. Rep. 2018;25:94–97. doi: 10.1016/j.gore.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Findlay B.L., Gargollo P.C., Granberg C.F. Use of Hyperthermic intraperitoneal chemotherapy (HIPEC) in pediatric sarcoma for maximal oncologic control. Urology. 2020;141:139–142. doi: 10.1016/j.urology.2020.04.032. [DOI] [PubMed] [Google Scholar]