

Abstract
INTRODUCTION
Research‐oriented autopsy cohorts provide critical insights into dementia pathobiology. However, different studies sometimes report disparate findings, partially because each study has its own recruitment biases. We hypothesized that a straightforward metric, related to the percentage of research volunteers cognitively normal at recruitment, would predict other inter‐cohort differences.
METHODS
The National Alzheimer's Coordinating Center (NACC) provided data on N = 7178 autopsied participants from 28 individual research centers. Research cohorts were grouped based on the proportion of participants with normal cognition at initial clinical visit.
RESULTS
Cohorts with more participants who were cognitively normal at recruitment contained more individuals who were older, female, had lower frequencies of apolipoprotein E ε4, Lewy body disease, and frontotemporal dementia, but higher rates of cerebrovascular disease. Alzheimer's disease (AD) pathology was little different between groups.
DISCUSSION
The percentage of participants recruited while cognitively normal predicted differences in findings in autopsy research cohorts. Most differences were in non‐AD pathologies.
Highlights
Systematic differences exist between autopsy cohorts that serve dementia research.
We propose a metric to use for gauging a research‐oriented autopsy cohort.
It is essential to consider the characteristics of autopsy cohorts.
Keywords: Alzheimer's disease neuropathologic change, cerebrovascular, dementia with Lewy bodies, epidemiology, frontotemporal dementia, frontotemporal lobar degeneration with TDP‐43, infarction, limbic‐predominant age‐related TDP‐43 encephalopathy neuropathologic change, neuropathology, plaques, prevalence, stroke, synuclein, tangles, TAR DNA‐binding protein 43
1. INTRODUCTION
Neuropathologic assessments provide gold‐standard diagnostic information for dementia‐related research, including documentation of the presence and severity of neurodegenerative diseases. Autopsy results can be correlated with cognitive, behavioral, genetic, imaging, and therapeutic data but the interpretation and generalization of autopsy data can be challenging; results from different cohorts often show contrasting findings. Such variation is not entirely random. Instead, the study designs and variability among different cohorts tend to follow certain patterns. Most autopsy cohorts fall somewhere along a spectrum of characteristics between two study designs: many autopsy cohorts strongly emphasize recruitment of participants with dementia, and on the other end of the spectrum are cohorts with sampling strategies that aim to be highly representative of a target population. 1 , 2 , 3
As implied by the term itself, clinic‐based cohorts are usually associated with a particular individual or group of neurologic clinic(s) and/or hospital(s). These cohorts are essential for studying rare dementia subtypes. Such samples have provided the bases for many breakthroughs in genetically driven, early onset, and/or diseases associated with a single, rather than mixed, pathologies (see, e.g., Roggenbuck et al., 4 Ono et al.,5 Kertesz et al.,6 and Keren et al. 7 ). By contrast, population‐ and community‐based cohorts are used by researchers aiming to study samples that are more representative of a target population; such cohorts often focus on the prevalent subtypes of pathology, as well as dementia‐free aging (see Bennett et al., 8 Zaccai et al.,9 and Suemoto et al. 10 ). Strengths of these cohorts include the ability to generalize findings to the target population, 11 , 12 , 13 , 14 to study common conditions, to identify incident cases with onset during time on study, and then to study their disease‐associated longitudinal trajectories. 11 , 12 , 13 , 14
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources. Selection bias in autopsy cohorts is well recognized and has been addressed in several ways previously by dementia researchers, but there is a lack of research on biases in multi‐center or consortia studies, which much of Alzheimer's disease and related dementia research is based upon. We included all relevant citations.
Interpretation: We hypothesized that a straightforward metric, related to the percentage of research volunteers cognitively normal at recruitment, would predict other inter‐cohort differences and found that this metric did predict differences in findings in autopsy research cohorts.
Future directions: This article highlights that there are systematic differences between study designs in the various autopsy cohorts that serve Alzheimer's Disease Research Centers and dementia research. We propose a metric to use for gauging a research‐oriented autopsy cohort and predicting certain findings that can be used in future research to provide a proxy for determining where along the continuum from “clinic‐based” to “community‐based” a cohort may fall.
Aspects of selection bias in autopsy cohorts are well recognized and have been addressed in several ways by dementia researchers. 15 , 16 , 17 , 18 , 19 To address the possible confounding influence of selection bias in autopsy cohort studies, techniques such as inverse‐probability weighting have been developed. 17 Another body of research focuses on the lack of participant diversity in such cohorts, and seeks to identify barriers and facilitators to brain donation, 20 , 21 , 22 including barriers to completion of brain donation after participants have consented. 23 The focus of the current study aligns with another line of research, which seeks to understand how recruitment characteristics of longitudinal cohorts influence the frequency and types of neuropathologies observed in autopsied members of the cohort. 18 , 24 This research has focused on one or two cohorts, but much Alzheimer's disease and related dementia (ADRD) research is based on multi‐center or consortia studies. 25 , 26 , 27 , 28
A productive consortium in the field of dementia research is the National Alzheimer's Coordinating Center (NACC). The NACC team aggregates and provides extensive granular data from the US National Institute on Aging–funded Alzheimer's Disease Research Centers (ADRCs). 29 , 30 , 31 NACC also illustrates some of the challenges in terms of ascertaining potential selection bias in autopsy cohorts studies. In NACC data sets, research data are available but often with no description of the study designs that were used to recruit and follow the participants. Investigators know only that participants were recruited and followed by a particular, anonymized center. Contributory ADRCs vary in their recruitment practices and protocols while providing standardized assessment instruments across the different institutions.
Despite the limitations, NACC presents an extraordinary opportunity to test associations between recruitment practices and study findings. Here, we examined data from ADRCs that contribute data to NACC and follow participants to autopsy. We tested the aggregated data set to assess the relationships among recruitment characteristics, study populations, and neuropathologic findings.
2. METHODS
2.1. Participants, data source, and inclusion criteria
Data were obtained from NACC, which is the data repository for past and present ADRCs. Participants are assessed using the standardized Uniform Data Set (UDS) at their local ADRC, approximately annually. The UDS is a robust set of data including participant demographics, health history, physical and neurological exams, ADRD symptomatology, the Clinical Dementia Rating (CDR) dementia staging instrument plus NACC frontotemporal lobar degeneration (FTLD) behavior and language domains. 30 , 32 , 33 , 34 Standardized data collection on neuropathological features present at the time of death are available for participants who were assessed with the UDS and who consented to autopsy. 31 , 35 The NACC Neuropathology (NP) form is used by the ADRCs, and provides guidance based on established criteria for evaluation of presence of pathologic depositions of amyloid beta (Aβ), tau, TAR DNA‐binding protein 43 (TDP‐43), and alpha synuclein. Also operationalized are cerebrovascular injuries, as well as rare pathologies like Huntington's disease. Version 10 of the NACC NP form, implemented in January 2014, introduced the assessment of frontotemporal lobar degeneration with TDP‐43 (FTLD‐TDP) inclusions and more generally, the presence of TDP‐43 immunoreactive inclusions in the spinal cord, amygdala, hippocampus, entorhinal/inferior temporal cortex, and neocortex.
Participants who met the study's eligibility criteria were selected from the December 2022 data freeze, which included data from the participant's initial and most recent UDS visits, collected from September 2005 to December 2022. Additional details about the UDS are described elsewhere. 29 , 30 , 31 , 34 , 36 The analyzed sample included participants with autopsy data available and was further limited to ADRCs with ≥ 30 eligible autopsied participants. The final analytic sample included 7178 participants and 28 ADRC autopsy cohorts. To facilitate analyses, the included ADRCs were grouped based on the proportion of participants with normal cognition at initial UDS visit. Participants were considered to have normal cognition if they were diagnosed with normal cognition by a clinician at their initial UDS visit. Participants were considered impaired if they were diagnosed with either mild cognitive impairment (MCI) or dementia at their initial UDS visit. The “more normal” (M‐Norm; n = 8 ADRCs) group included ADRCs with > 30% normal to impaired participants. The “more impaired” (M‐Imp; n = 10 ADRCs) group included ADRCs with < 15% normal to impaired participants. A third “middle group” (Mid; n = 10 ADRCs) included the ADRCs with 15% to 30% normal to impaired participants. These cut‐points were chosen to produce three groups with approximately the same numbers of ADRCs in each group.
2.2. Statistical analyses
To compare the demographic characteristics, clinical profiles, and neuropathologic features between the M‐Norm, M‐Imp, and Mid groups, we report both descriptive statistics (mean proportion, min, max) and P‐values from trend test using regression models. The analysis was done at the center level; thus, all statistics are reported for the ADRCs within each group. Continuous measures were compared with linear models, while counts were compared using Poisson models with a log offset for the number of participants with autopsy in each ADRC. Robust standard errors were used for the calculation of P‐values, which allows non‐constant variance across groups and overdispersion of count outcomes. Participant characteristics examined included age at initial UDS visit, follow‐up time, age at death, sex, race, and the presence of the apolipoprotein E (APOE) ε4 allele. Etiologic diagnoses were assessed at both the first UDS and most recent UDS visits. Neuropathologic features investigated include Alzheimer's disease neuropathologic change (ADNC) score, Braak neurofibrillary tangle (NFT) stage, Consortium to Establish a Registry for Alzheimer's Disease (CERAD) neuritic plaque density, limbic‐predominant age‐related TDP‐43 encephalopathy neuropathologic change (LATE‐NC), Lewy bodies, FTLD‐TDP, frontotemporal lobar degeneration with tau pathology (FTLD‐tau), argyrophilic grains, infarcts or lacunes, microinfarcts, moderate to severe brain arteriolosclerosis, and hippocampal sclerosis (HS). In the current study, LATE‐NC was defined as the presence of TDP‐43 inclusions in the amygdala, hippocampus, or neocortex and the absence of an overall clinical diagnosis of frontotemporal dementia (FTD) and/or FTLD‐TDP pathology. LATE‐NC negativity was defined as having an absence of TDP‐43 inclusions in the amygdala, hippocampus, and neocortex. Demographics and clinical and neuropathological features were treated as means or mean proportions and compared among the three groups (M‐Norm, M‐Imp, and Mid). For neuropathological conditions that were added in NPv10, which was implemented January 2014 (ADNC score, Thal Aβ phase, FTLD‐tau, TDP‐43 pathologies including FTLD‐TDP, argyrophilic grains, and HS), proportions were based on the subset of participants who had NPv10+ data available (n = 4105 participants from 28 ADRCs). The level of statistical significance was set at P < 0.05.
3. RESULTS
After the application of exclusion criteria, the final analytic sample included 28 ADRCs (M‐Imp n = 10 centers, 3113 participants; Mid n = 10 centers, 1961 participants; M‐Norm n = 8 centers, 2104 participants), comprising 7178 individual research participants (Figure 1). The subset of the sample with NPv10–11 data included 4105 participants from 28 ADRCs (M‐Imp n = 10 centers, 1855 participants; Mid n = 10 centers, 1188 participants; M‐Norm n = 8 centers, 1062 participants; Figure 1). After this stratification, the groups were analyzed using three types of parameters for the purpose of comparisons: demographics (Table 1), clinical diagnoses (Table 2), and subsequent neuropathologic diagnoses (Table 3).
FIGURE 1.
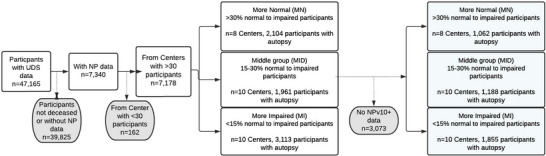
Study cohort exclusions, inclusions, sample sizes, and delineation of study groups. NP, National Alzheimer's Coordinating Center Neuropathology form; UDS, Uniform Data Set
TABLE 1.
Demographic characteristics among participants with NP data available.
| Center group, n | ||||
|---|---|---|---|---|
| Characteristic | More impairment (N/MCI&D < 15%) n = 10 centers, 3113 participants with autopsy | Middle group (N/MCI&D 15%–30%) n = 10 centers, 1961 participants with autopsy | More normal (N/MCI&D > 30%) n = 8 centers, 2104 participants with autopsy | P‐values |
| Number of unique 3‐digit zip codes in ADRC, mean (min, max) | 34.6 (0, 101) | 16.8 (0, 26) | 23.1 (0, 66) | 0.22 |
| Average number of UDS visits, mean (min, max) | 3.6 (2.5, 4.7) | 4.5 (3.4, 5.7) | 4.5 (3, 5.5) | 0.01 |
| Average follow‐up time (in years), mean (min, max) | 3.0 (1.5, 4.2) | 4.1 (2.9, 5) | 4.1 (2.5, 5.5) | <0.01 |
| % deceased participants with autopsy, mean % (min%, max%) | 60.0 (33.7, 86.3) | 60.5 (42.2, 76.0) | 62.8 (47.3, 82.2) | 0.68 |
| Age at baseline, mean (min, max) | 72.1 (66.2, 78.3) | 74.9 (69.6, 78.5) | 79.3 (75.1, 83.9) | <0.01 |
| Age at death, mean (min, max) | 77.1 (70.7, 83.8) | 80.3 (75.4, 84.4) | 85.1 (80.0, 89.8) | <0.01 |
| Sex, mean % (min%, max%) | ||||
| Male | 58.9 (51.2, 66.7) | 54.7 (47.3, 66.7) | 46.7 (38.8, 56.9) | <0.01 |
| Female | 41.1 (33.3, 48.9) | 45.4 (33.3, 52.7) | 53.3 (43.1, 61.2) | <0.01 |
| Race, mean % (min%, max%) | ||||
| White | 94.4 (89.1, 97.4) | 91.6 (85.4, 96.6) | 88.0 (43.9, 97.8) | 0.46 |
| Black or African American | 2.4 (0.6, 4.8) | 4.4 (0.5, 10.2) | 8.2 (0.0, 45.9) | 0.09 |
| Other | 2.1 (0.7, 3.4) | 3.5 (1.2, 8.8) | 3.7 (1.6, 9.2) | 0.32 |
| APOE ε4, mean % (min%, max%) | ||||
| ε4 carrier | 39.7 (22.2, 53.8) | 40.4 (16.7, 54.7) | 35.7 (26, 46.3) | 0.36 |
| No ε4 allele | 42.4 (14.3, 58) | 46.3 (22.5, 56.9) | 55.7 (40.8, 71.8) | 0.01 |
| Missing/unknown/ not assessed | 17.9 (1.2, 63.5) | 13.3 (0.0, 60.9) | 8.7 (0.5, 21.7) | 0.09 |
Note: number of participants missing data (percentages based on full sample—M‐Imp = 3113, Mid = 1961, M‐Norm = 2104); 3‐digit zip: M‐Imp = 545 (17.5%), Mid = 336 (17.1%), M‐Norm = 418 (20.8%); Race: M‐Imp = 49 (1.6%), Mid = 8 (0.4%), M‐Norm = 1 (0%); APOE ε4: M‐Imp = 442 (14.2%), Mid = 243 (12.4%), M‐Norm = 128 (6.4%). Bold values indicate a statistically significant difference in frequencies between the three groups (p<0.05).
Abbreviations: ADRC, Alzheimer's Disease Research Center; APOE, apolipoprotein E; Mid, Middle group; M‐Imp, More Impairment group; M‐Norm, More Normal group; N/MCI&D, proportion of participants with normal cognition to participants with mild cognitive impairment and dementia; NP, National Alzheimer's Coordinating Center Neuropathology form; UDS, Uniform Data Set.
TABLE 2.
Primary clinical diagnoses at initial and last standardized Uniform Data Set visit among participants with NP data available.
| Center group, n | ||||
|---|---|---|---|---|
| Characteristic | More impairment (N/MCI&D < 15%) n = 10 centers, 3113 participants with autopsy | Middle group (N/MCI&D 15‐30%) n = 10 centers, 1961 participants with autopsy | More normal (N/MCI&D > 30%) n = 8 centers, 2104 participants with autopsy | P‐values |
| Initial UDS visit | ||||
| Primary clinical diagnoses, mean % (min%, max%) | ||||
| AD | 51 (30.7, 69.2) | 54.5 (28.2, 82.8) | 41.3 (22.3, 57.1) | 0.16 |
| DLB | 8.8 (2.3, 20.1) | 4.2 (1.2, 7.4) | 1.6 (0.0, 3.8) | <0.01 |
| CVD | 0.8 (0.0, 3.3) | 1.5 (0.0, 5.5) | 1.3 (0.0, 3.4) | 0.12 |
| FTDa | 17.5 (3.1, 42.2) | 9.5 (0.0, 38.9) | 3.4 (0.0, 8.2) | <0.01 |
| Clinical diagnoses, b mean % (min%, max%) | ||||
| AD | 54.8 (37.9, 72.8) | 59.0 (44.7, 82.8) | 43.1 (23.5, 60.2) | 0.08 |
| DLB | 11.5 (3.1, 24.3) | 6.0 (1.2, 10.1) | 4.5 (2.1, 6.0) | <0.01 |
| CVD | 3.3 (0.0, 8.0) | 4.6 (0.9, 10.7) | 3.7 (1.3, 6.6) | 0.90 |
| FTD | ||||
| Any FTD a | 21.4 (3.6, 53.7) | 12.7 (1.2, 53.2) | 5.0 (1.9, 10.2) | <0.01 |
| bvFTD | 10.7 (2.6, 20.7) | 6.6 (0.0, 23.3) | 3.5 (1.3, 9.2) | <0.01 |
| PPA | 6.8 (1.0, 15.4) | 5.2 (0.0, 30.2) | 1.4 (0.0, 2.7) | <0.05 |
| Last UDS visit | ||||
| Primary clinical diagnoses, mean % (min%, max%) | ||||
| AD | 57.9 (34.3, 76.2) | 64.1 (37.1, 85.1) | 58.7 (43.2, 66.8) | 0.56 |
| DLB | 10.2 (3, 22.5) | 6.0 (2.2, 11.6) | 2.9 (0.0, 5.8) | <0.01 |
| CVD | 1.2 (0.0, 3.1) | 2.3 (0.0, 10.2) | 4.3 (1.0, 7.4) | <0.01 |
| FTD a | 16.9 (1.5, 40.7) | 10.1 (0.0, 37.6) | 4.0 (0.9, 8.2) | <0.01 |
| Clinical diagnoses, b mean % (min%, max%) | ||||
| AD | 62.3 (41.2, 80) | 69.2 (48.8, 88.5) | 61.9 (45.3, 69.8) | 0.65 |
| DLB | 15.0 (4.1, 30.8) | 10.2 (6.8, 15.4) | 6.6 (3.2, 9.8) | <0.01 |
| CVD | 5.0 (0.0, 11.9) | 8.9 (4.0, 19.1) | 10.0 (4.1, 16.7) | <0.01 |
| FTD | ||||
| Any FTD a | 21.6 (3.1, 53.9) | 13.2 (1.2, 53.5) | 5.0 (1.9, 9.6) | <0.01 |
| bvFTD | 10.7 (1.5, 22.0) | 6.4 (0.0, 20.1) | 3.3 (0.4, 8.2) | <0.01 |
| PPA | 7.3 (1.0, 16) | 4.8 (0.0, 29.7) | 1.1 (0.0, 2.9) | 0.03 |
Note: Number of participants missing clinical diagnosis at first UDS visit due to having normal cognition (not assessed; percentages based on full sample—M‐Imp = 3113, Mid = 1961, M‐Norm = 2104): More‐Imp = 264 (8.5%); Mid = 358 (18.3%); More‐Norm = 868 (43.1%). Number of participants missing clinical diagnosis at last UDS visit due to having normal cognition (not assessed; percentages based on full sample—M‐Imp = 3113, Mid = 1961, M‐Norm = 2104): M‐Imp = 161 (5.2%); Mid = 213 (10.9%); More‐Norm = 460 (22.8%). Bold values indicate a statistically significant difference in frequencies between the three groups (p<0.05).
Abbreviations: AD, Alzheimer's disease; ADRC, Alzheimer's Disease Research Center; ALS, amyotrophic lateral sclerosis; APOE, apolipoprotein E; bvFTD, behavioral variant frontotemporal dementia; CBD, corticobasal degeneration; CVD, cardiovascular disease; DLB, dementia with Lewy bodies; FTD, frontotemporal dementia; FTLD, frontotemporal lobar degeneration; Mid, Middle group; M‐Imp, More Impairment group; M‐Norm, More Normal group; MSA, multiple system atrophy; N/MCI&D, proportion of participants with normal cognition to participants with mild cognitive impairment and dementia; NP, National Alzheimer's Coordinating Center Neuropathology form; PPA, primary progressive aphasia; PSP, progressive supranuclear palsy; UDS, Uniform Data Set.
Includes MSA, PSP, CBD, FTLD with motor neuron disease (e.g., ALS, and other FTLD including bvFTD and PPA.
Rows are not mutually exclusive.
Due to the criteria used to define each group, we expected more participants in the “M‐Norm” group to be missing a primary clinical diagnosis (participants with positive biomarkers but no clinical symptoms of AD, Lewy body disease, or frontotemporal lobar degeneration should not have these etiologic diagnoses marked as “Present” by clinicians).
TABLE 3.
Neuropathological features.
| Center group, n | ||||
|---|---|---|---|---|
| Characteristic, mean % (min%, max%) | More impairment (N/MCI&D < 15%) n = 10 centers, 3113 participants | Middle group (N/MCI&D 15‐30%) n = 10 centers, 1961 participants | More normal (N/MCI&D > 30%) n = 8 centers, 2104 participants | P‐values |
| Alzheimer's disease pathology, mean % (min%, max%) | ||||
| Pathological NIA‐AA criteria b | ||||
| No ADNC | 14.2 (6.6, 25.7) | 11.1 (4.3, 17.4) | 13.0 (6.9, 19.5) | 0.47 |
| Low ADNC | 14.5 (0.0, 30.0) | 14.9 (7.5, 26.3) | 18.5 (10.0, 28.5) | 0.48 |
| Intermediate ADNC | 16.2 (10, 30.7) | 23.1 (10.1, 52.5) | 28.3 (9.8, 40) | <0.01 |
| High ADNC | 50.0 (34.7, 68.2) | 48.9 (23.2, 67.5) | 39.7 (28.8, 54.1) | <0.05 |
| Braak stage | ||||
| 0 | 9.5 (1.3, 27.1) | 3.7 (0.0, 12.6) | 4.2 (0.5, 10.4) | <0.01 |
| I | 7.7 (3.1, 17.7) | 6.6 (3.5, 11.4) | 6.6 (1.9, 13.2) | 0.49 |
| II | 10.1 (3.3, 18.9) | 10.6 (6.5, 14.8) | 11.6 (2.2, 19.2) | 0.27 |
| III | 9.5 (5.5, 20.6) | 9.4 (4.9, 16.4) | 13.4 (7.6, 31.6) | 0.01 |
| IV | 10.6 (7.3, 19.1) | 10.3 (7.2, 13.5) | 18.5 (4.7, 35.3) | 0.01 |
| V | 15.6 (5.8, 25.4) | 21.4 (12.7, 37) | 20.4 (5.7, 36.2) | 0.06 |
| VI | 34.5 (12.7, 51.8) | 36.7 (17.1, 51.8) | 24.6 (12.2, 56.6) | <0.01 |
| CERAD score | ||||
| None | 23.9 (9.5, 37.3) | 17.6 (12.2, 26.2) | 23.9 (12.1, 33.9) | 0.89 |
| Sparse | 10.3 (2.6, 19.5) | 8.8 (2.8, 18.0) | 17.4 (4.2, 45.4) | 0.01 |
| Moderate | 18.3 (9.5, 34.9) | 19.1 (3.8, 35.1) | 19.0 (5.8, 42.4) | 0.71 |
| Frequent | 46.7 (23.8, 73.4) | 54.2 (34.3, 69.8) | 39.7 (6.6, 59.0) | 0.16 |
| LATE‐NC, mean % (min%, max%) | ||||
| LATE‐NC stage a , b , c | ||||
| 1 | 3.3 (0.0, 12.9) | 4.2 (0.0, 12.5) | 3.1 (0.0, 6.8) | 0.93 |
| 2 | 9.7 (0.0, 20.8) | 14.5 (0.9, 21.0) | 11.4 (0.0, 27.8) | 0.23 |
| 3 | 1.5 (0.0, 4.3) | 3.4 (0.0, 9.2) | 2.6 (0.0, 10.2) | 0.10 |
| Lewy body pathology, mean % (min%, max%) | ||||
| Lewy bodies, n % | ||||
| None | 61.5 (43.9, 71.8) | 66.8 (49.8, 79.7) | 69.3 (54.3, 85.3) | 0.25 |
| Brainstem‐predominant | 3.3 (1.6, 5.3) | 3.0 (0.0, 9.8) | 4.8 (0.0, 8.2) | 0.21 |
| Limbic transitional/ amygdala‐predominant | 15.0 (8.7, 29.2) | 16.2 (0.0, 34.6) | 10.9 (0.9, 19.5) | 0.24 |
| Neocortical diffuse | 14.7 (7.3, 21.2) | 11.0 (2.9, 20.8) | 11.9 (5.3, 23.5) | 0.75 |
| FTLD‐TDP‐43 b | 7.9 (0.0, 20.8) | 4.3 (0.0, 14.6) | 2.5 (0.0, 5.0) | 0.01 |
| FTLD‐tau b | 14.4 (0.0, 39.0) | 10.2 (1.0, 29.5) | 15.9 (0.0, 54.6) | 0.97 |
| Argyrophilic grains b | 2.4 (0.0, 7.89) | 2.4 (0.0, 11.74) | 6.1 (0.0, 24.68) | 0.72 |
| Cerebrovascular pathology, mean % (min%, max%) | ||||
| Infarcts or lacunes | 15.2 (8.5, 30.1) | 15.6 (9.0, 31.6) | 23.2 (10.4, 41.8) | 0.03 |
| Microinfarcts | 17.2 (3.1, 32.1) | 19.6 (8.4, 37.8) | 21.4 (5.3, 31.7) | 0.13 |
| Mod/severe arteriolosclerosis | 46.5 (15.9, 76.7) | 41.0 (8.7, 72.4) | 22.0 (0.0, 45.7) | 0.09 |
| Hippocampal sclerosis b | 15.4 (5.5, 29.5) | 13.0 (3.1, 20.2) | 13.4 (7.1, 30.7) | 0.54 |
Note: number of participants missing data for NPv1–11 conditions (percentages based on full sample—M‐Imp = 3113, Mid = 1961, M‐Norm = 2104): Braak stage: M‐Imp = 87 (2.8%), Mid = 28 (1.4%), M‐Norm = 20 (1%); CERAD score: M‐Imp = 26 (0.8%), Mid = 8 (0.4%), M‐Norm = 0 (0%); Lewy bodies: M‐Imp = 18 (0.6%), Mid = 22 (1.1%), M‐Norm = 9 (0.4%); infarcts or lacunes: M‐Imp = 56 (1.8%), Mid = 4 (0.2%), M‐Norm = 1 (0%); microinfarcts: M‐Imp = 55 (1.8%), Mid = 3 (0.2%), M‐Norm = 0 (0%). Number of participants missing data for NP10+ conditions (percentages based on subset of participants with NPv10+—M‐Imp = 1855, Mid = 1188, M‐Norm = 1062): NIA‐AA ADNC score: M‐Imp = 94 (5.1%), Mid = 22 (1.9%), M‐Norm = 12 (1.1%); LATE‐NC: M‐Imp = 945 (50.9%), Mid = 468 (39.4%), M‐Norm = 473 (44.5%); FTLD‐TDP‐43: M‐Imp = 282 (15.2%), Mid = 229 (19.3%), M‐Norm = 171 (16.1%); FTLD‐tau: M‐Imp = 10 (0.5%), Mid = 26 (2.2%), M‐Norm = 17 (1.6%); argyrophilic grains: M‐Imp = 216 (11.6%), Mid = 205 (17.3%), M‐Norm = 124 (11.7%); hippocampal sclerosis: M‐Imp = 99 (5.3%), Mid = 7 (0.6%), M‐Norm = 8 (0.8%). Bold values indicate a statistically significant difference in frequencies between the three groups (p<0.05).
Abbreviations: AA, Alzheimer's Association; ADNC, Alzheimer's disease neuropathologic change; CERAD; Consortium to Establish a Registry for Alzheimer's Disease; FTD, frontotemporal dementia; FTLD‐tau, frontotemporal lobar degeneration with tau pathology; FTLD‐TDP, frontotemporal lobar degeneration with TDP‐43; LATE‐NC, limbic‐predominant age‐related TDP‐43 encephalopathy neuropathologic change; Mid, Middle group; M‐Imp, More Impairment group; M‐Norm, More Normal group; MSA, multiple system atrophy; N/MCI&D, proportion of participants with normal cognition to participants with mild cognitive impairment and dementia; NFT, neurofibrillary tangles; NIA, National Institute on Aging; NIAA, TDP‐43, TAR DNA‐binding protein 43.
LATE‐NC is defined as the presence of TDP‐43 inclusions in the amygdala, hippocampus, or neocortex; excludes any participants with FTLD‐TDP‐43 pathology and/or a clinical diagnosis of FTD at any visit; and includes participants with any Braak NFT stage.
Percentages based off the subset of participants who have NPv10+ data available where these conditions were first assessed (N—More impairment = 1855; N—Middle group = 1188; N—More normal = 1062).
Excludes any participants with FTLD‐TDP‐43 pathology and/or a clinical diagnosis of FTD at any visit.
We observed demographic and clinical diagnosis differences among groups. The M‐Norm group was older at both the initial UDS visit and at death (mean age at initial visit 79.3 years, range 75.1–83.9 years, P < 0.01; mean age at death 85.1 years, range 80.0–89.8 years, P < 0.01; Figure 2A), and had more females than the M‐Imp group and conversely fewer males than the M‐Imp group (mean% females: 53% M‐Norm vs. 41% M‐Imp, P < 0.01; mean% males: 47% M‐Norm vs. 59% M‐Imp, P < 0.01; Table 1; Figure 2B). Additionally, the M‐Norm group had significantly more participants without an APOE ε4 allele than the M‐Imp group (mean 56% vs. mean 42%, P = 0.01). We found no differences in the number of unique three‐digit ZIP codes at the ADRCs between the groups, but it is important to note that this variable can be blank if ZIP code is unknown, thus some centers do not report any three‐digit ZIP codes to NACC. In each table, we report the amount of missing data for the conditions listed. There were no noticeable differences in missingness among groups, thus we did not exclude missingness from denominators when reporting the descriptive statistics.
FIGURE 2.
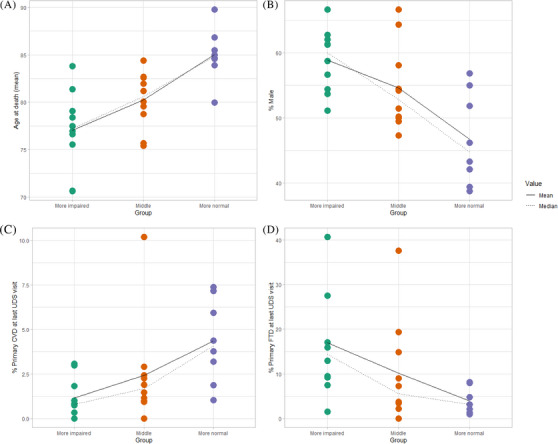
Participant characteristics, center averages, by group. The three groups were more normal (M‐Norm), more impaired (M‐Imp), or intermediate (Mid). Shown are the correlative results by group categories, for age of death (A), sex (B), final diagnosis of CVD (C), and final diagnosis of FTD (D). CVD, cerebrovascular disease; FTD, frontotemporal dementia; UDS, Uniform Data Set
In a sensitivity analysis we explored additional data that may provide insights regarding ADRC recruitment methods, which included the primary reason for coming to the ADRC, principal referral source, and the presumed disease status at enrollment (Table S1 in supporting information). While there were no differences in the primary reason for coming to the ADRC, the M‐Norm group was more likely to be referred by a non‐professional contact (self, relative, or friend) or other source, compared to the M‐Imp group, which was more likely to be referred by a professional contact (e.g., clinician, nurse, or other health‐care provider). Looking at presumed disease status at enrollment, the results align with the groupings we created for the study; the M‐Imp group was significantly more likely to be considered cases at enrollment while the M‐Norm group was made up of more controls than the other two groups.
In terms of the primary etiologic (clinical) diagnoses at the initial UDS visit, there were no significant differences in the percent of participants diagnosed with AD as primary diagnosis before death. However, the M‐Imp group had significantly more clinical dementia with Lewy bodies (DLB) than the M‐Norm group (mean 9% vs. 2%, P < 0.01) as well as more clinical FTD than the M‐Norm group (mean 18% vs. mean 3%, P ≤ 0.01; Table 2). The same differences were observed at the last UDS visit, and furthermore the M‐Imp group displayed less clinical cardiovascular disease (CVD) at the last UDS visit than the M‐Norm group (mean 1% vs. 4%, P = < 0.01; Table 4). Because participants can only have one primary diagnosis assigned at each UDS visit, but they can have multiple diagnoses marked as present, we assessed whether these diagnoses were present even if they were not primary. Similar patterns were observed; at the initial UDS visit, the M‐Imp group had significantly more DLB than the M‐Norm group (mean 12% vs. 5%, P < 0.01) as well as more FTD, specifically behavioral variant frontotemporal dementia (bvFTD) and primary progressive aphasia (PPA), than the M‐Norm group (mean 21% vs. mean 5%, P = < 0.01; Table 2, Table 4, and Figure 2D). The same differences were observed at the last UDS visit. Additionally, the M‐Imp group had significantly less clinical CVD than the M‐Norm group at the last UDS visit (mean 10% vs. 5%, P < 0.01, Table 2 and Figure 2C).
TABLE 4.
Summary of findings.
| Cohort recruitment criteria | Findings |
|---|---|
| More participants recruited while cognitively normal | Older participants (at intake and death) |
| Longer study follow‐up | |
| More females | |
| More CVD clinically | |
| More infarcts at autopsy | |
| More intermediate ADNC | |
| More participants recruited while cognitively impaired | Younger participants |
| More males | |
| More APOE ε4 (though not statistically significant at the p=0.05 level) | |
| More severe ADNC | |
| More clinical DLB at last UDS visit | |
| More clinical FTD at last UDS visit and FTLD‐TDP at autopsy | |
| (Percent with primary final clinical diagnosis of AD was not different according to cohort selection criteria) | |
Abbreviations: AD, Alzheimer's disease; ADNC, Alzheimer's disease neuropathologic change; APOE, apolipoprotein E; CVD; cardiovascular disease; DLB, dementia with Lewy bodies; FTD, frontotemporal dementia; FTLD‐TDP, frontotemporal lobar degeneration with TAR DNA‐binding protein 43, UDS, Uniform Data Set.
Some of the neuropathologic features were different between groups using the statistical testing methods we applied (Table 3). For example, there was an apparent shift between groups in ADNC severity—the M‐Imp tended to have more severe ADNC, whereas the M‐Norm tended to have more intermediate ADNC. Further, the M‐Imp group tended to have more FTLD‐TDP pathology than the M‐Norm group (mean 8% vs. 3%, P = 0.01), but not more FTLD‐tau. Importantly, there was also a pathologic phenomenon that was more severe in the M‐Norm group: the M‐Norm group had more infarcts and lacunes than M‐Imp (mean 23% vs. 15%, P = 0.03; Table 3).
Because the ADNC pathologies were all four‐ to six‐level ordered categories, we performed a sensitivity analysis that dichotomized the ADNC score, CERAD score, and Braak stage. After these ordinally leveled pathologies were dichotomized, there were no significant differences among the groups in ADNC severity (Table S2 in supporting information).
4. DISCUSSION
We separated autopsy cohorts (among 28 different ADRCs) into groups based on a single relatively straightforward parameter: global cognitive status at recruitment. The resulting groups of ADRCs had additional systematic differences from each other, in the demographic, clinical, and neuropathological features of the samples. ADRC autopsy cohorts with relatively high percentage of normal cognition at the time of recruitment (M‐Norm group) were on average older, more female, followed research volunteers longer, and had more frequent large vessel disease as seen through higher proportions of brain infarcts and CVD clinical diagnoses. By contrast, autopsy cohorts that had lower percentage of normal cognition at the time of recruitment (M‐Imp group) had more DLB, more FTD (and especially bvFTD), and the participants had higher frequency of APOE ε4 allele (Table 4). All of the centers are tasked with studying AD in some way or other, and indeed there was not a difference between our groups in the percent with clinical diagnoses of AD; however, the M‐Norm group tended to have more with final autopsy diagnoses of intermediate ADNC, whereas in the M‐Imp group the final diagnoses were more likely to be severe ADNC.
The current study highlights that there are systematic differences between study designs in the various autopsy cohorts that serve ADRCs and dementia research. The cut‐points we used to differentiate groups—related to the percentage of participants who were recruited while cognitively normal—were oriented to produce three groups with approximately the same numbers of ADRCs in each group. Notably, the overall percentage of cases with clinical AD and LATE‐NC were similar across these groupings, underscoring that AD research was a common denominator for these cohorts. However, the differences in ages, sex, and other clinical conditions were substantial. These distinctions underscored that an autopsy cohort that is useful in one context may be challenged to address other research questions.
The importance of understanding the connections between recruitment biases and study results is self‐evident, but two key reasons for focusing on these relationships stand out. First, there may be a tendency for researchers to place added emphases on the data they are most familiar with and the research volunteers who are under their care. This could lead to potential problems if epidemiologic inferences were being generated. For example, among the M‐Imp ADRC samples, the average proportion of cases with FTD was extremely high at 17.5% (range 3.1%–42.2%) and the autopsy‐confirmed FTLD‐TDP frequency was 7.9% (range 0%–20.8%). This contrasts strongly with the population prevalence of FTLD‐TDP—lifetime risk of FTLD‐TDP is 1:1000 or lower, or < 0.1% according to epidemiologic studies. 37 , 38 , 39 This > 100‐fold difference between cohort prevalence and population prevalence of FTLD‐TDP indicates that these cohorts are unique resources for the study of many or most aspects of FTLD‐TDP, but not its epidemiology. On the other hand, recruitment biases can also lead to other types of pitfalls, including underestimating the risk for a relatively unusual disease in a given age range. A possible example of this is in amyotrophic lateral sclerosis (ALS), which is sometimes considered rare in the aging population, but epidemiologic studies that included older participants found that ≈20% of persons with clinical ALS live beyond 80 years of age. 40 , 41 Our study confirms that recruitment methods have a strong influence on participant age and other demographics.
A second phenomenon that is highlighted by the comparison among dementia cohorts is that the (downstream) pathology differs based on (upstream) recruitment practices. As FTD/FTLD‐TDP cases were more common in M‐Imp centers, cerebrovascular disease (both clinically and pathologically) was more severe in M‐Norm centers. Moreover, a particular histopathologic observation at autopsy may have contrasting implications in different contexts. For instance, argyrophilic grains are an enigmatic tau‐immunoreactive phenotype seen at autopsy that can indicate a specific rare subtype of dementing illness (argyrophilic grain disease, or AGD) in clinic‐based cohorts. 42 , 43 , 44 , 45 In contrast, in community‐based cohorts, the observation of argyrophilic grains at autopsy has been associated with relatively intact cognition. 46 , 47 , 48 , 49 Indeed, in the present study there was a trend for argyrophilic grains to be commonly diagnosed in the M‐Norm ADRCs (average 6.1% of cases, range 0%–24.7% in the M‐Norm group) but in this context the presence of argyrophilic grains probably does not signal AGD.
Prior studies have highlighted the differences in results that can be observed in dementia research findings comparing clinic‐ or hospital‐based cohorts on the one hand, versus community‐ or population‐based autopsy cohorts on the other. Clinic‐based cohorts have reported a relatively higher frequency of dementia 1 , 50 and a higher rate of conversion of MCI to dementia. 51 In terms of neuropathologic correlation, several reports have indicated that there is overlap in terms of the findings between clinic‐ and community‐based cohorts in terms of ADNC. 3 , 52 However, clinic‐based cohorts have been reported to have more FTLD and other rare diseases, whereas community‐based cohorts have more CVD. 3 , 53 The findings in the present study are compatible with these results and indicate that the metric we are using (based on proportion of participants recruited while normal) may provide a proxy for determining where along the continuum from clinic‐based to community‐based a cohort may fall. The present study indicates that other criteria that may be contemplated include age of recruitment (higher in M‐Norm), sex predominance (more females in M‐Norm), and APOE genotype frequency (higher in M‐Imp).
Strengths of the present study included the analyses of granular data in terms of clinical and pathological parameters, and research participants that were followed longitudinally with yearly visits (average > 4 years on study). Because the question we were addressing involved a comparison between different institutions’ study designs, the NACC data set provides a unique opportunity to cross‐compare between dozens of different state‐of‐the‐art research centers. The data evaluated were multi‐modal including longitudinal assessments. Notably, each of the ADRCs used the same clinical and pathological data parameters so they could be directly compared to each other. To aid in reproducibility, we have included a list of the variables used in the analysis (Table S3 in supporting information).
There were also limitations and pitfalls to this study. Some degree of variance is implicit for a multi‐center study, with at least slight differences in clinician practices, diagnostic methods, and consensus dynamics. This is only partially addressed by the uniformity of the data elements (e.g., UDS data fields). However, this concern is also counterbalanced by the necessity of studying multiple centers to accomplish the aims of the present study. Although the number of included research centers was relatively high for a study of this nature, the categories (M‐Norm, Mid, and M‐Imp) nonetheless each were represented by 8, 10, and 10 centers, respectively, not a robust sample for statistical purposes. There was also as expected some variability in results rendering it difficult to reach statistical significance in delineating systematic differences. Comparing the number of 3‐digit ZIP codes, comparisons for this demographic variable are limited by the fact that some centers do not report any 3‐digit zip codes to NACC. While we have reported the amount of missing data in the tables, including the number of participants missing 3‐digit zip codes, and did not see a noticeable difference in missingness among groups, we did not perform an in‐depth analysis of the missing data, which is a limitation of the current study. Further, although we were assessing differences among the groups, there are meaningful similarities in these cohorts.
All 28 ADRCs have some overlapping biases including that ADRCs tend to recruit, retain, and achieve autopsy consent for White individuals of relatively high socioeconomic status; thus, there is a relative paucity of Blacks and Hispanics and participants lacking higher education. 9 As of the December 2022 data freeze, the average number of years of education for the participants in this analysis is 15.4 years, with 76% of participants having ≥ 13 years of education, and 33% having > 16 years of education. There was presumably a common high risk for AD pathology as reflected by the high APOE ε4 allele frequency—each group including M‐Norm averaged > one third of participants with APOE ε4 allele, whereas the frequency of APOE ε4+ participants is ≈25% in most populations. 54 , 55 Notably, NACC does not account for all ADRC‐specific recruitment biases when they provide results. It is known that some ADRCs exclude subjects at recruitment with various clinical features (e.g., parkinsonism or neuropsychiatric disease), 56 but those criteria are not centrally tabulated. Biases of studying amnestic (“Alzheimer's‐type”) dementia may overlook some other common dementia types such as Parkinson disease dementia, brain trauma–related pathologies, neurologic impairments related to infections and substance abuse, and subtypes of cerebrovascular disease.
5. CONCLUSION
Keeping in mind the limitations of the study samples, we found that using the percent of the research participants who were neurologically normal as a group criterion had potential utility in gauging a research‐oriented autopsy cohort and predicting certain findings. Many of the ADRCs recruited an intermediate number of neurologically normal participants—yet still more than a population‐based study design would entail. The implications are that some ADRCs are probably more appropriate to study subsets of dementia‐related conditions, and it is essential to consider the characteristics of the autopsy cohort when interpreting study findings. A list of recommendations for an autopsy cohort study is available in Table S4 in supporting information. Although autopsy results are the gold standard of disease diagnosis, sample selection and basic structure of autopsy cohort studies along the clinic versus population axis must be considered when making inferences related to disease prevalence, natural history, and the impact of risk factors.
CONFLICT OF INTERESTS STATEMENT
The authors have no competing interests to declare. Author disclosures are available in the supporting information.
CONSENT STATEMENT
ADRCs obtained written informed consent from their participants and maintain their own separate institutional review board review and approval from their institution prior to submitting data to NACC.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The NACC database is funded by NIA/NIH Grant U24 AG072122. NACC data are contributed by the NIA‐funded ADCs: P50 AG005131 (PI James Brewer, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG005136 (PI Thomas Grabowski, MD), P50 AG005138 (PI Mary Sano, PhD), P50 AG005142 (PI Helena Chui, MD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005681 (PI John Morris, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG008051 (PI Thomas Wisniewski, MD), P50 AG008702 (PI Scott Small, MD), P30 AG010124 (PI John Trojanowski, MD, PhD), P30 AG010129 (PI Charles DeCarli, MD), P30 AG010133 (PI Andrew Saykin, PsyD), P30 AG010161 (PI David Bennett, MD), P30 AG012300 (PI Roger Rosenberg, MD), P30 AG013846 (PI Neil Kowall, MD), P30 AG013854 (PI Robert Vassar, PhD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P30 AG019610 (PI Eric Reiman, MD), P50 AG023501 (PI Bruce Miller, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P30 AG028383 (PI Linda Van Eldik, PhD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), P30 AG035982 (PI Russell Swerdlow, MD), P50 AG047266 (PI Todd Golde, MD, PhD), P50 AG047270 (PI Stephen Strittmatter, MD, PhD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG049638 (PI Suzanne Craft, PhD), P30 AG053760 (PI Henry Paulson, MD, PhD), P30 AG066546 (PI Sudha Seshadri, MD), P20 AG068024 (PI Erik Roberson, MD, PhD), P20 AG068053 (PI Marwan Sabbagh, MD), P20 AG068077 (PI Gary Rosenberg, MD), P20 AG068082 (PI Angela Jefferson, PhD), P30 AG072958 (PI Heather Whitson, MD), P30 AG072959 (PI James Leverenz, MD). Dr. Nelson is supported by NIA/NIH grants R01 AG061111, RF1 NS118584. Dr. Katsumata is supported by NIA/NIH grants R56AG057191, R01AG057187, R21AG061551, R01AG054060, and the UK‐ADC P30AG028383 from the National Institute on Aging. No direct funding for this study to report.
Gauthreaux K, Kukull WA, Nelson KB, et al. Different cohort, disparate results: Selection bias is a key factor in autopsy cohorts. Alzheimer's Dement. 2024;20:266–277. 10.1002/alz.13422
REFERENCES
- 1. Gianattasio KZ, Bennett EE, Wei J, et al. Generalizability of findings from a clinical sample to a community‐based sample: a comparison of ADNI and ARIC. Alzheimers Dement. 2021;17(8):1265‐1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bennett DA, Schneider JA, Aggarwal NT, et al. Decision rules guiding the clinical diagnosis of Alzheimer's disease in two community‐based cohort studies compared to standard practice in a clinic‐based cohort study. Neuroepidemiology. 2006;27(3):169‐176. [DOI] [PubMed] [Google Scholar]
- 3. Massoud F, Devi G, Stern Y, et al. A clinicopathological comparison of community‐based and clinic‐based cohorts of patients with dementia. Arch Neurol. 1999;56(11):1368‐1373. [DOI] [PubMed] [Google Scholar]
- 4. Roggenbuck J, Palettas M, Vicini L, Patel R, Quick A, Kolb SJ. Incidence of pathogenic, likely pathogenic, and uncertain ALS variants in a clinic cohort. Neurol Genet. 2020;6(1):e390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ono R, Sakurai T, Sugimoto T, et al. Mortality risks and causes of death by dementia types in a Japanese cohort with dementia: NCGG‐STORIES. J Alzheimers Dis. 2023;92(2):487‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kertesz A, Mcmonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128(Pt 9):1996‐2005. [DOI] [PubMed] [Google Scholar]
- 7. Keren K, Busse M, Fritz NE, et al. Quantification of daily‐living gait quantity and quality using a wrist‐worn accelerometer in Huntington's disease. Front Neurol. 2021;12:719442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community‐based studies. Neurology. 2006;66(12):1837‐1844. [DOI] [PubMed] [Google Scholar]
- 9. Zaccai J, Ince P, Brayne C. Population‐based neuropathological studies of dementia: design, methods and areas of investigation–a systematic review. BMC Neurol. 2006;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suemoto CK, Ferretti‐Rebustini REL, Rodriguez RD, et al. Neuropathological diagnoses and clinical correlates in older adults in Brazil: a cross‐sectional study. PLoS Med. 2017;14(3):e1002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grodstein F, Leurgans SE, Capuano AW, Schneider JA, Bennett DA. Trends in postmortem neurodegenerative and cerebrovascular neuropathologies over 25 years. JAMA Neurol. 2023;80(4):370‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dugger BN, Hentz JG, Adler CH, et al. Clinicopathological outcomes of prospectively followed normal elderly brain bank volunteers. J Neuropathol Exp Neurol. 2014;73(3):244‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Polvikoski T, Sulkava R, Rastas S, et al. Incidence of dementia in very elderly individuals: a clinical, neuropathological and molecular genetic study. Neuroepidemiology. 2006;26(2):76‐82. [DOI] [PubMed] [Google Scholar]
- 14. Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59(11):1737‐1746. [DOI] [PubMed] [Google Scholar]
- 15. Yahagi‐Estevam M, Farias‐Itao DS, Leite REP, et al. The potential role of selection bias in the association between coronary atherosclerosis and cognitive impairment. J Alzheimers Dis. 2023:1307‐1316. [DOI] [PubMed] [Google Scholar]
- 16. Rennert L, Xie SX. Bias induced by ignoring double truncation inherent in autopsy‐confirmed survival studies of neurodegenerative diseases. Stat Med. 2019;38(19):3599‐3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haneuse S, Schildcrout J, Crane P, Sonnen J, Breitner J, Larson E. Adjustment for selection bias in observational studies with application to the analysis of autopsy data. Neuroepidemiology. 2009;32(3):229‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsuang D, Simpson KL, Li G, et al. Evaluation of selection bias in an incident‐based dementia autopsy case series. Alzheimer Dis Assoc Disord. 2005;19(2):67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weuve J, Proust‐Lima C, Power MC, et al. Guidelines for reporting methodological challenges and evaluating potential bias in dementia research. Alzheimers Dement. 2015;11(9):1098‐1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bonner GJ, Darkwa OK, Gorelick PB. Autopsy recruitment program for African Americans. Alzheimer Dis Assoc Disord. 2000;14(4):202‐208. [DOI] [PubMed] [Google Scholar]
- 21. Boise L, Hinton L, Rosen HJ, et al. Willingness to be a brain donor: a survey of research volunteers from 4 racial/ethnic groups. Alzheimer Dis Assoc Disord. 2017;31(2):135‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bilbrey AC, Humber MB, Plowey ED, et al. The impact of Latino values and cultural beliefs on brain donation: results of a pilot study to develop culturally appropriate materials and methods to increase rates of brain donation in this under‐studied patient group. Clin Gerontol. 2018;41(3):237‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glover CM, Shah RC, Bennett DA, Wilson RS, Barnes LL. Perceived impediments to completed brain autopsies among diverse older adults who have signed a uniform anatomical gift act for brain donation for clinical research. Ethn Dis. 2020;30(Suppl 2):709‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robinson AC, Chew‐Graham S, Davidson YS, et al. A comparative study of pathological outcomes in The University of Manchester longitudinal study of cognition in normal healthy old age and brains for dementia research cohorts. J Alzheimers Dis. 2020;73(2):619‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Franklin EE, Perrin RJ, Vincent B, Baxter M, Morris JC, Cairns NJ. Brain collection, standardized neuropathologic assessment, and comorbidity in Alzheimer's disease neuroimaging initiative 2 participants. Alzheimers Dement. 2015;11(7):815‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nelson PT, Brayne C, Flanagan ME, et al. Frequency of LATE neuropathologic change across the spectrum of Alzheimer's disease neuropathology: combined data from 13 community‐based or population‐based autopsy cohorts. Acta Neuropathol. 2022;144(1):27‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miyagawa T, Brushaber D, Syrjanen J, et al. Utility of the global CDR. Alzheimers Dement. 2020;16(1):106‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alafuzoff I, Gelpi E, Al‐Sarraj S, et al. The need to unify neuropathological assessments of vascular alterations in the ageing brain: multicentre survey by the BrainNet Europe consortium. Exp Gerontol. 2012;47(11):825‐833. [DOI] [PubMed] [Google Scholar]
- 29. Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the uniform data set. Alzheimer Dis Assoc Disord. 2007;21(3):249‐258. [DOI] [PubMed] [Google Scholar]
- 30. Besser L, Kukull W, Knopman DS, et al. Version 3 of the National Alzheimer's Coordinating Center's uniform data set. Alzheimer Dis Assoc Disord. 2018;32(4):351‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Besser LM, Kukull WA, Teylan MA, et al. The revised National Alzheimer's Coordinating Center's neuropathology form‐available data and new analyses. J Neuropathol Exp Neurol. 2018;77(8):717‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weintraub S, Besser L, Dodge HH, et al. Version 3 of the Alzheimer Disease Centers' neuropsychological test battery in the uniform data set (UDS). Alzheimer Dis Assoc Disord. 2018;32(1):10‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the uniform data set. Alzheimer Dis Assoc Disord. 2007;21(3):249‐258. [DOI] [PubMed] [Google Scholar]
- 34. Morris JC, Weintraub S, Chui HC, et al. The uniform data set (uds): clinical and cognitive variables and descriptive data from Alzheimer disease centers. Alzheimer Dis Assoc Disord. 2006;20(4):210‐216. [DOI] [PubMed] [Google Scholar]
- 35. Mock C, Teylan M, Beecham G, et al. The utility of the National Alzheimer's Coordinating Center's database for the rapid assessment of evolving neuropathologic conditions. Alzheimer Dis Assoc Disord. 2020;34(2):105‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beekly DL, Ramos EM, van Belle G, et al. The National Alzheimer's Coordinating Center (NACC) database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18(4):270‐277. [PubMed] [Google Scholar]
- 37. Knopman DS, Roberts RO. Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci. 2011;45(3):330‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Coyle‐Gilchrist ITS, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016;86(18):1736‐1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Onyike CU, Diehl‐Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry. 2013;25(2):130‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ryan M, Heverin M, Mclaughlin RL, Hardiman O. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 2019;76(11):1367‐1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mehta P, Kaye W, Raymond J, et al. Prevalence of amyotrophic lateral sclerosis ‐ United States, 2015. MMWR Morb Mortal Wkly Rep. 2018;67(46):1285‐1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saito Y, Ruberu NN, Sawabe M, et al. Staging of argyrophilic grains: an age‐associated tauopathy. J Neuropathol Exp Neurol. 2004;63(9):911‐918. [DOI] [PubMed] [Google Scholar]
- 43. Saito Y, Yamazaki M, Kanazawa I, Murayama S. Severe involvement of the ambient gyrus in a case of dementia with argyrophilic grain disease. J Neurol Sci. 2002;196(1‐2):71‐75. [DOI] [PubMed] [Google Scholar]
- 44. Togo T, Sahara N, Yen S‐H, et al. Argyrophilic grain disease is a sporadic 4‐repeat tauopathy. J Neuropathol Exp Neurol. 2002;61(6):547‐556. [DOI] [PubMed] [Google Scholar]
- 45. Wurm R, Klotz S, Rahimi J, et al. Argyrophilic grain disease in individuals younger than 75 years: clinical variability in an under‐recognized limbic tauopathy. Eur J Neurol. 2020;27(10):1856‐1866. [DOI] [PubMed] [Google Scholar]
- 46. Josephs KA, Whitwell JL, Parisi JE, et al. Argyrophilic grains: a distinct disease or an additive pathology? Neurobiol Aging. 2008;29(4):566‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sabbagh MN, Sandhu SS, Farlow MR, et al. Correlation of clinical features with argyrophilic grains at autopsy. Alzheimer Dis Assoc Disord. 2009;23(3):229‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodriguez RD, Suemoto CK, Molina M, et al. Argyrophilic grain disease: demographics, clinical, and neuropathological features from a large autopsy study. J Neuropathol Exp Neurol. 2016;75(7):628‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nelson PT, Abner EL, Schmitt FA, et al. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol. 2010;20(1):66‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Davis DH, Creavin ST, Yip JL, Noel‐Storr AH, Brayne C, Cullum S. Montreal cognitive assessment for the detection of dementia. Cochrane Database Syst Rev. 2021;7(7):CD010775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Farias ST, Mungas D, Reed BR, Harvey D, Decarli C. Progression of mild cognitive impairment to dementia in clinic‐ vs community‐based cohorts. Arch Neurol. 2009;66(9):1151‐1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Plassman BL, Khachaturian AS, Townsend JJ, et al. Comparison of clinical and neuropathologic diagnoses of Alzheimer's disease in 3 epidemiologic samples. Alzheimers Dement. 2006;2(1):2‐11. [DOI] [PubMed] [Google Scholar]
- 53. Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis. 2009:691‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Crean S, Ward A, Mercaldi CJ, et al. Apolipoprotein E epsilon4 prevalence in Alzheimer's disease patients varies across global populations: a systematic literature review and meta‐analysis. Dement Geriatr Cogn Disord. 2011;31(1):20‐30. [DOI] [PubMed] [Google Scholar]
- 55. Corbo RM, Scacchi R. Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a ‘thrifty’ allele? Ann Hum Genet. 1999;63(Pt 4):301‐310. [DOI] [PubMed] [Google Scholar]
- 56. Schmitt FA, Nelson PT, Abner E, et al. University of Kentucky Sanders‐Brown healthy brain aging volunteers: donor characteristics, procedures and neuropathology. Curr Alzheimer Res. 2012;9(6):724‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information