

Abstract

Understanding the transport and inhibition mechanisms of substrates by P-glycoprotein (P-gp) is one of the important approaches in addressing multidrug resistance (MDR). In this study, we evaluated a variety of rhodamine derivatives as potential P-gp inhibitors targeting CmABCB1, a P-gp homologue, with a focus on their ATPase activity. Notably, a Q-rhodamine derivative with an o,o′-dimethoxybenzyl ester moiety (RhQ-DMB) demonstrated superior affinity and inhibitory activity, which was further confirmed by a drug susceptibility assay in yeast strains expressing CmABCB1. Results from a tryptophan fluorescence quenching experiment using a CmABCB1 mutant suggested that RhQ-DMB effectively enters and binds to the inner chamber of CmABCB1. These findings underscore the promising potential of RhQ-DMB as a tool for future studies aimed at elucidating the substrate-bound state of CmABCB1.
Keywords: Multidrug resistance, ATP-Binding cassette transporter, Rhodamine, Inhibitor, Structure−activity relationship
Multidrug resistance (MDR) poses a significant challenge in cancer treatment, rendering cancer cells resistant to a broad spectrum of drugs with varying structures and mechanisms of action.1,2 According to recent studies on the MDR of cancer cells,3−5 the overexpression of ATP-binding cassette (ABC) transporters plays a pivotal role in facilitating MDR activity in cancer cells. The ABC transporter superfamily in humans is comprised of 48 members, of which three are primarily implicated in MDR.6 P-glycoprotein (P-gp), also known as MDR1 or ABCB1, is the earliest example identified to be associated with MDR.7,8 P-gp is capable of recognizing and transporting a wide range of substances, including toxic xenobiotics, endogenous metabolites, and chemotherapeutic drugs such as paclitaxel and doxorubicin.9,10 Thus, P-gp reduces the intracellular accumulation of these drugs, eventually causing drug resistance.11,12
For years, P-gp has been identified as a potential therapeutic target to overcome MDR. The strategy of combining P-gp modulators with anticancer drugs has been viewed as a promising avenue to counteract P-gp-mediated MDR.13 However, despite extensive research for development of an efficient P-gp modulators over the past several decades,14−17 there is still much room for improvement. One of the major obstacles is that the mechanisms governing substrate transport and inhibition, including (a) the structure of P-gp when bound to the substrate or postsubstrate efflux and (b) the substrate binding site and mode of transportation, remain elusive. On a positive note, advances in structural biology have begun to shed light on the three-dimensional structures of P-gps.17−22 Even the structure of human P-gp bound to a substrate has been revealed by a cryo-EM technique.17,23−25
In the transport cycle, the ABCB1 transporter initially assumes an inward-facing conformation, exposing the substrate-binding cavity (Figure 1a). This conformation facilitates binding of the substrate to the transmembrane domain. Following this, ATP binds to the nucleotide-binding domain, triggering a transition to an outward-facing conformation. In this state, the substrate-binding cavity collapses, resulting in the transport of the substrate out of the cell. However, comprehensive structural data including each inward- and outward-facing state have only been sporadically reported and often in conjunction with the use of different mutants. To deepen our understanding of the transport process, it is crucial to examine the structure of these states in wild-type P-gp or at least in the same mutant form. In line with our ongoing research into the molecular mechanisms of substrate transport by P-gp, we have elucidated the high-resolution inward- and outward-facing structures of Cyanidioschyzon marolae P-gp (CmABCB1), a eukaryotic homologue of ABCB1 with a similar amino acid sequence, structure, and substrate specificity to human P-gp, using X-ray crystallography.19,21 However, it is crucial to note that the substrate-bound state of CmABCB1 remains a puzzling aspect of our research. Addressing this gap is critical for a comprehensive understanding of the molecular dynamics of this transporter. To explore the substrate-bound state of CmABCB1, we focused on the development of inhibitors that can enter the inner chamber of CmABCB1 but are not readily effluxed (Figure 1b). Among the potential substrates of CmABCB1, we selected rhodamines, well-known transport substrates for various ABC transporters including human P-gp, for a structure–activity relationship study, which was informed by our two key precedents: (a) rhodamine 6G (1) has exhibited stimulation and inhibition activities toward CmABCB1,19 and (b) tryptophan quenching analysis revealed that several commercially available rhodamines can access the inner chamber of CmABCB1.26 In addition to these findings, our plan was also drawn from the pioneering work of Raub and Detty, who developed P-gp inhibitors based on the sulfur-rhodamine derivatives.14 In this study, we synthesized a variety of rhodamine derivatives and evaluated their affinity and inhibitory activity against CmABCB1, leading to the discovery of the novel Q-rhodamine (RhQ; Q for tetrahydroquinoline) derivative 2, RhQ-DMB, as a potent P-gp inhibitor, a tool for future studies aimed at elucidating the substrate-bound state of CmABCB1.
Figure 1.

(a) Mode of action of ABCB1. (b) Development of a CmABCB1 inhibitor.
Based on an established synthetic method, 19 rhodamine derivatives were synthesized as shown in Scheme 1.27 Aminophenols I and phthalic anhydride (3) were heated in o-dichlorobenzene at 175 °C to give the corresponding carboxylate II with a rhodamine skeleton in one step. The subsequent esterification with various alcohols was conducted under standard conditions using a condensation reagent (EDC·HCl, DMAP) or alkyl iodide under basic conditions (Cs2CO3). Purification by silica gel column chromatography (prewashed with MeOH, eluent: CHCl3/MeOH) afforded the target compound III. Comprehensive synthetic procedures for each rhodamine derivative are detailed in the Supporting Information.
Scheme 1. Synthesis of Rhodamine Derivatives.

Reagents and conditions: (a) o-dichlorobenzene, 175 °C; (b) alcohol, EDC·HCl, DMAP, CH2Cl2, rt; (c) alkyl iodide, Cs2CO3, DMF, rt.
The ATPase activities of the synthesized rhodamine derivatives were evaluated to establish a SAR. In general, ABC transporters hydrolyze ATP to ADP and phosphate while effluxing substrates out of the cell,28 a property also exhibited by CmABCB1.19 Our previous research has illuminated that CmABCB1, when in micelles, exhibits high thermal stability, making it amenable to purification followed by quantitative kinetic analyses of ATPase activity.19
CmABCB1 exhibits low basal ATPase activity in the absence of a transport substrate but shows higher ATPase activity when substrates are present, a phenomenon termed substrate-stimulated ATPase activity in human P-gp (Figure 2).19,29,30 During titration with P-gp substrates, ATPase activity initially increases (Figure 2a), reaching a maximum before declining at higher substrate concentrations (Figure 2b) to form a characteristic bell-shaped profile analyzed using modified Michaelis–Menten kinetics.31−34 The decrease in ATPase activity is attributed to substrate inhibition, which is a common feature of transporters and enzymes. Along these lines, we evaluated the affinity (Km) and inhibitory activity (Ki) of the substrates against CmABCB1. The parameters are derived from the equation described in the Supporting Information. Substrates with a low Km value induce the structural change of the transporter by entering its inner chamber, whereas ones with a low Ki value inhibit substrate transport and are not readily effluxed. Thus, the ideal candidate of the inhibitor consistent with our aim would possess low values of both Km and Ki.
Figure 2.

(a) The behavior of CmABCB1 at low substrate concentration showing substrate-stimulated ATPase activity. (b) The behavior of CmABCB1 at high substrate concentration showing substrate inhibition.
Upon screening rhodamine derivatives, RhQ-DMB (2) emerged as a potent inhibitor, exhibiting an affinity 5 times greater and an inhibitory activity 120 times higher than the reference compound (Figure 3), i.e., rhodamine 6G (1), as detailed below.
Figure 3.

Substrate-concentration dependence of the CmABCB1 ATPase activity. The ATPase activity was measured as a function of the rhodamine 6G (1) or RhQ-DMB (2) concentration with 5 mM ATP at 37 °C in a buffer solution of 50 mM Tris–HCl (pH 7.5 at 37 °C), 150 mM NaCl, and 0.05% (w/v) β-DDM for 30 min. Data are means ± SD (n = 3). The solid line is a fit of the equation to the data as described in the Supporting Information (eq 1, S4).
Table 1 shows the results of the affinity and inhibitory activity of rhodamine derivatives 1 and 4–8 with different substituents, mainly in the tricyclic xanthene core. The parent compound, rhodamine 6G (1), exhibited moderate affinity (Km = 2.9 μM) and inhibitory activity (Ki = 690 μM). By comparison, the corresponding butyl ester 4 showed a slight improvement in inhibitory activity and served as a benchmark for subsequent studies. N-Butyl derivative 5 showed improved inhibitory activity (Ki = 160 μM), although its affinity remained unchanged (Km = 3.2 μM). The introduction of a pyrrolidine ring (6) led to improvements in both affinity (Km = 1.1 μM) and inhibitory activity (Ki = 110 μM).
Table 1. Effect of the Xanthene Moiety on Affinity and Inhibitory Activity toward CmABCB1a.


Km and Ki values were obtained from t hree independent repeats except 6. Data of 6 were obtained from four independent repeats.
Further improvements were observed with Q-rhodamine 7, a pentacyclic rhodamine derivative with two fused piperidine rings, which exhibited significantly improved inhibitory activity (Ki = 14 μM), while maintaining an affinity (Km = 1.7 μM) comparable to that of 6. By comparison, X-rhodamine 8 displayed only moderate inhibitory activity with a Ki value of 43 μM.
Having identified the optimal structure for the xanthene moiety, we proceeded to investigate variations in the ester moiety, as detailed in Table 2. Given that the inner chamber of CmABCB1 is predominantly hydrophobic and rich in aromatic residues,19 we hypothesized that the incorporation of an aromatic ring could hopefully enhance the affinity and inhibitory activity through an aromatic interaction. This idea aligns with findings that aromatic moieties increase binding affinity toward other homologues of P-glycoprotein.35−37 Pleasingly, the introduction of both a benzyl group (9) and a naphthyl group (10) led to improvements in both affinity and inhibitory activity. Rhodamine derivative 11 with a phenethyl group, i.e., an aromatic substituent that is one carbon longer than a benzyl group, showed reduced affinity (Km = 1.4 μM). Next, the substituents of the benzyl group were further investigated (2, 12–21). It was found that the introduction of one substituent at the meta-position, whether an electron-donating or electron-withdrawing group, was effective with respect to affinity in all case: A benzyl group with methyl (12), dimethylamino (13), methoxy (14), trifluoromethyl (15), and nitro (16) groups led to improved affinity. In terms of inhibitory activity, derivatives 15 and 16 with an electron-withdrawing group showed moderate reduction (Ki = 18 μM for 15 and Ki = 12 μM for 16).
Table 2. Effects of the Ester Moiety on Affinity and Inhibitory Activity toward CmABCB1a.
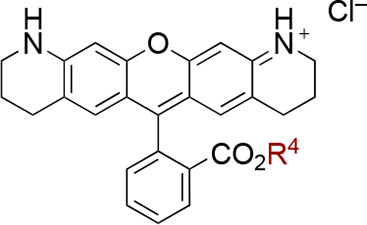

Km and Ki values were obtained from three independent repeats.
We subsequently evaluated the impact of varying the positions and numbers of substituents (2, 17–21), selecting the methoxy and trifluoromethyl groups as electron-donating and -withdrawing groups, respectively. Derivatives with one trifluoromethyl group in the ortho position (17) or two trifluoromethyl groups in the meta position (18) showed a clear reduction in affinity. The data for compound 17 present an unusual scenario where the value of Km exceeds Ki. This might indicate that substrate inhibition occurs notably earlier than the point at which the hyperbolic response curve approaches saturation, a phenomenon that is illustrated in Figure 2. By comparison, the methoxy group, depending on the number and position, had a positive effect on both affinity and inhibitory activity. Among several derivatives with o- (19), p- (20), m,m′- (21), and o,o′-dimethoxy group(s) (2), o,o′-dimethoxybenzyl derivative 2 (RhQ-DMB) showed well-balanced affinity and inhibitory activity with a 5-fold improvement in affinity (Km = 0.55 μM) and a 120-fold improvement in inhibitory activity (Ki = 5.8 μM) compared to the parent rhodamine 6G (1).
To gain insights into the mechanism of action of RhQ-DMB (2), we conducted drug susceptibility assays with Saccharomyces cerevisiae AD1–8u–, a drug-sensitive yeast which lacks seven major transporters of the ABC family associated with multidrug efflux (Figure 4).38 Rhodamines are well-known growth inhibitors in yeast; therefore, strains lacking CmABCB1 accumulate rhodamines even at low concentrations and thus show low resistance. In contrast, strains containing CmABCB1 show enhanced resistance due to the efflux of the rhodamines. Indeed, while rhodamine 6G (1) did not inhibit cell growth in the CmABCB1-positive strain up to 30 μM (Figure 4a, WT), CmABCB1-negative strains demonstrated reduced growth at significantly lower concentrations (1 μM) (Figure 4a, mock). This supports the idea that 1 is actively effluxed by CmABCB1. With these results in mind, we conducted further assays using RhQ-DMB (2) (Figure 4b). If 2 indeed functions as an inhibitor of CmABCB1, we would expect it to hinder cell growth once inside the cell, regardless of whether the transporter is present. Pleasingly, both CmABCB1-positive and -negative strains exhibited similar growth inhibition at concentrations above ca. 1 μM. This suggested that 2 was less likely to be effluxed by CmABCB1 and is in alignment with its high ATPase inhibitory activity.
Figure 4.

Drug susceptibility assay in S. cerevisiae AD1–8u– cells in YPD medium for 30 °C for 15–17 h. AD1–8u– cells expressing WT CmABCB1 (●) were grown in various concentrations of drugs. For each drug assayed, mock-transfected AD1–8u– cells (□) were also grown. Data are means ± SD (n = 3).
Our above findings confirmed that RhQ-DMB (2) effectively inhibits CmABCB1. However, while ATPase stimulation does point to potential substrate entry into the inner chamber, it does not definitively confirm that inhibition is occurring because of interactions within that space. Instead, the substrate might inhibit CmABCB1 by interacting with either intracellular or extracellular domains without actually entering the chamber. To better understand this situation, we have recently developed a method of tryptophan (Trp) fluorescence quenching to observe substrate binding within the CmABCB1 chamber.26,39,40 The method employs the 4WY/M391W mutant, a CmABCB1 mutant that four of six intrinsic Trp residues are removed and one extrinsic tryptophan residue is introduced at the top of the inner chamber, i.e., the probable substrate-binding site, enabling to verify substrate entry into the chamber via site-specific fluorescence quenching from the Trp391 residue.26 Note that the mutant has essentially the same structure and activity as the WT. We measured fluorescence quenching of the extrinsic Trp391 residue of the 4WY/M391W mutant in micelles after mixing with RhQ-DMB (2) (Figure 5a). The data showed concentration-dependent quenching of the tryptophan fluorescence in 4WY/M391W by 2 without a shift in the emission maxima.26 The plot in Figure 4b illustrates the quenching ratio of tryptophan emission at 340 nm as a function of the concentration of 2. The tryptophan quenching ratio reached over 30% at saturation levels of 2 (Figure 5b), suggesting that 2 enters the inner chamber in a manner consistent with other major substrates such as rhodamine 6G (1), nicardipine, and tetraphenylphosphonium as we reported previously.26
Figure 5.
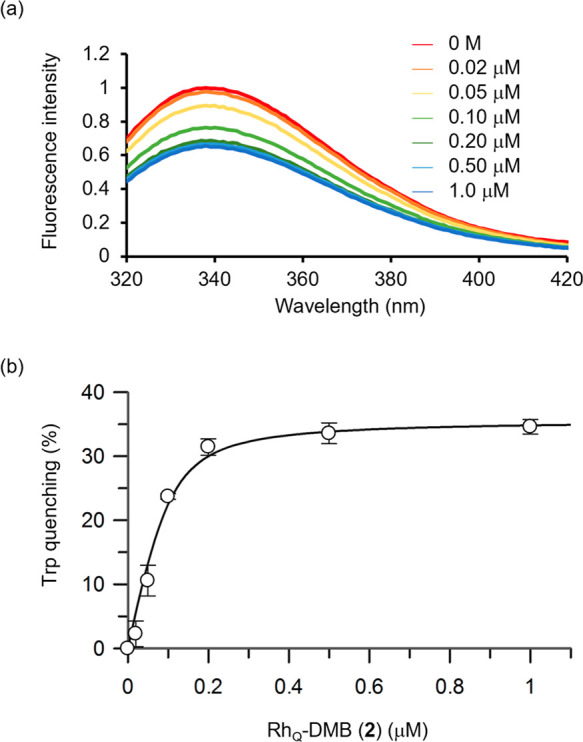
(a) Fluorescence emission spectra for the purified CmABCB1 mutant (4WY/M391W) (13 μg/mL) in a buffer solution of 20 mM Tris–HCl (pH 7.5 at 25 °C), 150 mM NaCl, and 0.05% (w/v) β-DDM containing RhQ-DMB (2) (0–1.0 μM). Fluorescence emission was recorded at 25 °C following excitation at 290 nm. (b) Quenching of the extrinsic tryptophan fluorescence of P-gp by the sequential addition of 2. Compound 2 was added at increasing concentrations to a 13 μg/mL solution of purified mutant CmABCB1 (4WY/M391W) in buffer at 25 °C. Fluorescence emission at 340 nm was recorded at 25 °C following excitation at 290 nm.
In this study, a variety of rhodamine derivatives were synthesized and evaluated for their affinity and inhibitory activity toward CmABCB1 contained in micelles to discover a novel inhibitor. The SAR study suggested that RhQ-DMB (2), a Q-rhodamine derivative bearing an o,o′-dimethoxybenzyl ester moiety, exhibited the most potent affinity and inhibitory activity (Km = 0.55 μM, Ki = 5.8 μM) against CmABCB1 as measured by an ATPase activity assay. The results of the drug susceptibility assay using yeast strains expressing CmABCB1 indicated that 2 was barely effluxed by CmABCB1. The tryptophan fluorescence quenching experiment showed a concentration-dependent quenching of tryptophan fluorescence, indicating that 2 enters the inner chamber. These results indicate that 2 has the desired characteristics of being able to enter the inner chamber but is hardly effluxed by CmABCB1. Future efforts will focus on elucidating the substrate-bound state of CmABCB1 through structural biology techniques, such as X-ray crystallography and cryo-electron microscopy, to provide a more comprehensive understanding of the functional dynamics. Simultaneously, further studies are being conducted, encompassing ADMET analysis and in vivo evaluations, to advance the development of ABCB1 inhibitors as a strategy to MDR.
Acknowledgments
We thank Yoshiki Inoue for technical support and advice for performing tryptophan quenching assay of CmABCB1. We also thank Dr. Tomohiro Yamaguchi and Dr. Yousuke Yamaoka for technical support and advice on preliminary investigation. This work was financially supported by Grants-in-Aid from the Japan Society for the Promotion of Science (JSPS) (21H02068, 21H05211, 23H02664, and 20H03222), BINDS from AMED (22ama121042j0001 and 22ama121034j0001), and the Kobayashi Foundation. S.M. and R.M. acknowledge support from JST SPRING (JPMJSP2110).
Glossary
Abbreviations
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- cryo-EM
cryogenic electron microscopy
- β-DDM
N-dodecyl-β-maltoside
- DMAP
4-(dimethylamino)pyridine
- DMF
dimethylformamide
- EDC·HCl
3-ethylcarbodiimide hydrochloride
- PCR
polymerase chain reaction
- Pi
inorganic phosphate
- SAR
structure activity relationship
- SD
standard deviation
- SDS
sodium dodecyl sulfate
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- Tris
tris(hydroxymethyl)aminomethane
- YPD
yeast extract–peptone–dextrose
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00526.
Cell lines and cell culture, preparation and spectra data, the results of ATPase assay, HPLC chromatogram of representative compounds, and the 1H NMR and 13C NMR spectra of new compounds (PDF)
Author Contributions
K.T. and H.K. conceived the idea and supervised the project. S.M. and R.T. synthesized the compounds and performed the ATPase assay. S.M. performed the drug-susceptibility assay and tryptophan quenching assay. R.M. and K.M. performed the purification of wild-type CmABCB1. S.M. and H.T. wrote the manuscript and K.T. and H.K. revised the manuscript. All authors approved the final version of the manuscript to be published.
The authors declare no competing financial interest.
Supplementary Material
References
- Gottesman M. M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- Holohan C.; Van Schaeybroeck S.; Longley D. B.; Johnston P. G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Shaffer B. C.; Gillet J.-P.; Patel C.; Baer M. R.; Bates S. E.; Gottesman M. M. Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resist. Updates 2012, 15, 62–69. 10.1016/j.drup.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S.; Huang X.; Wang Y.; He S.; Xu X.; Zhu X.; Yang X.; Ding Z.; Yao L.; Huang Y.; Wang C. A role for activator of G-protein signaling 3 (AGS3) in multiple myeloma. Int. J. Hematol. 2014, 99, 57–68. 10.1007/s12185-013-1484-8. [DOI] [PubMed] [Google Scholar]
- O’Donovan T. R.; O’Sullivan G. C.; McKenna S. L. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011, 7, 509–524. 10.4161/auto.7.5.15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M.; Rzhetsky A.; Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- Gottesman M. M.; Fojo T.; Bates S. E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Eckford P. D. W.; Sharom F. J. ABC Efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009, 109, 2989–3011. 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- Aller S. G.; Yu J.; Ward A.; Weng Y.; Chittaboina S.; Zhuo R.; Harrell P. M.; Trinh Y. T.; Zhang Q.; Urbatsch I. L.; Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Liu K. P-gp inhibition-based strategies for modulating pharmacokinetics of anticancer drugs: an update. Curr. Drug Metab. 2016, 17, 806–826. 10.2174/1389200217666160629112717. [DOI] [PubMed] [Google Scholar]
- Eadie L. N.; Hughes T. P.; White D. L. Interaction of the efflux transporters ABCB1 and ABCG2 with imatinib, nilotinib, and dasatinib. Clin. Pharmacol. Ther. 2014, 95, 294–306. 10.1038/clpt.2013.208. [DOI] [PubMed] [Google Scholar]
- Cui H.; Zhang A. J.; Chen M.; Liu J. J. ABC transporter inhibitors in reversing multidrug resistance to chemotherapy. Curr. Drug Targets 2015, 16, 1356–1371. 10.2174/1389450116666150330113506. [DOI] [PubMed] [Google Scholar]
- Binkhathlan Z.; Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. 10.2174/15680096113139990076. [DOI] [PubMed] [Google Scholar]
- Gannon M. K.; Holt J. J.; Bennett S. M.; Wetzel B. R.; Loo T. W.; Bartlett M. C.; Clarke D. M.; Sawada G. A.; Higgins J. W.; Tombline G.; Raub T. J.; Detty M. R. Rhodamine Inhibitors of P-Glycoprotein: An Amide/Thioamide “Switch” for ATPase Activity. J. Med. Chem. 2009, 52 (10), 3328–3341. 10.1021/jm900253g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Qin Z.; Zhang W.-D.; Cheng G.; Yehuda A. G.; Ashby C. R.; Chen Z.-S.; Cheng X.-D.; Qin J.-J. Medicinal chemistry strategies to discover P-glycoprotein inhibitors: An update. Drug Resist. Updat. 2020, 49, 100681. 10.1016/j.drup.2020.100681. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Xu H.; Ashby C. R.; Assaraf Y. G.; Chen Z.-S.; Liu H.-M. Chemical molecular-based approach to overcome multidrug resistance in cancer by targeting P-glycoprotein (P-gp). Med. Res. Rev. 2021, 41, 525–555. 10.1002/med.21739. [DOI] [PubMed] [Google Scholar]
- Urgaonkar S.; Nosol K.; Said A. M.; Nasief N. N.; Bu Y.; Locher K. P.; Lau J. Y. N.; Smolinski M. P. Discovery and characterization of potent dual P-glycoprotein and CYP3A4 inhibitors: Design, synthesis, cryo-EM analysis, and biological evaluations. J. Med. Chem. 2022, 65, 191–216. 10.1021/acs.jmedchem.1c01272. [DOI] [PubMed] [Google Scholar]
- Jin M. S.; Oldham M. L.; Zhang Q.; Chen J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 2012, 490, 566–569. 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodan A.; Yamaguchi T.; Nakatsu T.; Sakiyama K.; Hipolito C. J.; Fujioka A.; Hirokane R.; Ikeguchi K.; Watanabe B.; Hiratake J.; Kimura Y.; Suga H.; Ueda K.; Kato H. Structural basis for gating mechanisms of a eukaryotic Pglycoprotein homolog. Proc. Natl. Acad. Sci. USA 2014, 111, 4049–4054. 10.1073/pnas.1321562111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser L.; Zhou F.; Pluchino K. M.; Shiloach J.; Ma J.; Tang W.-k.; Gutierrez C.; Zhang A.; Shukla S.; Madigan J. P.; Zhou T.; Kwong P. D.; Ambudkar S. A.; Gottesman M. M.; Xia D. Structures of the multidrug transporter P-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity. J. Biol. Chem. 2017, 292, 446–461. 10.1074/jbc.M116.755884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodan A.; Yamaguchi T.; Nakatsu T.; Matsuoka K.; Kimura Y.; Ueda K.; Kato H. Inward- and outward-facing X-ray crystal structures of homodimeric P-glycoprotein CmABCB1. Nat. Commun. 2019, 10, 88. 10.1038/s41467-018-08007-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Chen J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. 10.1126/science.aar7389. [DOI] [PubMed] [Google Scholar]
- Alam A.; Kowal J.; Broude E.; Roninson I.; Locher K. P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. 10.1126/science.aav7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosol K.; Romane K.; Irobalieva R. N.; Alam A.; Kowal J.; Fujita N.; Locher K. P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. 10.1073/pnas.2010264117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri A.; Thonghin N.; Shafi T.; Prince S. M.; Col-lins R. F.; Ford R. C. Structure of ABCB1/P-Glycoprotein in the Presence of the CFTR Potentiator Ivacaftor. Membranes 2021, 11, 923. 10.3390/membranes11120923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y.; Yamaguchi T.; Otsuka T.; Utsunomiya Y.; Pan D.; Ogawa H.; Kato H. Structure-based alteration of tryptophan residues of the multidrug transporter CmABCB1 to asses substrate binding using fluorescence spectroscopy. Protein Sci. 2022, 31, e4331 10.1002/pro.4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan A.; Boden E. P.; Burns A.; McCloskey P. J.; Rishel M. J. Synthesis, chemical reactivity, and photophysical properties of 2′,7′ phenylated rhodamine dyes. Tetrahedron Lett. 2014, 55, 4222–4226. 10.1016/j.tetlet.2014.05.124. [DOI] [Google Scholar]
- Ambudkar S. V.; Cardarelli C. O.; Pashinsky I.; Stein W. D. Relation between the turnover number for vinblastine transport and for vinblastine-stimulated ATP hydrolysis by human P-glycoprotein. J. Biol. Chem. 1997, 272, 21160–21166. 10.1074/jbc.272.34.21160. [DOI] [PubMed] [Google Scholar]
- Horio M.; Gottesman M. M.; Pastan I. ATP-dependent transport of vinblastine in vesicles from human multidrug-resistant cells. Proc. Natl. Acad. Sci. U.S.A. 1988, 85, 3580–3584. 10.1073/pnas.85.10.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimoto Y.; Gatmaitan Z.; Hsu J.; Arias I. M. The function of Gp170, the multidrug resistance gene product, in rat liver canalicular membrane vesicles. J. Biol. Chem. 1989, 264, 11693–11698. 10.1016/S0021-9258(18)80120-X. [DOI] [PubMed] [Google Scholar]
- Litman T.; Zeuthen T.; Skovsgaard T.; Stein W. D. Structure-activity relationships of P-glycoprotein interacting drugs: kinetic characterization of their effects on ATPase activity. Biochim. Biophys. Acta 1997, 1361, 159–168. 10.1016/S0925-4439(97)00026-4. [DOI] [PubMed] [Google Scholar]
- Al-Shawi M. K.; Polar M. K.; Omote H.; Figler R. A. Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J. Biol. Chem. 2003, 278, 52629–52640. 10.1074/jbc.M308175200. [DOI] [PubMed] [Google Scholar]
- Aanismaa P.; Seelig A. P-Glycoprotein kinetics measured in plasma membrane vesicles and living cells. Biochemistry 2007, 46, 3394–3404. 10.1021/bi0619526. [DOI] [PubMed] [Google Scholar]
- Gatlik-Landwojtowicz E.; Aanismaa P.; Seelig A. Quantification and characterization of P-glycoprotein-substrate interactions. Biochemistry 2006, 45, 3020–3032. 10.1021/bi051380+. [DOI] [PubMed] [Google Scholar]
- Pearce H. L.; Safa A. R.; Bach N. J.; Winter M. A.; Cirtain M. C.; Beck W. T. Essential features of the P-glycoprotein pharmacophore as defined by a series of reserpine analogs that modulate multidrug resistance. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 5128–5132. 10.1073/pnas.86.13.5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Fukazawa N.; San-nohe K.; Sato W.; Yano O.; Tsuruo T. Structure-activity relationship of newly synthesized quinoline derivatives for reversal of multidrug resistance in cancer. J. Med. Chem. 1997, 40, 2047–2052. 10.1021/jm960869l. [DOI] [PubMed] [Google Scholar]
- Isca V. M. S.; Ferreira R. J.; Garcia C.; Monteiro C. M.; Dinic J.; Holmstedt S.; André V.; Pesic M.; dos Santos D. J. V. A.; Candeias N. R.; Afonso C. A. M.; Rijo P. Molecular Docking studies of royleanone diterpenoids from Plectranthus spp. as P-glycoprotein inhibitors. ACS Med. Chem. Lett. 2020, 11, 839–845. 10.1021/acsmedchemlett.9b00642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K.; Niimi M.; Niimi K.; Holmes A. R.; Yates J. E.; Decottignies A.; Monk B. C.; Goffeau A.; Cannon R. D. Functional expression of Candida albicans drug efflux pump Cdr1p in a Saccharomyces cerevisiae strain deficient in membrane transporters. Antimicrob. Agents Chemother. 2001, 45, 3366–3374. 10.1128/AAC.45.12.3366-3374.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Siemiarczuk A.; Sharom F. J. Intrinsic Fluorescence of the P-glycoprotein Multidrug Transporter: Sensitivity of Tryptophan Residues to Binding of Drugs and Nucleotides. Biochemistry 2000, 39, 14927–14938. 10.1021/bi0018786. [DOI] [PubMed] [Google Scholar]
- Vaiana A. C.; Neuweiler H.; Schulz A.; Wolfrum J.; Sauer M.; Smith J. C. Fluorescence Quenching of Dyes by Tryptophan: Interactions at Atomic Detail from Combination of Experiment and Computer Simulation. J. Am. Chem. Soc. 2003, 125, 14564–14572. 10.1021/ja036082j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.