

Abstract
Cancer patients by immune checkpoint therapy have achieved long-term remission, with no recurrence of clinical symptoms of cancer for many years. Nevertheless, more than half of cancer patients are not responsive to this therapy due to immune exhaustion. Here, we report a novel gene engineered exosome which is rationally designed by engineering PD1 gene and simultaneously enveloping an immune adjuvant imiquimod (PD1-Imi Exo) for boosting response of cancer immune checkpoint blockage therapy. The results showed that PD1-Imi Exo had a vesicular round shape (approximately 139 nm), revealed a significant targeting and a strong binding effect with both cancer cell and dendritic cell, and demonstrated a remarkable therapeutic efficacy in the melanoma-bearing mice and in the breast cancer-bearing mice. The mechanism was associated with two facts that PD1-Imi Exo blocked the binding of CD8+ T cell with cancer cell, displaying a PD1/PDL1 immune checkpoint blockage effect, and that imiquimod released from PD1-Imi Exo promoted the maturation of immature dendritic cell, exhibiting a reversing effect on the immune exhaustion through activating and restoring function of CD8+ T cell. In conclusion, the gene engineered exosome could be used for reversing T cell exhaustion in cancer immunotherapy. This study also offers a promising new strategy for enhancing PD1/PDL1 therapeutic efficacy, preventing tumor recurrence or metastasis after surgery by rebuilding the patients' immunity, thus consolidating the overall prognosis.
Keywords: Gene engineered exosomes, T cell exhaustion, PD1/PDL1, Imiquimod, Cancer immunotherapy
Graphical abstract
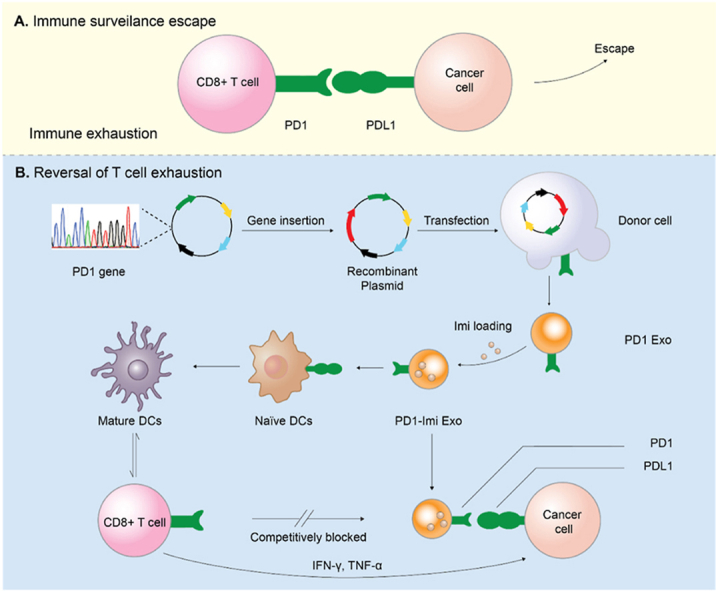
Illustration for rational design of PD1 gene engineered exosomes.(A) Immune surveillance escape. Cancer cells resist to the host immune antitumor response via overexpressing programmed cell death ligand 1 (PDL1) that exhausts antigen-specific CD8+ T cells through binding with programmed cell death protein 1 (PD1) receptor. (B) Reversal of T cell exhaustion. The immune activation effect was achieved through antagonizing the PD1/PDL1 axis by PD1 gene engineered exosome (PD1-Imi Exo), and maturation of immature dendritic cells by immune adjuvant imiquimod released from PD1-Imi Exo.
Highlights
-
•
PD1 gene engineered exosome rationally designed by enveloping imiquimod (PD1-Imi Exo).
-
•
Imiquimod promoting maturation of dendritic cells and further activating T cells.
-
•
PD1-Imi Exo reversing T cell exhaustion and competitively blocking PD1/PDL1 axis.
-
•
PD1-Imi Exo boosting cancer immunotherapy and preventing metastasis after surgery.
1. Introduction
Immune checkpoint therapy, which enhances anticancer immune response by modulating immune checkpoint signaling pathway in T cell, has made important clinical advances and provides a new weapon in the fight against cancer [1]. Although some adverse reactions have been identified with immune checkpoint therapy, patients have achieved lasting clinical benefits and some patients have achieved long-term remission, with no recurrence of clinical symptoms of cancer for many years [2]. Currently developed strategies for the immune checkpoint therapy consist of inhibition of cytotoxic T-lymphocyte associated protein 4 (CTLA4) [3,4], interception of CD47 signaling pathway [5,6], blockage of programmed death-1/programmed death-ligand 1 (PD1/PDL1) pathway, etc. Among which, the PD1/PDL1 pathway has demonstrated a landmark strategy through which cancer cell can be eliminated in approximately 30 % of patients with various types of cancer [7], such as melanoma, renal cell carcinoma, non-small cell lung cancer (NSCLC), and bladder cancer.
Nevertheless, more than half of cancer patients are not responsive to PD1/PDL1 therapy due to the loss of targeted tumor-associated antigens, inefficient maturation of dendritic cells (DCs), and poor activation and infiltration of cytotoxic T lymphocytes [[8], [9], [10]]. Consequently, how to improve the responsiveness of PD1/PDL1 therapy is not only an important scientific issue to be solved, but also a critical clinical problem to be solved urgently. Here, we report a kind of gene engineered exosome which is rationally designed for boosting response of cancer immune checkpoint blockage therapy by recombing PD1 gene into a natural exosome and by simultaneously enveloping an immune adjuvant imiquimod.
Increasing evidence indicates that CD8+ T cell is the most effective cytotoxic T lymphocyte, exerting a killing effect on cancer cell through cytokine secretion, cytotoxic granule release, and/or through Fas/FasL interaction [11]. Under normal circumstances, T cell plays an immune surveillance role and is able to correct the disorder of cell growth in time [12]. PD1 is expressed on the surface of T cell while PDL1 expressed on the surface of normal cell. The specific PD1/PDL1 binding can prevent normal cell from being killed by T cell. Once cancer cell occurs and its surface does not express PDL1, T cell can recognize and then kill it. However, cancer cells usually evolve to express PDL1 very quickly. By binding PD1 to PDL1, T cells escape immune surveillance, leading to persistent cancer growth [13].
In fact, PD1/PDL1 cancer immune checkpoint blockage therapy is a process that reactivates the killing effect of CD8+ T cells. The activation is predominantly induced by the cross-priming, in which tumor associated antigens are sampled and presented to naïve T cells by antigen presenting cells (APCs). DCs have been identified as the most potent APCs in cancer microenvironment in initiating, and in regulating innate and adaptive immunity. DCs capture tumor-associated antigens, form major histocompatibility complex (MHC)-antigen aggregates, and present the antigen to T cells, thus providing costimulatory/soluble factors to shape T cell anticancer responses [14]. However, the ability of DCs to activate T cells is largely dependent on its mature state. Due to immunosuppression in cancer microenvironment, DCs are usually in immature state, and hence their function for initiating immune response is significantly hindered [15].
Toll-like receptor (TLR) agonist has the potential as an adjuvant for inducing the maturation of DCs. Compared with traditional adjuvant such as aluminum adjuvant, TLR agonist has a higher immune stimulating effect and its action mechanism is well understood [16]. Imiquimod, an imidazoquinoline compound, is a typical TLR agonist [17], which is clinically used to treat genital warts, squamous cell carcinoma, basal cell carcinoma [18] and actinic keratosis. Recent studies have found that it is an effective adjuvant for activation of DCs [19]. However, due to its rapid distribution throughout the body after administration, it causes adverse reactions, which seriously hinders its clinical application [20].
Biomimetic nanomaterials have provided many new opportunities for drug delivery and cancer immunotherapy due to their uniqueness [[21], [22], [23], [24], [25]]. Exosome is a kind of biomimetic nanocarrier with broad development prospects [26]. Generally, exosomes are derived from extracellular vesicles released by eukaryotic cells, and their size is between 40 and 200 nm [27]. It has been found that exosomes have the ability to transfer membrane components and nucleic acids between cells, thus playing a role in intercellular communication [[28], [29], [30], [31]]. Exosomes' inherent biocompatibility, transport capacity, blood flow stability, and engineering ability make them promising vectors for delivering therapeutic genes, proteins, and small molecules to tumor sites [32]. In addition, exosomes are less immunogenic and less virulent than other non-viral vectors [[33], [34], [35]]. However, the lack of targeted orientation limits their use. In order to optimize the performance of exosomes and take advantage of their natural capabilities, scientists have striven through genetic or metabolic engineering methods to equip exosomes with new parts of the surface while preserving vesicle integrity [36].
In view of the unique advantages of exosome, we genetically engineered donor human embryonic kidney HEK 293T cells to stably express mouse PD1 and produce a gene recombination engineered PD1 exosome (PD1 Exo) in vitro. We then applied the exosome to cancer cell to restore the exhausted CD8+ T cell by blocking PDL1. Furthermore, PD1 Exo can also carry and transport the toll-like receptor 7 agonist imiquimod as an immune adjuvant (forming PD1-Imi Exo) to activate DCs in the cancer microenvironment, further enhancing the anticancer effect of CD8+ T lymphocytes in the surgical cancer microenvironment (Fig. 1).
Fig. 1.

Illustration for rational design of PD1 gene engineered exosomes.
(A) Immune surveillance escape. Cancer cells resist to the host immune antitumor response via overexpressing programmed cell death ligand 1 (PDL1) that exhausts antigen-specific CD8+ T cells through binding with programmed cell death protein 1 (PD1) receptor. (B) Reversal of T cell exhaustion. The immune activation effect was achieved through antagonizing the PD1/PDL1 axis by PD1 gene engineered exosome (PD1-Imi Exo), and maturation of immature dendritic cells by immune adjuvant imiquimod released from PD1-Imi Exo.
2. Materials and methods
2.1. Cell cultures
Human embryonic kidney 293T (HEK 293T) cells were purchased from the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing, China). Mouse dendritic (DC 2.4) cells were purchased from BeNa Culture Collection (Xinyang, Henan, China). Mouse melanoma cell lines (B16F10, B16F10-Luc, B16F10-OVA) and breast cancer 4T1 cells were purchased from American Type Culture Collection (ATCC, USA). HEK 293T cells, melanoma cells and DC 2.4 cells were cultured in Dulbecco's modified Eagle's medium (Macgene, Beijing, China) at 37 °C under 5 % CO2. 4T1 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Macgene, Beijing, China) at 37 °C under 5 % CO2. All these culture media were supplemented with 10 % fetal bovine serum (FBS; PAN, Beijing local agent, Germany), 100 U/mL penicillin, and 100 μg/mL streptomycin (Macgene, Beijing, China).
2.2. Antibodies
InVivoMab anti-mouse PDL1 (B7–H1) antibody was purchased from Bioxcell (Beijing, China). HSP70/HSPA1 rabbit pAb was purchased from ABclonal (Wuhan, China). Anti-PD1 antibody (# ab214421), and anti-TSG101 antibody (# ab125011) were purchased from Abcam (Shanghai local agent, USA). PE anti-mouse CD80, (# 104708), APC anti-mouse CD86 (# 105011), FITC anti-mouse CD11c (# 117305), PE anti-mouse CD8a (# 553032), FITC anti-mouse CD4 (# 557307), APC anti-mouse CD3 (# 100236), FITC anti-mouse CD3 (# 100204), PE/Cyanine7 anti-mouse CD4 (# 100528), PerCP/Cyanine5.5 anti-mouse CD8a (# 100734), PE anti-mouse/human CD44 (# 103024), PE anti-mouse CD40 (# 157505), APC anti-mouse CD62L (# 104412), PE anti-mouse TNF-α (# 506305) and brilliant violet 421 anti-mouse IFN-γ (# 505830) were purchased from Biolegend (Beijing local agent, USA). Anti-mouse CD3e (# 553057) was purchased from BD (Beijing local agent, USA). Recombinant murine IL-4 (# 214-14), recombinant murine IL-2 (# 212-12), and murine granulocyte-macrophage colony stimulating factor (# GM-CSF, 315-03) were purchased from PeproTech (Beijing local agent, USA). Anti-41BB antibody (# 18798T) was purchased from CellSignalingTechnology (Beijing local agent, USA). IgG/Alexa Fluor/647 41BB (# A0468) was purchased from Beyotime Biotechnology (Beijing local agent, China). Anti-PDL1 antibody (# 66248-1) was purchased from proteintech (Beijing local agent, USA). IgG/Alexa Fluor/488 PD1 (# ZF-0511), IgG/Alexa Fluor/594 PDL1 (# ZF-0513) were purchased from ZSGB-BIO (Beijing local agent, China).
2.3. Materials
Polyjet (# SL100688) was purchased from Unique Biotechnology Company (Beijing, China), Imiquimod (# 401020) was purchased from Sigma-Aldrich (Beijing local agent, China). Red blood cell lysis buffer (#R1010) was purchased from Solarbio (Beijing, China). Bovine serum albumin (BSA, # 36101ES60) was purchased from Yeasen (Shanghai, China). Bicinchoninic acid (BCA) (#P0012) was purchased from Beyotime Biotechnology (Shanghai, China). 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyaineiodide (DiR, # 40757ES25) was purchased from Yeasen Biotech (Shanghai, China). 1, 1‘-dioctadecyl-3,3,3′,3′ -tetramethylindocarbocyanine perchlorate (DiI, # CD4666) was purchased from Coolaber Science (Beijing, China). 4%–20 % precast gel (# DE101) was purchased from DiNing (Beijing, China). Radio immunoprecipitation assay (RIPA, #C1053), electrophoretic buffer (#B1005), electrotransfer buffer (#B1006), tris-buffered saline and Tween 20 (TBST, #B1009), and SDS denatured protein loading buffer (#B1012) were purchased from Applygen (Beijing, China). Polyvinylidene fluoride (PVDF, # ISEQ00010) membrane was purchased from Merck-Millipore (Beijing local agent, China). Hoechst 33,342 (#H21492) was purchased from Invitrogen (Shanghai local agent, China). CCK-8 (# MA0218) was purchased from Meilunbio (Dalian, China). IP-10(#E08183 m), and IP-12p70(#E04600 m) ELISA kits were purchased from Cusabio (Wuhan, China). Lipopolysaccharide (LPS) was purchased from Solarbio (Beijing, China). Erythrocyte lysis solution was purchased from Macgene (Beijing, China). D-luciferin (# 40901ES01) was purchased from Yeasen Biotech (Shanghai, China). OVA257-264 (# CP7214) was purchased from WSHTBio (Beijing, China). PD1 protein (# PD1-M5259), and PDL1 Protein (# PD1-M5220) were purchased from Acrobiosystem. Carboxyfluorescein succinimidyl ester (CFSE, #T6802) was from Targetmol (Boston, MA, USA).
2.4. Plasmid construction
pCMV-VSV-G (plasmid #8454), and gag/pol (plasmid #14887) were purchased from Addgene (Beijing local agent, China). Mouse PD1 gene (NM_008798.3) was synthesized and inserted into the VC141-1-VT vector by Cyagen Biosciences (Suzhou, China). The sequences were designed and analyzed with Snapgene (version 2.3.2, USA). The gene sequencing was completed by Tsingke Biotechnology (Beijing, China).
2.5. Animals
BALB/c and C57BL/6J mice (16–20 g, 6–7 weeks old) were purchased from Vital River Laboratory Animal Center (Beijing, China) and maintained in a specific pathogen free (SPF) environment.
2.6. Lentivirus packaging and PD1 gene recombination
For preparing the gene recombination PD1 expressing system, HEK 293T cells grown to 70 % confluence in a 75-mm culture flask were transfected with 8 μg recombined PD1 plasmid, 1.6 μg gag/pol plasmid, and 0.32 μg pCMV-VSV-G plasmid according to the manufacturer's instructions. Briefly, three plasmids were added into 400 μL serum-free DMEM with high glucose, and mixed by vortex as the system A. A volume of 10 μL PolyJet regent was diluted into 400 μL serum-free DMEM with high glucose, and gently mixed by vortex as the system B. Afterwards, the system A and system B were mixed immediately by vortex, and incubated for 15 min at room temperature to form the PolyJet/DNA complexes as the system C, which was then mixed gently with 10 mL fresh complete DMEM medium, and further cultured at 37 °C under 5 % CO2 for 12 h as the system D. After replacing with fresh complete DMEM medium at 12 h, the system D was further cultured for 48 h, and its supernatant was collected, and stored at 4 °C. The system D was added with the same volume of fresh complete DMEM medium and further cultured for another 24 h, and its supernatant was collected again. Finally, all the collected supernatants were combined together and concentrated by centrifugation at 10, 000×g at 4 °C for 1 h. The pellets were collected and suspended in 1 mL PBS (pH 7.4) as the system E, which was added into a 25-mm flask HEK 293T cells and further cultured at 37 °C under 5 % CO2. After selecting with 2 μg/mL puromycin for two weeks, the gene recombinant engineered HTK 293T cells were obtained as the gene recombination PD1 expressing system.
2.7. Exosomes preparation
For preparing exosome-depleted FBS, FBS was ultracentrifuged at 100,000g, 4 °C for 20 h, and then filtered through 100-nm filters. For isolating exosomes, the gene recombinant engineered HEK 293T cells were cultured in fresh DMEM medium with 10 % exosome-depleted FBS at 37 °C under 5 % CO2. After culturing for 48 h, the cell culture supernatant was collected. Exosomes were isolated by a standard differential centrifugation protocol. Briefly, the culture medium was centrifuged at 4 °C for 10 min at 300×g to remove the living cells, then centrifuged at 2,000×g for 10 min to remove the dead cells (Beckman Coulter, Optima L-XP), and filtrated by 0.22-μm filter to remove the cell debris. The final medium was then ultracentrifuged at 100,000×g at 4 °C for 70 min to precipitate the small vesicles that correspond to exosomes [37]. The pellets were collected, washed in a large volume of PBS (pH7.4) to eliminate contaminating proteins, and ultracentrifuged at 100,000×g at 4 °C for 70 min again. Finally, the resultant pellets were diluted in PBS (pH 7.4), and stored at −80 °C for further use [38,39]. The total protein concentration of exosomes was quantified by BCA protein assay kit according to the manufacturer's instructions. Typically, approximately 33 μg total protein of exosomes was collected from 1 bottle of T75 culture bottle.
2.8. Exosomes enveloping imiquimod
For enveloping imiquimod into the gene recombination engineered exosomes, imiquimod was dissolved in dimethyl sulfoxide (DMSO) (2 mg/mL). A volume of 500 μL imiquimod solution was added into 1 mg PD1 Exo (1 mg/mL total protein) suspensions, and mixed by vortex. The mixture was added into 1 mL electroporation buffer, followed by electroporation shock (400 V and 150 μF) in the 0.4 cm cuvette at 4 °C using Gene Pluser Xcell (Bio-Rad) system. The mixture was further incubated on ice for 30 min. For removing the unloaded imiquimod, the mixture was re-suspended in 10 mL PBS (pH7.4), and centrifuged at 100,000 g at 4 °C for 1 h to collect the pellets. The pellets (PD1-Imi Exo) were collected and suspended in 1 mL PBS (pH7.4) for further use. To quantify the loading rate, 1 mL PD1-Imi Exo was dispersed in 10-fold volume of DMSO, followed by a sonication for 10 min on water bath. An appropriate amount of imiquimod was the same processed and included as the control. The content of imiquimod was measured at 320 nm by spectroscopy [40].
2.9. PD1 concentration on PD1-Imi Exo
The concentration of PD1 protein was determined using a mouse PD1 ELISA kit (copies per ml) (Neobioscience), and the concentration of PD1-Imi Exo in the same solutions was determined via nanoparticle tracking analysis (particles per ml) (NanoSight NS300, Malvern Panalytical). The average number of PD1 molecules on each individual exosome was calculated to be 28.9 ± 14.9 copies [38].
2.10. Physiochemical characterizations
For measuring the encapsulation efficiency of PD1-Imi Exo, imiquimod was quantified at 320 nm with UV–visible spectrometry, and the encapsulation efficiency of imiquimod was calculated with the formula: EEimi = (Imitoal - Imifree)/Imitotal, where EEimi represents the encapsulation efficiency of imiquimod, Imitoal represents total amount of imiquimod added, and Imifree represents the cumulative imiquimod amount in above wash solutions. The encapsulation efficiency of Imi Exo was similarly measured.
For detecting the drug release rate, PD1-Imi Exo was put into dialysis bag (10 kDa), and immersed in 200 mL PBS (pH 7.4) solution, and magnetically stirred at 37 °C for 24 h. At predetermined time-points (0, 2, 4, 8, 12, and 24 h), a certain volume of release solution was sampled, and immediately replaced by the same volume of fresh PBS (pH7.4). The amount of released imiquimod was measured with UV–visible spectrometry as above, the cumulative release rate (Rimi%) was calculated with the formula: Rimi% = Rreleased/Rtotal, where Rreleased represents the cumulative released imiquimod in the release solution, and Rtoal represents total amount of imiquimod in the PD1-Imi Exo.
For observing the morphology, transmission electron microscopy (TEM, JEM-2100, FEI, OR, USA) was used to evaluate the particle sizes of Exo, PD1 Exo, and PD1-Imi Exo, respectively. Briefly, a sample of Exo, PD1 Exo or PD1-Imi Exo (0.5 mg/mL, protein weight) was dropped on a copper grid (25 mm2), stained with 10 μL 2 % uranyl acetate solution for 2 min. After drying the copper grid for 30 min, the sample was observed and photographed under TEM.
For studying the particle sizes, Exo, PD1 Exo, and PD1-Imi Exo were measured using Nanoparticle Tracking Analysis (NTA, NanoSight NS300 Malvern Instruments, UK), respectively. Besides, their zeta potentials were measured using dynamic light scattering (DLS) apparatus (Nano Series Zenith 4003 Zetasizer, Malvern Instruments, UK) at 25 °C.
2.11. Western blot
For identifying PD1 gene recombination engineered exosomes, Western blot assay was performed to evaluate the PD protein expression. Briefly, Exo, PD1 Exo, PD1-Imi Exo or gene recombinant HEK 293T cells were collected, and lysed in RIPA buffer (Applygen, Beijing, China) with protease phosphatase inhibitor (Beyotime, Shanghai, China) at 4 °C for 1 h. The lysed mixture was then centrifuged at 12000 rpm at 4 °C for 20 min for collecting its supernatant. Afterwards, the protein concentration in the supernatant was quantified by BCA assay kit, and further separated by 4%–20 % precast sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel. The separated proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (Merck-Millipore, Darmstadt, Germany) and blocked with 5 % bovine serum albumin (BSA) in TBS-T (Tris buffered saline with Tween 20) solution. After incubating with the appropriate primary antibodies at 4 °C overnight, the PVDF membranes was further incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at 25 °C for 1 h. Finally, the blots were observed by a MiniChemi610 imaging system (Sage Creation, Beijing, China).
2.12. CCK8 assay
For preliminarily evaluating the effect of Exo or PD1 Exo on the growth of melanoma B16F10 cells, a cell counting kit-8 (CCK-8) assay was performed to observe the survival rate of the cancer cells. Briefly, B16F10 cells were incubated in 96-well culture plates at 37 °C under 5 % CO2 overnight. Afterwards, various concentrations of Exo and PD1 Exo (5, 10, 20, 40, 80, 100 μg/mL, total protein weight) were added into the culture plates, and incubated at 37 °C under 5 % CO2 for another 24 h. Then, CCK-8 solution was added, and incubated for another 2 h. Finally, the absorbance of each well was measured using Infinite F50 microplate reader at 450 nm. Survival rate was calculated with the formula:
Where As represents the experiment group, Ac represents the control group, and Ab represents the blank group.
2.13. Targeting capability
For studying the targeting effect on cancer cells, Exo and PD1 Exo were labeled with fluorescent probe DiI according to the manufacturer's instructions. B16F10 cells (15 × 104 cells/well) were incubated in the confocal dishes at 37 °C under 5 % CO2 for 12h. A volume of 200 μL Exo (1 mg/mL protein) or PD1 Exo (1 mg/mL protein) was incubated with 1 μL DiI (5 mM) at 4 °C for 30 min. After filtrating by 40-μm nylon filter membrane to remove the unbounded DiI, the DiI labeled Exo or DiI labeled PD1 Exo was added into the confocal dish, and cultured at 37 °C under 5 % CO2 for another 4 h. After removing the culture medium, B16F10 cells were fixed with paraformaldehyde at room temperature for 10 min, and stained with Hoechst 33342 in the dark for 15 min. After washing with PBS (pH 7.4) for three times, the cells were observed under laser confocal microscope (BD, NJ, USA). To study the targeting effect on dendritic cells, similar processes were performed.
For understanding the targeting mechanism, B16F10 cells were pre-treated with anti-PDL1 (20 ng/mL) for 4 h to saturate PDL1 receptors on B16F10 cells, and then treated with DiI labeled PD1 Exo. The control group was the same pre-treated with PBS (pH 7.4), and then treated with DiI labeled PD1 Exo. After washing with PBS (pH 7.4) for three times, the treated cells were observed under laser confocal microscope (BD, NJ, USA). The uptake ratio was quantified by software Image J (NIH, USA).
2.14. Endocytosis mechanism
For displaying the potential endocytosis pathways of PD1 Exo, the uptake inhibitory tests were investigated in DC2.4 cells with no treated blank cells as control. DC2.4 cells were seeded in confocal dish at 1.5 × 105 cells per well and incubated overnight. Then, the medium was removed and cells were pre-incubated with 10 μg/mL chlorpromazine, 20 μM, 5-(N-ethyl-nisopropyl) amiloride (EIPA), 3 μg/mL, M-β-cyclodextrin for 2 h, respectively. After that, the medium was removed and replenished with DiI-labeled PD1 Exo (2 mg/mL) with corresponding endocytic inhibitors (the same concentration as above) for another 2 h incubation. Afterwards, the cells were stained with Hoechst 33342 and observed by laser confocal microscope (BD, NJ, USA). The uptake ratio was quantified by software Image J (NIH, USA).
2.15. Surface plasmon resonance
For measuring the specific binding ability between PD1 Exo and PD1, the study was performed by surface plasmon resonance (SPR BIAcore T200, GE Healthcare, MA, USA) with a CM5 sensor chip (GE Healthcare). Briefly, PDL1 protein (20 μg/mL) in acetate buffer (pH 5.5) was immobilized on the chip. Then, a sample of PD1 Exo (3.70, 1.85, 0.93, 0.46, 0.23, and 0.12, pM) or free PD1 protein (0.50, 0.25, 0.13, 0.06, and 0.03 M) was injected into the SPR system at a flow rate of 30 μL/min. The running buffer was PBS (pH 7.4). The association time was 60 s. The chip was regenerated for 30 s with NaOH (2.5 mM), and then equilibrated with running buffer for 10 s before the next injection.
2.16. Biodistribution in vivo
For analyzing the biodistribution in the melanoma-bearing mice, eight C57BL/6J mice were inoculated with 1 × 106 melanoma B16F10 cells in the right flank to establish the melanoma-bearing mice model. At day 7, the melanoma-bearing mice were divided into 2 groups (each 4 mice), and intravenously injected with 200 μL DiR-labeled Exo or PD1 Exo (1 mg/mL total protein) via the tail vein. At 6 h after dosing, three mice of each group were sacrificed, the main organs (heart, liver, spleen, lung, and kidney) and tumor tissues were collected to measure the fluorescence intensity of DiR. The other one of each group was survived and scanned by a Living Image system (PerkinElmer, MA, USA) at a fixed time-point (0, 1, 4, 6, 12 and 18 h).
2.17. Maturation of DCs in vitro and in vivo
For evaluating the maturation of immature DCs in vitro, murine bone marrow-derived dendritic cells (DCs) were extracted from the C57BL/6J mice and cultured with GM-CSF (20 ng/mL) and IL-4 (20 ng/mL) in RPMI 1640 medium. At day 7, the DCs were seeded into 24-well plate, and treated with PBS (pH7.4), Imi Exo (200 μg/mL total protein), LPS (1 μg/mL), PD1 Exo (200 μg/mL total protein), aPDL1 (20 μg/mL), and PD1-Imi Exo (200 μg/mL total protein), respectively. After incubation at 37 °C under 5 % CO2 for 24 h, DCs were collected, and stained with PE anti-mouse CD40, PE anti-mouse CD80, APC anti-mouse CD86, FITC anti-mouse CD11c by following the guides of kit manuals, and measured by flow cytometry (Becton Dickinson, San Jose, USA). In addition, the supernatant of each culture well was collected for evaluating cytokines (IP-10 and IL-12p70), and quantified by ELISA kits.
For evaluating the maturation of immature DCs in vivo, C57BL/6J mice were randomly divided into five groups (each 3 mice) and intravenously injected with PBS (pH7.4), Imi Exo (25 mg/kg total protein), PD1 Exo (25 mg/kg total protein), anti-PDL1 (2 mg/kg), and PD1-Imi Exo (25 mg/kg total protein), respectively. Three days later, the same dosing scheme was repeated once. At day 7, the serum of each mouse was sampled for quantify cytokines (IP-10 and IL-12p70) by ELISA kits, and then, axillary lymph nodes and inguinal lymph nodes of the mice were dissected after sacrificing animals, prepared into single cell suspensions, and stained with PE anti-mouse CD80, APC anti-mouse CD86, FITC anti-mouse CD11c, and measured by flow cytometry.
2.18. Immune activation in the melanoma-bearing mice
For analyzing the immune activation effect in vivo, C57BL/6J mice were inoculated with 1 × 106 B16F10-OVA cells in the right flank to establish melanoma-bearing mice. The melanoma-bearing mice were then randomly divided into five groups (each 3 mice), and intravenously injected with PBS, Imi Exo (25 mg/kg total protein), PD1 Exo (25 mg/kg total protein), anti-PDL1 (2 mg/kg), and PD1-Imi Exo (25 mg/kg total protein) at a fixed time-point (at day 7, 10, 13, 16, 19, and 21), respectively. At day 23, all mice were intravenously injected with the OVA257-264 peptide-pulsed CFSEhigh splenocyte suspensions (as below).
For preparing the OVA257-264 peptide-pulsed CFSEhigh splenocyte suspensions, the spleens of additional health C57BL/6J mice (without dosing) were harvested after sacrificing, and prepared into single cell suspensions. The single cell suspensions were equally divided into two parts: the first half of the splenocyte single cell suspensions was incubated with 10 μg/mL OVA257-264 peptide (peptide corresponding to the 257–264 region of OVA) at 37 °C for 30 min, washed with PBS (pH 7.4), and centrifuged at 500×g at 4 °C for 5 min twice. The pellets were collected, and resuspended in PBS (pH 7.4) to obtain a final concentration of 2 × 107 cells/mL as the system A. A volume of 5 μM CFSE solution was added into the system A at a 1:1 vol ratio (v/v), mixed by vortex, and incubated at room temperature in the dark for 15 min as the system B. Afterwards, the system B was washed with RPMI medium, centrifuged at 500×g at 4 °C for 5 min to collect the pellets, which were resuspended in sterile PBS (pH7.4) at a final concentration of 1 × 107 cells/100 μL as the system C, i,e., the OVA257-264 peptide-pulsed CFSEhigh splenocyte suspensions. Besides, the other half of the splenocyte single cell suspensions was used to prepare the non-pulsed CFSElow splenocyte suspensions with a similar process, except for addition of OVA257-264 peptide, and used as the system D. Furthermore, the system C and system D were gently mixed together at a 1:1 ratio (v/v) as the system E. Finally, the system E was intravenously injected into above drug-treated (PD1-Imi Exo or other control) melanoma-bearing mice at day 23 (100 μL/mouse), respectively. At day 25, the mice were sacrificed, and the spleens were collected to prepare the splenocyte single cell suspensions, respectively. The splenocyte single cell suspensions were measured by flow cytometry on the FITC channel for observing CFSE fluorescence intensity. The specific killing ratio was calculated using the formula:
Where and represent the fluorescence intensities of the splenocyte single cell suspensions from the drug-treated group of system C and the drug-treated group of system D, respectively; and represent the fluorescence intensities of the splenocyte single cell suspensions from the PBS-treated group of system C and the PBS-treated group of system D, respectively.
2.19. Anti-relapse efficacy in the melanoma-bearing mice
For evaluating the anti-relapse effect, C57BL/6J mice were inoculated with 1 × 106 B16F10-luc cells in the right flank and randomly divided into five groups (each 6 mice). At day 9, the tumor mass was incompletely excised by surgery operation, and left a residual mini-tumor mass (1 % of original tumor volume) to mimic the local recurrence of melanoma post-surgery. At a fixed time-point (day 10, 13, 16, 19 and 22), the incomplete-resection melanoma-bearing mice were intravenously injected with PBS (pH7.4), Imi Exo (25 mg/kg total protein), PD1 Exo (25 mg/kg total protein), anti-PDL1 (2 mg/kg), and PD1-Imi Exo (25 mg/kg total protein), respectively.
Tumor volume of each mouse was monitored at a fixed time-point (day 12, 15, 18, 21 and 24), and calculated with the following equation: volume = length × width2 × 0.5. Body weight of each mouse was weighed and recorded (at day 9, 14, 19, and 24). At day 27, serum of each mouse was sampled for routine blood test to measure the contents and the proportions of lymphocyte, monocyte, and granulocytes, respectively.
The inhibition rate based on the treatment was calculated with the formula:
| Rinhibit = (1−Vexp/Vcontrol) × 100% |
Where Rinhibit represents tumor inhibition rate; Vexp represents the tumor volume of experimental group; and Vcontrol represents the tumor volume of control group.
The results demonstrated that the Rinhibit values of PD1 Exo, Imi Exo and PD1-Imi Exo were 35.49 %, 15.48 %, and 74.57 %, respectively.
The synergistic coefficient of treatment efficacy was defined with a simplified Bliss-Babu formula:
| Sc = 2 × RPD1-Imi Exo / (RPD1 Exo + RImi Exo) |
Where, Sc represents the synergistic coefficient of treatment efficacy. RPD1-Imi Exo represents the inhibition rate of PD1-Imi Exo; RPD1 Exo represents tumor inhibition rate of PD1 Exo. If Sc > 1, the two drugs exhibit a synergistic effect.
At day 27, the mice were sacrificed, and the partial tumor masses were collected to prepare the single cell suspensions, respectively. Afterwards, the suspensions were stained with PE anti-mouse CD8a, FITC anti-mouse CD4, and APC anti-mouse CD3, and measured by flow cytometry to evaluate the activation status of CD8+ T cells, and part of the suspensions was stained with corresponding antibodies to evaluate the cytokine IFN-γ and TNF-α release. Besides, the rest partial tumor masses were prepared into paraffin sections, and observed by fluorescence microscopy through multiplex immunofluorescence staining (CD4+, CD8+ and 41BB). The percentages of CD4+T and CD8+T cells were quantified by using software Image J to measure the positive signals on the immunofluorescence staining images.
For understanding the effects of various formulations on the major organs, the heart, liver, spleen, lung and kidney of the melanoma-bearing mice were collected, fixed in 4 % paraformaldehyde, embedded with paraffin, sliced and stained with hematoxylin-eosin (HE) dye for pathological observation.
For evaluating the status of memory T lymphocytes, the spleens of the melanoma-bearing mice were collected, and prepared into single cell suspensions, stained with FITC anti-mouse CD3, PE/Cyanine7 anti-mouse CD4, PerCP/Cyanine5.5 anti-mouse CD8a, PE anti-mouse/human CD44, and APC anti-mouse CD62L, and then measured by flow cytometry.
2.20. In vivo bioluminescence imaging
For evaluating the anti-relapse efficacy by direct bioluminescence imaging, the incomplete-resection melanoma-bearing mice were the same divided into five groups (each 3 mice), and the same treated as above. At day 18, D-luciferin in Dulbecco's PBS (pH 7.4; 15 mg/mL; 10 μL/g) was intraperitoneally injected into each mouse, and 10 min later, the melanoma-bearing mice were observed by the IVIS spectrum in vivo imaging system, respectively.
2.21. Anti-metastasis efficacy in the melanoma-bearing mice
For studying the anti-metastasis efficacy, C57BL/6J mice were inoculated with 1 × 106 B16F10-luc cells in the right flank, and the same treated as above. At day 27, 2 × 105 B16-luc cells were intravenously injected into the mice, respectively. At day 41, D-luciferin in Dulbecco's PBS (pH 7.4; 15 mg/mL; 10 μL/g) was intraperitoneally injected into each mouse, and 10 min later, melanoma metastasis nodules on lungs was observed by using the IVIS spectrum in vivo imaging system, respectively. Afterwards, the mice were sacrificed, and the lungs were collected to count the numbers of melanoma metastasis nodules.
2.22. Therapeutic efficacy in the breast cancer-bearing mice
For observing the therapeutic efficacy in the breast cancer-bearing mice, BALB/C mice were inoculated with 1 × 106 4T1 cells in the right flank, and randomly divided into five groups (each 6 mice). At day 6, the mice were intravenously injected with PBS, Imi Exo (25 mg/kg total protein), PD1 Exo (25 mg/kg total protein), anti-PDL1 (2 mg/kg) and PD1-Imi Exo (25 mg/kg total protein) at a fixed time-point (at day 6, 9, 12, 15 and 18), respectively. The tumor volume of each mouse was monitored at a fixed time-point (at day 6, 9, 12, 15 and 18), and calculated with the following equation: volume = length × width2 × 0.5. At day 21, the mice were sacrificed, and the tumor masses were dissected and weighed.
For evaluating the survival time of the mice after treatment, the breast cancer-bearing mice were the same divided into five groups (each 6 mice), and the same treated as above. The survival time of each mouse was also recorded until 50 days, and analyzed by Kaplan-Meier survival curves.
2.23. Statistical analysis
Prism 7 software (GraphPad, La Jolla, CA, USA) was used for statistical analyses. The statistical analysis for two groups was performed by two-tailed unpaired Student's t-test. The statistical analysis for three and more groups was performed by one-way ANOVA, followed by Tukey's test. The statistical analysis for Kaplan-Meier survival plot was performed by log-rank test. The results are presented as mean ± standard deviation.
3. Results
3.1. Design and characterization of imiquimod-enveloped gene engineered exosome (PD1-Imi Exo)
For confirming the overexpression of PDL1 in tumor cells, the study was performed on melanoma B16F10 cells, breast cancer 4T1 cells, and human embryonic kidney 293T cells (tool cells) by flow cytometry, respectively. The results showed that the expression of PDL1 was significantly higher in B16F10 cells or in 4T1 cells compared with that in HEK 293T cells (Fig. S1).
For developing the PD1 engineered exosome, mouse PD1 overexpressing plasmid was design and synthesized (Fig. 2A and B, Fig. S2, Fig. S3) and transfected into donor cells (HEK293T). After infecting and selecting by puromycin, the HEK 293T cells which stably expressed PD1 protein were obtained (Fig. 2C), the PD1-expressed exosomes (PD1 Exo) were collected from culture medium, and enriched by ultracentrifugation. Imiquimod was enveloped into PD1 Exo by electroporation for preparing PD1-Imi Exo. After optimizing the encapsulation condition, and quantifying evaluation by ultraviolet visible spectrophotometry (Fig. S4A, Fig. S4B), the enveloping content of imiquimod was approximately 12 % (μg/ml). In the accelerated release conditions in PBS (pH 7.4), the cumulative release of imiquimod from PD1 Exo was 50 % within 24 h (Fig. S5). Nevertheless, either Imi Exo or PD1-Imi Exo remained to be stable in a 14-day static storage as it kept a stable particle size and a stable encapsulation efficiency (Fig. S6).
Fig. 2.

Gene recombination engineering and characterization
(A) Design for lentivirus packaging PD1 gene. WPRE, Woodchuck hepatitis posttranscriptional regulatory element; CMV, cytomegalovirus promoter; Puro, puromycin; T2A, 2A self-cleaving peptides; EGFP, enhanced green fluorescent protein. (B) Verification of PD1 gene precise insertion. The images were two end partial DNA sequences of PD1 gene (from 5′ promoter to PD1, upper side; and from 3′ PD1 to WPRE, lower side) in the lentiviral plasmid. (C) Expression of PD1 gene in tool cells. After selecting by puromycin, the expressions of green fluorescence protein were observed in tool cells (HEK 293T cells) at day 1 and more evidently at day 6 under the inverted fluorescent microscope (n = 3). (D–F) Morphology of PD1 Exo. The study was performed by transmission electron microscopy (n = 3). (D) Exo; (E) PD1Exo; (F) PD1-Imi Exo. (G–I) Particle size of PD1 Exo. The study was performed by nanoparticle tracking analysis (NTA). (G) Size of Exo (110.1 ± 49.1 nm); (H) Size of PD1 Exo (130.0 ± 51.5 nm); (I) Size of PD1-Imi Exo (139.4 ± 51.5 nm). (J) Storage stability. Hydrodynamic sizes of PD1 Exo and Exo in PBS pH7.4 for 14 days (n = 3). (K) Zeta potential of PD1 Exo. The study was performed by dynamic laser scattering analysis (n = 3). (L) Western blot images of PD1 engineered exosomes. GAPDH, glyceraldehyde-3-phosphate dehydrogenase (internal reference protein); HSP70, heat shock protein 70; TSG101, tumor susceptibility gene-expressed protein 101; calnexin, calcium-binding protein located on the endoplasmic reticulum membrane; cell lysate, lysate of the PD1 engineered HEK293T cells.
The transmission electron microscopy (TEM) analysis showed that PD1 Exo and PD1-Imi Exo were round vesicular in shape, displaying a similar appearance to natural exosomes (Exo) (Fig. 2D–E, Fig. 2F). The nanoparticle tracking analysis (NTA) results showed that the particle sizes of Exo, PD1 Exo and PD1-Imi Exo were 110.0 ± 49.1 nm, 130.0 ± 51.5 nm and 139.4 ± 51.5 nm, respectively, indicating a gradual increase in diameter after PD1 recombining and imiquimod enveloping (Fig. 2G–H, Fig. 2I).
Meanwhile, both PD1 Exo and Exo were well dispersed in phosphate-buffered saline (PBS, pH 7.4) and were stable for a longer storage time at freezing condition (−80 °C) (Fig. 2J). The zeta potential results showed a decreased value after PD1 expressed on the exosomes (Fig. 2K). Western blot analysis showed that the typical biomarkers (HSP70, TSG101) of exosomes were clearly observed in three formulations. Besides, PD1 was detected in PD1 Exo, and PD1-Imi Exo, except for Exo. In addition, calnexin, a protein located in endoplasmic reticulum (ER), was detected in whole PD1 donor cell lysates, but not found in the isolated exosomes (Fig. 2L), indicating the purity of the separated exosomes. These results also confirmed a successful preparation of the PD1-Imi Exo.
3.2. Enhanced targeting capability and binding effect
The reason for the unsatisfactory effect of PD1 blockade therapy may be related to the off-target binding with tumor site and insufficient lymphocytic infiltration of the tumor microenvironment. The present rational design can facilitate the PD1 Exo binding to tumor cells and antigen-presenting cells (APCs) through PDL1.
For confirming the interaction between PD1-Imi Exo and PDL1 on melanoma cells, the study was performed by confocal laser scanning microscopy. The results showed that the PD1-Imi Exo obviously interacted with the PDL1 protein in melanoma cells (Fig. S7).
Besides, DiI-labeled PD1 Exo and Exo were incubated with melanoma cells and observed by confocal laser scanning microscopy (CLSM). The results showed that the phagocytosis of PD1 Exo by melanoma cells was obviously higher than that of Exo by melanoma cells (Fig. 3A and B), demonstrating an enhanced targeting effect on melanoma cells. Furthermore, when melanoma cells were saturated with anti-PDL1 in advance, the uptake of PD1 Exo was remarkably decreased (Fig. S8), indicating that the targeting mechanism of PD1 Exo was associated with the specific PD1-PDL1 binding. Similarly, PD1 Exo was also obviously phagocytosed more by dendritic cells compared with Exo (Fig. 3C and D), demonstrating an enhanced targeting effect on dendritic cells as well.
Fig. 3.

Targeting capability and binding effect.
(A–B) Targeting capability of PD1 Exo on melanoma cells. The study was performed melanoma cells using confocal laser scanning microscopy. The nuclei of melanoma B16F10 cells were counter-stained with Hoechst (blue) (n = 4). (A) Fluorescence images; (B) Quantification on image A. (C–D) Targeting capability of PD1 Exo on dendritic cells. The study was further performed on dendritic cells using confocal laser scanning microscopy. The nuclei of dendritic DC2.4 cells were counter-stained with Hoechst (blue) (n = 4). (C) Fluorescence images; (D) Quantification on image C. (E–F) Endocytosis mechanism of PD1 Exo on DCs. The study was performed on dendritic cells (DCs) using confocal laser scanning microscopy. The nuclei of dendritic DC2.4 cells were counter-stained with Hoechst (blue) (n = 3). (E) Fluorescence images; (F) Quantification on image E. (G–H) Binding between PD1 and PDL1. The binding effect between PD1 Exo and free PDL1 protein was observed by surface plasmon resonance (SPR) analysis (n = 3). (G) Binding profiles between free PD1 protein and free PDL1 protein; (H) Binding profiles between PD1 Exo and free PDL1 protein. (I–K) Accumulation in cancer tissue. The accumulations in various tissues were observed by imaging DiR labeled exosomes in melanoma-bearing mice (n = 3). (I) Real-time in vivo images post injection; (J) Fluorescence images of ex vivo tissues; (K) Accumulation comparison of ex vivo tissues. The statistical analysis was performed by two-tailed unpaired Student's t-test. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, ns, not significant. Data are represented as mean ± standard deviation.
For displaying the endocytosis mechanism of PD1 Exo by DCs,C-β-cyclodextrin (β-CD, caveolae-mediated endocytosis inhibitor), chlorpromazine (CPZ, clathrinmediated endocytosis inhibitor), and ethylisopropylamiloride (EIPA, inhibits micropinocytosis inhibitor) were used to pretreat DC2.4 cells, respectively. The results showed that PD1 Exo were endocytosed by DCs mainly through the clathrin-mediated endocytosis and macropinocytosis (Fig. 3E and F).
The binding affinity between PD1 Exo and free PDL1 protein was performed by surface plasmon resonance (SPR) assay for further elucidating the targeting mechanism of PD1 Exo. The results showed that free PD1 protein and free PDL1 protein exhibited an evident binding, demonstrating a concentration-dependent pattern, but then a dissociation (Fig. 3G). In contrast, PD1 Exo and free PDL1 protein displayed a significant binding, revealing a binding a concentration-dependent pattern as well, but then a keeping association state (Fig. 3H). Moreover, PD1 Exo (affinity constant Kd = 1 nM) had a stronger binding affinity than free PD1 (Kd = 0.38 μM), and indicated a lasting binding at comparable situation.
For detecting the targeted delivery of PD1 Exo, DiR-labeled PD1 Exo and Exo were injected into the melanoma cancer-bearing nude mice via tail vein, the mice were separately imaged at 0 h, 1h, 4 h, 6 h, 12 h and 18 h for observing the accumulation of formulations in tumor site and in major organs (Fig. 3I). After sacrificing animals, the ex-tissues were imaged and the fluorescence intensity was also quantified. The results showed that DiR-labeled PD1-Exo in tumor site was significantly increased compared with DiR-labeled Exo. Besides, DiR-labeled PD1-Exo was mainly distributed in liver, spleen, and kidney, except for heart and kidney (Fig. 3J and K).
The cell viability was performed by cell counting kit-8 (CCK8) assay for evaluating cytotoxic effects of Exo and PD1 Exo on melanoma cells. The results showed that the survival rates of melanoma cells were above 90 % when Exo or PD1 Exo reached up to a higher concentration (100 μg/ml) (Fig. S9).
3.3. Dendritic cell maturation and immune activation effect
Effective T cell activation depends on the maturity of APCs. PD1-Imi Exo was applied to mouse bone marrow-derived dendritic cells (DCs) for observing its role in promoting DCs maturation. The results from FCM assay showed that PD1-Imi Exo significantly increased the expression of CD40+, CD80+, CD86+ cells compared with the controls (Imi Exo, PD1 Exo, anti-PDL1 and PBS), and the effect was comparable to the positive control (LPS), exhibiting an evident effect in promoting the maturation of DCs (Fig. 4A and B, Fig. S10). Meanwhile, PD1-Imi Exo significantly increased the secretion of cytokines (IP-10, IL-12p70) in the treated DCs as well (Fig. 4C and D). Those data confirmed that PD1-Imi Exo was able to effectively promote maturation of DC.
Fig. 4.

Dendritic cells maturation and immune activation effect. (A–B) Maturation of bone marrow-derived dendritic cells (BMDCs). BMDCs were treated with various formulations, and CD80+ and CD86+ expressions were measured by flow cytometry (FCM). (A) FCM image showing CD80+ and CD86+ expressions. (B) Quantification on image A (n = 3). 1, PBS (pH7.4); 2, Imi Exo; 3, LPS; 4, PD1 Exo; 5, Anti-PDL1; 6, PD1-Imi Exo. (C–D) Secretions of cytokines. BMDCs were treated with various formulations, and the cytokines were detected by enzyme linked immunosorbent assay (ELISA) (n = 3). (C) IP-10 level; (D) IL-12p70 level. 1, PBS (pH7.4); 2, Imi Exo; 3, LPS; 4, PD1 Exo; 5, Anti-PDL1; 6, PD1-Imi Exo. (E–F) Antigen-specific killing effect in melanoma-bearing mice. Antigen-specific killing effect was detected by FCM and indicated by the ratio of CFSEhigh to CFSElow labeled splenocytes (n = 3). (E) FCM images for CFSEhigh and CFSElow spleen cells; (F) Quantification for antigen specific killing effect. CFSE, carboxyfluorescein succinimidyl amino ester. 1, PBS (pH7.4); 2, Imi Exo; 3, PD1 Exo; 4, Anti-PDL1; 5, PD1-Imi Exo. (G) Illustration for mechanism of immune activation. As a Toll like-receptor (TLR) agonist, the released imiquimod (Imi) from PD1-Imi Exo is involved in accelerating the maturation of naïve dendritic cell (DC), playing the role of an immune adjuvant. Mature DC highly expressed costimulatory molecules (CD80, CD86, etc.) and release cytokines (IP-10, IL-12p70) to further induce the proliferation and differentiation of activated CD8+ T cell, thus fully triggering the subsequent immune killing effect by secreting cytokines (TNF-α, IFN-γ).
The statistical analysis was performed by two-tailed unpaired Student's t-test. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, ns, not significant. Data are represented as mean ± standard deviation.
The antigen-specific killing capacity of PD1-Imi Exo was used as an indicator of the immune activation effect, and evaluated in the melanoma B16-OVA-bearing mice. After treatment with various formulations, the mice were intravenously injected with an equal number of OVA peptide-treated splenocytes (CFSEhigh) and non-treated splenocytes (CFSElow). After sacrificing animals at 48 h, the spleens were excised and detected by FCM. The results showed that PD1-Imi Exo caused increased cell lysis in contrast to other treatments, demonstrating that the PD1-Imi Exo significantly enhanced the killing ability of OVA-specific CD8+ T cells in vivo (Fig. 4E and F).
Immune adjuvant imiquimod released from PD1-Imi Exo played a role in accelerating DCs maturation. The mechanism could be explained by the following descriptions. DCs were derived from hematopoietic bone marrow progenitor cells, which were initially transformed into immature DCs. Such cells possessed the properties of high endocytosis activity but low T cell activation potential. They constantly sampled the stimulus imiquimod via pattern recognition receptors (Toll-like receptors, TLRs), and engulfed the tumor associated antigen, then processed into peptide-MHC I molecular complex on the cell surface. Accordingly, immature DCs were activated into mature DCs and began to migrate to the lymph nodes, and upregulated cell surface receptor (such as CD80, CD86) and cytokines (IP-10, IL-12p70). The mature DCs became highly efficient in activating T cells by inducing the proliferation and differentiation of activated CD8+ T cells, thus fully triggering the subsequent immune killing effect by secreting cytokines (TNF-α, IFN-γ, verified below) (Fig. 4G).
3.4. Therapeutic efficacy in the cancer-bearing mice
The effect of PD1-Imi Exo on DCs maturation in vivo was further evaluated. After treated with various formulations, the animals were sacrificed, the serum samples and the lymph nodes were collected for assessment. The results from ELISA that PD1-Imi Exo significantly increased the serum levels of IP-10 and IL-12p70 (Fig. S11A, Fig. S11B) as compared with other controls. In addition, Imi Exo was also able to evidently pull up the expressions of costimulatory marker CD80 and CD86, and the secretions of IP-10 and IL-12p70. Besides, the results from FCM showed that PD1-Imi Exo significantly upregulated the expressions of costimulatory marker CD80 and CD86, compared with other controls (Fig. S11C).
For evaluating the postoperative anti-relapse efficacy after surgery, PD1-Imi Exo was performed on the incomplete-resection model of the melanoma B16F10-luc-bearing mice (Fig. 5A). Tumor growth were monitored by measuring tumor volume using a vernier caliper and by bioluminescence imaging (Fig. 5B and C, Fig. S12). The results showed that, compared with the controls, PD1-Imi Exo demonstrated the most significant postoperative anti-relapse efficacy. The results indicated that the Sc value of PD1 and imiquimod was 2.93, showing an evident synergistic effect.
Fig. 5.

Therapeutic efficacy in the cancer-bearing mice.
(A) Dosing scheme in the melanoma-bearing mice. In the dosing scheme A1, approximately 99 % of the cancer tissue was removed by surgery at day 9 since the inoculation of melanoma cells, and then mice were divided into groups, and treated by various formulations for evaluating the anti-relapse efficacy in the melanoma-bearing mice. In the dosing scheme A2, full scheme A1 was performed and all the mice were survived until the experimental end-point, and melanoma cells were intravenously injected into the mouse of each group once again at day 27 via tail vein for observing the anti-metastasis efficacy in the melanoma-bearing mice. (B–C) Efficacy in the melanoma-bearing mice. The tumor growth profile of each mouse was recorded by monitoring tumor volume every 3 days using a vernier caliper. (B) Individual tumor growth profile; (C) Average tumor growth profile (n = 6). (D) Effect on body weight. The preliminary safety was evaluated by recording body weight of mice after various treatments (n = 6). (E) Anti-metastatic evaluation. The anti-metastatic effect was evaluated by observing the numbers of metastatic melanoma nodules on lungs (n = 6). (F) Dosing scheme in the breast cancer-bearing mice. At day 6 since the inoculation of breast cancer 4T1 cells, the mice were treated by various formulations according to dosing scheme. (G–H) Efficacy in the breast cancer-bearing mice. The tumor growth profile of each mouse was recorded by monitoring tumor volume every 3 days using a vernier caliper. (G) Individual tumor growth profile; (H) Average tumor growth profile (n = 6). (I) Inhibitory effect on breast cancer masses. The ex vivo breast cancer masses were weighed after various treatments (n = 6). (J) Survival analysis. Kaplan-Meier survival curves were plotted for breast cancer-bearing mice after various treatments until 50 days (n = 6).
1, PBS (pH7.4); 2, Imi Exo; 3, PD1 Exo; 4, Anti-PDL1; 5, PD1-Imi Exo. The statistical analysis was performed by one-way ANOVA, followed by Tukey's test (Fig. C and Fig. H), by log-rank test (Fig. J) or by two-tailed unpaired Student's t-test (others).
*p < 0.05; **p < 0.01; ****p < 0.0001. Data are presented as mean ± standard deviation.
Besides, the preliminary safety of PD1-Imi Exo was also monitored. The results from body weight monitoring showed that the treatment of PD1-Imi Exo did not affect the body weight of the mice (Fig. 5D). Meanwhile, the results from blood indicator assay showed that the treatment of PD1-Imi Exo did not evidently affect the contents and ratios of lymphocytes, lymphocytes and monocytes (Fig. S13). In addition, the results from hematoxylin and eosin (HE) staining showed that the treatment of PD1-Imi Exo did not obviously affect the major organs of the animals (Fig. S14)
The anti-metastatic efficacy of PD1-Imi Exo was performed on the melanoma B16F10-luc bearing mice. At day 41, tumor masses and lung metastases were observed by bioluminescence imaging in vivo. After sacrificing animals, the lungs were collected, and metastatic nodules were calculated. The results showed that, compared with the controls, PD1-Imi Exo treatment had the most significant efficacy in inhibiting lung metastasis of luc-labeled melanoma cells, both by observing bioluminescent signals in vivo and by observing metastatic nodules on ex vivo lung tissues (Fig. S15A, Fig. S15B, Fig. 5E).
The anticancer efficacy of PD1-Imi Exo was further performed on the breast cancer 4T1-bearing mice for evaluating the tumor growth inhibition, induction of apoptosis, and survival time (Fig. 5F), respectively. The tumor growth inhibition was evaluated by measuring tumor volume using a vernier caliper and by weighing tumor masses after sacrificing animals. The induction of apoptosis was estimated by a TUNEL assay (terminal deoxynucleotidyl transferase dUTP nick-end labeling). And the survival curve was observed using Kaplan-Meier plotting. The results showed that, compared with the controls, PD1-Imi Exo has the most significant efficacy in suppressing the tumor volume, in inhibiting the tumor weight (Fig. 5G–H, Fig. 5I), and in inducing apoptosis of cancer cells in tumor masses (Fig. S16A, Fig. S16B). Furthermore, the treatment of PD1-Imi Exo has the most evident efficacy in prolonging the survival time of the cancer-bearing mice, leading to a full survival of animals in the observation period of 50 days (Fig. 5J).
3.5. Immune activation mechanism in the cancer-bearing mice
Tumor T cell infiltration is essential for the tumor immunotherapy. To assess whether PD1-Imi Exo treatment reverse T cell exhaustion and maintain their antitumor function, the serum samples were collected, and after sacrificing animals, the tumor masses and spleens were harvested for assessments.
The results from FCM analysis showed that, compared with the controls, PD1-Imi Exo significantly enhanced the infiltration of cytotoxic CD8+ T lymphocytes in tumor masses (Fig. 6A and B), and remarkably increased the effector memory T cells in spleens (Fig. 6C and D, Fig. S17).
Fig. 6.

Immune activation mechanism in the cancer-bearing mice (A–B) Activation of CD3+ CD8+ T cells. Infiltrating CD3+ CD8+T lymphocytes were isolated from cancer masses in the melanoma-bearing mice after various treatments, and analyzed by flow cytometry (FCM, n = 5). (A) FCM images; (B) Quantification for CD3+ CD8+ cells. (C-D) Immune memory. Immune memory effect (Tem, CD44+ CD62L-) was evaluated on the splenocytes from melanoma-bearing mice after various treatments, and analyzed by FCM (n = 5). (C) FCM images; (D) Qualification for Tem cells. Tem, effector memory T cell. (E–G) CD8+ and CD4+ expression in tumors. CD8+, CD4+ markers evaluation was performed by immunofluorescence staining on melanoma masses, and quantified by software Image J (n = 6). (E) Immunofluorescence images; (F) Quantification for CD8+ markers. (G) Quantification for CD4+ markers. Red spots, CD8+ T cells; green spots, CD4+ T cells. Scale bar, 100 μm. (H–I) IFN-γ and TNF-α levels in tumors. IFN-γ and TNF-α levels in the isolated tumor masses after various treatments were analyzed by flow cytometry (n = 5). 1, PBS (pH7.4); 2, Imi Exo; 3, PD1 Exo; 4, Anti-PDL1; 5, PD1-Imi Exo. The statistical analysis was performed by two-tailed unpaired Student's t-test. *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001. Data are presented as mean ± standard deviation, ns, not significant.
The results from immunostaining assay also demonstrated that, compared with the controls, PD1-Imi Exo obviously enhanced the infiltration of CD8+ cytotoxic T lymphocytes, and CD4+ T cells (Fig. 6E and F, Fig. 6G), leading to obviously upregulated biomarker of 41BB (an activated immune checkpoint molecule) in tumor mass (Fig. S18), Moreover, as expected by our design, PD1-Imi Exo exhibited the capability in directly activating both native T cells and exhausted T cells. The results from ELISA showed that, compared with the controls, PD1-Imi Exo significantly increased secretions of interferon-gamma (IFN-γ) and tumor necrosis factor (TNF-α) (Fig. 6H and I), demonstrating an excellent anti-tumor potential.
4. Discussion
PD1/PDL1 immune checkpoint blockage therapy has made significant progress in the treatment of refractory malignances, It has been applied in the treatment of more than 15 malignancies such as melanoma and breast cancer, significantly improving the survival of patients [41,42]. However, the evidence gained from the results of basic research and clinical application demonstrates that PD1/PDL1 therapy also exhibits extensive immune resistance [43,44], with only part of patients benefited. Most likely reasons are due to the loss of targeted tumor-associated antigens [45], inefficient maturation of dendritic cells (DCs), and/or poor activation and infiltration of cytotoxic T lymphocytes which severely hamper its clinical prognosis. In this study, a novel gene engineered exosome by incorporating PD1 gene and simultaneously enveloping an immune adjuvant imiquimod (PD1-Imi Exo) was rationally designed for boosting response of cancer immune checkpoint blockage therapy by reversing T cell exhaustion through activating DCs and further restoring the function of T cells (Fig. 1).
Commercially available PDL1 monoclonal antibodies (anti-PDL1) are usually prepared by inducing immunized animals in vivo, for example, by purifying the mouse ascites after intraperitoneal inoculation of hybridoma cells. Such preparation needs a complicated process, a higher purification requirement, and a longer production cycle. Moreover, PDL1 monoclonal antibodies exhibit widespread immune resistance in clinical therapy, resulting in immune escape in more than half of cancer patients [46]. In this study, PD1-Imi Exo carries not only PD1 protein, but also immune adjuvant imiquimod, which plays a double immunotherapy-enhancing effect. On the one hand, imiquimod promotes the maturation of dendritic cells and then activates T cell function. On the other hand, PD1-Imi Exo could competitively block the binding of PD1 on T cell to PDL1 on tumor cell, thereby restoring the killing function of T cell. Besides, PD1-Imi Exo is an endogenous vesicle, which easily enters the tumor microenvironment and has stronger binding affinity with tumor cell compared with free PDL1 antibody, acting stronger immune check point blocking effect. In addition, the formulation is relatively simple, and it deserves further development for clinical application.
For verifying the preparation of gene engineered exosome, DNA sequencing confirms that PD1 gene-containing recombinant plasmid is successfully synthesized. Besides, the fluorescence indicator by GFP-gene expressed protein demonstrates that the recombinant plasmid has been successfully transfected into tool cells. The characterization reveals that gene engineered exosome are a negatively charged, uniformly dispersed, and spherical vesicles (130 nm) with good stability. The end product of the recombinant exosomes is further identified by the expression of typical exosome biomarkers (HSP70, TSG101) and PD1 protein, and can be significantly distinguished from the negative control endoplasmic retinal protein (calnexin) [47]. In addition, the enveloped immune adjuvant imiquimod can be identified and quantified by the ultravioletvisible spectrophotometry (Fig. 2).
The characterization for targeting capability verify that gene engineered exosome are able to target on cancer cells and on dendritic cells as well through the specific binding effect of PD1/PDL1. These could be explained by the fact that PDL1 protein is overexpressed on cancer cells and on immune cells like dendritic cells [48]. PD1 Exo enters cells through the clathrin-mediated endocytosis and macropinocytosis pathways. The binding affinity of PD1 Exo with PDL1 protein from SPR assay exhibits that PD1 Exo displays a more persistent and stronger binding with PDL1 protein. In contrast, free PD1 protein binds to PDL1 protein but rapidly dissociates. The exact reason remains unclear, but it is speculated that this phenomenon may be related to more PD1 binding sites on the exosome surface, leading to a stronger binding of PD1 Exo with PDL1 protein. The in vivo targeting observations by the real-time in vivo and ex vivo IVIS imaging further reveals that PD1 Exo is able to be accumulated into tumor tissues for a longer time, as compared to the negative control natural exosomes (Exo). In addition to the enhanced permeability and retention effect of nanosized exosomes [47], above mentioned targeting binding could explain such a tumor tissue targeting capability (Fig. 3).
Toll-like receptors (TLRs) are a class of proteins that play a key role in the innate immune system [49]. They are single-pass membrane-spanning receptors usually expressed on sentinel cells such as macrophages and dendritic cells, which recognize structurally conserved molecules derived from microbes. Once these microbes have reached physical barriers such as the skin or intestinal tract mucosa, they are recognized by TLRs, which activate immune cell responses. During the last 10 years, more than a dozen TLRs have been reported, and new information about the multiple roles of TLRs has rapidly increased. Among them, TLR 7/8 agonists have great potential as adjuvants in cancer immunotherapy and are localized to the endosomal compartments of human immune cells including DCs [50,51], monocytes, macrophages, lymphocytes, Langerhans cells, and NK cells. In this study, immune adjuvant imiquimod was used as a TLR 7 agonists for activating DCs [52,53]. The mechanism of imiquimod released from the gene engineered exosome could be explained by its action in promoting maturation of DCs, as dendritic cells are the most powerful antigen presenting cell while the function is strongly dependent on maturity. Once immature DCs have come into contact with TLR 7 agonist imiquimod and presentable antigen, they become activated into mature DCs, begin to migrate to a lymph node under the action of chemokines, and present the major MHC-I peptide to T-cell receptor (TCR) under the involvement of intrinsic antigen [54]. Meanwhile, the mature DCs increase the expressions of co-stimulatory molecules (CD80, CD86) and the secretions of immunomodulatory cytokines (IP-10 and IL-12p 70), thus enhancing the T cell proliferation and differentiation into effector CD8+ T cells, and then eliciting a stronger CTL cell response for antitumor efficacy. Moreover, the gene engineered exosome reinvigorate the exhausted T cells by blocking the PD1/PDL1 axis, together with the enhanced function of CD8+ T cell by the mature DCs, more immune cytokines such as interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α) are produced to restore the dormant or exhausted T cells, thereby boosting the immune responsiveness in killing cancer cells (Fig. 4).
By taking advantage of the rational design, PD1-Imi Exo offer potent anticancer efficacy in diverse animal models, including the melanoma and the breast cancer. As we know, the postoperative recurrence and metastasis of cancer brings great challenges for the clinical treatment of malignant tumor. In our project, PD1-Imi Exo demonstrates an excellent anti-relapse and an anti-metastatic efficiency in the melanoma incomplete-resection model, as it displays the most effective effect in inhibiting tumor growth, and in decreasing the number of metastatic pulmonary nodules, even more effective than commercially available PDL1 antibody, thus verifying a remarkable boosted responsiveness for immune checkpoint blockage therapy. Similarly, PD1-Imi Exo treatment also shows optimal therapeutic effect in inhibiting tumor growth, and especially in prolonging the survival rate in the breast cancer-bearing mice. In addition, due to the natural safety of exosomes, intravenous administration of PD1-Imi Exo has no significant harmful effects on body weight, major organs, and blood indexes of mice, while these results are critical for its future clinical transformation (Fig. 5). While administering in vivo, PD1 engineered exosomes produced in 150 ml culture medium are sufficient for a single dose of 1 mouse, displaying that this yield also has the industrialization potential.
CD8+ T cells are the primary anti-tumor defenders and the most essential weapons for killing tumor. In the environment of cancer, CD8+ T cells are constantly exposed to antigen and inflammatory signals, leading to a deterioration of T cell function, i.e., exhaustion. The exhausted T cells are characterized by low proliferation in response to antigen stimulation, loss of immune surveillance function (cytokine production and killing function), expression of multiple inhibitory receptors such as PD1, and metabolic alterations from oxidative phosphorylation to glycolysis [55,56]. In our study, the boosted immune response by PD1-Imi Exo can be explained by the fact that the cytotoxic CD8+ T cells are successfully activated by integrating the reversal of immunosuppression with the activation of domino T cells (a cascade T cells) in tumor site. Specifically, PD1-Imi Exo achieves an evident synergistic effect of tumor treatment through two pathways: (i) PD1 on the PD1-Imi Exo reverses T cell exhaustion by exerting immune checkpoint inhibition; (ii) adjuvant imiquimod released from the PD1-Imi Exo further activates T cells by stimulating the maturation of DC cells, and both together promote the effect of immunotherapy. Furthermore, PD1-Imi Exo also improve the memory T cell in spleens in inducing the long-term memory. The primary function of memory cells is that when tumor cells reform, cytotoxic T cells are reactivated by tumor-associated antigens to augment the immune response. In addition, these effects are further confirmed in the cancer-bearing animals, including increased expression of CD8+ T cells, induction of cancer cell apoptosis, and increased expression of cytokines (IFN-γ and TNF-α) (Fig. 6).
5. Conclusion
In sum, a gene engineered exosome was rationally designed by incorporating PD1 gene and simultaneously enveloping an immune adjuvant imiquimod (PD1-Imi Exo) for boosting response of cancer immune checkpoint blockage therapy. PD1-Imi Exo had a vesicular round shape (approximately 139 nm), and exhibited significant targeting and strong binding effects on cancer cells and on dendritic cells. Furthermore, it demonstrated a remarkable therapeutic efficacy in melanoma-bearing mice and in breast cancer-bearing mice, indicating an evidently boosted immune response. The action mechanism could be explained by two facts that PD1-Imi Exo blocked the binding of CD8+ T cell with cancer cell, displaying a PD1/PDL1 immune checkpoint blockage therapy, and that imiquimod released from PD1-Imi Exo promoted the maturation of immature dendritic cell, exhibiting a restoring effect on the immune exhaustion through activating and restoring killing function of CD8+ T cell. In conclusion, the gene engineered exosome could be used for boosting response of cancer immune checkpoint blockage therapy. This study also offers a promising new strategy for enhancing PD1/PDL1 therapeutic efficacy, preventing tumor recurrence or metastasis after surgery by rebuilding the patients' immunity, thus consolidating the overall prognosis.
Ethics approval and consent to participate
All animal experiments were carried out in accordance with guidelines, and approved by the Committee for Experimental Animal Welfare of Biomedical Ethics of Peking University (LA2021252).
CRediT authorship contribution statement
Peishan Li: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation. Ying Xie: Writing – original draft, Methodology, Investigation. Jinling Wang: Writing – original draft, Validation, Methodology, Investigation, Formal analysis, Data curation. Chunjie Bao: Formal analysis, Data curation. Jialun Duan: Methodology, Investigation. Yixuan Liu: Methodology, Conceptualization. Qian Luo: Writing – original draft, Formal analysis. Jiarui Xu: Methodology. Yuxin Ren: Methodology. Min Jiang: Methodology, Formal analysis. Jianwei Li: Formal analysis. Haitao Guo: Methodology. Huihui Zhao: Methodology. Guiling Wang: Formal analysis. Yanqin Liang: Validation, Project administration, Formal analysis. Wanliang Lu: Writing – review & editing, Validation, Supervision, Resources, Project administration, Investigation, Funding acquisition, Conceptualization.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82173752 and No. 81874303).
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioactmat.2024.01.008.
Contributor Information
Yanqin Liang, Email: liangyq@bici.org.
Wanliang Lu, Email: luwl@bjmu.edu.cn.
Appendix A. Supplementary data
The following is the supplementary data to this article:
References
- 1.Liu R., Xie Y., Xu J.-R., Luo Q., Ren Y.-X., Chen M., Duan J.-L., Bao C.-J., Liu Y.-X., Li P.-S., Li J.-W., Wang G.-L., Lu W.-L. Engineered stem cell biomimetic liposomes carrying levamisole for macrophage immunity reconstruction in leukemia therapy. Chem. Eng. J. 2022;447 [Google Scholar]
- 2.Zou W., Wolchok J.D., Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016;8(328):328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dovedi S.J., Elder M.J., Yang C., Sitnikova S.I., Irving L., Hansen A., Hair J., Jones D.C., Hasani S., Wang B., Im S.A., Tran B., Subramaniam D.S., Gainer S.D., Vashisht K., Lewis A., Jin X., Kentner S., Mulgrew K., Wang Y., Overstreet M.G., Dodgson J., Wu Y., Palazon A., Morrow M., Rainey G.J., Browne G.J., Neal F., Murray T.V., Toloczko A.D., Dall'Acqua W., Achour I., Freeman D.J., Wilkinson R.W., Mazor Y. Design and efficacy of a monovalent bispecific PD-1/CTLA4 antibody that enhances CTLA4 blockade on PD-1(+) activated T cells. Cancer Discov. 2021;11(5):1100–1117. doi: 10.1158/2159-8290.CD-20-1445. [DOI] [PubMed] [Google Scholar]
- 4.Marable J., Ruiz D., Jaiswal A.K., Bhattacharya R., Pantazes R., Agarwal P., Suryawanshi A.S., Bedi D., Mishra A., Smith B.F., Sandey M. Nanobody-based CTLA4 inhibitors for immune checkpoint blockade therapy of canine cancer patients. Sci. Rep. 2021;11(1) doi: 10.1038/s41598-021-00325-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu T., Liu H., Liang Z., Wang F., Zhou C., Zheng X., Zhang Y., Song Y., Hu J., He X., Xiao J., King R.J., Wu X., Lan P. Tumor-intrinsic CD47 signal regulates glycolysis and promotes colorectal cancer cell growth and metastasis. Theranostics. 2020;10(9):4056–4072. doi: 10.7150/thno.40860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishiga Y., Drainas A.P., Baron M., Bhattacharya D., Barkal A.A., Ahrari Y., Mancusi R., Ross J.B., Takahashi N., Thomas A., Diehn M., Weissman I.L., Graves E.E., Sage J. Radiotherapy in combination with CD47 blockade elicits a macrophage-mediated abscopal effect. Nat. Cancer. 2022;3(11):1351–1366. doi: 10.1038/s43018-022-00456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin Y.X., Wang Y., Ding J., Jiang A., Wang J., Yu M., Blake S., Liu S., Bieberich C.J., Farokhzad O.C., Mei L., Wang H., Shi J. Reactivation of the tumor suppressor PTEN by mRNA nanoparticles enhances antitumor immunity in preclinical models. Sci. Transl. Med. 2021;13(599) doi: 10.1126/scitranslmed.aba9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duan X., Chan C., Lin W. Nanoparticle-mediated immunogenic cell death enables and potentiates cancer immunotherapy. Angew Chem. Int. Ed. Engl. 2019;58(3):670–680. doi: 10.1002/anie.201804882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X., Liu Y., Xue C., Hu Y., Zhao Y., Cai K., Li M., Luo Z. A protein-based cGAS-STING nanoagonist enhances T cell-mediated anti-tumor immune responses. Nat. Commun. 2022;13(1):5685. doi: 10.1038/s41467-022-33301-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang N., Liu C., Li Y., Huang D., Wu X., Kou X., Wang X., Wu Q., Gong C. A cooperative nano-CRISPR scaffold potentiates immunotherapy via activation of tumour-intrinsic pyroptosis. Nat. Commun. 2023;14(1):779. doi: 10.1038/s41467-023-36550-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halle, Stephan Halle, Olga Foerster, Reinhold Mechanisms and dynamics of T cell-mediated cytotoxicity in vivo. Trends Immunol. 2017;38(6):432–443. doi: 10.1016/j.it.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 12.Li X., Khorsandi S., Wang Y., Santelli J., Huntoon K., Nguyen N., Yang M., Lee D., Lu Y., Gao R., Kim B.Y.S., de Gracia Lux C., Mattrey R.F., Jiang W., Lux J. Cancer immunotherapy based on image-guided STING activation by nucleotide nanocomplex-decorated ultrasound microbubbles. Nat. Nanotechnol. 2022;17(8):891–899. doi: 10.1038/s41565-022-01134-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu H., Hu M., Liu M., An S., Guan K., Wang M., Li L., Zhang J., Li J., Huang L. Nano-puerarin regulates tumor microenvironment and facilitates chemo- and immunotherapy in murine triple negative breast cancer model. Biomaterials. 2020;235 doi: 10.1016/j.biomaterials.2020.119769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Binnewies M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., Coussens L.M., Gabrilovich D.I., Ostrand-Rosenberg S., Hedrick C.C., Vonderheide R.H., Pittet M.J., Jain R.K., Zou W., Howcroft T.K., Woodhouse E.C., Weinberg R.A., Krummel M.F. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24(5):541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watts C., West M.A., Zaru R. TLR signalling regulated antigen presentation in dendritic cells. Curr. Opin. Immunol. 2010;22(1):124–130. doi: 10.1016/j.coi.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Steinhagen F., Kinjo T., Bode C., Klinman D.M. TLR-based immune adjuvants. Vaccine. 2011;29(17):3341–3355. doi: 10.1016/j.vaccine.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Mercier I., Poujol D., Sanlaville A., Sisirak V., Gobert M., Durand I., Dubois B., Treilleux I., Marvel J., Vlach J., Blay J.-Y., Bendriss-Vermare N., Caux C., Puisieux I., Goutagny N. Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res. 2013;73(15):4629–4640. doi: 10.1158/0008-5472.CAN-12-3058. [DOI] [PubMed] [Google Scholar]
- 18.Oldfield V., Keating G.M., Perry C.M. Imiquimod: in superficial basal cell carcinoma. Am. J. Clin. Dermatol. 2005;6(3):195–200. doi: 10.2165/00128071-200506030-00006. [DOI] [PubMed] [Google Scholar]
- 19.Nuhn L., De Koker S., Van Lint S., Zhong Z., Catani J.P., Combes F., Deswarte K., Li Y., Lambrecht B.N., Lienenklaus S., Sanders N.N., David S.A., Tavernier J., De Geest B.G. Nanoparticle-conjugate TLR7/8 agonist localized immunotherapy provokes safe antitumoral responses. Adv. Mater. 2018;30(45) doi: 10.1002/adma.201803397. [DOI] [PubMed] [Google Scholar]
- 20.Chen Q., Xu L., Liang C., Wang C., Peng R., Liu Z. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat. Commun. 2016;7 doi: 10.1038/ncomms13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan J., Bao C., Xie Y., Guo H., Liu Y., Li J., Liu R., Li P., Bai J., Yan Y., Mu L., Li X., Wang G., Lu W. Targeted core-shell nanoparticles for precise CTCF gene insert in treatment of metastatic breast cancer. Bioact. Mater. 2022;11:1–14. doi: 10.1016/j.bioactmat.2021.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li P., Liu L., Lu Q., Yang S., Yang L., Cheng Y., Wang Y., Wang S., Song Y., Tan F., Li N. Ultrasmall MoS2 nanodots-doped biodegradable SiO2 nanoparticles for clearable FL/CT/MSOT imaging-guided PTT/PDT combination tumor therapy. ACS Appl. Mater. Interfaces. 2019;11(6):5771–5781. doi: 10.1021/acsami.8b18924. [DOI] [PubMed] [Google Scholar]
- 23.Théry C., Ostrowski M., Segura E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009;9(8):581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 24.Meng Q.F., Zhao Y., Dong C., Liu L., Pan Y., Lai J., Liu Z., Yu G.T., Chen X., Rao L. Genetically programmable fusion cellular vesicles for cancer immunotherapy. Angew Chem. Int. Ed. Engl. 2021;60(50):26320–26326. doi: 10.1002/anie.202108342. [DOI] [PubMed] [Google Scholar]
- 25.Cheng K., Kalluri R. Guidelines for clinical translation and commercialization of extracellular vesicles and exosomes based therapeutics. Extracell. Vesicle. 2023;2 [Google Scholar]
- 26.Bai J., Duan J., Liu R., Du Y., Luo Q., Cui Y., Su Z., Xu J., Xie Y., Lu W. Engineered targeting tLyp-1 exosomes as gene therapy vectors for efficient delivery of siRNA into lung cancer cells. Asian J. Pharm. Sci. 2020;15(4):461–471. doi: 10.1016/j.ajps.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalluri R., LeBleu V.S. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478) doi: 10.1126/science.aau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mondal J., Pillarisetti S., Junnuthula V., Saha M., Hwang S.R., Park I.K., Lee Y.K. Hybrid exosomes, exosome-like nanovesicles and engineered exosomes for therapeutic applications. J Control Release. 2022;353:1127–1149. doi: 10.1016/j.jconrel.2022.12.027. [DOI] [PubMed] [Google Scholar]
- 29.Tian Y., Li S., Song J., Ji T., Zhu M., Anderson G.J., Wei J., Nie G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35(7):2383–2390. doi: 10.1016/j.biomaterials.2013.11.083. [DOI] [PubMed] [Google Scholar]
- 30.Herrmann I.K., Wood M.J.A., Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021;16(7):748–759. doi: 10.1038/s41565-021-00931-2. [DOI] [PubMed] [Google Scholar]
- 31.Haney M.J., Zhao Y., Fallon J.K., Yue W., Li S.M., Lentz E.E., Erie D., Smith P.C., Batrakova E.V. Extracellular vesicles as drug delivery system for treatment of neurodegenerative disorders: optimization of the cell source. Adv. Nanobiomed Res. 2021;1(12) doi: 10.1002/anbr.202100064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elsharkasy O.M., Nordin J.Z., Hagey D.W., de Jong O.G., Schiffelers R.M., Andaloussi S.E.L., Vader P. Extracellular vesicles as drug delivery systems: why and how? Adv. Drug Deliv. Rev. 2020;159:332–343. doi: 10.1016/j.addr.2020.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Shrivastava S., Ray R.M., Holguin L., Echavarria L., Grepo N., Scott T.A., Burnett J., Morris K.V. Exosome-mediated stable epigenetic repression of HIV-1. Nat. Commun. 2021;12(1):5541. doi: 10.1038/s41467-021-25839-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu M., Khan A.R., Ji J., Lin G., Zhao X., Zhai G. Crosslinked self-assembled nanoparticles for chemo-sonodynamic combination therapy favoring antitumor, antimetastasis management and immune responses. J. Control Release. 2018;290:150–164. doi: 10.1016/j.jconrel.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Liu M., Du H., Khan A.R., Ji J., Yu A., Zhai G. Redox/enzyme sensitive chondroitin sulfate-based self-assembled nanoparticles loading docetaxel for the inhibition of metastasis and growth of melanoma. Carbohydr. Polymers. 2018;184:82–93. doi: 10.1016/j.carbpol.2017.12.047. [DOI] [PubMed] [Google Scholar]
- 36.Richter M., Vader P., Fuhrmann G. Approaches to surface engineering of extracellular vesicles. Adv. Drug Deliv. Rev. 2021;173:416–426. doi: 10.1016/j.addr.2021.03.020. [DOI] [PubMed] [Google Scholar]
- 37.Kojima R., Bojar D., Rizzi G., Hamri G.C., El-Baba M.D., Saxena P., Ausländer S., Tan K.R., Fussenegger M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson's disease treatment. Nat. Commun. 2018;9(1):1305. doi: 10.1038/s41467-018-03733-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen G., Huang A.C., Zhang W., Zhang G., Wu M., Xu W., Yu Z., Yang J., Wang B., Sun H., Xia H., Man Q., Zhong W., Antelo L.F., Wu B., Xiong X., Liu X., Guan L., Li T., Liu S., Yang R., Lu Y., Dong L., McGettigan S., Somasundaram R., Radhakrishnan R., Mills G., Lu Y., Kim J., Chen Y.H., Dong H., Zhao Y., Karakousis G.C., Mitchell T.C., Schuchter L.M., Herlyn M., Wherry E.J., Xu X., Guo W. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560(7718):382–386. doi: 10.1038/s41586-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melo S.A., Luecke L.B., Kahlert C., Fernandez A.F., Gammon S.T., Kaye J., LeBleu V.S., Mittendorf E.A., Weitz J., Rahbari N., Reissfelder C., Pilarsky C., Fraga M.F., Piwnica-Worms D., Kalluri R. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523(7559):177–182. doi: 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L., He Y., He T., Liu G., Lin C., Li K., Lu L., Cai K. Lymph node-targeted immune-activation mediated by imiquimod-loaded mesoporous polydopamine based-nanocarriers. Biomaterials. 2020;255 doi: 10.1016/j.biomaterials.2020.120208. [DOI] [PubMed] [Google Scholar]
- 41.Gettinger S., Horn L., Jackman D., Spigel D., Antonia S., Hellmann M., Powderly J., Heist R., Sequist L.V., Smith D.C., Leming P., Geese W.J., Yoon D., Li A., Brahmer J. Five-year follow-up of nivolumab in previously treated advanced non-small-cell lung cancer: results from the CA209-003 study. J. Clin. Oncol. 2018;36(17):1675–1684. doi: 10.1200/JCO.2017.77.0412. [DOI] [PubMed] [Google Scholar]
- 42.Borghaei H., Gettinger S., Vokes E.E., Chow L.Q.M., Burgio M.A., de Castro Carpeno J., Pluzanski A., Arrieta O., Frontera O.A., Chiari R., Butts C., Wójcik-Tomaszewska J., Coudert B., Garassino M.C., Ready N., Felip E., García M.A., Waterhouse D., Domine M., Barlesi F., Antonia S., Wohlleber M., Gerber D.E., Czyzewicz G., Spigel D.R., Crino L., Eberhardt W.E.E., Li A., Marimuthu S., Brahmer J. Five-year outcomes from the randomized, phase III trials checkmate 017 and 057: nivolumab versus docetaxel in previously treated non-small-cell lung cancer. J. Clin. Oncol. 2021;39(7):723–733. doi: 10.1200/JCO.20.01605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeng H., Chen W., Zheng R., Zhang S., Ji J.S., Zou X., Xia C., Sun K., Yang Z., Li H., Wang N., Han R., Liu S., Li H., Mu H., He Y., Xu Y., Fu Z., Zhou Y., Jiang J., Yang Y., Chen J., Wei K., Fan D., Wang J., Fu F., Zhao D., Song G., Chen J., Jiang C., Zhou X., Gu X., Jin F., Li Q., Li Y., Wu T., Yan C., Dong J., Hua Z., Baade P., Bray F., Jemal A., Yu X.Q., He J. Changing cancer survival in China during 2003-15: a pooled analysis of 17 population-based cancer registries. Lancet Glob Health. 2018;6(5):e555–e567. doi: 10.1016/S2214-109X(18)30127-X. [DOI] [PubMed] [Google Scholar]
- 44.Robert C., Ribas A., Schachter J., Arance A., Grob J.J., Mortier L., Daud A., Carlino M.S., McNeil C.M., Lotem M., Larkin J.M.G., Lorigan P., Neyns B., Blank C.U., Petrella T.M., Hamid O., Su S.C., Krepler C., Ibrahim N., Long G.V. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019;20(9):1239–1251. doi: 10.1016/S1470-2045(19)30388-2. [DOI] [PubMed] [Google Scholar]
- 45.Schumacher T.N., Schreiber R.D. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 46.Vanhersecke L., Brunet M., Guégan J.P., Rey C., Bougouin A., Cousin S., Moulec S.L., Besse B., Loriot Y., Larroquette M., Soubeyran I., Toulmonde M., Roubaud G., Pernot S., Cabart M., Chomy F., Lefevre C., Bourcier K., Kind M., Giglioli I., Sautès-Fridman C., Velasco V., Courgeon F., Oflazoglu E., Savina A., Marabelle A., Soria J.C., Bellera C., Sofeu C., Bessede A., Fridman W.H., Loarer F.L., Italiano A. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat. Cancer. 2021;2(8):794–802. doi: 10.1038/s43018-021-00232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yong T., Zhang X., Bie N., Zhang H., Zhang X., Li F., Hakeem A., Hu J., Gan L., Santos H.A., Yang X. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat. Commun. 2019;10(1):3838. doi: 10.1038/s41467-019-11718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X., Wang C., Wang J., Hu Q., Langworthy B., Ye Y., Sun W., Lin J., Wang T., Fine J., Cheng H., Dotti G., Huang P., Gu Z. PD-1 blockade cellular vesicles for cancer immunotherapy. Adv. Mater. 2018;30(22) doi: 10.1002/adma.201707112. [DOI] [PubMed] [Google Scholar]
- 49.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449(7164):819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 50.Heil F., Hemmi H., Hochrein H., Ampenberger F., Kirschning C., Akira S., Lipford G., Wagner H., Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 51.Diebold S.S., Kaisho T., Hemmi H., Akira S., Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 52.Min Y., Roche K.C., Tian S., Eblan M.J., McKinnon K.P., Caster J.M., Chai S., Herring L.E., Zhang L., Zhang T., DeSimone J.M., Tepper J.E., Vincent B.G., Serody J.S., Wang A.Z. Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat. Nanotechnol. 2017;12(9):877–882. doi: 10.1038/nnano.2017.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janeway C.A., Jr., Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76(2):275–285. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 54.Mellman I., Steinman R.M. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106(3):255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 55.Wherry E.J., Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015;15(8):486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsui C., Kretschmer L., Rapelius S., Gabriel S.S., Chisanga D., Knöpper K., Utzschneider D.T., Nüssing S., Liao Y., Mason T., Torres S.V., Wilcox S.A., Kanev K., Jarosch S., Leube J., Nutt S.L., Zehn D., Parish I.A., Kastenmüller W., Shi W., Buchholz V.R., Kallies A. MYB orchestrates T cell exhaustion and response to checkpoint inhibition. Nature. 2022;609(7926):354–360. doi: 10.1038/s41586-022-05105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.