

SUMMARY
Target deconvolution is a crucial but costly and time-consuming task that hinders large-scale profiling for drug discovery. We present a matrix-augmented pooling strategy (MAPS) which mixes multiple drugs into samples with optimized permutation and delineates targets of each drug simultaneously with mathematical processing. We validated this strategy with thermal proteome profiling (TPP) testing of 15 drugs concurrently, increasing experimental throughput by 60x while maintaining high sensitivity and specificity. Benefiting from the lower cost and higher throughput of MAPS, we performed target deconvolution of the 15 drugs across 5 cell lines. Our profiling revealed drug-target interactions can differ vastly in targets and binding affinity across cell lines. We further validated BRAF and CSNK2A2 as potential off-targets of bafetinib and abemaciclib, respectively. This work represents the largest thermal profiling of structurally diverse drugs across multiple cell lines to date.
eTOC Blurb:
Ji et al develop a high-throughput strategy and algorithms for large-scale derivatization-free drug target deconvolution. The strategy is validated experimentally on large set of diverse drugs across multiple cell lines revealing drug-protein interactions that differ across cell lines.
Graphical Abstract
INTRODUCTION
Bioactive compounds often modulate cellular processes by interacting with proteins whereas drug off-targets can contribute to both toxicity and therapeutic effects 1,2. Various cell types have their own unique developmental trajectory with specific transcriptional programs and respond differently to environmental challenges; as such, drug responses may also vary in different cell types 3. Identifying the binding targets of bioactive compounds (i.e., target deconvolution) is conventionally addressed by affinity-based and activity-based chemical proteomics approaches which often require tedious chemical derivatization which in itself may not be feasible depending on synthetic challenges and compound availability. Derivatization-free methods with mass spectrometry (MS) readouts like DARTS 4, LiP-MS5,6, SPROX 7, and TPP (Jafari et al. 2014) are more scalable yet are significantly limited in throughput due to instrumentation time.
Thermal proteome profiling (TPP) or MS-based cellular thermal shift assay (CETSA) 8–10 is a recently developed technique enabling unbiased target identification 11 in relevant biological contexts, such as lysates, intact cells 8, cell surface 12, tissue and whole blood samples 13 and was recently adapted for protein complex profiling based on thermal proximity co-aggregation phenomenon (TPCA) 14. TPP requires multiplexing samples processed by serial of denaturing temperatures with isobaric tandem mass tags (TMT) for MS analysis. The classic 1D protocol subjects both drug-treated and untreated samples to multiple denaturing temperatures while the 2D protocol introduces additional concentration series for each temperature condition 15,16 which further increases demand for MS instrumentation time.
Previous efforts to increase throughput reduced the number of samples required for testing a single drug, e.g., PISA 17 and iTSA 18. Inspired by pooled coronavirus testing 19,20 where samples are mixed for RT-PCR assays, we proposed a Matrix-Augmented Pooling Strategy (MAPS) for large-scale proteome-wide profiling of protein-chemical interactions with derivatization-free MS-based approaches. In brief, MAPS increases analytical throughput by mixing multiple drugs into samples in specific combinations followed by mathematical processing to directly delineate the targets of each drug without further profiling; this increases throughput by one to two orders of magnitude compared with current experimental techniques. Here, we developed MAPS for profiling 15 drugs each time with TPP using the iTSA format where a single denaturing temperature is used 12,13. We assessed the performance of MAPS-iTSA using the subset of profiled drugs which cognate targets were successfully identified by TPP previously. Staurosporine, a broad-spectrum kinase inhibitor, was subsequently included among the pooled drugs to increase complexity of data for target delineation. We validated that MAPS-iTSA permits 15x and 60x analysis throughput over iTSA and the classic 1D TPP respectively, with minimal compromise in sensitivity while reducing reagent consumption. MAPS-iTSA identified potential off-targets for 5 drugs with ambiguous mechanism-of-action; two of them were validated with molecular docking, molecular dynamics stimulation, intracellular engagement assays and in vitro enzymatic assays. In addition, MAPS-iTSA revealed differentiated targets and responses across multiple cell lines. In summary, MAPS is a viable strategy for increasing throughput of derivatization-free target deconvolution with mass spectrometry (MS), and offers an effective and powerful approach for unveiling new targets and mechanisms of bioactive compounds to expediate drug discovery.
RESULTS
Overview of MAPS-iTSA.
The concept and workflow for MAPS were summarized in Fig. 1a while the details of the algorithms can be found in methods. First, a binary sensing matrix was derived to guide mixing of drugs across samples which was optimized with a genetic algorithm to minimize correlation among the mixtures and to increase the information entropy obtained. As a proof-of-concept, we applied MAPS to 15 drugs including 10 whose targets were previously validated by TPP (termed verification set) and 5 drugs with unclear mechanism-of-action, (termed exploratory set, see methods). Drugs in the exploratory set were designed for specific targets, but recent studies suggest their effects are due to unconfirmed off-targets 21,22.
Fig. 1. Workflow and effective verification of MAPS-iTSA.
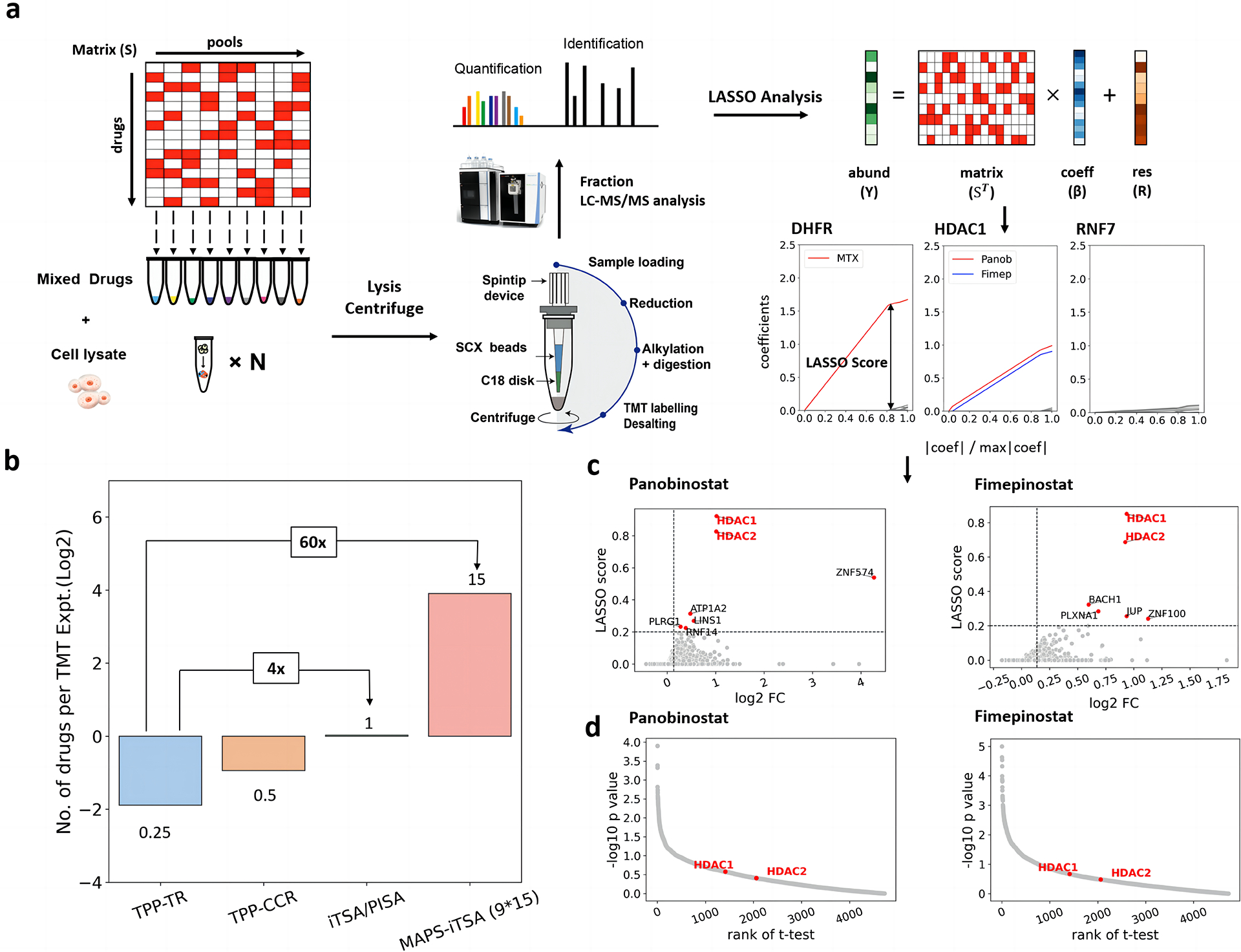
a. Illustrative application of the MAPS-iTSA workflow involves target deconvolution. The 9×15 binary sensing matrix is optimized for mixing the testing drugs. The value of 1 means put the corresponding drug into the tube containing cell lysate. Subsequently, the mixtures are incubated under specific temperature (e.g., 52 °C) followed by protein extraction, labelling and quantification. The normalized vector of the concentrations of the target protein is used for the LASSO analysis. Through LASSO analysis, the coefficient values quantify the contributions of the drugs to the variation in thermal stability of the protein. By increasing the penalty, the coefficients computed for the various drugs will progressively decrease to zero. For each protein, the LASSO score of each drug is defined as the corresponding coefficient value when only three non-zero values remained. b. Bar charts comparing the number of drugs that can be analyzed by a TMT experiment for TPP-TR, TPP-CCR, iTSA, PISA and pooled TPP/iTSA. c. LASSO score calculated from the path of coefficients compared with the fold changes reveal that HDAC1 and HDAC2 are the targets of panobinostat and fimepinostat. d. Scatter plots of the p-values and the ranking of the true targets indicate t-test cannot handle two drugs share the same targets.
Accordingly, we designed a 9×15 sensing matrix with an in-house script (see and provided in supplementary materials, Fig. S1a and Fig. S1b), which means 15 drugs were pooled across 9 tubes for target deconvolution (Table S1 and Table S2). Our script dictates each drug is found in at least 3 tubes. After mixing of drugs, samples were subjected briefly to a denaturing temperature and soluble proteins were quantified by protein mass spectrometry using SISPROT with TMT multiplexing reagents 23,24. Finally, drug-target interactions were deconvoluted based on quantified protein abundances and the sensing matrix with a LASSO regression algorithm. For every protein detected, the LASSO algorithm computed a LASSO path and corresponding LASSO score for each drug. The LASSO score of each drug was defined by the regression coefficient when the penalty of LASSO making only k regression coefficients was nonzero (Fig. S1c), where k was optimized to three in our experiments (see methods and Fig. S1d for details). This MAPS strategy with 9×15 matrix offers a 15x throughput compared with iTSA or PISA, which requires a TMT experiment for each drug. Compared with the classic temperature-response TPP with four TMT experiments (two replicates each for drug and control), it trades some sensitivity (some targets may not exhibit shifts at the selected temperature) for 60x throughput (Fig. 1b).
Validation of MAPS-iTSA and LASSO algorithm.
Briefly, MAPS-iTSA was first performed on the K562 cell line with 15 drugs from the verification set and exploratory set for preliminary assessment of its sensitivity. Subsequently, in an attempt to increase the complexity of data obtained, we elected to replace parthenolide in the exploratory set with staurosporine, a broad-spectrum kinase inhibitor which were conducted for five different cell lines. Parthenolide is chosen for replacement as we failed to detect robust differentiated response for any protein.
First, the performance of MAPS-iTSA with LASSO regression algorithm for delineating targets of drugs pooled were evaluated in K562 cell lysate. Encouragingly, eight of nine drugs except olaparib in the verification drug set have targets identified within the top five hits of each drug, where the targets of six drugs were identified as the top hit. For example, for the LASSO path of DHFR, the coefficient trendline of methotrexate is much higher than other drugs, implying that DHFR is the target of methotrexate. In contrast, the LASSO paths of most proteins are disordered with low LASSO score (Fig. 1a). Univariate difference analysis algorithms (e.g., t-Test) can be applied in classical iTSA for target identification. However, in MAPS-iTSA, it was observed that the LASSO score performs better, especially when multiple drugs share the same target which can be explained by the outstanding variable selection characteristic of LASSO. Both panobinostat and fimepinostat in the verification set target HDAC1 and HDAC2. The LASSO paths of the two drugs stand out significantly for HDAC1/2 indicating that both drugs bind HDAC1/2, which is also reflected in the LASSO score in the scatter plots (Fig. 1c). In comparison, t-test performed for samples with and without panobinostat or fimepinostat ranks HDAC1 low as a target based on p-value (Fig. 1d). In addition, the cognate target(s) of each drug were not identified for other drugs profiled indicating there was no “cross-contaminant” of targets (Fig. 2a). Thus, MAPS with LASSO algorithm has high sensitivity and high specificity with increased experimental throughput. All corresponding scatter plots of LASSO scores against fold changes were summarized in Fig. S2.
Fig. 2. Cell type specific drug target verification.
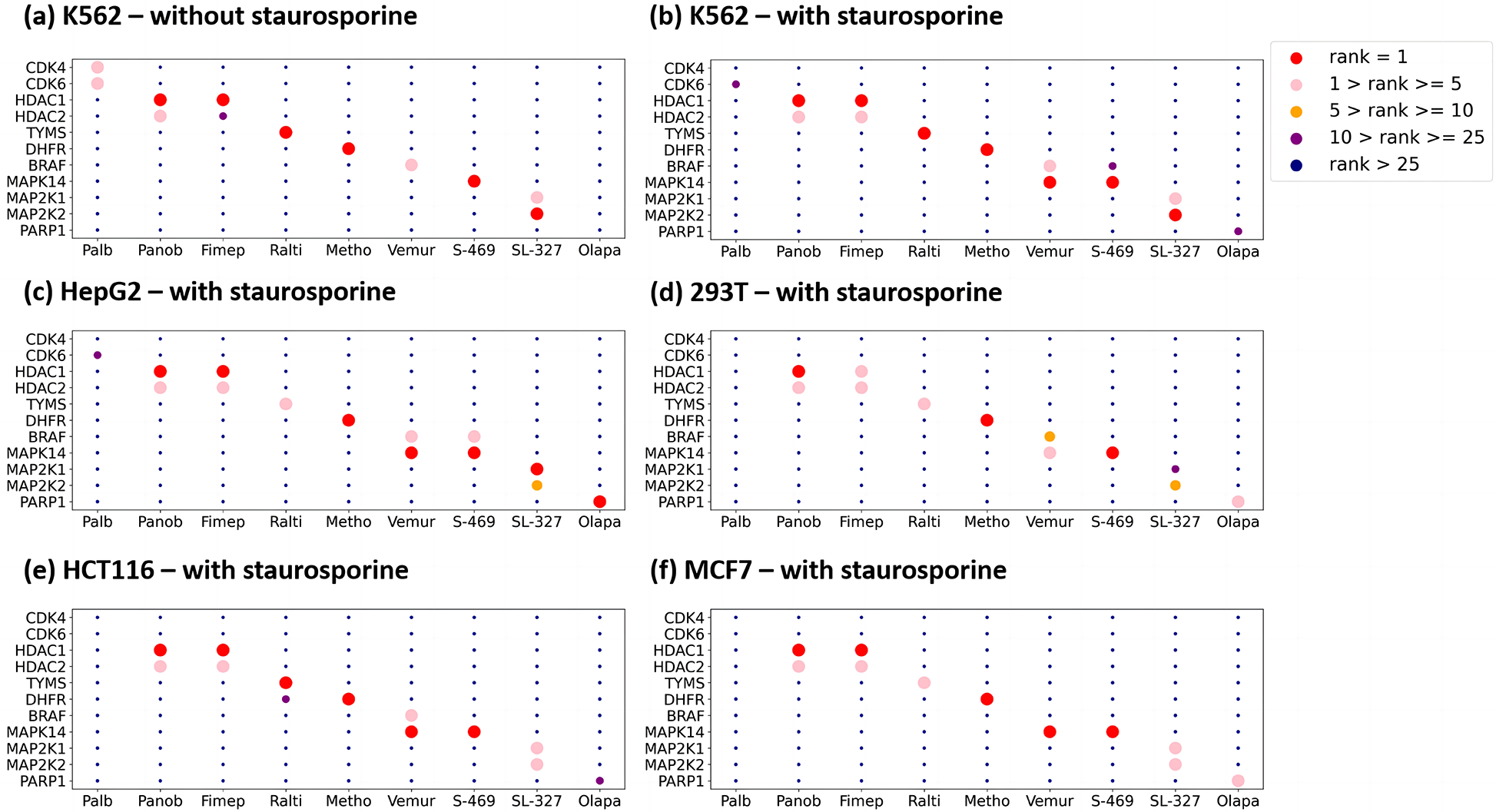
In each subplot, the x-axis represents the drugs, the y-axis represents the targets, and the color of the points represent the ranking of the target deconvolution based on LASSO score.
Staurosporine, a broad-spectrum kinase inhibitor, binds many kinases and ATP-binding proteins which also perturbs many downstream substrate proteins. We subsequently included staurosporine into our verification drug set to increase data complexity and difficulty in the delineation of targets. Encouragingly, cognate targets of all drugs in the verification set, except for olaparib and palbociclib, were identified as top hits (Fig.2b–2f). Palbociclib inhibits CDK4 and CDK6 which are also targeted by abemaciclib (exploratory drug set) and staurosporine, suggesting that three or more drugs sharing the same targets could lead to false negatives. As such, we designed a structure-based target assumption strategy to minimize mixing of drugs potentially sharing similar targets (supplementary materials), although it may still fail when structurally diverse drugs share the same targets.
Olaparib is the only drug in the verification set that we fail to identify its cognate target, PARP1, in the two previous experiments using K562 cells. To investigate the possibility of a cell type effect, we repeat the last MAPS-iTSA experiment (with staurosporine) using 293T, HCT116, MCF7 and HepG2 cell lysates. Interestingly, PARP1 was revealed as the top hit in HepG2 and among the top 5 targets for olaparib in 293T and MCF7 (Fig. 2a). Therefore, PARP1 was most likely missed in our earlier experiments due to cell-type specific effect. This differential response could be due to post-translational modifications of PARP1 and its interactions with metabolites and other proteins in different cell type.
Next, we assumed all kinases and ATP-binding proteins are true targets of staurosporine to further evaluate the performance of MAPS-iTSA. Kinases and ATP- binding proteins identified in the five different cell types were visualized in Fig. 3a. Generally, over 30 true targets were identified for each cell type which account for over 70% of targets identified in most cell types (Fig. 3b). As a comparison, MAP-iTSA identified 41 kinase identified in 293T cells, comparable to 47 kinases (p-value < 0.05, Fig. S1f) identified by classical iTSA performed in this work for staurosporine in 293T cell line, but offering 15x higher throughputs.
Fig. 3. Target deconvolution result of staurosporine.
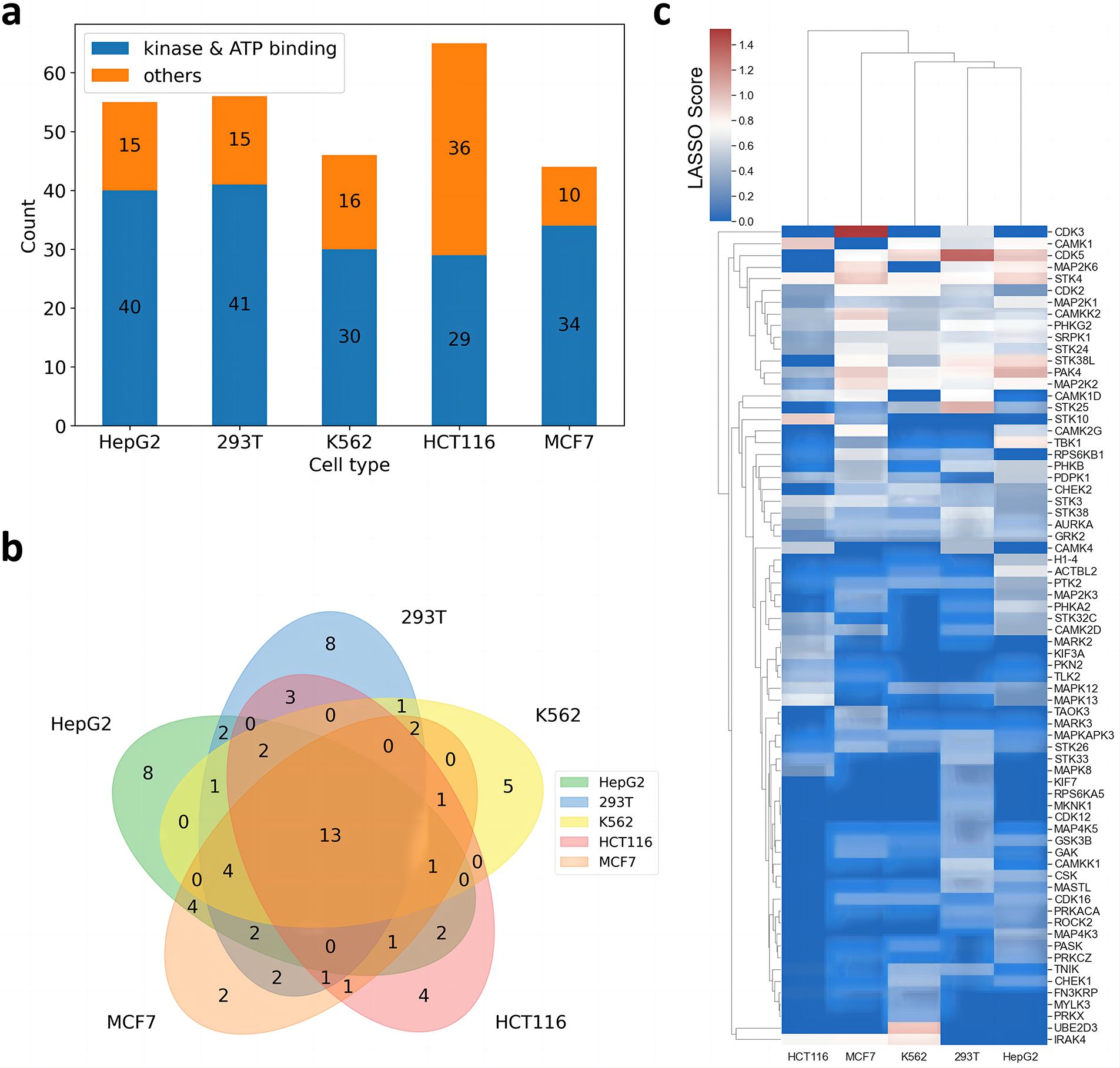
a. Bar charts of the number of identified targets in different cell types. b. Venn plot of the identified kinases or ATP binding proteins of the different cell types. c. Correlation based heatmap of the identified kinases or ATP binding proteins, the color scale indicates the LASSO score.
We observed the identified kinases and ATP-binding proteins vary across cell type. For example, only 13 targets were identified across all the five cell types while 25 targets were uniquely identified in one cell type only that accounted for 18.6% and 35.8% of all the identified kinases and ATP-binding proteins, respectively. This together with our observation regarding PARP1 suggests that drug binding can vary substantially in terms of targets and affinity across cell type. This may arise from specific proteoforms of targets due to isoforms and/or posttranslational modification present in each cell type or cellular state. Altogether, 70 kinases and ATP-binding proteins were identified. Comparing the identified targets among different cell types via a heatmap revealed kinases of the same class generally have highly correlated characteristics in various cell types, such as CDK2/3/5 and MAPK8/12/13 (Fig. 3c).
Off-target exploration.
An exploratory set of 5 drugs namely bafetinib, abemaciclib, CCT137690, belumosudil and OTS964, were included in all our experiments across 5 cell lines. The first four drugs have very different efficacy from other drugs targeting the same proteins in 578 cancer cell lines (Corsello et al. 2020) while the cognate target for otherwise toxic OTS964 is dispensable for cancer cell viability (Lin et al. 2019). Our MAPS-iTSA analysis uncovers many new potential off-targets of these drugs including several that were observed across multiple cell types as well as new targets unique to specific cell types (Fig. 4 and Fig. S3). First, we confirmed AURKA as the cognate target of CCT137690, which was identified in all cell types. We also validated CDK11 as an off-target of OTS964 as reported previously 22 although it was identified only in two out of five cell types profiled (Fig. 4a). On the other hand, MAPS-iTSA verified STK3, STK4 and AURKA as targets of belumosudil across cell lines profiled but could not identified ROCK1 and ROCK2 as targets of belumosudil in any of the cell lines profiled (Fig. 4b). This together with the observed disparity in toxicity profile of belumosudil to other ROCK inhibitors (Fig. S4a) suggests the two proteins may not be the functional targets of belumosudil (Corsello et al. 2020).
Fig. 4. Cell type specific off target exploration.
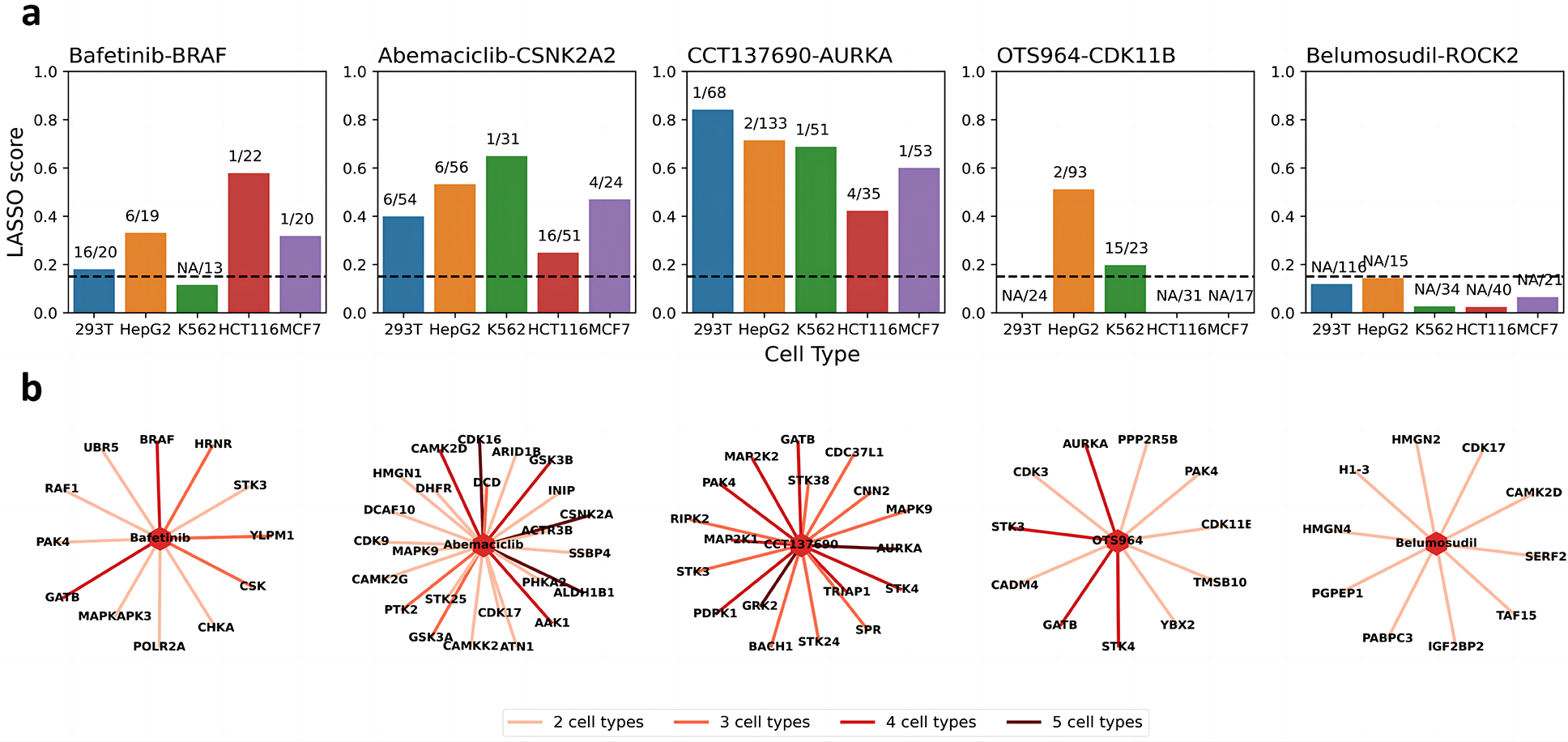
a. Bar charts of LASSO score accompanied ranks of the drug-target pairs. The horizontal dotted line indicates the threshold. The labels are the ranks of the target of all identified significant proteins. NA means the LASSO score is below the threshold. b. Targets of the exploratory drugs identified in multiple cell types.
Furthermore, our analysis suggests that bafetinib, an experimental drug targeting BCR-ABL and LYN for treatment of lymphocytic leukemia 25, can bind BRAF to different degrees in four cell types. (Fig. 4a, 4b). Molecular docking reveals bafetinib could bind BRAF in a similar mode as belvarafenib, another BRAF inhibitor 26 (Fig. 5a). Molecular dynamics (MD) simulations and MMPBSA free energy analysis 27 on protein-ligand pairs reveals that their binding is strong and stable (Fig. 5b). Moreover, the anti-proliferative efficacy of bafetinib across 578 cancer cell lines is similar to RAF inhibitors but differs from BCR-ABL/LYN inhibitors (Fig. S4a) (Corsello et al. 2020). Thus, our profiling strongly supports the notion that BRAF, not BCR-ABL and LYN, is likely the direct and functional target of bafetinib.
Fig. 5. Off target explanation and validation.
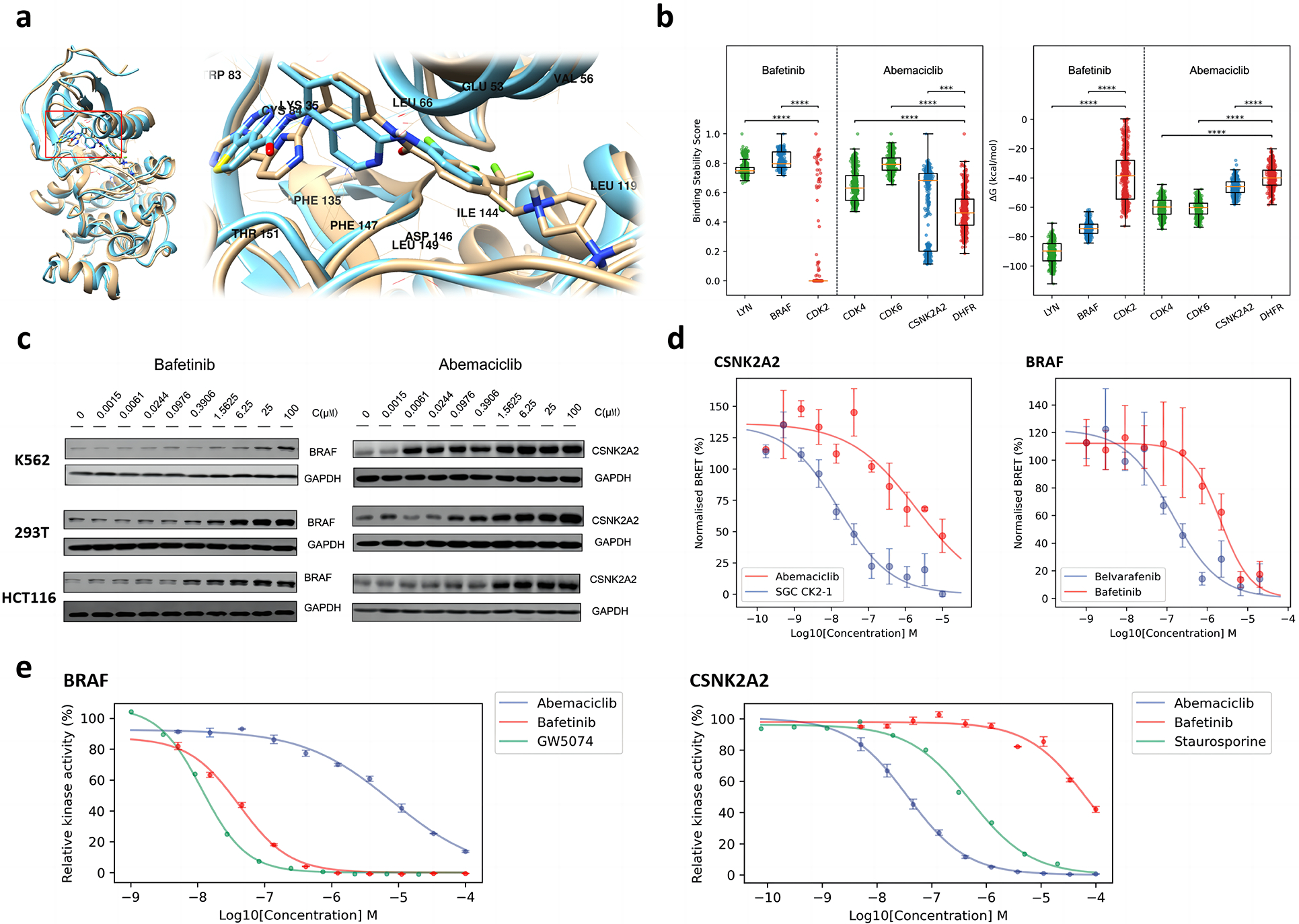
a. Binding mode comparison between bafetinib and belvarafenib in complex with BRAF. The structure closest to the average structure (measured by RMSD) of bafetinib-BRAF MD simulations is displayed in brown; The co-crystal structure of belvarafenib-BRAF (PDB ID: 6XFP) is displayed in cyan. b. Comparison of binding stability scores (left) and MMPBSA binding free energies (right) of the last 150 ns of MD simulation trajectories (frame interval is 1 ns). Positive controls are displayed in green; Negative controls are displayed in red; Identified off-targets are displayed in blue. Significance levels of z-tests: ***, p <= 0.001; ****, p <= 0.0001. c. Western blot of dose-dependent CETSA of abemaciclib-CSNK2A2 and bafetinib-BRAF. The changing of blots is consistent to the LASSO score of MAPS-iTSA. d. Dose-response curve for NanoBERT in-vitro off-target validation of bafetinib-BRAF and abemaciclib-CSNK2A2. Known inhibitors are taken as positive control while their cross matching is taken as negative control. e. Dose-response curve for kinase hotpots in-vitro off-target validation of bafetinib-BRAF and abemaciclib-CSNK2A2. Relative kinase activities are obtained by normalization based on blank controls.
Intensity and ranking of LASSO scores suggests binding of bafetinib to BRAF is absent in K562 but varies in affinity across the other four cell types (Fig. 4a). To validate this, we performed isothermal dose-response (ITDR) CETSA in HCT116, 293T and K562, to quantify the thermal stability of BRAF against a bafetinib concentration gradient (Fig. 5c). We observed a dose-dependent stabilization of BRAF by bafetinib at concentrations slightly lower in HCT116 over 293T. On the other hand, stabilization of BRAF in K562 can only be observed at 100 μM of bafetinib which is 5x more than the concentration used in our MAPS-iTSA experiment. These observations are consistent with the LASSO score and rank of BRAF in these cell types. Recent study reveals high KRAS activity possibly enhance the engagement between inhibitor and BRAF 2, suggesting the strong stabilization of BRAF by bafetinib observed for HCT116 could be due to presence of KRAS harboring activating G13D mutation in that cell line.
Abemaciclib, presently in clinical trials for treatment of breast and non-small cell lung cancer 28, is an inhibitor of CDK4 and CDK6. Interestingly, we note that GSK3A, GSK3B, and CSNK2A2 outrank CDK4 and CDK6 in our analysis. Recent studies have shown that abemaciclib can inhibit GSK3A, GSK3B and activate WNT signaling with MIB/MS competition assay 29,30. In fact, dose-response curve data had revealed that the binding of abemaciclib to GSK3A/3B is stronger than for CDK4/6, which is consistent with our analysis. In addition, our analysis further suggests that abemaciclib also targets CSNK2A2 (Fig. 4a, 4b) which we verified with the MD simulations and MMPBSA analysis (Fig. 5b). LASSO scores also suggest abemaciclib binds CSNK2A2 strongly in K562 but relatively weakly in HCT116 which we validated with ITDR CETSA. Thus, CSNK2A2 was stabilized at very low nanomolar concentrations of abemaciclib in K562, while it was stabilized at higher micromolar concentration in HCT116, with the concentration in 239T ranking in between. (Fig. 5c). These results are consistent with analysis by MAPS-iTSA which reveals that CSNK2A2 is an off-target of abemaciclib with low nanomolar affinity in specific cell types. Thus, ITDR validation of two drugs demonstrated that LASSO score correlates with differential drug-target interaction and binding affinity across cell types.
Experimental validation of off-targets.
Next, NanoBRET assays were performed to assess intracellular engagement of bafetinib and abemaciclib with BRAF and CSNK2A2 respectively. The NanoBRET signal was measured in the presence of increasing concentrations of test drugs. The curve-fit results show a concentration-dependent decrease in the NanoBRET signal, indicating a reduction in tracer-protein interaction in the presence of the bafetinib and abemaciclib for their respective off-targets (Fig. 5d, observed raw data were summarized in Table S3). The IC50 of abemaciclib for CSNK2A2 is calculated as 2.010 μM while that for SGC CK2–1, a known CSNK2 inhibitore, is 0.016μM. The IC50 of bafetinib for BRAF was calculated as 2.138 μM compared to 0.134M for belvarafenib, the known BRAF inhibitor with the similar binding site in docking simulation. These results validated the intracellular binding of bafetinib and abemaciclib to their respective off-targets identified by MAPS-iTSA. We also performed the same assay on BRAF V600E and observed the mutant was engaged more strongly by bafetinib (IC50 0.217 μM vs 2.138 μM, Fig. S4b).
Next, plausible inhibitory effect of abemaciclib and bafetinib on the respective kinase enzymatic activity of CSNK2A and BRAF were assessed with hotspot target validation assay. We observed a dose-dependent reduction in kinase activity of CSNK2A in the presence of abemaciclib and the kinase activity of BRAF in the presence of bafetinib (Fig. 5e, observed raw data were summarized in Table S4). The calculated IC50 of abemaciclib for CSNK2A2 was calculated as 36.40 nM, which is lower than positive control staurosporine (500.76 nM). The calculated IC50 of bafetinib for BRAF was calculated as 41.61 nM, which is at the same magnitude as the known BRAF inhibitor GW5074 (11.66 nM). Thus, both drugs bind their respective identified off-targets in cells and could inhibit their enzymatic activities in vitro, validating that MAP-iTSA could identify off-targets with increased experimental throughput.
DISCUSSION
We performed extensive testing of MAPS with iTSA using 15 drugs across 5 cell lines of different cell type origin, validating that it is an effective and efficient approach to increase the throughput of drug target deconvolution experiments with minimal compromise in sensitivity and specificity. Factoring in the cell type effect of olaparib, all drugs in our verification set had their cognate targets identified among the first 5 hits, with targets of seven drugs consistently identified as top hit across the majority of cell lines profiled. Thus, coverage and specificity are high, but we observed sensitivity was compromised for CDK4 and CDK6 when they were targeted by 3 drugs (palbociclib, abemaciclib and staurosporine) in the same test set. This situation can be avoided with increased sample size in large scale screening (see supporting information and Fig. S5 for methods and analysis) as well as pre-filtering based on potential targets of compounds profiled.
Taking advantage of the increased throughput offered by MAPS, we profiled 15 drugs across 5 cell lines. While cognate targets of drugs in our verification set exhibited very consistent signatures across the cell lines profiled, we noticed relatively more variation in target profiles of drugs in our exploration set in both the proteins identified and stabilization signal. Drugs in the exploration set were chosen as they divert substantially from drugs targeting the same proteins in their anti-proliferative activity across a panel of cancerous cell lines. For example, ITDR assay validated BRAF and CSNK2A2 as targets of bafetinib and abemaciclib, respectively, but also revealed that their interactions can vary by a few orders of magnitude in affinity across different cell types. This suggests that target profiling of drugs in relevant cell types is needed for better assessment of drug action, efficacy, and potential side effects, and should constitute an important consideration in precision medicine.
While we had validated MAPS with iTSA, MAPS strategy is also theoretically suitable for other derivatization-free methods to increase experimental throughput. The response vector Y in MAPS represents the corresponding response of protein changing caused by the mixtures. As long as the response can be measured numerically, continuously and robustly, the LASSO analysis should be feasible. Moreover, the throughput could be increased further with higher multiplex reagents (e.g., TMT16 or TMT18, see support information and Fig. S5) or label-free quantitative MS approaches, enabling large-scale many-vs-many chemical-protein interaction screening with endogenous proteins in diverse cellular contexts to expediate drug discovery and development.
Limitations of the study
We recognize several limitations in our study that should be acknowledged. Firstly, our MAPS method, designed for quick off-target screening, is constrained by its theoretical framework. Consequently, it has the potential to generate both false positives and false negatives, especially when multiple drugs within a sensing matrix have a shared target. Secondly, although we conducted multiple orthogonal experiments to confirm that bafetinib and abemaciclib target BRAF and CSNK2A2, respectively, it is crucial to interpret these findings cautiously. Further validation is necessary before considering the clinical applicability of these results. Another important caveat to note regarding this work is the inclusion of staurosporine. Its purpose in this study is to increase the challenge when evaluating the performance of MAPS. However, it is not advisable to study drugs with a wide range of targets in practical applications of MAPS, as this significantly increases the risk of false positives for other drugs.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Chris Tan (christan@sustech.edu.cn).
Materials availability
Antibodies, reagents and cell lines used for biological studies were obtained from commercial or internal sources described in the key resources table. Where available these may be shared by the lead contact. This study did not generate new unique reagents.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-rabbit antibodies | Beyotime | Cat# A0208 |
| Rabbit Monoclonal [SU3404] to B-Raf | GXP | Cat# GXP 52349 |
| Anti-CSNK2A2 Antibody | GXP | GXP123212 |
| GAPDH antibody | Proteintech | Cat# 10494-1-AP |
| Chemicals, peptides, and recombinant proteins | ||
| Palbociclib | MCE | HY-50767 |
| Panobinostat | SANTA | SC-208148 |
| Raltitrexed | MCE | HY-10821 |
| Methotrexate | Sigma-Aldrich | M8407-100MG |
| Vemurafenib | MCE | HY-12057 |
| Fimepinostat | MCE | HY-13522 |
| Olaparib | MCE | HY-15307 |
| Bafetinib | MCE | HY-50868 |
| SCIO-469 | GLPBIO | GC34072 |
| OTS964 | MCE | HY-12467 |
| SL-327 | APExBIO | A1894 |
| Abemaciclib | MCE | HY-16297A |
| CCT137690 | MCE | HY-10804 |
| Belumosudil | MCE | HY-15307 |
| Staurosporine | MCE | HY-15141 |
| Experimental models: Cell lines | ||
| K562 | National Collection of Authenticated Cell Cultures | TCHu191 |
| HEK 293T | National Collection of Authenticated Cell Cultures | GNHu17 |
| MCF7 | National Collection of Authenticated Cell Cultures | TCHu74 |
| HCT116 | National Collection of Authenticated Cell Cultures | TCHu99 |
| HepG2 | National Collection of Authenticated Cell Cultures | TCHu72 |
| Deposited data | ||
| Proteome experimental data | ProteomeXchange | PXD043882 |
| Software and algorithms | ||
| Proteome Discoverer | Thermo Fisher Scientific | v2.4 |
| AutoDock Vina | The Scripps Research Institute | v1.2.3 |
| Amber | UC San Francisco | v2021 |
| UCSF Chimera | UC San Francisco | v1.14 |
| Python script: MAPS-iTSA workflow | github.com/hcji/MAPS-iTSA | v1.0 |
Data and code availability
All data is available in the main text or the supplementary materials. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX31,33 partner repository with the dataset identifiers PXD043882.
The python script code used to perform the analyses is accessible as a GitHub repository at https://github.com/hcji/MAPS-iTSA.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Study Participant Details
Cell Culture
The five cell lines used in this study were acquired from the National Collection of Authenticated Cell Cultures, Chinese Academy of Sciences, and were all authenticated using short tandem repeat (STR) analysis. All cell lines were of human origin. Among them, K562, MCF7, and HEK 293T cell lines were derived from female donors, while HCT116 and HepG2 cell lines were derived from male donors. The culture conditions for each cell line were as follows: K562 cells were cultivated in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and incubated in a humidified incubator at 37°C with 95% humidity and 5% CO2. HEK 293T and MCF7 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS, and maintained at 37°C with 5% CO2. HCT116 cells were grown in McCoy’s 5A culture medium, while HepG2 cells were cultured in MEM culture medium, both supplemented with 10% FBS, and both maintained at 37°C with 5% CO2.
| Cell line | Organism | Gender | Culture medium | Fetal bovine serum (FBS) | Cultivation environment |
|---|---|---|---|---|---|
| K562 | Human | Female | RPMI 1640 medium | ||
| HEK 293T | Human | Female | Dulbecco’s Modified Eagle Medium (DMEM) | ||
| MCF7 | Human | Female | Dulbecco’s Modified Eagle Medium (DMEM) | 10% | 37°C with 95 % humidity and 5 % CO2 |
| HCT116 | Human | Male | McCoy’s 5A | ||
| HepG2 | Human | Male | Minimum Eagle’s Medium MEM | ||
METHOD DETAILS
Selection of exploratory drugs
Drugs in exploratory set have high probabilities of off-targets based on published works. Most of these drugs are selected based on Corsello’s work 21, which measured the antiproliferative profiles of 4518 oncology and non-oncology drugs across 578 cancerous cell lines, which constituted a 4518×578 numeric matrix. Whereafter, u-map algorithm is used for dimensionality reduction and visualization (Fig. S4a). Each point in the scatter plots represent a kind of drug, and the proximity between two points reflect the similar antiproliferative profiles of cell lines. We selected five drugs with dissimilar profiles to majority of drugs targeting the same designed targets, which suggests that their anti-cancer effect could be due to off-target effects. In addition, OTS964, an inhibitor designed for TOPK, was included as CDK11 was recently revealed to be the functional target of the experimental drug 22.
Sensing matrix generation
Sensing matrix is designed to guide the mixing of drugs into each sample. The sensing matrix design is a combinatorial optimization problem, of which the objective is to ensure reliable and robust deconvolution. Supposing the task is testing 15 drugs with a TMT-11 experiment, the shape of the sensing matrix should be 9×15 (with two TMT channels for 37°C control sample). Each row represents a pool, and each column represents a drug. If a drug is added to a pool, the value of the corresponding coordinate is 1, otherwise the value is 0. The correlation among vectors of drugs will affect target deconvolution. For example, if the correlation between the vector of two drugs is 1, it means the two drugs are found in the same set of samples, and it will be impossible to distinguish the proteins targeted by each drug. On the contrary, if their correlation is 0, the distinction between them will be simple. The goal of optimizing the sensing matrix is to attain minimal correlation between the vectors of any two drugs in a test set. Since it is time-consuming to use an exhaustive method for this, we adopted a genetic algorithm approach (Fig. S1a).
First, we appoint the immutable parameter , which indicates that each drug has to exist in at least three samples. Then, the adapting matrix is randomly initialized with values between 0 and 1. The dimension of is the same as the sensing matrix . The criterion which transforms to is to change the top values of each column to 1, and change the other values to 0. Next, a genetic algorithm approach is used to optimize , in order to minimum in the following equation:
| (1) |
where is the sensing matrix transformed from is used for calculating the pairwise correlation of the rows (drugs). is the identity matrix (a square matrix such that all the entries in the main diagonal are 1, and the rest of the entries are all 0), which is used to remove the self-correlation values. is the sum values of each row of . The formal item, , is used to minimize the correlation between the vectors, while the latter item, , is used to ensure each sample shares the same number of drugs. Lastly, the final sensing matrix is obtained by the transformation of the optimized . The optimization in this work was performed through genetic algorithm with 500 iterations, which was visualized in Fig. S1b.
Preparation of cell lysate
K562 cells were harvested in 50 ml centrifuge tube, washed twice with ice-cold PBS where supernatant was discarded after centrifugation at 900 rpm for 5 min. HEK293T, HCT116, MCF7, HepG2 cells were washed twice with ice-cold PBS in 15 cm dish, and the cells were collected with a scraper and washed again with ice-cold PBS. All cells were washed with ice-cold PBS before lysed in a lysis buffer containing 50 mM HEPES, pH 7.5, 5 mM β-glycerohosphate, 0.1 mM activated Na3VO4, 10 mM MgCl2, and 1 mM TCEP, with EDTA-free protease inhibitors added freshly to the lysis buffer before use. Cells were freeze-thaw three times by alternating the immersion of tubes between liquid nitrogen and a 37°C water bath. The cell mixture was then pulled through a needle (25” gauge needle, 1ml volume) 10 times to facilitate cell lysis by mechanical crushing. Next, centrifugation is performed at 21000 rcf at 4°C for 20 minutes to collect the supernatant and protein concentration is determined with BCA reagent.
TMT-labeled proteomics analysis
The compounds were purchased from MCE company, and each compound is dissolved in DMSO at 10 mM and stored in −80°C refrigerator. Prior experiment, each compound was diluted to 400 μM using ultra-pure water with ultrasound treatment to facilitate dissolution 32. According to the sensing matrix, e.g.,9*15 matrix, there are five drugs mixed in each TMT label samples. 10 μL for each drug (400 μM) was mixed into PCR tubes, each of which were added 50 μL diluent buffer. Each PCR tube was then topped up with diluent buffer to 100 μL attaining a concentration of 40 μM for each drug in a pool. The control samples did not contain any drugs but with 2% DMSO identical to other samples.
We had performed extensive testing of MAPS with iTSA using 15 drugs across 5 cell lines of different cell type origins. An equal volume of cell lysate and drug mixture was mixed and incubated at room temperature for 10 minutes, then heated at 52°C for 3 minutes before cooled rapidly to 4°C on a PCR machine. The control samples were subjected to same treatment but heated at 37 °C for 3 minutes instead. We have nine PCR tubes heated at 52°C and two tubes heated at 37°C as controls. The samples were centrifuged at 21,000 rcf for 20 minutes at 4°C and the supernatant was collected.
We used the SISPROT workflow 24 to prepare collected samples for MS analysis. Proteins were denatured in a solution of 0.1% formic acid (FA) to acidify the sample. The SISPROT digestion device was filled with C18 disk (3 M Empore) and SCX/SAX mixed beads (1 mg, SCX: SAX = 1:1, Biosystems) into a standard 200 μL pipet tip 34. SISPROT tip was washed with methanol and 100 mM PCB potassium citrate buffer (pH 3) with 10 mM PCB to balance the tip. We processed 10 μg of protein from 37°C sample but obtained same volume (as the 37°C sample) of drug-treated samples for processing with acidification (final concentration is 0.1% (v/v) formic acid) and then loaded each sample on a SISPROT tip. Each tip was washed with 20% (v/v) acetonitrile (ACN) in 8 mM potassium citrate buffer (pH 3) and ACN and incubate with 10 mM Tris(2-carboxyethyl) phosphine hydrochloride (TCEP) for 15 min at room temperature. The tips were then washed with 50 mM Tris-HCl, pH 8, and digestion solution (0.1 μg/μL trypsin (Promega) in 10 mM iodoacetamide, 100 mM Tris-HCl, pH 8) was added with incubation at 37°C for 60 min. After digestion, peptides were washed with 500 mM NaCl from mixed beads to C18 before TMT-11 labeling for 30–60 min. After labeling, 1% (v/v) FA was added and the peptides were washed with 80 % (v/v) ACN, 0.5% (v/v) acetic acid for collection. All labeled peptide samples were mixed and lyophilized.
Peptide fractionation was performed after mixing of 11 samples labelled with TMT reagents. We used a 200 μL pipette tip filled with C18 membrane as a carrier for fractionation. The C18 device was activated with methanol and 80 % (v/v) ACN, 0.5% (v/v) acetic acid, followed by 1% (v/v) FA to wash off the excess methanol solution. The lyophilized samples were diluted with 1% (v/v) FA and loaded onto a C18 apparatus. The C18 device was washed with 1% (v/v) FA solution and 5 mM ammonium formate, respectively. The collection bottles were then replaced with the collection devices with (3%, 5%, 7%, 9%, 11%, 13%, 15 %, 17%, 19%, 21%, 23%, 24%, 26%, 28%, 30%, 35%, 40%, 80%) ACN in 5 mM ammonium formate, pH 10. The effluent collected from 18 peptide fractions are (3%, 15%, 26%), (5%, 17%, 28%), (7%, 19%, 30%), (9%, 21%, 35%), (11%, 23%, 40%), (13%, 24%, 80%) ACN, which were then mixed together to get 6 fractions. Finally, the 6 fractions were lyophilized to dryness for MS analysis.
Each sample was diluted with 10 μL 0.1% (v/v) FA, and ultrasound-treated to improve solubility of peptide. The samples were centrifuged at 14000 rcf 4°C for 5 min, and 9 μL of each sample was taken and placed in the injection vial of the mass spectrometer, for analysis by mass spectrometry. Samples were analyzed by LC–MS/MS (LC: Thermo Fisher Scientific U3000 HPLC system; MS: Thermo Fisher Scientific Orbitrap Exploris™ 480). The analytical condition were LC: column: integrated spray tip (100 μm i.d. × 20 cm) packed with 1.9 μm/120 Å ReproSil-Pur C18 beads (Dr. Maisch GmbH); solvent system: solvent A (0.1% formic acid in water) and solvent B (80% ACN, 0.1 % formic acid in water); gradient program: A 135 min gradient separation was configured as: 4%–8% buffer B in 2 min, 8%–25% buffer B for 105 min, 25%–40% buffer B for 15 min, 40%–100% buffer B in 6 min, followed by a 7 min wash with 1% buffer B; flow rate: 0.5 μL min−1 at 0–123 min, 0.7 μL min−1 at 123–128 min, 0.7 μL min−1 at 128–135. MS detection: scan mode: DDA (data-dependent acquisition); resolution MS1 scan resolution was 60,000 and MS/MS scan resolution was 30000 with turbo-TMT option. Full scan and top 50 MS/MS scans were acquired per cycle; Scan range: 350(m/z)-1200(m/z); Maximum Injection Time: 45ms; Exclusion duration: 45s; Isolation window: 0.7(m/z); HCD Collision Energy: 36%.
Obtained raw data files were analyzed with Proteome Discoverer (PD, version 2.4, Thermo Fisher Scientific). Software parameter setting: Human FASTA database from UniProt (reviewed database entries, downloaded on March 22, 2021); Mass tolerances for precursor was set at 20 ppm; fragment ions: 0.6 Da; Maximum missed cleavage for trypsin digestion: 2 missed cleavage; Methionine oxidation, asparagine and glutamine deamidation were set as variable modification and carbamoylmethylation was set as fixed modification. FDR control is set to 1% at the PSM and peptide levels. The co-isolation threshold for report ion quantification is set to 50%.
Statistical analysis with LASSO algorithm
Let be the vector for intensity of a detected protein across samples obtained from MS analysis and as the vector of relative abundances of a given protein obtained by dividing the intensity value of the protein in each sample by the maximum value in . Given as the sensing matrix for samples and drugs, let as the coefficient vector for drugs, and is the residual vector for samples such that
| (2) |
Accordingly,
| (3) |
Since is a matrix with binary values, if a drug contributes to the increase in , the corresponding value will be positive. Otherwise, the value will be close to zero. In order to solve , the non-negative LASSO algorithm is used with the equation as follow:
| (4) |
where and are the index of matrix , i.e., the index of samples and drugs, respectively; is the penalty which serves to shrink coefficients reducing the coefficients of some drugs to zero, where larger forces more coefficients to zero. In other words, the non-negative LASSO algorithm selects highly-correlated coefficients and compress the rest to zero, removing coefficients of drugs not related to the abundance of protein. It conforms to the theoretical requirement and practical situation of MAPS-iTSA experiments where only a maximum of one or two drugs could interact with each protein.
A series of increasing values are selected to gradually reduce the coefficient of each drug. The progression of these values is known as the LASSO path. As the LASSO path progresses, the coefficient for each drug will eventually decrease to zero. For each protein, we select value in the LASSO path giving non-zero coefficients, and the corresponding coefficient for each drug is defined as the LASSO score for the protein (Fig. S1c):
| (5) |
where is the index of the drug. The is optimized to 3 based on the number and true positive rate of identified targets of staurosporine in HegG2 cell line (Fig. S1d). The LASSO score threshold of significant is optimized to 0.15 based on the same criterion (Fig. S1e). These two parameters can be adjusted according to the need for different experiments for a balance of precision and recall rates. Each protein detected in our MS analysis will have a score from the LASSO analysis. For each drug, all the proteins can be ranked by their LASSO scores of that drug where high-scoring proteins are potential targets.
Western blot
We performed isothermal dose-response (ITDR) CETSA in HCT116, 293T and K562. BRAF (bafetinib), CSNK2A2(abemaciclib) and GAPDH proteins were detected by Western blot. 10 drug concentrations were prepared with 4-fold serial dilution (starting from 200 μM) that were mixed with equal volume of cell lysate for 10 minutes prior heating. Subsequently, the samples underwent heating at specific temperatures: 52°C for 3 minutes in a PCR machine for the BRAF-bafetinib group, and 50°C for 3 minutes in a PCR machine for the CSNK2A2-abemaciclib group. Following the heating step, the samples were rapidly cooled to 4°C. The concentration of supernatant was determined using BCA protein quantitation kit (Thermo Scientific, USA). Equal volumes of protein were subjected to 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (PVDF) (Millipore, USA). After being blocked in 5% (W/V) BSA dissolved by 1xTBST buffer at room temperature for 2h, the membranes were incubated with BRAF, CSNK2A2 and GAPDH antibody at 4°C overnight, and the corresponding horseradish peroxidase-conjugated secondary antibodies were incubated at 4°C for 2h. Finally, western blot analysis was performed by enhanced chemiluminescence (ECL) (BIO-RAD, USA).
Molecular docking and dynamics simulation
All protein structure files were obtained from Protein Data Bank (PDB), including LYN (PDB ID: 5XY1), BRAF (6XFP), CDK2 (1B39), CDK4 (2W96), CDK6 (5L2S), CSNK2A2 (6QY9), and DHFR (4KEB). The missing loops of CDK2, CDK4, CDK6 and LYN were modeled using Modeller v9.2435. The structure files of bafetinib and abemaciclib were obtained from PDB entries 2E2B and 5L2S, respectively. The protein-ligand complex structure of positive controls for each ligand (LYN for bafetinib, CDK4 and CDK6 for abemaciclib) were prepared by aligning the proteins to their homologous proteins in PDB entries 2E2B and 5L2S. Other protein-ligand complex structures were obtained through unbiased molecular docking with the entire protein contained in the search box using AutoDock Vina v1.2.336. The search exhaustiveness was set to 10,000 to sufficiently sample ligand binding modes. The binding modes with the highest score were selected for MD simulations.
All MD simulations and preparations were conducted using the Amber 21 program suite37. The ff19SB force field was used for protein parameterizations38. The parameters of ligands were prepared using the AM1-BCC method39 and the gaff2 force field40. Missing parameters were obtained using the Amber/parmchk2 program. Topology and coordinate files for the protein-ligand complexes were prepared using the Amber/tleap program, with protein-ligand complexes solvated in an octahedral box of OPC water molecules41 with thickness extending 8 Å from the protein surface. The complexes were neutralized by adding Na+ or Cl− counter ions.
The Amber/pmemd.cuda program was used for all MD simulations42. A 10 Å cutoff was used for nonbonded interactions and short-range electrostatic corrections. Long-range electrostatic interactions were handled by the particle mesh Ewald (PME) method. The hydrogen atom 43 bond lengths were fixed with the SHAKE algorithm. Minimization was performed in two steps to relieve any possible atomic overlaps. The first step involved relaxing only water molecules, while the second step minimized the whole system. Langevin dynamics with a 1 ps−1 collision frequency were used to gradually increase the system temperature from 0 to 300 K over 200 ps. The systems were first equilibrated for 100 ns under constant pressure and temperature (NPT) to adjust the system density; then, an additional 100 ns simulations were performed under constant volume and temperature (NVT) conditions. Each simulation was repeated three times with a different random seed, starting from identical minimized structures. A 2-fs integration time step was utilized with trajectory snapshots extracted every 1 ns.
All simulation trajectories were visualized using the MD Movie feature of UCSF Chimera v1.1444. Protein and ligand RMSD with reference to the minimized structure were calculated using the rmsd command of the Amber/cpptraj program45. A modified version stability score based on the score used in previous studies was applied to determine the binding stability of the protein-ligand pairs during simulation46. The equilibrated contact pairs are defined as the heavy atom pairs within protein and ligand that are within the distance of 7 Å in the average structure of the last 150 ns of the simulation trajectory, which are obtained using the nativecontacts command of the Amber/cpptraj program. The stability score of the ith frame SSi is the fraction of the total amount of equilibrated contact pairs that remain within 7 Å distances.
The Amber/MMPBSA.py program was used to calculate the binding free energies (ΔG) of protein-ligand pairs47. MMPBSA calculations were conducted on the last 150 ns of each simulation trajectory. The ionic strength was set at 0.100 M. Because both bafetinib and abemaciclib are highly charged molecules, the internal dielectric constant was set to 4, which is suitable for charged protein-ligand complex systems. Entropy was not taken into consideration in MMPBSA calculations.
NanoBRET target engagment assay
The NanoBRET target engagement assay was performed according to the manufacturer’s instructions 48. In brief, HEK293 cells were transfected with C-terminally tagged CSNK2A2 NanoLuc fusion vector (#NV1191, Promega) and BRAF NanoLuc fusion vectors (created by the Company). After 24 h cells were counted and diluted to 2×105 cells/mL in assay medium (Optimem + 4 % FBS). K-05 kinase tracer (#N2482, Promega) for CSNK2A2, K-10 kinase tracer for BRAF were added to cells in a final concentration of 1 μM before 40 μL/well were added to a 384 well plate containing pre-plated compounds in triplicates. After 2 h incubation 20 μL/well of substrate and extracellular NanoLuc inhibitor mix (1:166, 1:500 in Optimem) were added. Donor (460 nm) and acceptor (610 nm) signals were measured after 10 min incubation at room temperature with a PheraSTAR FSX plate reader. Data were analysed by calculating the ratio of acceptor to donor signal and subtracting the background (transfected cells w/o tracer). Data were then normalised to DMSO and fitted with a three-parameter nonlinear regression model in Prism.
Kinase hotspot assay
Reaction Biology Corporation performed BRAF and CNS2K2A2 kinase panels using their “HotSpot” assay platform 49. A reaction buffer containing specific kinase/substrate pairs and cofactors was prepared in 20 mM Hepes pH 7.5, 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/ml BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO. (For detailed information about individual components of the kinase reaction, see Supplementary Table). Compounds were delivered into the reaction, followed ~20 min later by addition of a mixture of ATP (MCE) and 33P ATP (PerkinElmer) to a final concentration of 10 μM. The reactions were carried out at 25°C for 120 minutes, following which they were spotted onto P81 ion exchange filter paper (Whatman). Filters were extensively washed in 0.75% phosphoric acid to remove unbound phosphate. Using background data subtracted from inactive control reactions, the kinase activity was calculated based on the percent remaining kinase activity in the test samples and the dimethyl sulfoxide reaction as a control.
| CAT# | HGNC | Substrates | Compounds |
|---|---|---|---|
| BRAF | BRAF | MEK1 (K97R), 3 μM ATP 10 μM | Bafetinib |
| CK2a2 | CSNK2A2 | Peptide substrate, [RRRDDDSDDD], 20 μMATP 10 μM | Abemaciclib |
Drug grouping for sensing matrix generation
The advantage of MAPS-iTSA is reduced time and cost to increase the throughput of drug target deconvolution experiment. However, multiple drugs within a TMT experiment targeting same protein could decrease the sensitivity of the strategy. One solution is distributed for drugs that potentially share similar target into different experimental batches or TMT experiments. Hereby, we introduce an enhanced pooled strategy using in-silico drug target prediction algorithm to further optimize grouping of drugs (Fig. S5a). The results indicate this strategy can reduce the co-targets of drugs within the same group (Fig. S5b).
In this work, we only tested 15 drugs so this procedure is not involved. But it would be useful if anyone applies MAPS-iTSA on more drugs in the future. MAPS-iTSA/iTSA uses a similarity-based method for predicting the potential targets of the test drugs, which is based on the observation that structurally similar bioactive molecules are more likely to share similar targets. The drug target interaction data from DrugBank is used as reference. The prediction methods are as follow: First, Morgan fingerprints of the test drugs and the other drugs recorded in DrugBank are calculated with RDKit package. Then, the structural related drugs of each testing drug are retrieved from the database. The similarity is calculated by the dice function:
| (6) |
where and are the molecular fingerprints of two drugs. The threshold is set as 0.6. Finally, all the targets of the structurally related drugs are treated as the potential targets of the test drug (Fig. S5a). This relaxed criterion aims to reduce the false negative error, because false positive error has less influence on the drug grouping.
MAPS-iTSA/iTSA uses the predicted targets of the test drugs to group them into different experimental batches. The goal of the optimization is to minimize the probability that the drugs in a group share the same targets. Here we define the optimizing function as equation 3
| (7) |
| (8) |
where is a set of drugs in the group, is the number of drugs in each group, is the set of predicted targets of the drug in the group, and is the Jaccard function defined by:
| (9) |
Genetic algorithm is used for achieving this goal. Suppose that the number of drugs is , and the number of experimental batches is . First, the drugs tested are randomly permuted. Second, the drugs located at are assigned to group 1, and the drugs located at to are assigned to group 2, and so on. Then, is calculated by the equation 7. Finally, minor adjustments are given to the drug permutation for obtaining a better . The iteration is proceeding until a local minimum is achieved. The final grouping manner should be used for MAPS-iTSA/iTSA workflow (Fig. S5c).
We tested this drug grouping strategy with 180 randomly picked drugs from DrugBank database. Their targets are predicted by the proposed method with leave-one-out test. Then, the drugs were grouped into 12 groups optimally, and each group consists of 15 drugs where a 9×15 sensing matrix is applied. The optimized curve is shown in Fig. S5d. We calculated the number of the co-targets in each group with the true targeted recorded in the database, and compare it with 1,000 times randomly placing of the drugs. Fig. S5b is the distribution plot of the number of the total co-targets of the randomly placing, and the red line indicates the optimized permutation, which is less than most of the randomized permutation. Considering the optimization was based on the predicted targets instead of the true targets, the grouping strategy is effective for reducing the co-targets.
Extended feasibility analysis with TMT16 and TMT18
We present the MAPS-iTSA scheme for 9 pools×15 drugs as example in this work. However, this strategy should also feasible for deconvolving the targets of more drugs with TMT16 and TMT18. Therefore, it would be helpful to estimate the theoretical maximum number of drugs that could be deconvolved by MAPS.
The core restricted criterion of the number of test drugs is: How many pools of a drug shared with another drug. For example, drug A in the sensing matrix is put into three pools. If drug B shares one of the pools with t drug A, it may cause very small effect on target deconvolution of both drugs. If drug B share two of the pools with drug A, it increases the false positive rate. If drug B shares all the three pools with drug A, target deconvolution will be impossible. Therefore, we calculate the average shared pools and the maximum shared pools between each pair of drugs, along with the number of test drugs. Since the sensing matrix generation is based on GA, a algorithm with stochasticity, each point of the scatter is the average value with 10-replicates computation. From the scatter plots, the maximum shared pools of 9-pools-15-drugs is around 1.75. From our results, MAPS-iTSA works well in this condition. Taking this criterion, the number of test drugs of TMT-16 should be greater than 32, and the number of test drugs of TMT-18 should be greater than 40 (Fig. S5e). However, the inference will be confirmed with experimental verification in the future.
QUANTIFICATION AND STATISTICAL ANALYSIS
All analyses were performed using python. All results can be reproduced with the provided data and scripts. All relevant statistical details are included in the figure captions, and text. Additional details for each experiment type are included in the Method Details section of the STAR Methods.
Supplementary Material
Highlights:
MAPS strategy boosts throughput of derivatization-free target deconvolution by 15x.
MAPS profiling across multiple cell lines reveals cell-specific drug targets.
BRAF and CSNK2A2 were validated as target of bafetinib and abemaciclib respectively.
SIGNIFICANCE.
Derivatization-free drug target deconvolution methods generally require more MS instrumentation time to survey the entire proteome compared to both affinity-based and activity-based techniques that enrich proteins for MS analysis. Approaches that avoid time-consuming derivatization, applicable to most chemicals while being lean on MS instrument time could enable large-scale target deconvolution studies. To circumvent the current high demand for MS instrument time of derivatization-free techniques, we (1) proposed an unprecedented strategy in derivatization-free proteome-wide methods to increase their throughput, (2) developed the necessary enabling algorithms and (3) validated the concept experimentally with thermal proteome profiling of 15 drugs simultaneously across 5 cell lines. Our current setup has a 60x throughput compared to classical TPP and a 15x throughput compared to the current fastest format reported while maintaining similar performance. We subsequently performed cell type specific target deconvolution of 15 drugs with MAPS-based TPP assay. Our results revealed drug-target interactions can differ vastly across different cell lines in term of proteins and binding affinity. Furthermore, we uncovered potential off-targets for five drugs previously uncharacterized by TPP and validated the off-targets for two of them. This work presents the largest target profiling of structurally diverse drugs across multiple cell lines to date.
ACKNOWLEDGMENTS
This work is financially supported by grants from Shenzhen Innovation of Science and Technology Commission Grant (Nos. JCY20200109140814408), National Natural Science Foundation of China (Nos. 22074060, 22103036, 22150610470), National Institutes of Health (No. GM130367) and the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement No 875510. The JU receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA and Ontario Institute for Cancer Research, Royal Institution for the Advancement of Learning McGill University, Kungliga Tekniska Hoegskolan, Diamond Light Source Limited.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Harrison RK (2016). Phase II and phase III failures: 2013–2015. Nat Rev Drug Discov 15, 817–818. 10.1038/nrd.2016.184. [DOI] [PubMed] [Google Scholar]
- 2.Vasta J, Michaud A, Zimprich C, Thomas M, Wilkinson J, Crapster A, and Robers M (2022). Protomer Selectivity of RAF Inhibitors Within the RAS/RAF Signalosome. 10.21203/rs.3.rs-2175742/v1. [DOI] [PubMed] [Google Scholar]
- 3.Handley A, Schauer T, Ladurner AG, and Margulies CE (2015). Designing Cell-Type-Specific Genome-wide Experiments. Mol Cell 58, 621–631. 10.1016/j.molcel.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 4.Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, Wang J, Wu RP, Gomez F, Loo JA, et al. (2009). Target identification using drug affinity responsive target stability (DARTS). Proceedings of the National Academy of Sciences 106, 21984–21989. 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng Y, De Franceschi G, Kahraman A, Soste M, Melnik A, Boersema PJ, de Laureto PP, Nikolaev Y, Oliveira AP, and Picotti P (2014). Global analysis of protein structural changes in complex proteomes. Nat Biotechnol 32, 1036–1044. 10.1038/nbt.2999. [DOI] [PubMed] [Google Scholar]
- 6.Schopper S, Kahraman A, Leuenberger P, Feng Y, Piazza I, Müller O, Boersema PJ, and Picotti P (2017). Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nat Protoc 12, 2391–2410. 10.1038/nprot.2017.100. [DOI] [PubMed] [Google Scholar]
- 7.West GM, Tang L, and Fitzgerald MC (2008). Thermodynamic Analysis of Protein Stability and Ligand Binding Using a Chemical Modification- and Mass Spectrometry-Based Strategy. Anal Chem 80, 4175–4185. 10.1021/ac702610a. [DOI] [PubMed] [Google Scholar]
- 8.Molina DM, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, and Nordlund P (2013). Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science (1979) 341, 84–87. 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 9.Savitski MM, Reinhard FBM, Franken H, Werner T, Savitski MF, Eberhard D, Molina DM, Jafari R, Dovega RB, Klaeger S, et al. (2014). Tracking cancer drugs in living cells by thermal profiling of the proteome. Science (1979) 346, 1255784. 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- 10.Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundbäck T, Nordlund P, and Molina DM (2014). The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 9, 2100–2122. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- 11.Petrilli WL, Adam GC, Erdmann RS, Abeywickrema P, Agnani V, Ai X, Baysarowich J, Byrne N, Caldwell JP, Chang W, et al. (2020). From Screening to Targeted Degradation: Strategies for the Discovery and Optimization of Small Molecule Ligands for PCSK9. Cell Chem Biol 27, 32–40.e3. 10.1016/j.chembiol.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Kalxdorf M, Günthner I, Becher I, Kurzawa N, Knecht S, Savitski MM, Eberl HC, and Bantscheff M (2021). Cell surface thermal proteome profiling tracks perturbations and drug targets on the plasma membrane. Nat Methods 18, 84–91. 10.1038/s41592-020-01022-1. [DOI] [PubMed] [Google Scholar]
- 13.Perrin J, Werner T, Kurzawa N, Rutkowska A, Childs DD, Kalxdorf M, Poeckel D, Stonehouse E, Strohmer K, Heller B, et al. (2020). Identifying drug targets in tissues and whole blood with thermal-shift profiling. Nat Biotechnol 38, 303–308. 10.1038/s41587-019-0388-4. [DOI] [PubMed] [Google Scholar]
- 14.Tan CSH, Go KD, Bisteau X, Dai L, Yong CH, Prabhu N, Ozturk MB, Lim YT, Sreekumar L, Lengqvist J, et al. (2018). Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science (1979) 359, 1170–1177. 10.1126/science.aan0346. [DOI] [PubMed] [Google Scholar]
- 15.Becher I, Werner T, Doce C, Zaal EA, Tögel I, Khan CA, Rueger A, Muelbaier M, Salzer E, Berkers CR, et al. (2016). Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat Chem Biol 12, 908–910. 10.1038/nchembio.2185. [DOI] [PubMed] [Google Scholar]
- 16.Kurzawa N, Becher I, Sridharan S, Franken H, Mateus A, Anders S, Bantscheff M, Huber W, and Savitski MM (2020). A computational method for detection of ligand-binding proteins from dose range thermal proteome profiles. Nat Commun 11, 1–8. 10.1038/s41467-020-19529-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaetani M, Sabatier P, Saei AA, Beusch CM, Yang Z, Lundström SL, and Zubarev RA (2019). Proteome Integral Solubility Alteration: A High-Throughput Proteomics Assay for Target Deconvolution. J Proteome Res 18, 4027–4037. 10.1021/acs.jproteome.9b00500. [DOI] [PubMed] [Google Scholar]
- 18.Ball KA, Webb KJ, Coleman SJ, Cozzolino KA, Jacobsen J, Jones KR, Stowell MHB, and Old WM (2020). An isothermal shift assay for proteome scale drug-target identification. Commun Biol 3, 75. 10.1038/s42003-020-0795-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mallapaty S (2020). The mathematical strategy that could transform coronavirus testing. Nature 583, 504–505. 10.1038/d41586-020-02053-6. [DOI] [PubMed] [Google Scholar]
- 20.Mutesa L, Ndishimye P, Butera Y, Souopgui J, Uwineza A, Rutayisire R, Ndoricimpaye EL, Musoni E, Rujeni N, Nyatanyi T, et al. (2021). A pooled testing strategy for identifying SARS-CoV-2 at low prevalence. Nature 589, 276–280. 10.1038/s41586-020-2885-5. [DOI] [PubMed] [Google Scholar]
- 21.Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, Humeidi R, Peck D, Wu X, Tang AA, et al. (2020). Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer 1, 235–248. 10.1038/s43018-019-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin A, Giuliano CJ, Palladino A, John KM, Abramowicz C, Yuan M. Lou, Sausville EL, Lukow DA, Liu L, Chait AR, et al. (2019). Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med 11, eaaw8412. 10.1126/scitranslmed.aaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao Y, Chen P, Ke M, Chen X, Ji S, Chen W, and Tian R (2021). Fully Integrated and Multiplexed Sample Preparation Technology for Sensitive Interactome Profiling. Anal Chem 93, 3026–3034. 10.1021/acs.analchem.0c05076. [DOI] [PubMed] [Google Scholar]
- 24.Chen W, Wang S, Adhikari S, Deng Z, Wang L, Chen L, Ke M, Yang P, and Tian R (2016). Simple and Integrated Spintip-Based Technology Applied for Deep Proteome Profiling. Anal Chem 88, 4864–4871. 10.1021/acs.analchem.6b00631. [DOI] [PubMed] [Google Scholar]
- 25.Kadia T, Delioukina ML, Kantarjian HM, Keating MJ, Wierda WG, Burger JA, Wieland S, and Levitt D (2011). A Pilot Phase II Study of the Lyn Kinase Inhibitor Bafetinib in Patients with Relapsed or Refractory B Cell Chronic Lymphocytic Leukemia. Blood 118, 2858–2858. 10.1182/blood.V118.21.2858.2858. [DOI] [Google Scholar]
- 26.Yen I, Shanahan F, Lee J, Hong YS, Shin SJ, Moore AR, Sudhamsu J, Chang MT, Bae I, Dela Cruz D, et al. (2021). ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature 594, 418–423. 10.1038/s41586-021-03515-1. [DOI] [PubMed] [Google Scholar]
- 27.Wang C, Nguyen PH, Pham K, Huynh D, Le T-BN, Wang H, Ren P, and Luo R (2016). Calculating protein-ligand binding affinities with MMPBSA: Method and error analysis. J Comput Chem 37, 2436–2446. 10.1002/jcc.24467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chong Q-Y, Kok Z-H, Bui N-L-C, Xiang X, Wong AL-A, Yong W-P, Sethi G, Lobie PE, Wang L, and Goh B-C (2020). A unique CDK4/6 inhibitor: Current and future therapeutic strategies of abemaciclib. Pharmacol Res 156, 104686. 10.1016/j.phrs.2020.104686. [DOI] [PubMed] [Google Scholar]
- 29.Cousins EM, Goldfarb D, Yan F, Roques J, Darr D, Johnson GL, and Major MB (2018). Competitive Kinase Enrichment Proteomics Reveals that Abemaciclib Inhibits GSK3β and Activates WNT Signaling. Molecular Cancer Research 16, 333–344. 10.1158/1541-7786.MCR-17-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hafner M, Mills CE, Subramanian K, Chen C, Chung M, Boswell SA, Everley RA, Liu C, Walmsley CS, Juric D, et al. (2019). Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem Biol 26, 1067–1080.e8. 10.1016/j.chembiol.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma J, Chen T, Wu S, Yang C, Bai M, Shu K, Li K, Zhang G, Jin Z, He F, et al. (2019). iProX: an integrated proteome resource. Nucleic Acids Res 47, D1211–D1217. 10.1093/nar/gky869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huber KVM, Olek KM, Müller AC, Tan CSH, Bennett KL, Colinge J, and Superti-Furga G (2015). Proteome-wide drug and metabolite interaction mapping by thermal-stability profiling. Nat Methods 12, 1055–1057. 10.1038/nmeth.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen T, Ma J, Liu Y, Chen Z, Xiao N, Lu Y, Fu Y, Yang C, Li M, Wu S, et al. (2022). iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res 50, D1522–D1527. 10.1093/nar/gkab1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xue L, Lin L, Zhou W, Chen W, Tang J, Sun X, Huang P, and Tian R (2018). Mixed-mode ion exchange-based integrated proteomics technology for fast and deep plasma proteome profiling. J Chromatogr A 1564, 76–84. 10.1016/j.chroma.2018.06.020. [DOI] [PubMed] [Google Scholar]
- 35.Webb B, and Sali A (2016). Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics 54. 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trott O, and Olson AJ (2009). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem, NA-NA. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, and Woods RJ (2005). The Amber biomolecular simulation programs. J Comput Chem 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian C, Kasavajhala K, Belfon KAA, Raguette L, Huang H, Migues AN, Bickel J, Wang Y, Pincay J, Wu Q, et al. (2020). ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J Chem Theory Comput 16, 528–552. 10.1021/acs.jctc.9b00591. [DOI] [PubMed] [Google Scholar]
- 39.Jakalian A, Bush BL, Jack DB, and Bayly CI (2000). Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J Comput Chem 21, 132–146. . [DOI] [PubMed] [Google Scholar]
- 40.Wang J, Wolf RM, Caldwell JW, Kollman PA, and Case DA (2004). Development and testing of a general amber force field. J Comput Chem 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 41.Izadi S, Anandakrishnan R, and Onufriev AV (2014). Building Water Models: A Different Approach. J Phys Chem Lett 5, 3863–3871. 10.1021/jz501780a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Grand S, Götz AW, and Walker RC (2013). SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput Phys Commun 184, 374–380. 10.1016/j.cpc.2012.09.022. [DOI] [Google Scholar]
- 43.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera?A visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 45.Roe DR, and Cheatham TE (2013). PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 46.Zhao S, Ni F, Qiu T, Wolff J, Tsai S-C, and Luo R (2020). Molecular Basis for Polyketide Ketoreductase–Substrate Interactions. Int J Mol Sci 21, 7562. 10.3390/ijms21207562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, and Roitberg AE (2012). MMPBSA.py : An Efficient Program for End-State Free Energy Calculations. J Chem Theory Comput 8, 3314–3321. 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 48.Robers MB, Dart ML, Woodroofe CC, Zimprich CA, Kirkland TA, Machleidt T, Kupcho KR, Levin S, Hartnett JR, Zimmerman K, et al. (2015). Target engagement and drug residence time can be observed in living cells with BRET. Nat Commun 6, 10091. 10.1038/ncomms10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anastassiadis T, Deacon SW, Devarajan K, Ma H, and Peterson JR (2011). Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 29, 1039–1045. 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the main text or the supplementary materials. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX31,33 partner repository with the dataset identifiers PXD043882.
The python script code used to perform the analyses is accessible as a GitHub repository at https://github.com/hcji/MAPS-iTSA.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.