
Figure 1.

Schematic overview of SpaCAE augmentation-encoding-decoding (A–C) processes and potential biological applications of SpaCAE in downstream SRT analysis (D, E). (A) The three data sources for SpaCAE inputs are STR expression matrix, spatial coordinates and hematoxylin and eosin (H&E) staining image. (B) Weighted graph (i.e. 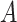 ) construction from spatial multi-modal data and spatial expression augmentation from aggregating expression information from neighborhood spots. SpaCAE integrates spatial information into gene expression to augment the shared expression between spots by aggregating the gene expression from their K spatial neighbors through weighted graph, which forms the spots’ spatial contrastive structure (e.g.
) construction from spatial multi-modal data and spatial expression augmentation from aggregating expression information from neighborhood spots. SpaCAE integrates spatial information into gene expression to augment the shared expression between spots by aggregating the gene expression from their K spatial neighbors through weighted graph, which forms the spots’ spatial contrastive structure (e.g. 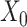 →
→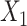 ). (C) SpaCAE model. SpaCAE uses robust graph convolutional encoder and deep contrastive variational autoencoder to generate the low-dimensional representation (i.e.
). (C) SpaCAE model. SpaCAE uses robust graph convolutional encoder and deep contrastive variational autoencoder to generate the low-dimensional representation (i.e. 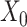 →
→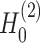 ,
, 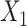 →
→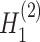 ). The input data are the original gene expression matrix (i.e.
). The input data are the original gene expression matrix (i.e. 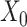 ), the augmented gene expression matrix (i.e.
), the augmented gene expression matrix (i.e. 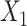 ) and weighted spatial graph
) and weighted spatial graph 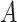 from (B). Based on the original and low-dimensional data, SpaCAE performs data reconstructions via graph deconvolutional decoder and deep contrastive variational autoencoder. SpaCAE iteratively learns the graph convolutional encoder
from (B). Based on the original and low-dimensional data, SpaCAE performs data reconstructions via graph deconvolutional decoder and deep contrastive variational autoencoder. SpaCAE iteratively learns the graph convolutional encoder 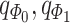 and graph deconvolutional decoder
and graph deconvolutional decoder 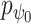 ,
, 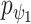 by minimizing the sum of reconstruction losses and contrastive losses (see Methods). When SpaCAE reaches convergence, the optimum is achieved for further downstream analyses. (D, E) Biological applications for SpaCAE including data denoising and spatial domain identification. (D) The reconstructed augmented spatial expression
by minimizing the sum of reconstruction losses and contrastive losses (see Methods). When SpaCAE reaches convergence, the optimum is achieved for further downstream analyses. (D, E) Biological applications for SpaCAE including data denoising and spatial domain identification. (D) The reconstructed augmented spatial expression 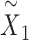 can be employed to denoise data. (E) The low-dimensional representation (i.e. (
can be employed to denoise data. (E) The low-dimensional representation (i.e. (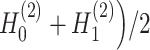 ) can be applied to detect spatial domains.
) can be applied to detect spatial domains.