

Summary
Proteins are essential biomolecules and central to biotechnological applications. In many cases, assembly into higher-order structures is a prerequisite for protein function. Under conditions relevant for applications, protein integrity is often challenged, resulting in disassembly, aggregation, and loss of function. The stabilization of quaternary structure has proven challenging, particularly for trimeric and higher-order complexes, given the complexity of involved inter- and intramolecular interaction networks. Here, we describe the chemical bicyclization of homotrimeric protein complexes, thereby increasing protein resistance toward thermal and chemical stress. This approach involves the structure-based selection of cross-linking sites, their variation to cysteine, and a subsequent reaction with a triselectrophilic agent to form a protein assembly with bicyclic topology. Besides overall increased stability, we observe resistance toward aggregation and greatly prolonged shelf life. This bicyclization strategy gives rise to unprecedented protein chain topologies and can enable new biotechnological and biomedical applications.
Keywords: Aggregation, biocatalysis, bioconjugation, chemical biology, cross-linking, crystallography, enzymes, INCYPRO, protein engineering, structural stabilization
Graphical abstract
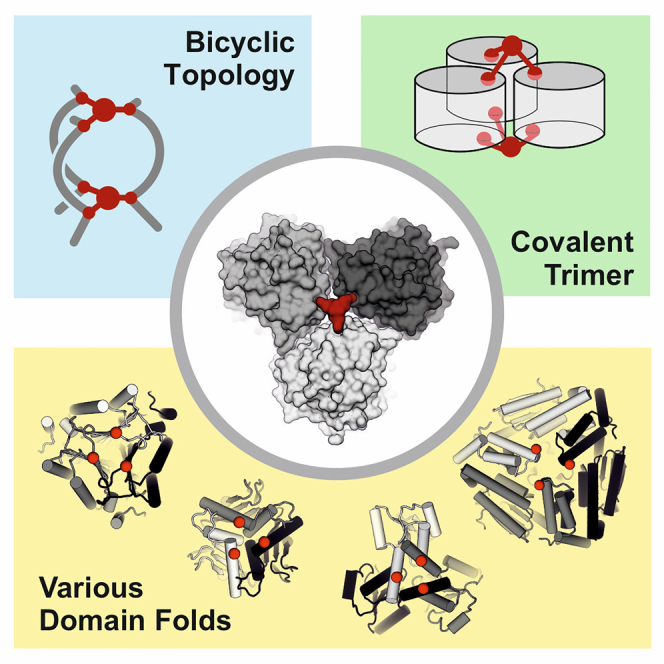
Highlights
-
•
Triselectrophiles cross-link homotrimeric proteins, forming bicyclic topologs
-
•
Bicyclic enzyme exhibits activity under denaturing conditions
-
•
Approach is rapidly applicable to a structurally diverse range of complexes
The bigger picture
The cyclization of small molecules, peptides, and macromolecules is a fundamental strategy to design precise three-dimensional molecular shapes tailored to chemical function. Proteins, however, typically consist of linear polypeptide strands, where protein folding and assembly are governed by non-covalent interactions. Protein engineering to form precise interlinked and cyclized protein chains has the potential to expand the biotechnological applications of proteins beyond their natural environments. Here, we construct a covalent bicyclic enzyme, capable of withstanding an exceptional degree of chemical and thermal stress. Such molecules can offer new avenues for sustainable industrial chemistry and biocatalysis, functioning in chemical environments that were previously inaccessible with biological components.
Modulating the stability of protein complexes is crucial for their use in biotechnological applications, such as biocatalysis, offering more sustainable alternatives to traditional chemical transformations. Protein stabilization is typically a labor-intensive process, reliant on extensive screening. Here, we apply a structure-based approach, which utilizes three-pronged reactive cross-linkers to chemically modify and tether a diverse range of trimeric protein complexes. The resulting abiological and cyclic chain connectivity offers robust enzymatic activity and complex stability under challenging thermal and chemical conditions.
Introduction
Proteins represent a versatile molecular platform, essential to life on earth and a rich source of inspiration for the development of novel biotechnological tools.1,2,3,4,5 Self-association of proteins into homomeric complexes is widespread, facilitating an evolutionarily relevant increase in morphological complexity.6 However, applications such as catalysis in abiotic environments often involve extreme conditions that challenge the structural integrity of proteins and disrupt complex assemblies. Although considerable efforts have enhanced the thermal and chemical stability of protein folds (tertiary structure),7,8,9,10,11 the stabilization of protein complexes and their supramolecular assembly (quaternary structure) remains a challenge. Such approaches have mainly focused on homodimeric protein complexes. For example, homodimeric isologous interfaces (in which each interaction surface is identical)12 have been stabilized or artificially engineered via systematic mutagenesis13 and computational approaches.14,15 The introduction of inter-molecular disulfide bridges or non-canonical amino acids, and the design of fusion proteins, have been utilized to covalently link monomer units within dimeric complexes.16,17,18,19,20 In contrast, many higher-order complexes, such as homotrimers, possess heterologous interfaces (i.e., where the interacting surfaces differ), which present a bigger challenge for engineering due to the inherent necessity for more mutations and the risk of rearrangement into polymeric aggregates.21 General approaches toward the efficient stabilization of these complexes are currently lacking.
Bioconjugation strategies have emerged as powerful techniques to enhance and alter protein functionality, providing access to powerful labeling tools, screening platforms, and novel therapeutic entities, such as antibody-drug conjugates.22,23,24 The use of bioconjugation approaches for protein structure stabilization is uncommon, and has focused on either tertiary structure stabilization25,26,27 or inter-chain cross-linking to trap weak complexes and probe for unknown protein-protein interactions.28,29,30 Herein, we aimed to employ biocompatible, multivalent modifiers for the stabilization of protein complexes. We have previously reported the in situ cyclization of proteins (INCYPRO) for the stabilization of tertiary structures,26,27,31 which utilizes C3-symmetric triselectrophiles32,33,34 that react with three spatially aligned cysteine side chains on the protein surface. Given the C3 symmetry of protein homotrimers, we considered whether it is possible to cross-link homotrimeric complexes with these triselectrophiles, thereby constructing protein conjugates with high tolerance toward thermal and chemical stress. Our approach aimed to fundamentally alter the chain topology of trimeric complexes, converting three protein monomers into a single, covalently interlinked chain with bicyclic topology. This enabled the construction of a stress-resistant enzyme capable of catalysis under denaturing conditions. To further challenge the concept, we explored a suite of structurally diverse trimeric complexes, successfully generating a range of stabilized trimeric complexes and obtaining four additional precisely bicyclized protein trimers with substantially increased thermal stability.
Results and discussion
Construction of covalently cross-linked protein trimers
We initially selected Pseudomonas fluorescens esterase (PFE) as a target for complex stabilization. PFE is a well-characterized homotrimeric hydrolase, which has served previously as a platform for protein engineering efforts.35,36 Based on an earlier reported crystal structure of PFE (Protein Data Bank; PDB: 1va4),37 we searched for cross-linking sites on the two faces of the trimer (Figure 1A) to enable covalent linkage by a triselectrophile (Figure 1B). Given the C3-symmetry of the PFE complex, the variation of a single residue results in three equivalent modified sites per trimer. We considered suitable Cα–Cα distances of 8–15 Å (Figure S1) between modified residues and identified two solvent exposed residues (T3 and Q174) close to the central C3 axis as potential cross-linking sites (Figure 1A). Cysteine residues were introduced to the wild-type enzyme (denoted p1) at these two positions, furnishing two variants for mono-cross-linking (p2, T3C; p3: Q174C) and one for bis-cross-linking, combining both variations (p4, Figure 1A). Notably, the latter generates an interlinked bicyclic topology upon modification. The selected variation sites are distant from the catalytic pocket and therefore not expected to directly interfere with enzymatic activity (Figure S1). Each PFE monomer bears one native cysteine which is, however, buried and not accessible for electrophilic modification.
Figure 1.

Covalent multimerization and bicyclization
(A) Crystal structure of Pseudomonas fluorescens esterase (p1, PDB: 1va4), including modification sites (red, T3, and Q174) with amino acid variations and corresponding protein (and complex) names. For sequences and design details see Figure S1.
(B) Cysteine-selective cross-linking using chloroacetamide or iodoacetamide-based cross-linkers (Ta-Cl3, Ta-I3).
(C) Scheme of trimer chain topology. Mass spectra (ESI-TOF) of cross-linked protein trimers including the charge state of the most abundant signal (see Tables S2–S5 for complete lists of MS signals).
The chemical cross-linking of the PFE variants was initially pursued with the chloroacetamide bearing triselectrophile N,N′,N″-((1,3,5-triazinane-1,3,5-triyl)tris(2-oxoethane-2,1-diyl))tris(2-chloroacetamide) (Ta-Cl3) (Figure 1B) and analyzed using sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE). We observed only limited reactivity, however (Figure S2), particularly for variant p2 (T3C), failing to form detectable levels of product. To address this issue, we envisioned the use of a more reactive triselectrophilic cross-linker, retaining the triazinane core but replacing chloroacetamide with the more reactive and cysteine-specific iodoacetamide. The corresponding cross-linker N,N′,N″-((1,3,5-triazinane-1,3,5-triyl)tris(2-oxoethane-2,1-diyl))tris(2-iodoacetamide) (Ta-I3) (Figure 1B), was obtained via the Finkelstein reaction, employing Ta-Cl3 and NaI (see experimental procedures).
SDS-PAGE indicated the formation of a defined covalently linked multimeric species for each of the variants (p2, p3, and p4) upon incubation with Ta-I3 (Figure S3). Interestingly, all cross-linked variants showed a slower mobility than expected for their molecular weight (MW = 90 kDa, apparent MW = 100–150 kDa, Figure S3), which we suspected to be due to their unusual chain topology (Figure 1C). The terminally linked p23Ta exhibits an extended star-like topology, whereas p33Ta is cross-linked via a central cysteine (C174), resulting in a branched topology. Trimer p43Ta2 is cross-linked by two entities and therefore forms a compact bicyclic structure. To further analyze the reaction products, high-resolution mass spectrometry (MS) was performed confirming the formation of the expected covalently linked trimers: mono-cross-linked p23Ta and p33Ta, as well as bis-cross-linked p43Ta2 (Table S1).
We subsequently performed time-of-flight (TOF) MS under denaturing conditions. The resulting spectra revealed a distinct distribution of charged states for each covalent protein trimer (Figure 1C; Tables S2–S4). The N-terminally linked, star-shaped p23Ta experiences extensive protonation (z(max) = 91, ca. 30 charges per monomer), with an average protonation per monomer comparable to the three unmodified variants (ca. 37 charges, Table S5). Branched p33Ta exhibits reduced protonation levels (z(max) = 61, ca. 20 charges per monomer). This trend continues for the presumably most compact bicyclized trimer p43Ta2 (Figure S4), resulting in a considerably lower number of charges (z(max) = 42, ca. 14 charges per monomer). Analogous behavior is observed in SDS-PAGE (Figure S3) where the three covalent trimers show subtle differences in mobility (migration p43Ta2 > p33Ta > p23Ta), again suggesting the highest degree of compactness for bicyclic p43Ta2 in the unfolded state.
Robust catalysis by triselectrophile-linked enzyme complexes
We next investigated the impact of cross-linking on thermal stability using differential scanning fluorimetry (DSF), revealing enhanced stability for all cross-linked variants when compared with their unmodified precursors (Figure 2A). The most pronounced improvement was observed for bicyclic p43Ta2 (ΔTi = 8.4°C). In addition, we tested protein resistance toward chemical stress, employing guanidine hydrochloride (GuHCl) as a chaotropic denaturant. For all proteins, we observed decreasing Ti values with increasing GuHCl concentrations in line with the expected increased unfolding propensity under increasingly chaotropic conditions (Figure S5; Table S6). The sensitivity toward GuHCl, however, varies considerably between the different topologies. The wild-type protein p1 and the non-cross-linked variants p2, p3, and p4 exhibit strong sensitivity toward GuHCl with the absence of a defined melting curve at 1.5 M GuHCl (Figures 2B and S5). Cross-linking increases thermal stability in presence of GuHCl in all three cases (Figures 2C and S5), with the most stable cross-linked trimer p43Ta2 retaining a cooperative unfolding profile up to 1.5 M GuHCl.
Figure 2.

Cross-linking increases protein stability and enzymatic activity under stress
(A) Melting temperatures (Ti) of unreacted and cross-linked variants.
(B and C) Thermal denaturation profiles of p1 and p43Ta2, respectively, in the presence of varying concentrations of guanidine hydrochloride (GuHCl). For thermal denaturation curves of p2, p3, p4, p23Ta, and p33Ta see Figure S5.
(D) Relative initial rates of enzymatic reaction for p1 and the three covalent trimers (relative to initial rate of p43Ta2 at the given GuHCl concentration). The absolute value for each initial rate is provided (for details see Table S7). The errors account for 1σ (n = 3).
(E and F) Reaction course of substrate conversion by p1 and p43Ta2 in the presence of varying concentrations of GuHCl. For activity curves of p2, p4, p4, p23Ta, and p33Ta see Figure S6. All activity measurements were performed with 2 nM PFE trimer (50 mM HEPES, 50 mM NaCl, pH 7.5). The errors account for 1σ (n = 3).
Given the resistance of the covalently linked trimers to chemical denaturation, we tested if this translates into increased enzymatic activity under these conditions. PFE esterase activity was analyzed by monitoring the conversion of p-nitrophenyl acetate to p-nitrophenolate (p-NP, Figure S6).38 In the absence of denaturant, all proteins show comparable activity (Figure 2D) indicating that enzyme activity has not been affected by cysteine variation and cross-linking. As expected, all enzymes experience a reduction in activity with increasing concentrations of GuHCl (Figure 2D; Table S7). However, cross-linking considerably enhanced resistance to the denaturant, with highest residual activities observed for bicyclic PFE trimer p43Ta2 (Figure 2D). Although unmodified p1 only facilitates product formation up to 1 M GuHCl (Figure 2E), p43Ta2 remains active in the presence of up to 2 M GuHCl (Figure 2F).
Bicyclic p43Ta2 resists aggregation
Bicyclic trimer p43Ta2 experienced the largest stabilizing effect, surpassing both single-cross-linked trimers (p23Ta and p33Ta). To further investigate this pronounced stabilization, p43Ta2 was studied in more detail. Circular dichroism (CD) spectra under native conditions revealed a spectrum analogous to the wild-type enzyme p1, suggesting an unchanged overall fold (Figure 3A). Assessing the thermal denaturation behavior via CD (λ = 220 nm, Figure 3B), melting temperatures were obtained (p1 Tm = 69.5°C; p43Ta2 Tm = 77.0°C), which are in the range of above DSF results (Figure 2A). The slightly higher difference suggested a time dependence of stability, with a slower temperature ramp rate applied for CD compared with DSF measurements. We noted the complete absence of CD signal at high temperature (Figure S7) and the distinctive spike in the associated high tension (HT) values (Figure 3B, overlaid), both suggesting that the predominant inactivation pathway involves protein aggregation and subsequent precipitation.39
Figure 3.

Bicyclic p43Ta2 resists aggregation
(A) Circular dichroism (CD) spectra of p1 and p43Ta2 measured at 20°C at a 4 μM trimer concentration (in 50 mM potassium chloride, 50 mM potassium phosphate, pH 8).
(B) Thermal denaturation CD profiles of p1 (Tm = 69.5°C) and p43Ta2 (Tm = 77.0°C) measured at 220 nm. HT values suggest precipitation close to the midpoint temperature (Tm derived from the maximum of the CD first derivative curve as presented in Figure S7).
(C) Top: PFE trimer structure (PDB: 1va4) with approximate distances. Bottom: temperature-dependent size distribution derived from DLS experiment for p1 (gray) and p43Ta2 (orange). Measured at a 15 μM trimer concentration (in 50 mM HEPES, 50 mM NaCl, pH 8). The errors account for 1σ (n = 6). Above 69°C, it was not possible to reliably fit size distribution data for p1. For p43Ta2 data at higher temperatures, see Figure S8.
To compare the aggregation behaviors of p1 and p43Ta2, dynamic light scattering (DLS) measurements were performed, confirming for both the presence of homogeneous, well-dispersed trimers at ambient temperatures (up to T = 40°C, Figure 3C). Notably, the observed radius (r ∼ 4 nm) corresponds to the size of the PFE trimer in the crystal structure (Figure 3C). For p1 (gray), temperature-dependent DLS measurements indicated the appearance of higher-order oligomeric structures around 45°C (Figure S8). Initially, species with a radius of ca. 30 nm (T = 50°C) and 60 nm (T = 60°C) were detected. At 69°C, we mainly observed the presence of aggregates >1 μm, which corresponds to the CD-derived melting temperature of p1 (Tm = 69.5°C). Above 69°C, it was not possible to reliably fit size distribution data for p1, most likely due to severe precipitation.
In comparison, bicyclic p43Ta2 (orange) remained broadly monodisperse up to 65°C (Figure S8) suggesting a delay of aggregation onset by ca. 20°C. At 69°C, p43Ta2 shows species with increased size around 30 nm, analogous to p1 at 50°C (Figure 3C). Eventually, p43Ta2 also showed additional higher-order oligomeric states (60 nm at 70°C and >1 μm at 74°C, Figure S8). To further assess the effect of aggregation on long-term stability, we tested storage at 50°C, a temperature below the melting temperatures of both proteins (p1 Tm = 69.5°C; p43Ta2 Tm = 77.0°C). After 24 h incubation at 50°C, both enzymes retained their activity (Figure S9). After longer storage (more than 1 day), however, p1 lost activity, whereas p43Ta2 kept its activity for multiple weeks. At this temperature, unmodified p1 already showed the formation of oligomeric structures (Figure 3C), which could lead to aggregation and loss of enzymatic activity over longer time periods.
Crystal structure of bicyclic p43Ta2
In pursuit of a crystal structure of p43Ta2, conditions were screened to obtain suitable crystals for X-ray diffraction analysis. A dataset was collected, determining the crystal structure of p43Ta2 at a resolution of 2.5 Å (PDB: 8pi1, space group C121, Table S8) following molecular replacement with the PFE trimer as a search model (derived from PDB: 1va4). The obtained structure harbors five protomers of p43Ta2 per asymmetric unit (Figure 4A). Between both introduced cross-linking sites (C3 and C174) additional well-defined electron density was observed that matches the molecular structure of the cross-link (Figure 4B), thereby validating the expected site-specific modification and bicyclization of the p4 trimer.
Figure 4.

Crystal structure of bicyclic p43Ta2
(A) The asymmetric unit contains five p43Ta2 protomers (space group: C121, for details see Table S8).
(B) 2Fo−Fc electron density map around Ta cross-linked via cysteines C3 and C174, respectively, in the three monomers in p43Ta2.
(C) Overlayed crystal structures of p1 (gray, PDB: 1va4) and p43Ta2 (orange, backbone atom RMSD: 0.18 Å).
(D) Surface representation of p43Ta2 showing protein subunits (orange) and Ta cross-links (red).
Overall, the crystal structure of p43Ta2 (orange) aligns closely with the previously reported structure of wild-type PFE (p1, gray, Figure 4C, backbone atom root-mean-square deviation [RMSD]: 0.18 Å), demonstrating minimal perturbation of the macromolecular structure despite the modification of six residues within the cross-linked trimer. On one face of the trimer (top view), the cross-link connects the three N-terminal cysteines (C3) located in a short β strand. On the other side (bottom view), the cross-linked central cysteines (C174) are located at the beginning of an α helix (Figure 4C). Interestingly, at both sites, the cross-link is located within the cavity formed at the junction of the three protein monomers (Figure 4D).
Bicyclization of structurally diverse homotrimeric proteins
The bicyclization of trimeric PFE provides a unique architecture that has not been reported for protein complexes before. To assess the applicability of this approach to other homotrimeric proteins, we searched the RCSB PDB for C3-symmetric homotrimers with 50–400 residues per monomer and identified ca. 4,000 structures. To simplify protein production and characterization, this pool was reduced by selecting for proteins with a hexahistidine tag, at least one tryptophan, and confirmed expression in E. coli. In addition, we allowed a maximum of one cysteine residue per monomer to minimize off-target reactivity. These filter criteria produced an initial set of 119 trimers (Table S9), of which 18 structurally diverse protein complexes spanning 14 unique folds, as classified by their class, architecture, topology, and homologous superfamily (CATH) domain identities,40 were selected after visual inspection. To enable bicyclization, two residues per monomer were substituted for cysteine, with distances of 8–15 Å between Cα atoms of the resulting cysteine triplet, producing a total of 18 designs (Tables S10 and S11). Protein variants were expressed in BL21(DE3) cells in an IPTG inducible pET28(+) vector, retaining the purification tags from the original PDB sequences. Expression and purification were successful for 17 of the 18 variants (>0.5 mg protein from 1.5 L culture).
Cross-linking reactions with Ta-I3 were performed in analogy to the bicyclization of p4. Initially, stabilization effects were analyzed by thermal denaturation experiments using DSF to compare Ti values prior to and after cross-linking. Of the 17 obtained proteins, 10 showed a substantial increase in thermostability upon Ta-I3 treatment (ΔTi > 5°C, Figures S10 and S11) with 7 of them surpassing the stabilization effects observed for PFE (ΔTi > 10°C). The ten proteins that showed clearly increased thermal stability upon cross-linker treatment were analyzed by high-performance liquid chromatography (HPLC) coupled with MS to assess their cross-linking status. For five, we did not obtain interpretable spectra, which may suggest inhomogeneity or incompleteness of the cross-linking reaction or could be a result of the inherent technical difficulties of MS analysis of large, interlinked protein chains. For the remaining five variants, we confirmed covalent trimer formation (Figures S12–S16). One of these, however, only reacted with one cross-linker (Figure S12). For the other four complexes (Figure 5), we confirmed the formation of the bis-crosslinked trimers (monomer3Ta2, Figures S13–S16), additionally validated by SDS-PAGE (Figure S17).
Figure 5.

Stabilization of different homotrimeric proteins via bicyclization
(A) Characterization of l43Ta2 (Ip1913, PDB: 3fnj).
(B) Characterization of b43Ta2 (BH3489, PDB: 1vmf).
(C) Characterization of a43Ta2 (Au4130, PDB: 3c6v).
(D) Characterization of e43Ta2 (ECHD, PDB: 5c9g). Reported crystal structures and cysteine variation sites with average distances (davg) between Cα atoms of cross-linking residues are shown. Calculated and found molecular weights (MWs) and CD-derived thermal denaturation curves of unmodified (black) and corresponding cross-linked trimers (orange) are provided, including the resulting difference in melting temperatures (ΔTm). Tm values (Table S12) represent the maximum of the first derivative (corresponding HT curves and CD spectra are available in Figure S18).
The four confirmed bicyclic proteins originate from a diverse set of structural classes with MWs ranging from 34 to 91 kDa, and distances between introduced cross-linking sites ranging from 10 to 15 Å (Figure 5). To further validate their thermal stability, CD thermal denaturation experiments were performed providing increases in melting temperatures (ΔTm = 6°C–39°C) in line with DSF-derived values. The smallest bicyclic trimer, l43Ta2 (MW = 34 kDa, Figure 5A), was derived from lp_1913,41 a rhodanese-like domain from Lactobacillus plantarum, and revealed a considerably increased melting temperature (ΔTm = 19°C). Bicyclization of the BH349842 variant b4 resulted in b43Ta2 (MW = 50 kDa, Figure 5B) experiencing the smallest stabilization effect (ΔTm = 6°C) among the four tested variants. The heterologous interfaces of this highly compact trimeric structure are large relative to the size of the trimer, which may explain the moderate stability enhancement upon cross-linking. Thermal denaturation of l43Ta2 and b43Ta2, as well as their monomeric precursors (I4 and b4), produced an unexpected reduction in ellipticity at 220 nm; however, this wavelength remained the most effective way of assaying unfolding by CD. As the third example, bicyclic trimer a43Ta2 (Figure 5C) was derived from Au4130,43 an uncharacterized enzyme from Aspergillus fumigatus containing a tautomerase-3 domain. It showed the most pronounced thermo-stabilization effect upon cross-linking (ΔTm = 39°C). Notably, the un-cross-linked protein itself was the least stable of the tested targets (Tm = 41°C). The fourth bicyclic trimer e43Ta2 (Figure 5D), originated from ECHD, a putative enoyl-coenzyme A (coA) hydratase variant,44 shares the most structural similarity with PFE among the four additional proteins. However, the sequence similarity is minimal (22% sequence identity), and their structural domains are non-homologous. Bicyclization of e4 again resulted in a substantially increased thermal stability (ΔT m = 17°C).
Conclusions
We report the bicyclization and stabilization of trimeric proteins, which have been considered particularly difficult targets for stabilization efforts. In the case of PFE, bicyclization enhanced enzyme activity under chemical stress and reduced the formation of higher-order aggregates, as well as precipitation. This suggests that the cross-links tether the quaternary structure, thereby preventing exposure of the hydrophobic interior, which, in turn, results in a reduced tendency to nucleate initial, soluble aggregation states. This may also be supported by the polar nature of the cross-link conveying additional solubility. Most notably, the increased thermal stability and solubility of p43Ta2 results in an exceptional longevity with full enzyme activity after more than 3 weeks of storage in buffer at 50°C.
Crucially, we show the bicyclization strategy to be applicable across diverse structural domains. The protein conjugates we present here constitute an abiological topology and were obtained by converting protein complexes into quasi-tertiary structures with augmented connectivity. This topology results in increased compactness in the denatured state and may thereby mimic an intermediate state in the protein folding trajectory. Such systems can therefore shed new light on protein folding and its modulation.2,45,46,47 Considering the crucial role of proteins in emergent biotechnological and biomedical applications, the reported chemically modified complexes constitute an attractive modality for applications in sustainable biocatalysis, protein therapy, and diagnostics.
Experimental procedures
Further experimental procedures can be found in the supplemental information.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Tom Grossmann (t.n.grossmann@vu.nl).
Materials availability
All materials generated in this study are available from the lead contact with a completed materials transfer agreement and reasonable compensation by the requestor for its production, processing, and shipping.
Synthesis of Ta-I3
A round bottom flask was charged with 100 mg (0.206 mmol, 1 equiv) of Ta-Cl3 (obtained as earlier reported)27 and suspended in 2.1 mL acetone. Sodium iodine 559 mg (3.70 mmol, 18 equiv) was added, and the reaction mixture was stirred vigorously at room temperature for 16 h. 2 mL of water was added, and the clear yellowish solution was directly subjected to reversed-phase chromatography (water + 0.1% formic acid (FA)/acetonitrile (ACN) + 0.1% FA 90/10 to 10/90 in 15 min). Pure fractions were combined and lyophilized to give 120 mg (0.157 mmol) of a white lyophilizate with 76% yield. 1H NMR (600 MHz, MeOD) δ 5.37 (s, 6H), 4.28 (s, 6H), 3.80 (s, 6H). 13C NMR (151 MHz, MeOD) δ 171.86, 169.79, 56.93, 42.59, -2.45. High-resolution MS (Agilent 6230 ESI-TOF HPLC-MS) calculated for C15H21I3N6O6Na+ [M + Na]+ = 784.8549, found 784.8510.
Cross-linking reactions
Protein cross-linking was carried out by mixing 15 μM of protein (trimer concentration, i.e., monomer concentration of 45 μM) with 1 mM of Ta-Cl3 or 0.3 mM of Ta-I3 (freshly prepared in buffer at a concentration of 5 or 1 mM, respectively) in 50 mM HEPES, 50 mM NaCl buffered at pH 8, in a total reaction volume of 80–300 μL. Prior to cross-linking, protein stocks were concentrated to at least 65 μM (trimer concentration) in purification buffer (50 mM HEPES, 150 mM NaCl, at pH 8). This buffer also contained 0.5 mM TCEP to ensure reduction of cysteine side chains prior to cross-linking. The subsequent dilution of the protein upon mixing with the cross-linker resulted in dilution of the TCEP concentration to a maximum of 115 μM. Reactions were incubated at 20°C (except where indicated) overnight (Ta-Cl3), or 2–4 h (Ta-I3), and excess cross-linker was subsequently removed by buffer exchange with an Amicon centrifugal concentrator. Cross-linked samples for crystallography were further purified by an additional size exclusion chromatography step, as in the supplemental information section protein expression and purification. Cross-linking reactions for the determination of predominant charge states of PFE variants p23Ta, p33Ta, and p43Ta2 were quenched by addition of formic acid to a final concentration of 1% (v/v).
Thermal denaturation (DSF and CD)
Thermal stability was initially assessed by DSF using a Tycho NT.6 instrument. The fluorescence ratio of 350/330 nm was measured between 35°C–95°C (with a temperature ramp rate of 30°C/min) at a protein trimer concentration of 5 μM (50 mM HEPES, 50 mM NaCl, pH 8) and a given concentration of GuHCl. The fixed temperature ramp rate of this instrument is 30°C/min, from which we determined a comparative inflection midpoint (Ti). This value is likely to overestimate absolute stability; therefore, thermal stability was further validated by subsequent CD experiments at reduced ramp rate (1°C/min). Samples for CD were measured at a trimer concentration of 3–5 μM (50 mM potassium phosphate, 50 mM potassium chloride, pH 8) in a 1 mM quartz cuvette in a JASCO J1500 instrument. Thermal melts were recorded at 220 nm from 5°C to 95°C with a ramp rate of 1°C/min to determine the equilibrium unfolding midpoint (Tm). Full spectra were also recorded at 5°C and 95°C between 200 and 260 nm with a bandwidth of 1 nm, averaging four spectra accumulations. HT voltage was maintained below 500 V at every wavelength for all measurements. CD measurements are reported as mean residual ellipticity (MRE), converted by the following equation, in which n is the number of peptide bonds in the given protein.
Relative DSF and CD measurements for unmodified and cross-linked variants were in good accordance (ΔTi vs. ΔTm), however absolute Tm values were approximately 5°C–10°C lower, likely due to the slower temperature ramp.
Dynamic light scattering
Temperature-dependent dynamic light scattering (DLS) measurements were obtained using a Prometheus Panta (Nanotemper) instrument at a trimer concentration of 15 μM (50 mM HEPES, 50 mM NaCl, pH 8). Measurements were taken at 0.17°C intervals with a temperature ramp rate of 1°C/min. The determined autocorrelation function at each given temperature was fitted to obtain a size distribution using Panta Control (version 1.6.3) software. Six size distribution fits within a +0.95°C range of the temperature were averaged (e.g., data for T = 50°C represent an average of six measurements between 50.00°C and 50.95°C). The cumulant radii (indicating the predominant particle size) for both samples at every measured temperature between 20°C and 95°C were also calculated using Panta Control software, presented in Figure S8.
Crystallography
Initial screening of crystallization conditions was performed using the JSCG core suite I–IV at 20°C. Screens were assembled in 96-well plates using the sitting-drop vapor diffusion method, with a drop volume of 200 nL dispensed by a Mosquito crystallization robot (SPT Labtech). A protein concentration of 20 mg/mL was mixed in a 1:1 ratio with each crystallization solution. Refinement screens (drop size 1 μL) from initial hits identified two optimal crystallization solutions for growth of p43Ta2 crystals: solution I (100 mM N-cyclohexyl-2-aminoethanesulfonic acid, pH 10, 25% [w/v] PEG3000) and crystallization solution II (210 mM lithium acetate, 19% [w/v] PEG3350). Obtained crystals were collected, cryo-protected by soaking in 15%–20% (v/v) glycerol, and flash frozen in liquid nitrogen. Diffraction data were obtained at the i04 beamline at the Diamond Light Source (United Kingdom). Data were integrated using xds48 and two datasets were scaled using xscale. The crystal structure was solved by molecular replacement using Phaser within the CCP4i2 software suite49 and PDB: 1va4 as a model. Structure building and refinement was performed using Coot, Refmac5, and PDBRedo. X-ray collection and refinement statistics are provided in Table S8. The crystal structure of bicyclic p43Ta2 has been deposited to the PDB: 8pi1.
Acknowledgments
We thank David J. Hamilton for support with compound analytics. We thank the user support team, as well as the beamline staff of I04 at the Diamond Light Source (Oxfordshire, UK) for their support. Funding was provided by the European Research Council ERC proof-of-concept, no. 839088 (T.N.G.) and EU Commission in the framework of the Horizon Europe – EIC Transition Open programme, no. 101057978 (Incircular B.V., S.N.).
Author contributions
S.N., S.H., and T.N.G. conceived and designed the project. P.P. and M.I. performed the chemical synthesis. Protein expression and cross-linking was performed by G.H.H., S.K., P.P., I.D., and S.N. MS characterization data processing was performed by A.H.L., R.S., and A.M.R. Enzyme activity measurements, as well as DSF and CD analysis, were performed by G.H.H., S.K., and J.O. Protein crystallography and data processing was performed by S.K., N.M.P., and S.H. All authors analyzed the results. All authors discussed the results and commented on the manuscript. G.H.H., S.N., S.H., and T.N.G. prepared the manuscript.
Declaration of interests
G.H.H., S.K., P.P., I.D., S.N., S.H., and T.N.G. are listed as inventors on a patent application related to the cross-linking of protein complexes. S.N., S.H., and T.N.G. are co-founders and shareholders of Incircular BV, commercializing the corresponding bioconjugation technology. S.H. and T.N.G. are advisers of Incircular BV.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: November 2, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chempr.2023.10.003.
Contributor Information
Saskia Neubacher, Email: saskia@incircular.com.
Sven Hennig, Email: s.hennig@vu.nl.
Tom N. Grossmann, Email: t.n.grossmann@vu.nl.
Supplemental information
Data and code availability
The crystal structure of p43Ta2 (bicyclic PFE) has been deposited to the PDB with accession number PDB: 8pi1. All other data supporting this study are available in the manuscript and supplemental information.
References
- 1.Bornscheuer U.T., Huisman G.W., Kazlauskas R.J., Lutz S., Moore J.C., Robins K. Engineering the third wave of biocatalysis. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 2.Horne W.S., Grossmann T.N. Proteomimetics as protein-inspired scaffolds with defined tertiary folding patterns. Nat. Chem. 2020;12:331–337. doi: 10.1038/s41557-020-0420-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benítez-Mateos A.I., Roura Padrosa D., Paradisi F. Multistep enzyme cascades as a route towards green and sustainable pharmaceutical syntheses. Nat. Chem. 2022;14:489–499. doi: 10.1038/s41557-022-00931-2. [DOI] [PubMed] [Google Scholar]
- 4.Arnold F.H. Directed evolution: bringing new chemistry to life. Angew. Chem. Int. Ed. Engl. 2018;57:4143–4148. doi: 10.1002/anie.201708408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwizer F., Okamoto Y., Heinisch T., Gu Y., Pellizzoni M.M., Lebrun V., Reuter R., Köhler V., Lewis J.C., Ward T.R. Artificial metalloenzymes: reaction scope and optimization strategies. Chem. Rev. 2018;118:142–231. doi: 10.1021/acs.chemrev.7b00014. [DOI] [PubMed] [Google Scholar]
- 6.Hagner K., Setayeshgar S., Lynch M. Stochastic protein multimerization, activity, and fitness. Phys. Rev. E. 2018;98 doi: 10.1103/PhysRevE.98.062401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reetz M.T. The importance of additive and non-additive mutational effects in protein engineering. Angew. Chem. Int. Ed. Engl. 2013;52:2658–2666. doi: 10.1002/anie.201207842. [DOI] [PubMed] [Google Scholar]
- 8.Magliery T.J. Protein stability: computation, sequence statistics, and new experimental methods. Curr. Opin. Struct. Biol. 2015;33:161–168. doi: 10.1016/j.sbi.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman A.M., McNaughton B.R. Scratching the surface: resurfacing proteins to endow new properties and function. Cell Chem. Biol. 2016;23:543–553. doi: 10.1016/j.chembiol.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aalbers F.S., Fürst M.J.L.J., Rovida S., Trajkovic M., Gómez Castellanos J.R., Bartsch S., Vogel A., Mattevi A., Fraaije M.W. Approaching boiling point stability of an alcohol dehydrogenase through computationally-guided enzyme engineering. eLife. 2020;9 doi: 10.7554/eLife.54639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujita D., Suzuki R., Fujii Y., Yamada M., Nakama T., Matsugami A., Hayashi F., Weng J.-K., Yagi-Utsumi M., Fujita M. Protein stabilization and refolding in a gigantic self-assembled cage. Chem. 2021;7:2672–2683. doi: 10.1016/j.chempr.2021.08.005. [DOI] [Google Scholar]
- 12.Nooren I.M.A., Thornton J.M. Diversity of protein-protein interactions. EMBO J. 2003;22:3486–3492. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bosshart A., Panke S., Bechtold M. Systematic optimization of interface interactions increases the thermostability of a multimeric enzyme. Angew. Chem. Int. Ed. Engl. 2013;52:9673–9676. doi: 10.1002/anie.201304141. [DOI] [PubMed] [Google Scholar]
- 14.Meng Q., Capra N., Palacio C.M., Lanfranchi E., Otzen M., Van Schie L.Z., Rozeboom H.J., Thunnissen A.W.H., Wijma H.J., Janssen D.B. Robust ω-transaminases by computational stabilization of the subunit interface. ACS Catal. 2020;10:2915–2928. doi: 10.1021/acscatal.9b05223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuhlman B., O’Neill J.W., Kim D.E., Zhang K.Y.J., Baker D. Conversion of monomeric protein L to an obligate dimer by computational protein design. Proc. Natl. Acad. Sci. USA. 2001;98:10687–10691. doi: 10.1073/pnas.181354398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson C.R., Sauer R.T. Striking stabilization of Arc repressor by an engineered disulfide bond. Biochemistry. 2000;39:12494–12502. doi: 10.1021/bi001484e. [DOI] [PubMed] [Google Scholar]
- 17.Li J.C., Liu T., Wang Y., Mehta A.P., Schultz P.G. Enhancing protein stability with genetically encoded noncanonical amino acids. J. Am. Chem. Soc. 2018;140:15997–16000. doi: 10.1021/jacs.8b07157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang H., Sandberg W.S., Terwilliger T.C. Genetic fusion of subunits of a dimeric protein substantially enhances its stability and rate of folding. Proc. Natl. Acad. Sci. USA. 1993;90:7010–7014. doi: 10.1073/pnas.90.15.7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akanuma S., Matsuba T., Ueno E., Umeda N., Yamagishi A. Mimicking the evolution of a thermally stable monomeric four-helix bundle by fusion of four identical single-helix peptides. J. Biochem. 2010;147:371–379. doi: 10.1093/jb/mvp179. [DOI] [PubMed] [Google Scholar]
- 20.Dellarole M., Sánchez I.E., Freire E., De Prat-Gay G. Increased stability and DNA site discrimination of “single chain” variants of the dimeric β-barrel DNA binding domain of the human papillomavirus e2 transcriptional regulator. Biochemistry. 2007;46:12441–12450. doi: 10.1021/bi701104q. [DOI] [PubMed] [Google Scholar]
- 21.Hashimoto K., Panchenko A.R. Mechanisms of protein oligomerization, the critical role of insertions and deletions in maintaining different oligomeric states. Proc. Natl. Acad. Sci. USA. 2010;107:20352–20357. doi: 10.1073/pnas.1012999107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider A.F.L., Kithil M., Cardoso M.C., Lehmann M., Hackenberger C.P.R. Cellular uptake of large biomolecules enabled by cell-surface-reactive cell-penetrating peptide additives. Nat. Chem. 2021;13:530–539. doi: 10.1038/s41557-021-00661-x. [DOI] [PubMed] [Google Scholar]
- 23.Kale S.S., Bergeron-Brlek M., Wu Y., Kumar M.G., Pham M.V., Bortoli J., Vesin J., Kong X.D., Machado J.F., Deyle K., et al. Thiol-to-amine cyclization reaction enables screening of large libraries of macrocyclic compounds and the generation of sub-kilodalton ligands. Sci. Adv. 2019;5:eaaw2851. doi: 10.1126/sciadv.aaw2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prescher J.A., Dube D.H., Bertozzi C.R. Chemical remodelling of cell surfaces in living animals. Nature. 2004;430:873–877. doi: 10.1038/nature02791. [DOI] [PubMed] [Google Scholar]
- 25.Moore E.J., Zorine D., Hansen W.A., Khare S.D., Fasan R. Enzyme stabilization via computationally guided protein stapling. Proc. Natl. Acad. Sci. USA. 2017;114:12472–12477. doi: 10.1073/pnas.1708907114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pelay-Gimeno M., Bange T., Hennig S., Grossmann T.N. In situ cyclization of native proteins: structure-based design of a bicyclic enzyme. Angew. Chem. Int. Ed. Engl. 2018;57:11164–11170. doi: 10.1002/anie.201804506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neubacher S., Saya J.M., Amore A., Grossmann T.N. In situ cyclization of proteins (INCYPRO): cross-link derivatization modulates protein stability. J. Org. Chem. 2020;85:1476–1483. doi: 10.1021/acs.joc.9b02490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cigler M., Müller T.G., Horn-Ghetko D., von Wrisberg M.K., Fottner M., Goody R.S., Itzen A., Müller M.P., Lang K. Proximity-triggered covalent stabilization of low-affinity protein complexes in vitro and in vivo. Angew. Chem. Int. Ed. Engl. 2017;56:15737–15741. doi: 10.1002/anie.201706927. [DOI] [PubMed] [Google Scholar]
- 29.Zhou L., Chai F., He Y., Zhou Z., Guo S., Li P., Sun Q., Zu X., Liu X., Huang Q., et al. Homodimerized cytoplasmic domain of PD-L1 regulates its complex glycosylation in living cells. Commun. Biol. 2022;5:887. doi: 10.1038/s42003-022-03845-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen T.A., Gronauer T.F., Nast-Kolb T., Sieber S.A., Lang K. Substrate profiling of mitochondrial caseinolytic protease P via a site-specific photocrosslinking approach. Angew. Chem. Int. Ed. Engl. 2022;61 doi: 10.1002/anie.202111085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiehstaller S., Hutchins G.H., Amore A., Gerber A., Ibrahim M., Hennig S., Neubacher S., Grossmann T.N. Bicyclic engineered sortase A performs transpeptidation under denaturing conditions. Bioconjug. Chem. 2023;34:1114–1121. doi: 10.1021/acs.bioconjchem.3c00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Timmerman P., Beld J., Puijk W.C., Meloen R.H. Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. Chembiochem. 2005;6:821–824. doi: 10.1002/cbic.200400374. [DOI] [PubMed] [Google Scholar]
- 33.Heinis C., Rutherford T., Freund S., Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009;5:502–507. doi: 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- 34.Chen S., Bertoldo D., Angelini A., Pojer F., Heinis C. Peptide ligands stabilized by small molecules. Angew. Chem. Int. Ed. Engl. 2014;53:1602–1606. doi: 10.1002/anie.201309459. [DOI] [PubMed] [Google Scholar]
- 35.Yin D.L., Bernhardt P., Morley K.L., Jiang Y., Cheeseman J.D., Purpero V., Schrag J.D., Kazlauskas R.J. Switching catalysis from hydrolysis to perhydrolysis in pseudomonas fluorescens esterase. Biochemistry. 2010;49:1931–1942. doi: 10.1021/bi9021268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt M., Hasenpusch D., Kähler M., Kirchner U., Wiggenhorn K., Langel W., Bornscheuer U.T. Directed evolution of an esterase from Pseudomonas fluorescens yields a mutant with excellent enantioselectivity and activity for the kinetic resolution of a chiral building block. Chembiochem. 2006;7:805–809. doi: 10.1002/cbic.200500546. [DOI] [PubMed] [Google Scholar]
- 37.Cheeseman J.D., Tocilj A., Park S., Schrag J.D., Kazlauskas R.J. Structure of an aryl esterase from Pseudomonas fluorescens. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1237–1243. doi: 10.1107/S0907444904010522. [DOI] [PubMed] [Google Scholar]
- 38.Drienovská I., Gajdoš M., Kindler A., Takhtehchian M., Darnhofer B., Birner-Gruenberger R., Dörr M., Bornscheuer U.T., Kourist R. Folding assessment of incorporation of noncanonical amino acids facilitates expansion of functional-group diversity for enzyme engineering. Chemistry. 2020;26:12338–12342. doi: 10.1002/chem.202002077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muraoka T., Sadhukhan N., Ui M., Kawasaki S., Hazemi E., Adachi K., Kinbara K. Thermal-aggregation suppression of proteins by a structured PEG analogue: importance of denaturation temperature for effective aggregation suppression. Biochem. Eng. J. 2014;86:41–48. doi: 10.1016/j.bej.2014.03.001. [DOI] [Google Scholar]
- 40.Knudsen M., Wiuf C. The CATH database. Hum. Genomics. 2010;4:207–212. doi: 10.1186/1479-7364-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seetharaman J., Chen Y., Forouhar F., Sahdev S., Janjua H., Xiao R., Ciccosanti C., Foote E.L., Acton T.B., Rost B., et al. 2008. Crystal structure of the full-length lp_1913 protein from Lactobacillus plantarum, Northeast Structural Genomics Consortium Target LpR140. [DOI] [Google Scholar]
- 42.Joint Center for Structural Genomics (JCSG) 2004. Crystal structure of a YBJQ-like fold protein of unknown function (BH3498) from Bacillus halodurans at 1.46 A resolution. [DOI] [Google Scholar]
- 43.Singer A.U., Binkowski T.A., Skarina T., Kagan O., Edwards A.M., Joachimiak A., Savchenko A., Midwest Center for Structural Genomics (MCSG) 2008. Crystal structure of AU4130/APC7354, a probable enzyme from the thermophilic fungus Aspergillus fumigatus. [DOI] [Google Scholar]
- 44.Szlachta K., Cooper D.R., Chapman H.C., Cymbrowski M.T., Stead M., Hillerich B.S., Ahmed M., Bonanno J., Seidel R., Almo S.C., et al. 2015. Crystal structure of a putative enoyl-CoA hydratase/isomerase family protein from Hyphomonas neptunium. [DOI] [Google Scholar]
- 45.Jiang Y., Neti S.S., Sitarik I., Pradhan P., To P., Xia Y., Fried S.D., Booker S.J., O’Brien E.P. How synonymous mutations alter enzyme structure and function over long timescales. Nat. Chem. 2023;15:308–318. doi: 10.1038/s41557-022-01091-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newberry R.W., Raines R.T. Secondary forces in protein folding. ACS Chem. Biol. 2019;14:1677–1686. doi: 10.1021/acschembio.9b00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gan Q., Ferrand Y., Bao C., Kauffmann B., Grélard A., Jiang H., Huc I. Helix-rod host-guest complexes with shuttling rates much faster than disassembly. Science. 2011;331:1172–1175. doi: 10.1126/science.1200143. [DOI] [PubMed] [Google Scholar]
- 48.Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Potterton L., Agirre J., Ballard C., Cowtan K., Dodson E., Evans P.R., Jenkins H.T., Keegan R., Krissinel E., Stevenson K., et al. CCP4i2: the new graphical user interface to the CCP4 program suite. Acta Crystallogr. D Struct. Biol. 2018;74:68–84. doi: 10.1107/S2059798317016035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystal structure of p43Ta2 (bicyclic PFE) has been deposited to the PDB with accession number PDB: 8pi1. All other data supporting this study are available in the manuscript and supplemental information.