

Abstract
Virilizing ovarian tumors are rare but a clinically important diagnosis in a patient presenting with hyperandrogenism. Workup of hyperandrogenism is challenging with a broad range of differentials, including adrenal and ovarian pathology, tumoral or nontumoral in nature. Baseline follicular-phase 17-hydroxyprogesterone (17OHP) measurement is part of the investigation algorithm, and elevated levels are often associated with nonclassic congenital adrenal hyperplasia (NCCAH), which can have its first presentation in adolescence or adulthood. This case describes a young adult woman of reproductive age presenting with menstrual irregularity, raised testosterone, and 17OHP. After extensive workup and serial follow-up, she was found to have a Sertoli-Leydig cell tumor of the left ovary and underwent successful laparoscopic salpingo-oophorectomy with normalization of her menstrual irregularity and biochemical resolution of her testosterone and 17OHP levels.
Keywords: Sertoli-Leydig cell tumor, oligomenorrhea, hyperandrogenism, 17OH progesterone, testosterone, nonclassic CAH
Introduction
Approaching a patient with hyperandrogenism is a common diagnostic case encountered in endocrinology practice. Initial evaluation includes baseline hormonal evaluation with gonadotrophins, estradiol, testosterone, dehydroepiandrosterone sulfate (DHEAS), 17-hydroxyprogesterone (17OHP), prolactin, and thyroid function tests. Differentials are broad, including adrenal and ovarian pathology. Identifying the source of androgen excess can often be challenging due to significant clinical and biochemical overlap in presentation. Marked elevation in 17OHP is usually suggestive of congenital adrenal hyperplasia (CAH) from 21-hydroxylase deficiency. A significantly elevated testosterone level greater than or equal to 6.9 nmol/L (199.0 ng/dL) [1] or signs of overt virilization, especially that of rapid onset, would warrant urgent investigation for a neoplastic cause. Localization also relies on imaging, which is associated with its own pitfalls such as the presence of adrenal incidentalomas that may confound the diagnosis, and “false-negative” ovarian imaging due to occult ovarian tumors or the operator-dependent nature of certain imaging modalities.
Case Presentation
A 22-year-old Chinese woman was referred from the gynecologist for evaluation of oligomenorrhea with elevated serum testosterone and 17OHP. Menarche occurred at age 12 years. Her previous menstrual pattern was regular cycles every 30 days, of 7 days’ duration. For the past year prior to presentation, she had spotting of small amounts 2 to 3 times a week continuously.
She did not notice an increase in hair growth. From a young age, her voice had always been deeper compared to her peers. She did not experience weight gain, thin skin, easy bruising, or proximal weakness. She did not have symptoms of hyperthyroidism or hypothyroidism.
She had no significant medical history in childhood and adolescence.
She is the youngest in the family, with 3 older siblings who are well. There was no family history of consanguinity, abnormal uterine bleeding, or early menopause. Her paternal grandmother and father have hyperthyroidism, and her mother has Sjögren syndrome. There was no family history of adrenal tumors or CAH.
She was not sexually active. She denied taking long-term medications including traditional medications, supplements, and topical or oral testosterone products.
Her blood pressure was 128/70 mm Hg with no postural drop. She weighed 50.6 kg and was 1.67 m tall with a body mass index of 18.1. Her Ferriman-Gallwey score was 0. She had mild facial acne and a slightly deeper voice than the average female. She did not appear cushingoid. Her abdomen was soft, with no palpable masses. External genitalia was unambiguously female, but there was mild clitoral enlargement measuring 10 mm × 5 mm. She did not have frontal balding, hair growth in androgen-dependent areas, or breast atrophy. There was no acanthosis nigricans. There was no goiter and she was clinically euthyroid. Examination of the cardiac, respiratory, and neurological system was normal.
Initial investigations by the referring gynecologist revealed an elevated total testosterone level of 5.35 nmol/L (154.3 ng/dL) and 17OHP of 18.9 nmol/L (623.7 ng/dL). A transabdominal pelvic ultrasound showed a normal uterus with an endometrial thickness of 6.2 mm. The left ovary had a clear cystic space measuring 18.2 × 16 mm with volume of 21.6 cm3. The right ovary was morphologically normal with a volume of 6 cm3 (Fig. 1). The initial impression of the left ovarian cyst was that of a simple physiological cyst.
Figure 1.

Transabdominal ultrasound of the uterus and ovaries.
Diagnostic Assessment
Full blood count, renal function, liver function, and thyroid function were normal. Aldosterone was 242 pmol/L (8.72 ng/dL) with plasma renin activity of 3.1 ng/mL/h. DHEAS was normal at 6.5 μmol/L (247 μg/dL). Androstenedione was elevated at 8.6 nmol/L (246 ng/dL). Serum testosterone levels remained persistently elevated over time, even when analyzed on an alternative platform (Table 1). Free testosterone via equilibrium dialysis was more than 6 times above the upper limit of normal.
Table 1.
Trend of testosterone levels over time across various platforms
| November 2020 | December 2020 | February 2021 | May 2021 | Reference range | |
|---|---|---|---|---|---|
|
Testosterone
(Our institution, chemiluminescent microparticle immunoassay, Abbott platform) |
8.43 nmol/L (243.1 ng/dL) | 5.35 nmol/L (154.3 ng/dL) | 8.31 nmol/L (239.7 ng/dL) | 5.27 nmol/L (152 ng/dL) | 0.48-1.85 nmol/L (13.8-53.4 ng/dL) |
| Serum total testosterone (Mayo Clinic, LC-MS/MS) | 191 ng/dL (6.62 nmol/L) | 8-60 ng/dL (0.28-2.08 nmol/) | |||
| Serum free testosterone (Mayo Clinic, equilibrium dialysis) | 6.88 ng/dL (0.239 nmol/L) | 0.06-1.06 ng/dL (0.0021-0.037 nmol/L) |
Abbreviation: LC-MS/MS, liquid chromatography with tandem mass spectrometry.
After adrenocorticotropin (ACTH) stimulation (250 mcg of intravenous cosyntropin) (Table 2), 17OHP decreased from 18.7 nmol/L (617.1 ng/dL) at baseline, to 14.1 nmol/L (465.3 ng/dL) at 60 minutes. Peak ACTH-stimulated 17OHP value was lower than 31 nmol/L (1023 ng/dL) [2] and not in keeping with nonclassic CAH (NCCAH). 17OHP was evaluated using radioimmunoassay (Tecan MG12181) at our institution. Stimulated peak cortisol response of 469 nmol/L (17 μg/dL) was slightly suboptimal (using 500 nmol/L as the cutoff, analyzed using an Abbott Alinity chemiluminescent microparticle immunoassay). Baseline ACTH level was normal. As our patient had no features of adrenal insufficiency or exogenous steroid use, she was started on sick day hydrocortisone 10 mg every morning and 5 mg every evening when necessary.
Table 2.
17-Hydroxyprogesterone and cortisol levels after cosyntropin stimulation
| 0 min | 30 min | 60 min | Reference range | |
|---|---|---|---|---|
| 17OHP | 18.7 nmol/L (617.1 ng/dL) | 17.1 nmol/L (564.3 ng/dL) | 14.1 nmol/L (465.3 ng/dL) |
Basal:
Follicular phase: 0.3-3.3 nmol/L (9.91-109.05 ng/dL) Luteal phase: 2.9-15.2 nmol/L (95.83-502.30 ng/dL) Post-cosyntropin stimulation [2]: <30 nmol/L (991.38 ng/dL): likely unaffected or heterozygote >31-300 nmol/L (1024.43-9913.8 ng/dL): suggestive of NCCAH>300 nmol/L (9913.8 ng/dL): suggestive of classic CAH |
| Cortisol | 251 nmol/L (9.1 μg/dL) | 439 nmol/L (15.9 μg/dL) | 469 nmol/L (17 μg/dL) | |
| 3.1 pmol/L (14.1 pg/mL) | — | — | 0.0-10.2 pmol/L (0.0-46.32 pg/mL) |
Abbreviations: 17OHP, 17-hydroxyprogesterone; ACTH, adrenocorticotropin; CAH, congenital adrenal hyperplasia; NCCAH, nonclassic congenital adrenal hyperplasia.
Magnetic resonance imaging (MRI) scan of the adrenals was normal. There was no adrenal enlargement or tumors. With no convincing evidence of an adrenal androgen-secreting tumor, main differentials at this point included alternative adrenal pathology such as NCCAH and adrenal enzymatic deficiencies; or ovarian causes including virilizing ovarian tumors (VOTs), ovarian hyperthecosis, or polycystic ovary syndrome.
A progesterone challenge test was performed with 1 week of medroxyprogesterone 10 mg. This resulted in withdrawal bleeding on day 2 with corresponding laboratory tests and imaging performed the day of withdrawal bleeding (Table 3). Testosterone measured 2.88 nmol/L (83.1 ng/dL), significantly reduced from previous measurements of 5.27 to 8.31 nmol/L (152.0-239.68 ng/dL), while basal 17OHP was 10.7 nmol/L (353.1 ng/dL), compared to 18.7 to 18.9 nmol/L (617.96-624.57 ng/dL) prior. Repeat transabdominal pelvic ultrasound showed resolution of the simple left ovarian cyst.
Table 3.
Postprogesterone challenge investigations—day 2 menses following progesterone challenge test withdrawal bleed
| Investigation | Day 2 | Reference range | ||
|---|---|---|---|---|
| E2 | 112 pmol/L (30.5 pg/mL) | 83-423 pmol/L (22.61-115.22 pg/mL) | ||
| LH | 5.5 IU/L | 2.4-12.6 IU/L | ||
| FSH | 6.4 IU/L | 3.5-12.5 IU/L | ||
| Testosterone | 2.88 nmol/L (83.1 ng/dL) | 0.48-1.85 nmol/L (13.84-53.56 ng/dL) |
||
| Cosyntropin stimulation test (250 mcg IV cosyntropin) | 0 min | 30 min | 60 min | |
| 17OHP | 10.7 nmol/L (353.1 ng/dL) | 10.5 nmol/L (347.0 ng/dL) | 10.4 nmol/L (343.7 ng/dL) | Follicular phase: 0.3-3.3 nmol/L (9.91-109.05 ng/dL) Luteal phase: 2.9-15.2 nmol/L (95.83-502.30 ng/dL) |
| Cortisol | 396 nmol/L (14.4 μg/dL) | 464 nmol/L (16.2 μg/dL) | 552 nmol/L (20.0 μg/dL) | >500 nmol/L (18.23 μg/dL) is considered adequate |
Abbreviations: 17OHP, 17-hydroxyprogesterone; E2, estradiol; FSH, follicle-stimulating hormone; IV, intravenous; LH, luteinizing hormone.
Testosterone after low-dose dexamethasone suppression test (LDDST) (Table 4) remained persistently elevated at 8.28 nmol/L (238.8 ng/dL) at 48 hours, compared to 5.63 nmol/L (162.4 ng/dL) at 0 hours. Lack of suppression of testosterone after LDDST by more than 40% [3] was worrisome for a tumoral cause of testosterone excess.
Table 4.
Low-dose dexamethasone suppression test with 17-hydroxyprogesterone, cortisol, and testosterone levels at baseline and 48 hours
| 0 h | 48 h | Reference range | Interpretation | |
|---|---|---|---|---|
| 17OHP | 12.9 nmol/L (425.7 ng/dL) | 15.7 nmol/L (518.1 ng/dL) | Follicular phase: 0.3-3.3 nmol/L (9.91-109.05 ng/dL) Luteal phase: 2.9-15.2 nmol/L (95.83-502.30 ng/dL) |
No reduction in 17OHP |
| Cortisol | 326 nmol/L (11.8 μg/dL) | 15 nmol/L (0.54 μg/dL) | Normal response with appropriate suppression. No evidence of Cushing syndrome | |
| Testosterone | 5.63 nmol/L (162.4 ng/dL) | 8.28 nmol/L (238.8 ng/dL) | No reduction in testosterone, which is concerning for a tumoral source of testosterone excess |
Abbreviation: 17OHP, 17-hydroxyprogesterone.
Rare forms of NCCAH such as 11-β hydroxylase deficiency were also considered due to the raised 17OHP and initial suboptimal cortisol response to cosyntropin stimulation. As urine steroid profile was not readily available locally and regionally, a serum steroid profile (Table 5) analyzed via liquid chromatography with tandem mass spectrometry (LC-MS/MS) was sent off. This ruled out the diagnosis with normal levels of steroid precursors and metabolites.
Table 5.
Serum steroid profile showing normal levels of steroid precursors and metabolites
| Analyte | Result | Reference range | Comment |
|---|---|---|---|
| 17OHP | 228 ng/dL (6.9 nmol/L) | Follicular <80 ng/dL (2.42 nmol/L) Luteal <285 ng/dL (8.62 nmol/L) |
Unable to ascertain whether patient in follicular or luteal phase due to oligomenorrhea |
| Deoxycorticosterone | Below detection limit | CAH due to 17-α hydroxylase deficiency unlikely | |
| 11-Deoxycortisol | 31 ng/dL | <10-79 ng/dL | CAH due to 11-β hydroxylase unlikely |
| 21-Deoxycortisol | Below detection limit | <5 ng/dL, applies to all age groups | CAH due to 21-hydroxylase deficiency unlikely |
Abbreviations: 17OHP, 17-hydroxyprogesterone; CAH, congenital adrenal hyperplasia.
Repeat cosyntropin stimulation test showed adequate peak cortisol response (see Table 3) with no evidence of adrenal insufficiency. She had not required any stress-dose hydrocortisone.
With the LDDST suggesting tumoral origin, a clearly normal MRI of the adrenals, and suppression of testosterone with progesterone, we strongly suspected an ovarian origin despite the 2 previous normal ultrasounds. MRI of the pelvis showed an enlarged left ovary with a heterogeneous T2-weighted, hypointense, lobulated component measuring 3.0 × 1.9 cm (Fig. 2). Given the clinical context, this lesion was likely to be the offending androgen-secreting tumor.
Figure 2.
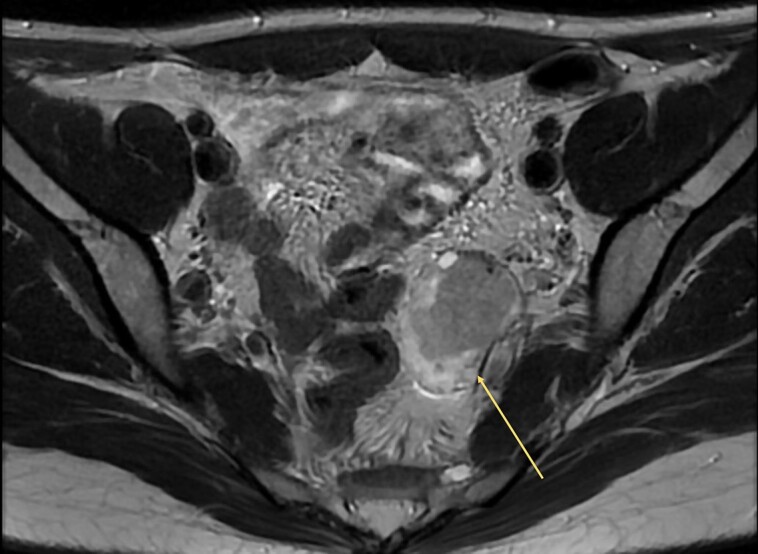
Magnetic resonance imaging of the pelvis (arrow showing a Sertoli-Leydig cell tumor within the left ovary).
A transrectal ultrasound performed preoperatively also confirmed the 3.2 × 2.0 × 2.2-cm hypoechoic solid mass with rich vascularity, which likely represented a Sertoli-Leydig cell tumor (SLCT).
Staging 2-[18F]fluoro-2-deoxyglucose (FDG) positron emission tomography computed tomography scan did not show any suspicious FDG-avid foci or nodal or distant metastases, apart from the minimally FDG-avid left ovarian mass with low metabolic activity, suggestive of lower aggressive potential.
Treatment
The patient underwent a left salpingo-oophorectomy. Intraoperative findings revealed an enlarged left ovary measuring 4 cm (Fig. 3), with histology and immunohistochemistry stains confirming a well-differentiated SLCT (Fig. 4). The tumor measured 27 mm, confined to the left ovary (pT1a). There was no evidence of lymphovascular or perineural invasion. Cytology for peritoneal washings were negative for malignancy. This was consistent with a well-differentiated FIGO stage IA tumor with no high-risk features.
Figure 3.
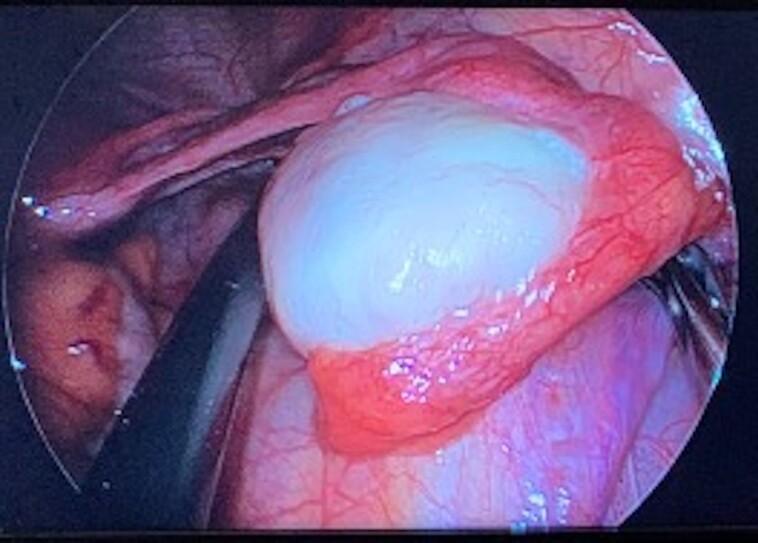
Intraoperative image of the left ovary during laparoscopy.
Figure 4.

A and B, Gross, and C to E, microscopic images of the ovary.
Outcome and Follow-up
There was complete normalization of testosterone and 17OHP on postoperative day 1 (Table 6).
Table 6.
Comparison of preoophorectomy and postoophorectomy 17-hydroxyprogesterone and testosterone values
| Investigation | Baseline pre operation | Postoperative d 1 | Reference range |
|---|---|---|---|
| Testosterone, total | 5.6-8.2 nmol/L (161.5-236.5 ng/dL) | 1.0 nmol/L (28.8 ng/dL) | 0.48-1.85 nmol/L (13.84-53.56 ng/dL) |
| 17OHP | 12.9 nmol/L (425.7 ng/dL) | 1.8 nmol/L (59.4 ng/dL) | Follicular phase: 0.3-3.3 nmol/L (9.91-109.05 ng/dL) Luteal phase: 2.9-15.2 nmol/L (95.83-502.30 ng/dL) |
Postoperative values are shown in bold font.
Abbreviation: 17OHP, 17-hydroxyprogesterone.
She was reviewed 3, 6, and 12 months postoperatively in the outpatient clinic and remains well, with normal testosterone and 17OHP values. Her menstrual cycle had returned 1 week after the operation and is now regular. She reported improvement in acne and no new symptoms of virilization. She will continue to be monitored regularly for clinical and biochemical features of recurrence.
Discussion
VOTs are rare, and SLCTs form the majority at 75%. They occur most frequently in women of reproductive age between the second and third decade [4], in keeping with patient demographics. Thirty percent present with typical features of secondary amenorrhea, hirsutism, and virilization [5], although up to 50% may be completely asymptomatic.
Imaging is often nondiagnostic as many VOTs can be less than 1 to 3 cm in size [4]. On ultrasonography, they usually have a solid cystic (58%) or purely solid (38%) appearance, with a minority (4%) being purely cystic [6]. In event of nondiagnostic imaging, selective venous sampling of the ovarian and adrenal veins could be considered for localization [7].
Limitations of evaluation in our patient include the initial diagnostic dilemma due to 2 normal transabdominal ultrasounds of the ovaries, causing us to pursue other differentials such as adrenal pathology due to the raised 17OHP.
Raised basal 17OHP suggested the possibility of 21-hydroxylase deficiency NCCAH. However, the 17OHP did not rise in response to cosyntropin stimulation. A similar case report by Wong et al [8] showed that their case of SLCT also had raised serum 17OHP. They had used the 24-hour urinary steroid profile (USP) and assessed for an adrenal and ovarian specific 17OHP metabolite pattern. In our case, we did not have access to 24-hour USP.
In a case series of ovarian steroid cell tumors [8], 81% presented with elevated baseline 17OHP, with a small minority having a response to ACTH stimulation [9]. Possible hypotheses include the expression of ACTH receptor on tumor cells, or spurious increase in response to ACTH stimulation due to cross-reactivity of immunoassays with cortisol or other precursors. This raises awareness to not overlook ovarian causes of elevated 17OHP. Careful assessment such as the use of cosyntropin stimulation and other adjunctive investigations are important to prevent misdiagnosis as NCCAH and delay in treatment. In our patient, the 17OHP analyzed on LC-MS/MS was still marginally raised but significantly lower than that via immunoassay, which may suggest other androgen interference with immunoassay.
Currently, steroid profiling is not commonly available in most clinical laboratories. In the future, there may be an increasing role for the use of pattern of metabolites in serum or USP to differentiate between an ovarian and adrenal source of 17OHP, such as 11-oxopregnanetriol, which is an adrenal-specific metabolite produced by the enzymatic action of 11-β hydroxylase on 17OHP [8]. This would be elevated in adrenal sources of 17OHP such as CAH, but normal in ovarian sources of 17OHP, which would aid in localization and confirming of diagnosis.
In our assessment, we used the progesterone challenge test to assess for bleeding and hormones in the follicular phase. The suppression of luteinizing hormone with exogenous progesterone would have driven the suppression of testosterone. Unfortunately, we did not have a testosterone level just prior to progesterone administration. However, the testosterone was consistently 5.6 to 8.2 nmol/L (161.5-236.5 ng/dL), and suppressed to 2.88 nmol/L (83.1 ng/dL). Shortly after, it had increased back to 5.63 nmol/L (162.4 ng/dL). In other literature, progesterone has not been described as a test. It is possible that using the progesterone challenge test is a simple and available alternative to a gonadotropin-releasing hormone stimulation test.
Learning Points
Virilizing SLCTs of the ovary can cause elevated 17OHP in addition to elevated testosterone
Elevated 17OHP may mislead the path of diagnosis to NCCAH. Consider analysis via LC-MS/MS and a cosyntropin stimulation test as the gold standard
Ultrasonography is operator-dependent. Where transvaginal ultrasound is contraindicated or unsuitable, consider MRI or transrectal ultrasound over transabdominal ovarian ultrasound earlier in the diagnostic algorithm
The low-dose dexamethasone suppression test and steroid profile are useful in the workup of hyperandrogenism
Suppression of testosterone with the progesterone challenge test may point to an ovarian source of testosterone
Acknowledgments
We would like to acknowledge Dr Jason Chng, Dr Lee Ming, Dr Ravichandran A/L Nadarajah, and Dr James Lim, who contributed to the care of this patient.
Abbreviations
- 17OHP
17-hydroxyprogesterone
- ACTH
adrenocorticotropin
- CAH
congenital adrenal hyperplasia
- DHEAS
dehydroepiandrosterone sulfate
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- LDDST
low-dose dexamethasone suppression test
- MRI
magnetic resonance imaging
- NCCAH
nonclassic congenital adrenal hyperplasia
- SLCT
Sertoli-Leydig cell tumor
- USP
urinary steroid profile
- VOT
virilizing ovarian tumor
Contributor Information
Ya Yuan Nicole Chong, Division of Endocrinology, Division of Medicine, National University Hospital, Singapore 119228, Singapore.
Jing Lin Jeslyn Wong, Division of Gynaecologic Oncology, Department of Obstetrics and Gynaecology, National University Hospital, Singapore 119228, Singapore.
Xiao Hong Joella Ang, Department of Obstetrics and Gynaecology, Singapore General Hospital, Singapore 169608, Singapore.
Yuan Ling Amanda Lim, Division of Endocrinology, Division of Medicine, National University Hospital, Singapore 119228, Singapore.
Contributors
All authors made individual contributions to the authorship and were involved in the care and diagnostic workup of the patient. Y.Y.N.C. and Y.L.A.L. were involved in the diagnosis and management of this patient and manuscript submission. J.L.J.W. and X.H.J.A. were involved in the management of this patient. All authors reviewed and approved the final draft.
Funding
No public or commercial funding.
Disclosures
None declared.
Informed Patient Consent for Publication
Signed consent obtained directly from the patient.
Data Availability Statement
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.
References
- 1. Derksen J, Nagesser SK, Meinders AE, Haak HR, van de Velde C. Identification of virilizing adrenal tumors in hirsute women. N Eng J Med. 1994;331(15):968‐973. [DOI] [PubMed] [Google Scholar]
- 2. Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline [published correction appears in J Clin Endocrinol Metab. 2010 Nov; 95(11):5137] [published correction appears in J Clin Endocrinol Metab. 2021 Jun 16; 106(7):e2853]. J Clin Endocrinol Metab. 2010;95(9):4133‐4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaltsas GA, Isidori AM, Kola BP, et al. The value of the low-dose dexamethasone suppression test in the differential diagnosis of hyperandrogenism in women. J Clin Endocrinol Metab. 2003;88(6):2634‐2643. [DOI] [PubMed] [Google Scholar]
- 4. Young RH, Scully RE. Ovarian Sertoli-Leydig cell tumors. A clinicopathological analysis of 207 cases. Am J Surg Pathol. 1985;9(8):543‐569. [DOI] [PubMed] [Google Scholar]
- 5. Huang LJ, Shi LY, Duan J. Clinicopathological analysis of ovarian Sertoli-Leydig cell tumor with postmenopausal vaginal bleeding as the first symptom. Medicine (Baltimore). 2021;100(13):e24922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Young RH. Sertoli-Leydig cell tumors of the ovary: review with emphasis on historical aspects and unusual variants. Int J Gynecol Pathol. 1993;12(2):141‐147. [DOI] [PubMed] [Google Scholar]
- 7. Levens ED, Whitcomb BW, Csokmay JM, Nieman LK. Selective venous sampling for androgen-producing ovarian pathology. Clin Endocrinol (Oxf). 2009;70(4):606‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wong FCK, Chan AZ, Wong WS, et al. Hyperandrogenism, elevated 17-hydroxyprogesterone and its urinary metabolites in a young woman with ovarian steroid cell tumor, not otherwise specified: case report and review of the literature. Case Rep Endocrinol. 2019;2019:9237459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kale K, Chauhan AR, Kalappa S. Virilization secondary to androgen-secreting tumor of the ovary: a report of three cases and review of literature. J Obstet Gynaecol India. 2019;69(Suppl 1):56‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.