

Abstract
Background
Formylated peptide receptor (FPR)-1 is a G-coupled receptor that senses foreign bacterial and host-derived mitochondrial formylated peptides (FPs), leading to innate immune system activation.
Aim
We sought to investigate the role of FPR1-mediated inflammation and its potential as a therapeutic target in inflammatory bowel disease (IBD).
Methods
We characterized FPR1 gene and protein expression in 8 human IBD (~1000 patients) datasets with analysis on disease subtype, mucosal inflammation, and drug response. We performed in vivo dextran-sulfate sodium (DSS) colitis in C57/BL6 FPR1 knockout mice. In ex vivo studies, we studied the role of mitochondrial FPs and pharmacological blockade of FPR1 using cyclosporin H in human peripheral blood neutrophils. Finally, we assess mitochondrial FPs as a potential mechanistic biomarker in the blood and stools of patients with IBD.
Results
Detailed in silico analysis in human intestinal biopsies showed that FPR1 is highly expressed in IBD (n = 207 IBD vs 67 non-IBD controls, P < .001), and highly correlated with gut inflammation in ulcerative colitis (UC) and Crohn’s disease (CD) (both P < .001). FPR1 receptor is predominantly expressed in leukocytes, and we showed significantly higher FPR1+ve neutrophils in inflamed gut tissue section in IBD (17 CD and 24 UC; both P < .001). Further analysis in 6 independent IBD (data available under Gene Expression Omnibus accession numbers GSE59071, GSE206285, GSE73661, GSE16879, GSE92415, and GSE235970) showed an association with active gut inflammation and treatment resistance to infliximab, ustekinumab, and vedolizumab. FPR1 gene deletion is protective in murine DSS colitis with lower gut neutrophil inflammation. In the human ex vivo neutrophil system, mitochondrial FP, nicotinamide adenine dinucleotide dehydrogenase subunit-6 (ND6) is a potent activator of neutrophils resulting in higher CD62L shedding, CD63 expression, reactive oxygen species production, and chemotactic capacity; these effects are inhibited by cyclosporin H. We screened for mitochondrial ND6 in IBD (n = 54) using ELISA and detected ND6 in stools with median values of 2.2 gg/mL (interquartile range [IQR] 0.0–4.99; range 0–53.3) but not in blood. Stool ND6 levels, however, were not significantly correlated with paired stool calprotectin, C-reactive protein, and clinical IBD activity.
Conclusions
Our data suggest that FPR1-mediated neutrophilic inflammation is a tractable target in IBD; however, further work is required to clarify the clinical utility of mitochondrial FPs as a potential mechanistic marker for future stratification.
Keywords: IBD, neutrophils, FPR1, mitochondria, DAMPs
Graphical Abstract
Graphical Abstract.

Key messages.
What is already known?
Despite advances in immune-suppressive treatments for inflammatory bowel disease (IBD), many patients fail to achieve complete mucosal healing. In the inflamed IBD mucosa, there are increased levels of damage-associate molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) that can drive the persistence of inflammation in IBD.
What is new here?
We provide evidence to show the importance of formylated peptide receptor (FPR)-1-mediated neutrophilic inflammation in IBD.
High expressions of FPR1 are associated with active IBD and treatment resistance.
Loss of FPR1 and/or pharmacologic inhibition of FPR1 reduce inflammation in mouse and human experimental models.
Mitochondrial DAMPs, ND6, are released during active gut inflammation and can activate FPR1 receptor.
How can this study help patient care?
FPR1-mediated inflammation can be therapeutically targeted in IBD as an adjunctive approach in IBD with potential stratification with mitochondrial DAMP biomarkers.
Introduction
Ulcerative colitis (UC) and Crohn’s disease (CD) are immune-mediated conditions with complex and overlapping pathogenic factors that can initiate and perpetuate a nonresolving pattern of mucosal inflammation.1 Most current therapies inhibit the downstream inflammatory response, yet complete mucosal healing is difficult to achieve and is seen in ~50% of treated severe inflammatory bowel disease (IBD). There remains a need to identify new therapeutic targets as part of a wider strategy to achieve deep mucosal healing and remission in IBD. In this context, there is an increasing focus on the upstream inflammatory factors, which can potentiate the abnormal gut inflammatory process observed in IBD. Here, exogenous pathogen-associated molecular patterns (PAMPs) by binding to germ-line encoded pathogen recognition receptors and endogenous damage-associated molecular patterns (DAMPs) that are endogenous host molecules released during tissue injury can act as danger signals that activate the innate immune system.2 Several lines of data suggest that high levels of and the persistence of PAMPs and DAMPs may be an important hitherto under-recognized contributory driver to the failure of IBD-associated inflammation to resolve completely in response to medical therapies.3 Presently, there is an increasing focus on targeting DAMP-mediated inflammation in many human inflammatory diseases.4
We recently showed that mitochondrial DAMPs (mtDAMPs), particularly mitochondrial DNA (mtDNA), are increased in IBD with a significant correlation with disease activity and severity.5 Several lines of evidence show that uncontrolled extracellular release of mtDAMPs can drive the development of inflammation and auto-immunity.6,7 MtDAMPs express at least 2 critical inflammatory molecular signatures: Mitochondrial N-formyl peptides (mtFPs) and mtDNA. In the latter, mtDNA shares similar immune-activating properties as bacterial DNA due to their shared ancestry. While the effects of mtDNA via a complex network of intracellular nucleic acid receptors such as TLR9, STING, and AIM3 can result in a graduated immune response involving different immune cell types,8 we recently showed that circulating blood mtFPs (FMMYALF, FMTPMRK, FMNPLAQ, FMNFALI, FMTMHTT) could be detected in severe IBD, by using a targeted liquid chromatography–mass spectrometry (LC-MS) approach screen.5 Of these 5 mtFPs, FMMYALF or nicotinamide adenine dinucleotide dehydrogenase subunit-6 from hereon, ND6 was the most abundant in our IBD subset, and interestingly, also the most pro-inflammatory mtFP.9
Mitochondrial FPs have long been considered an important chemoattractant for neutrophils.10 Recently, blood mitochondrial ND6 has been shown to be elevated in human diseases, pertinently in systemic inflammatory response syndrome, severe coronavirus disease 2019 (COVID-19), stroke, and rheumatoid arthritis.11–14 The uncontrolled release of mitochondrial ND6 can result in the rapid triggering of inflammation via formylated peptide receptor (FPR)-1, a G protein-coupled chemoattractant receptor.10,15 FPR1 is highly expressed in neutrophils and also on monocytes, macrophages, dendritic cells, and epithelial cells.16 Of interest, FPR1 recognizes both N-formyl peptides that are contained in bacteria or mitochondria,17 and other relevant DAMP ligands including cathepsin G, annexin A1 (ANXA), and FAM19A4.18–20 FPR1-mediated signaling, therefore, is relevant in human inflammatory diseases and is an attractive druggable pathway.15 In addition to more established FPR1 inhibitors such as cyclosporin H (CsH),21 there are now several small-molecule antagonists that are more potent and specific for FPR1 that may have promise in inflammatory diseases.22–24 Hence, in our study, we sought to first characterize the importance of FPR1 in gut inflammation and IBD, and, second, we explore the potential of measuring mitochondrial ND6 levels as a biomarker that may facilitate future stratification for FPR1 blockade as a therapeutic option in IBD.
Methods
Patients and Healthy Donors
For immunohistochemistry (IHC) work, colonic sections from 17 patients diagnosed with IBD (9 CD and 8 UC) from Western General Hospital, Edinburgh, UK, were obtained via NHS Lothian Bioresource (South East Scotland Ethics Reference 16/ES/0084). Each patient had an age- and sex-matched non-IBD uninflamed section for comparison. Human peripheral blood samples were collected from healthy individuals in the Centre for Inflammation Research Blood Donor Register under the provision of Ethics Reference 21-EMREC-041. For blood and stool ND6 work, biological samples were obtained from IBD patients and non-IBD controls at the Western General Hospital, Edinburgh, as part of the GI-DAMPs study (South East Scotland Ethics Reference 18/ES/0090). All clinical and NHS laboratory data were entered in a coded-anonymized fashion linked to the study patient ID in the Edinburgh Gut Research Unit RedCap Database (2020 to present).
Gene Expression Analysis
Details of publically available IBD gene datasets accessed are shown in Table 1 and accessed via http://www.ncbi.nlm.nih.gov/geo/. The gene expression units are (log2) normalized gene expression. Four of the 6 microarray datasets (data available under Gene Expression Omnibus [GEO] accession numbers GSE59071, GSE73661, GSE92415, and GSE206285) were already in Robust Microarray Analysis (RMA) normalized format (log2 normalized). GSE16879 and GSE23597 were log2 RMA normalized for our data analysis. For GSE11223 and GSE20881, gene expression was normalized to Agilent Stratagene Universal Human Reference. The difference in log2 fold change was calculated using linear models for microarray data (LIMMA; https://bioconductor.org/packages/release/bioc/html/limma.html). For single-cell RNA sequencing data analysis, we accessed raw sequencing reads of scRNA-seq samples, as well as UMI tables are available on the GEO under GEO Series accession number GSE134809, which was previously published by Martin et al.28
Table 1.
Summary of all IBD Gene Expression Omnibus (GEO) microarray/gene expression databases accessed for FPR1 gene analysis.
| GEO dataset | Description | Year |
|---|---|---|
|
GSE11223 GSE20881 |
Transcriptional profiling of colon epithelial biopsies from ulcerative colitis patients and healthy control donors.25 Colon biopsies from Crohn’s patients and healthy controls29 |
2008 |
| GSE16879 | Mucosal expression profiling in patients with inflammatory bowel disease before and after first infliximab treatment (anti-TNF)30 | 2009 |
| GSE23597 | Expression data from colonic biopsy samples of infliximab-treated UC patients (anti-TNF)31 | 2011 |
| GSE59071 | Mucosal gene expression profiling in patients with inflammatory bowel disease32 | 2015 |
| GSE73661 | The effect of vedolizumab (anti-α4β7-integrin) therapy on colonic mucosal gene expression in patients with ulcerative colitis (UC)33 | 2016 |
| GSE92415 | Characterization of molecular response to golimumab in ulcerative colitis by mucosal biopsy mRNA expression profiling: results from PURSUIT-SC induction study (anti-TNF)34 | 2018 |
|
GSE206285 UNIFI |
Efficacy and safety of ustekinumab treatment in patients with UC35 | 2022 |
Abbreviations: FPR1, formylated peptide receptor-1; IBD, inflammatory bowel disease.
Immunohistochemistry
IHC was performed on formalin-fixed paraffin-embedded patient and mice sections. In brief, sections were dewaxed in xylene and rehydrated in graded alcohols. Heat-mediated antigen retrieval was achieved using either Citrate Buffer pH6 (2 mM sodium citrate and 8 mM citric acid) or Tris/EDTA buffer pH 9 (1 mM ethylenediaminetetraacetic acid [EDTA] and 5 mM Tris Base). Endogenous peroxidase activity was blocked using 3% hydrogen peroxide and nonspecific binding was blocked using 2% horse serum diluted in 1× Tris buffer saline (TBS) (0.1 M Tris/HCL, 1.5 M NaCl). Ly6G (Sigma-Aldrich), neutrophil elastase (Novus Biologicals), and FPR1 (Sigma-Aldrich) antibodies were incubated overnight at 4°C at 1:1000 and 1:500, respectively. A negative control was included with the absence of the primary antibody. Subsequently, sections were incubated with the secondary antibody ImmPRESS detection kit (Vector Laboratories). Finally, the detection of the secondary antibody was achieved using 3,3-diaminobenzidine tetrahydrochloride (DAB, Dako UK Ltd.), counterstained using hematoxylin and Scott’s tap water, and dehydrated through a series of graded alcohols. Samples were then placed in xylene and mounted using a DPX-mounting medium.
Immunofluorescence
Dual immunofluorescence for FPR1 and neutrophils was performed on formalin-fixed paraffin-embedded patient samples as follows: Sections were dewaxed in xylene for 3 × 10 minutes and rehydrated through a series of graded alcohols. Heat-mediated antigen retrieval was performed using Tris/EDTA buffer pH 9. Sections were blocked in 2% fetal calf serum (FCS) diluted in TBS and incubated overnight in neutrophil elastase antibody (Novus Biologicals) and FPR1 antibody (Sigma-Aldrich) at 1:1000 and 1:500, respectively, at 4°C. A negative control was included with the absence of both primary antibodies. Alexa Fluor 488 and Alexa Fluor 647 secondary antibodies (ThermoFisher, UK) were combined at 1:500 in 1× TBS and incubated for 1 hour at room temperature. Sections were thoroughly washed and mounted using VECTASHIELD anti-fade mounting media with 4ʹ,6-diamidino-2-phenylindole (DAPI; Vector Laboratories) and stored in the dark at 4°C until analyzed.
Tissue Imaging
Brightfield images were obtained and visualized for analysis using the Carl Zeiss Zen 2 Blue edition program (Zeiss). IHC for neutrophil infiltration and the presence of FPR1 + infiltrating cells were counted. Immunofluorescence staining was captured using the Zeiss LSM 780 Confocal, visualized using Carl Zeiss ZEN 2 blue edition software (Zeiss), and categorized based on the absence and/or presence of FPR1 and/or neutrophil antibody. Tissue staining was analyzed in 3 representative 0.6 mm × 0.6 mm areas within the lamina propria and scored by 2 independent observers.
Neutrophil Isolation
Human peripheral blood was collected from healthy volunteers under local Ethics Approval Reference 21-EMREC-041. Blood was collected into 3.8% sodium citrate and centrifuged for 20 minutes at 350×g before discarding the plasma. Peripheral blood mononuclear cells and polymorphonuclear cells were isolated from red blood cells using 6% dextran sedimentation. A discontinuous (72.9, 63.0, and 49.5%) Percoll gradient was then used to separate the polymorphonuclear fraction from the PBMCs. Isolated cell fractions were then washed and resuspended in their appropriate culture media for further experimental analysis. A neutrophil preparation of >95% purity, as determined via cytospin centrifugation followed by Diff-Quik (Gentaur Molecular Products) staining, was deemed acceptable for use within our study.
Flow Cytometry
Isolated neutrophils were resuspended in PBS free from calcium and magnesium ions (PAA) at 10 × 106/mL and stimulated for 2 hours with 100 nM fMLF (Sigma-AldrichK) or 100 nM fMMYALF (GenScript) or pretreated for 10 minutes with 2.5 µM FPR1 antagonist CsH (Enzo Scientific) before stimulation. Neutrophils were incubated for 1 hour at 4°C with antibodies to Brilliant Violet 421 anti-human CD45 (Clone: HI30), APC/Fire 750 anti-human CD11b (activated) (Clone: CBRM1/5), Alexa Fluor 488 anti-human CD16 (Clone: 3G8), PE anti-human CD62L (Clone: DREG-56), and APC anti-human CD63 (Clone: H5C6) (BioLegend). Samples were washed and resuspended in 2% FCS and incubated briefly with DAPI (1:1000) before analysis using a BD Bioscience LSR Fortessa flow cytometer and FlowJo software (version 10.1).
ROS Production
ROS production was determined using a lumino-based approach by measuring chemiluminescence. In brief, 12.5 × 106 neutrophils/mL were pretreated with/without 2.5 µM CsH for 10 minutes at 37°C before being incubated for 10 minutes with luminol (150 µM) and HRP (18.75 U/mL) at 37°C in a 96-well round-bottom plate. Neutrophils were transferred to a precoated (1% fat-free milk in PBS) 96-well chemiluminescence white plate with/without 100 nM fMLF or 100 nM fMMYALF. Light emission production was recorded immediately using a plate reader and Synergy H1 plate reader (BioTek Instruments).
Stool Supernatant Preparation
Stored stool samples were defrosted and 2 supernatants were made from each sample. One was diluted 1:50 with extraction buffer using an Easy Extract device (Firefly Scientific) and vortexed for 3 minutes. For the second supernatant (used for ND6 detection), this process was repeated but 1× PBS without Ca/Mg was used as the diluent. Supernatant samples were then stored at –80°C until use.
Plasma Preparation for ELISA
Blood samples were obtained from patients as part of the GI-DAMPs study. Samples were taken in EDTA tubes and processed within 6 hours. Whole blood was centrifuged at 1000×g for 10 minutes, and plasma was transferred to sterile tubes. Plasma was further centrifuged at 3000×g for 10 minutes and divided into 1 mL aliquots for storage at –80°C, until use.
ELISA
A human MT-ND6 ELISA kit (MyBioSource) was used for both plasma and stool supernatant ND6 quantification. This kit is not optimized for use with stool samples. Prepared plasma samples were defrosted and diluted 1:2 with sample diluent as per product instructions. Prepared stool supernatants were defrosted and diluted at 1:50 with sample diluent. A Calprotectin ELISA kit (CalproLab) was used for the quantification of calprotectin in stool supernatants. Supernatants were defrosted and diluted 1:100 with sample dilution buffer as per manufacturer instructions.
Statistical Analysis
Analysis was performed using the Mann–Whitney and Wilcoxon signed-rank tests with 2-tailed P values in nonparametric continuous datasets. FlowJo (Tree Star) was used to analyze flow cytometry data to assess the percentage of positively/negatively labeled neutrophils and statistical analysis was performed using 1-way analysis of variance (ANOVA) with Bonferroni correction and Dunnett’s test. ROS assay readouts. All statistical analyses were performed using GraphPad Prism 9 (La Jolla, CA). For microarray data, analysis of normalized gene expression was carried out, and to correct for multiple testings, the false discovery rate was calculated for P values using GraphPad Prism 9.
Results
Formylated Peptide Receptor-1 Is Highly Expressed in Inflamed Intestinal Tissue in Human IBD
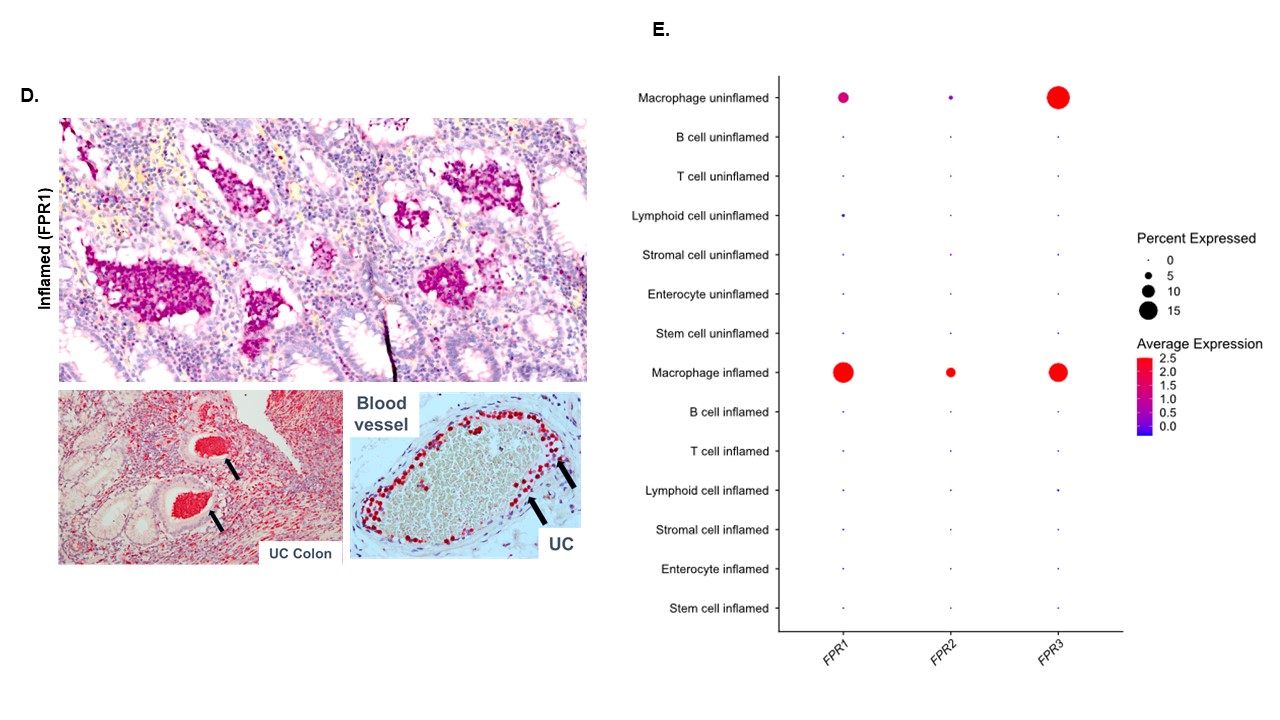
We performed in silico analysis of our previously published colonic gene microarray dataset (99 CD, 129 UC, and 56 non-IBD controls); data available at GEO http://www.ncbi.nlm.nih.gov/geo/; [accessed September 2022] accession25; (GSE11223 and GSE20881). Overall, we showed that FPR1 gene is highly expressed in the IBD colon compared to non-IBD controls (n = 207 [124 UC and 83 CD] vs. n = 67 colonic biopsies in each respective group; P = .0018) (Figure 1A). We analyzed and presented FPR1 gene expressions from colonic biopsies (as each patient has more than one colonic biopsy) to allow paired analyses with gut inflammation status and other relevant genes of interest. Here, FPR1 gene expression was higher in colonic biopsies from inflamed gut mucosa in UC and CD compared to noninflamed respective sections (both P < .0001; Figure 1B). As our microarray gene expression data were obtained from whole pinch gut biopsies, we performed paired analyses in each colonic biopsy sample and showed negative FPR1 correlation with epithelial gene markers EpCAM and CHD1, suggesting that the epithelial FPR1 gene is less dominant (Supplementary Figure 1A and B). Mitochondrial and bacterial FPs also bind to formylated peptide receptors 2 and 3 (FPR2 and FPR3) where FPR1 shares high homology with. In contrast with FPR1, both FPR2 and FPR3 were not differentially expressed in inflamed versus noninflamed IBD mucosa (Supplementary Figure 1C and D). FPR2 receptor has a low affinity for FPs and instead recognizes lipoxin A, and is more important in the resolution of inflammation.26 The function of FPR3 is unclear; of interest, FPR3 does not interact with FPs or ligands for FPR1 or FPR2.27 Using IHC, we demonstrated that inflamed mucosa in both UC and CD have significantly higher FPR1+ve lamina propria immune cell infiltration compared with noninflamed IBD gut (P < .0001 and .0002, respectively) (Figure 1D and E). Immunofluorescence co-staining with elastase identified these immune cells as predominantly neutrophils (Figure 1D and F). In UC, transmigrating neutrophils across gut endothelial vessels and crypt abscesses (typically a collection of dead neutrophils in the gut lumen) are notably FPR1+ (Supplementary Figure 1C). FPR1–3 are expressed in many cell types but FPR1 expression is highest in neutrophils.16 We accessed the publically available single-cell RNA sequencing data as published by Martin et al.28 to determine the cell types that expressed FPR1-3 in the gut epithelial, immune, and stromal compartments out with the neutrophil population. Of interest, we found the highest expressions of FPR1–3 in the macrophage population with no difference in inflamed and noninflamed CD gut (data available under GEO accession number GSE134809 and in Supplementary Figure 1D).
Figure 1.

(A) Overall in silico analysis of FPR1 in colonic pinch biopsies using Gene Expression Omnibus (GEO) GSE11223 and GSE20881 comparing IBD (n = 207 colonic pinch biopsies; comprising UC and CD [n = 124 and 83, respectively]) versus non-IBD controls (n = 67; P = .0018). (B) FPR1 gene expression in UC noninflamed versus inflamed pinch biopsies (n = 57 and 67, respectively), and CD noninflamed versus inflamed pinched biopsies (n = 41 and 42, respectively; both P < .0001) Mann–Whitney test. FPR1 gene expression expressed as relative units to Stratagene Universal Human Reference Manual: Universal Human Reference RNA (chem-agilent.com). (C) Representative immunohistochemistry sections of inflamed and noninflamed colonic sections of IBD (n = 17 CD and 24 CD, respectively), FPR1 is marked by horse-radish peroxide red (HRP) and neutrophils, elastase (DAB stained). (D) Quantification of FPR1+ve cells in CD and UC—average count/mm2 of colonic section. Mann–Whitney statistics. **P = .0002, ***P < .0001. (E) Representative immunofluorescence of UC and CD colonic sections—DAPI, FPR1, neutrophil elastase, and merged images. CD, Crohn’s disease; DAB, 3,3-diaminobenzidine tetrahydrochloride; DAPI, 4ʹ,6-diamidino-2-phenylindole; FPR1, formylated peptide receptor-1; IBD, inflammatory bowel disease; UC, ulcerative colitis.
Formylated Peptide Receptor-1 Expression Is Associated With Multiple Biologic Treatment Resistance in IBD
We further analyzed 6 independent IBD microarray gene expression GEO datasets of the gut (data available under GEO accession numbers GSE59071, GSE206285 [UNIFI], GSE73661, GSE16879, GSE92415 and GSE23597—comprising 858 IBD and 85 non-IBD patients) (Table 1). In 3 datasets (except for GSE16879 and GSE92415, P = .056 and .587, respectively, Figure 2D and E) with non-IBD groups for comparison, FPR1 expression was significantly higher in IBD (Figure 2A–E). In agreement with our data, GSE59071 comprising UC subjects showed higher FPR1 expression in inflamed gut mucosa compared with noninflamed UC mucosa (P < .001, n = 73 vs. 23 patients, respectively) (Figure 2A). We further investigated if FPR1 intestinal mucosal gene expression is associated with therapeutic response of biologic treatment in 6 IBD datasets, namely—ustekinumab (GSE206285 [UNIFI]), infliximab (GSE16879 and GSE23597), vedolizumab (GSE73661), and golimumab (GSE92415) (all data available in the aforementioned GEO series accession numbers). There is a consistent pattern of higher FPR1 gene expression in the nonresponders with significant associations seen in ustekinumab, infliximab, and vedolizumab therapy in UC (all P < .05; Figure 3A–D; Table 2). Taken together, we demonstrate consistently high FPR1 in the inflamed IBD gut, mainly on neutrophils in the lamina propria of actively inflamed IBD mucosa with an association with poor response to several current biologic treatments that target different inflammatory mechanisms in IBD.
Figure 2.

(A) FPR1 gene expression in inflamed and noninflamed UC tissue from Gene Expression Omnibus (GEO) dataset GSE59071. (B–E) FPR1 gene expressions from GEO datasets GSE206285, GSE73661, GSE16879, and GSE92415, respectively. The gene expression units are (log2) normalized gene expression in Robust Microarray Analysis (RMA) normalized format. Statistics: Mann–Whitney test with false discovery rate (FDR) P-value correction. FPR1, formylated peptide receptor-1; UC, ulcerative colitis.
Figure 3.

(A–E) FPR1 gene expression in responders and nonresponders to (A) ustekinumab GSE206285; (B, C) infliximab GSE16879 and GSE23596, respectively; (D) vedolizumab GSE73661; and (E) golimumab GSE92415. Statistics: Mann–Whitney test and gene expression units are (log2) normalized gene expression in Robust Microarray Analysis (RMA) normalized format. Statistics: Mann–Whitney test with false discovery rate (FDR) P-value correction. FPR1, formylated peptide receptor-1; IBD, inflammatory bowel disease; UC, ulcerative colitis.
Table 2.
Difference in log2 FPR1 expression in IBD, data available under Gene Expression Omnibus (GEO) databases above accessed for FPR1 gene analysis.
| GSE_number | Inflammation status | ∆Log2 FPR1 |
|---|---|---|
| GSE59071 | UC inflamed vs. UC noninflamed | 1.66 |
| GSE59071 | UC inflamed vs. non-IBD | 1.67 |
| GSE206285 | UC vs. non-IBD | 0.28 |
| GSE73661 | UC vs. non-IBD | 1.26 |
| GSE16879 | IBD vs. non-IBD | 0.43 |
| GSE92415 | UC vs. non-IBD | 0.07 |
| Drug response | ||
| GSE206285 | Responder vs. nonresponder | –0.16 |
| GSE73661 | Responder vs. nonresponder | –1.40 |
| GSE16879 | Responder vs. nonresponder | –0.64 |
| GSE92415 | Responder vs. nonresponder | –0.07 |
| GSE23597 | Responder vs. nonresponder | –0.40 |
Abbreviations: FPR1, formylated peptide receptor-1; IBD, inflammatory bowel disease; UC, ulcerative colitis.
Genetic Deletion of FPR1 Reduces the Severity of Mouse Experimental DSS Colitis
Constitutive gene deletion of FPR1 in mice does not result in overt spontaneous clinical phenotype; however, in systemic Listeria monocytogenes infection, FPR1 deficiency results in increased bacterial burden and mortality.36 In sterile lung injury models, FPR1 deficiency resulted in lower levels of neutrophilic inflammation.37,38 We investigated the effects of experimental colitis induced by dextran-sulfate sodium (2% DSS) over 7 days in FPR1-deficient and wild-type C57/BL6 mice. Here, we found that FPR1-gene deletion is protective in DSS colitis, with lower weight loss, histological and clinical evidence of colitis, and neutrophil infiltration of the colonic mucosa (Figures 4A–F). This is of interest, given the importance of FPR1-mediated signaling in response to bacterial formylated peptides (fMLF) that are abundant in the colon and likely an important host defense against gut luminal bacteria. This line of data points toward a key role in FPR1-mediated inflammatory signaling in mouse DSS colitis and raises the potential to target this pathway in IBD.
Figure 4.

(A) Percentage of weight loss in FPR1–/– and wild-type C57/BL6 in 2% dextran-sulfate sodium (DSS) in drinking water ad libitum. (B) Representative H&E cross-section of distal colon in FPR1–/– and wild-type following 7 days of DSS colitis. Bar is 500 µM. (C) Percent of inflamed distal colonic mucosa (ulcerated and loss of colonic epithelium/preserved noninflamed colonic epithelium with preserved crypt architecture) in FPR1–/– and wild type following 7 days of DSS colitis. (D) Colon length in FPR1–/– and wild type following 7 days of DSS colitis. (E) Quantification of LyG6+ve cells in the distal colon of FPR1–/– and wild type following 7 days of DSS colitis—average count/mm2 of colonic section. (F) Representative immunohistochemistry sections of distal colonic lamina propria of FPR1–/– and wild type following 7 days of DSS colitis neutrophils Ly6G (DAB stained). FPR1, formylated peptide receptor-1; IBD, inflammatory bowel disease.
Mitochondrial ND6 Activates Peripheral Blood Human Neutrophils Via FPR1
As we recently detected circulating blood mitochondrial ND6 in our IBD cohort, we synthesized mitochondrial ND6 and investigated its effects on human neutrophil activation and used the dose range as published by Rabiet et al.9 Using peripheral blood leukocytes from healthy donors, CD45+ cells were selected and gated, followed by CD16+, a marker of functional and non-apoptotic neutrophils (Figure 5A) and CD11b+, a neutrophil migration marker (Figure 3B). Following a 2-hour stimulation, ND6 (10 nM) increased CD11b+ expressing migratory neutrophils with similar effects seen with bacterial FP, fMLF (10 nM) (Figure 5B). CD11b+CD16+ neutrophils were further gated for and assessed based on their CD62L and CD63 cell surface expression. Following ND6 stimulation, neutrophils shedded CD62L (P < .001) and significantly increased their surface expression of CD63 (P < .001), a marker of full neutrophil activation when combined with loss of CD62L (Figure 5C). CsH, a potent FPR1 inhibitor, inhibited the effects of neutrophil activation. Prior to stimulation, human neutrophils were pretreated for 10 minutes with 2.5 µM CsH, which significantly reduced ND6 and fMLF-induced CD11b+CD63+CD62L– neutrophil surface expression when compared to similar groups not treated with CsH (P < .001) (Figure 5D). Both ND6 and fMLF increased the transmigration of neutrophils toward these respective stimuli that are again blocked by CsH (Figure 5E). FPR1 engagement stimulates the effector function of neutrophils as evidenced by the production of extracellular reactive oxygen species (ROS). ND6 (10 nM) stimulation of peripheral human neutrophils over 30 minutes resulted in a significant increase of ROS production with a similar magnitude seen in fMLF-treated neutrophils as a positive control (Figure 5F–H).
Figure 5.

(A) Neutrophils were isolated from healthy human peripheral blood and separated using density gradient centrifugation. Purified neutrophils were labeled for multicolor flow cytometry. Expression of DAPI (dead cells), CD45 (general leukocytes), CD16 (neutrophils), CD11b (activated neutrophils), CD62L (primed neutrophils), and CD63 (activated neutrophils) were analyzed and a representative gating strategy was applied to identify activated neutrophils. (B) Quantification of CD11b+ neutrophils. (C) Quantification of CD63+/CD62L– neutrophils. (D) Percentage of activated CD11b+/CD62L–/CD63+ neutrophils in response to fMLF/synthetic ND6 stimulation and/or cyclosporin H (CsH) treatment. (E) Number of migrated neutrophils in response to fMLF/synthetic ND6 stimulation and/or CsH treatment. (F) Extracellular neutrophil ROS production (with HRP to detect extracellular ROS) in response to fMLF/synthetic ND6 stimulation. (G) Extracellular neutrophil ROS production (with HRP to detect extracellular ROS) in response to fMLF/synthetic ND6 stimulation with CsH treatment. (h) Extracellular neutrophil ROS production (with HRP to detect extracellular ROS) in response to fMLF/synthetic ND6 stimulation and CsH treatment (Figure H is Figure F and G combined). Data are means ± standard error (SEM) from n = 3 experiments performed in triplicate. Two-tailed t-test, Mann–Whitney, and 1-way ANOVA with Bonferroni correction and Dunnet’s tests were considered significant if P < .05 with an asterisk (*) indicating P < .05, double asterisks (**) indicating P < .001, and triple asterisks (***) indicating ***P < .0001. ANOVA, analysis of variance; DAPI, 4ʹ,6-diamidino-2-phenylindole; FPR1, formylated peptide receptor-1; HRP, horse-radish peroxide red; IBD, inflammatory bowel disease; UC, ulcerative colitis.
Mitochondrial ND6 Is Present in Stools in IBD but Not in Circulation
We performed an initial screen for ND6 in blood plasma using an ELISA approach in our patient cohort with highly active disease (n = 16, with extensive evidence of endoscopic moderate to severe colitis and/or stool calprotectin of >500 μg/g and/or CRP >30 mg/L) and 16 non-IBD controls. Of interest, ND6 was undetectable with standard curve dilution to the picogram range (data not shown). Given the higher likelihood of mitochondrial DAMP release from IBD gut mucosa into stools, we further investigated for the presence of mitochondrial ND6 here. Our ELISA approach can detect measurable ND6 levels in stool supernatants extracted using a method optimized for stool calprotectin measurement. Calprotectin (s100a8/9) is a neutrophilic protein that is released during uncontrolled cell death and is a widely used biomarker for gut inflammation in the clinic. Here, the overall median stool ND6 was 2.2 ng/mL (IQR 0.0–4.99; range 0–53.3; Table 3). We first tested if stool ND6 levels were associated with the clinical severity of IBD inflammation but found no difference between the groups with active versus highly active disease (median 1.6 vs. 3.2 ng/mL, P = .51; Figure 6A). In addition, there was no statistical difference in stool ND6 levels between IBD patients with active disease and those in remission (median 2.2 vs. 4.8 ng/mL, respectively; P = .78; Figure 6B). In each stool sample, we performed paired stool calprotectin s100a8/0 ELISA measurements. Here, stool calprotectin levels were statistically higher in the active IBD versus remission groups (median 1163.0 vs. 160.6 μg/g; P = .0008) and compared to non-IBD stools (median 1163.0 vs. 86.8 μg/g; P = .0007; Figure 6C). We further investigated whether subgroups of IBD patients with more active disease, using blood C-reactive protein (CRP) measurement at a cut level of 10 mg/L. Here, IBD patients with CRP >10 mg/L have higher stool ND6, but this was not statistically significant (median 5.9 vs. 9.6 ng/mL; P = .46; Figure 6D). There was no significant correlation between stool ND6 with blood C-reactive and calprotectin s100a8/0 (both r = –0.09, P = .5). These lines of data suggest that although ND6 is present in stool supernatants, our ELISA data using methodology optimized for stool calprotectin did not show an association with clinically active IBD states.
Table 3.
Characteristics of IBD patient cohort for stool ND6 analysis.
| Active (n = 45) | Remission (n = 9) | Non-IBD (n = 5) | |
|---|---|---|---|
| UC | 24 | 3 | |
| CD | 18 | 5 | |
| IBD-U | 3 | 1 | |
| Age (years) | 37.6 (2.0) | 42.6 (5.3) | 48.0 (3.9) |
| Female/male | 23/22 | 3/6 | 3/2 |
| C-reactive protein (mg/dL) | 31.7 (8.7) | 3.6 (0.9) | NA |
| Stool calprotectin (μg/g) | 1255.0 (147.4) | 227.9 (97.2) | 115.4 (44.7) |
| No. of in-patients for active IBD treatment (%) | 35 (77%) |
Abbreviations: CD, Crohn’s disease; FPR1, formylated peptide receptor-1; IBD, inflammatory bowel disease; UC, ulcerative colitis.
Continuous data are presented as mean ± standard error of the mean (SEM).
Figure 6.

(A) Stool ND6 ELISA in IBD patients with active vs. highly active disease (n = 16 and 27, respectively). (B) Stool ND6 ELISA in IBD patients with active disease, in remission and non-IBD subjects (n = 45, 9, and 5, respectively). (C) Stool calprotectin s100a8/9 in IBD patients with active disease, in remission and non-IBD subjects (n = 45, 9, and 6, respectively). (D) Stool ND6 ELISA in IBD patients with active disease stratified according to C-reactive protein < or >10 mg/L (n = 22 and 21, respectively). (E) Correlation analyses of paired stool ND6 and blood C-reactive protein levels; 42 paired measurements. (F) Correlation analyses of paired stool ND6 and calprotectin s100a8/9 levels; 53 paired measurements. Data presented as mean ± standard error of the mean (SEM). Mann–Whitney statistical test between groups. Spearman correlation paired analyses. Significance level P < .05; **P = .0008; ***P = .0007. FPR1, formylated peptide receptor-1; IBD, inflammatory bowel disease; NS, not significant.
Discussion
In our study, we presented several lines of evidence to support the important role of FPR1-mediated inflammation in IBD. We found that FPR1 is highly expressed in neutrophilic inflammation in the IBD gut mucosa. Importantly, high FPR1 expression is associated with treatment resistance to several current IBD therapies with different mechanisms of action, namely anti-TNF (infliximab), anti-α4β7 (vedolizumab), and anti-IL23p40 (ustekinumab). This is further supported by recent studies that showed an upregulation of FPR1 in UC patients who do not achieve mucosal healing.39,40
Genetic deletion of FPR1 was protective in acute DSS colitis and in vitro blockade of FPR1 receptor against mitochondrial ND6 using CsH-reduced human peripheral blood neutrophil activation. Given the unique organ juxtaposition with gut bacteria where FPR1 can sense bacterial FPs in this rich environment, it is of interest to investigate if loss of FPR1 will result in worse or better colitis outcomes. Here using experimental mouse DSS colitis, we showed that genetic deletion of FPR1 resulted in less inflammation and neutrophil recruitment in the gut. This is of key interest as this suggests that mtDAMP (vis à vis PAMP) may play a relatively more important functional role, at least in the acute murine colitis setting. Although our findings agree with recent studies showing a protective effect of FPR1-gene deletion in murine colitis,41,42 the dominant mechanistic context of FPR1-mediated signaling in governing neutrophil trafficking and survival specifically in the gut and IBD has not been fully elucidated. Overall, it is noteworthy that FPR1 gene deletion in other injury/inflammatory models of the lung and brain is also protective and associated with lower inflammation.37,38,43FPR1 knockout mice develop normally and do not display signs of spontaneous colitis, but they display an increased bacterial burden and mortality in models of systemic L. monocytogenes infection.36,44 A recent study shows that FPR1 may have an additional role in regulating host metabolism with an effect of gut microbiome and luminal fMLF activation of FPR1—thus implicating a role in homeostasis.45
In the context of IBD, FPR1 has recently been identified as a key driver gene in the inflammatory process associated with IBD in a functional genomic predictive network study.46 Our combined human IBD and mouse in vivo data indicate that the FPR1 blockade could be particularly relevant in patients who do not respond to conventional advanced medical therapies, and hence offer a novel angle to address the current therapeutic ceiling in IBD. Collectively, they suggest that FPR1 blockade is a tractable approach in IBD, and there is now a need to investigate this in more detail in human studies. The recent development of inhibitory small molecules that may target FPR1,22–24 including the early-phase clinical trial EudraCT Number: 2021-000035-31.23
In our previous study, we have shown that blood and stool mitochondrial DNA are elevated in active IBD.5 Hence, we further investigated if mitochondrial FP, ND6 can serve as a mechanistic biomarker to identify patients with a potential dominant ND6 DAMP-mediated inflammatory endophenotype within IBD. Recently, Kwon et al. showed that high levels of blood ND6 in patients admitted with septic shock in the intensive care unit (ICU) were independently associated with increased infection and mortality.11 A further human study in intracerebral hemorrhage showed that blood ND6 levels correlated with the severity of tissue damage.13 Using a similar ELISA methodology, we could not detect circulating blood ND6 in the initial screening cohort of 16 patients with highly active IBD. In the significantly more unwell and compromised ICU patient cohort by Kwon et al., blood ND6 levels were measured at a range of 0.5–5 ng/mL; this magnitude difference is in stark contrast to our undetectable levels in blood. Notwithstanding, we found detectable levels of ND6 in stool supernatants in IBD with a range of 0–53.3 ng/mL, which is much lower. There were no associations with disease activity and no correlation with the gut inflammation biomarker, calprotectin s100a8/9. Our initial data suggest that blood and stool ND6 measurements are not useful as potential biomarkers to stratify IBD patients.
There are limitations in our studies. First, while it is pertinent that ND6 is not linked to IBD disease severity, our study is not geared toward the testing of the clinical utility of ND6 as a biomarker; and further testing in a much larger IBD cohort is required. These data, although negative, provide a useful basis for further work to explore DAMP-based biomarkers with the potential to stratify patients in future FPR1 interventional drug studies. We present our data as a key comparator to the currently used “DAMP” biomarker in IBD. Second, the role of circulating ND6 formed the focus of our investigation based on data from other studies. However, in IBD, mitochondrial ND6 is likely to impart its effects in the local gut environment. Notwithstanding this, we do not know the degradation profile of mitochondrial ND6 that may contribute to the absent/low signal in blood using our ELISA approach. Third, from a conceptual angle, many other ligands can activate neutrophils, and we do not know the relative importance of all these factors compared to ND6. Finally, we have used CsH as an FPR1 antagonist. Although this is widely used, CsH has off-target effects.47,48 More specific pharmacologic agents or experiments from FPR1–/– mouse neutrophils can provide clearer data.
There remains a significant unmet need in improving the medical management of IBD. Despite more treatment options, there is a “therapeutic ceiling” of 50%, particularly in severe IBD. Recent attention has increasingly turned to exploring adjunctive therapeutic options that may augment current therapies in IBD. Targeting DAMP/PAMP-mediated inflammation in this context, specifically FPR1 neutrophilic-mediated inflammation, has been explored in inflammatory diseases of the lungs, liver, and brain.13,37,43,49 Recent studies have now established neutrophils as a major component in complex inflammatory gene modules that are associated with medical treatment failure in IBD.35,50 Our data suggest that FPR1 is an attractive drug target in IBD; however; FPR1-inhibition strategies in clinical trial settings need careful appraisal. Such approaches are likely implemented in a time-defined window (at peak inflammation in acute severe IBD flare-up) and potentially as an adjunct to rescue therapy in conjunction with established IBD medical management.
Supplementary Material
{kind=link}
{kind=link}
Contributor Information
Milly J McAllister, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Rebecca Hall, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Robert J Whelan, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Lena J Fischer, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Cher S Chuah, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Peter D Cartlidge, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Broc Drury, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Duncan G Rutherford, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Rodger M Duffin, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Jennifer A Cartwright, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
David A Dorward, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Adriano G Rossi, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Gwo-tzer Ho, Edinburgh IBD Science Unit, Centre for Inflammation Research, Queens Medical Research Unit, University of Edinburgh, Edinburgh, Scotland, UK.
Funding
This work was funded by Guts UK Charity (M.M., G.T.H.), Crohn’s Colitis UK (L.F., G.T.H.), Wellcome Trust (J.A.C.), Chief Scientist Office Scotland (D.G.R.), Medical Research Council (R.J.W.), and Helmsley Charitable Trust (A.G.R., B.D., R.H., C.S.C., and G.T.H.).
Conflicts of Interest
None declared.
Data Availability
Microarray data available via access to Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo) microarray/gene expression databases GSE11223, GSE20881, GSE16879, GSE23597, GSE59071, GSE73661, GSE92415, and GSE206285. Single-cell RNA sequencing data is available via Gene Expression Omnibus (GEO) GSE134809.
References
- 1. Ho GT, Cartwright JA, Thompson EJ, Bain CC, Rossi AG.. Resolution of inflammation and gut repair in IBD: translational steps towards complete mucosal healing. Inflamm Bowel Dis. 2020;26(8):1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–305. [DOI] [PubMed] [Google Scholar]
- 3. Boyapati RK, Rossi AG, Satsangi J, Ho G-T.. Gut mucosal DAMPs in IBD: from mechanisms to therapeutic implications. Mucosal Immunol. 2016;9(3):567–582. [DOI] [PubMed] [Google Scholar]
- 4. Gong T, Liu L, Jiang W, Zhou R.. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20(2):9–112. [DOI] [PubMed] [Google Scholar]
- 5. Boyapati RK, Dorward DA, Tamborska A, et al. . Mitochondrial DNA is a pro-inflammatory damage-associated molecular pattern released during active IBD. Inflamm Bowel Dis. 2018;24(10):2113–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Q, Raoof M, Chen Y, et al. . Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crow MK. Mitochondrial DNA promotes autoimmunity. Science. 2019;366(6472):1445–1446. [DOI] [PubMed] [Google Scholar]
- 8. Emming S, Schroder K.. Tiered DNA sensors for escalating responses. Science. 2019;365(6460):1375–1376. [DOI] [PubMed] [Google Scholar]
- 9. Rabiet MJ, Huet E, Boulay F.. Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur J Immunol. 2005;35(8):248–2495. [DOI] [PubMed] [Google Scholar]
- 10. Carp H. Mitochondrial N-formyl methionyl proteins as chemoattractants for neutrophils. J Exp Med. 1982;155(1):264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kwon WY, Suh GJ, Jung YS, et al. . Circulating mitochondrial N-formyl peptides contribute to secondary nosocomial infection in patients with septic shock. Proc Natl Acad Sci U S A. 2021;118(17):e2018538118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuley R, Duvvuri B, Wallin JJ, et al. . Mitochondrial N-formyl methionine peptides contribute to exaggerated neutrophil activation in patients with COVID-19. Virulence. 2023;14(1):2218077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Z, Li Y, Han J, et al. . Formyl peptide receptor 1 signalling potentiates inflammatory brain injury. Sci Transl Med. 2021;13(605). [DOI] [PubMed] [Google Scholar]
- 14. Duvvuri B, Baddour AA, Deane KD, et al. . Mitochondrial N-formyl methionine peptides associate with disease activity as well as contribute to neutrophil activation in patients with rheumatoid arthritis. J Autoimmun. 2021;119(1):102630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dorward DA, Lucas CD, Chapman GB, Haslett C, Dhaliwal K, Rossi AG.. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol. 2015;185(5):1172–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weiss E, Kretschmer D.. Formyl-peptide receptors in infection, inflammation, and cancer. Trends Immunol. 2018;39(10):815–829. [DOI] [PubMed] [Google Scholar]
- 17. Boulay F, Tardif M, Brouchon L, Vignais P.. The human N-formylpeptide receptor characterization of two cDNA isolates and evidence for a new subfamily of G-protein-coupled receptors. Biochemistry. 1990;29(50):11123–11133. [DOI] [PubMed] [Google Scholar]
- 18. Wang W, Li T, Wang X, et al. . FAM19A4 is a novel cytokine ligand of formyl peptide receptor 1 (FPR1) and is able to promote the migration and phagocytosis of macrophages. Cell Mol Immunol. 2015;12(5):615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun R, Iribarren P, Zhang N, et al. . Identification of neutrophil granule protein cathepsin G as a novel chemotactic agonist for the G protein-coupled formyl peptide receptor. J Immunol. 2004;173(1):428–436. [DOI] [PubMed] [Google Scholar]
- 20. Leoni G, Alam A, Neumann PA, et al. . Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. J Clin Invest. 2013;123(1):443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wenzel-Seifert K, Seifert R.. Cyclosporin H is a potent and selective formyl peptide receptor antagonist comparison with N-t-butoxycarbonyl-L-phenylalanyl-L-leucyl-L-phenylalanyl-L-leucyl-L-phenylalanine and cyclosporins A, B, C, D, and E. J Immunol. 1993;150(10):4591–4599. [PubMed] [Google Scholar]
- 22. Yang SC, Wang YH, Ho CM, et al. . Targeting formyl peptide receptor 1 with anteiso-C13-surfactin for neutrophil-dominant acute respiratory distress syndrome. Br J Pharmacol. 2023;180(16):2120–2139. [DOI] [PubMed] [Google Scholar]
- 23. Murphy CK, Dixit B, Oleson FB, Dolle RE, Farquhar R, McCormick BA.. Development of ADS051, an oral, gut-restricted, small molecule neutrophil modulator for the treatment of neutrophil-mediated inflammatory diseases. FEBS Open Biol. 2023;13(8):1434–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Forsman H, Wu Y, Martensson J, et al. . AZ2158 is a more potent formyl peptide receptor 1 inhibitor than the commonly used peptide antagonists in abolishing neutrophil chemotaxis. Biochem Pharmacol. 2023;211(1):115529. [DOI] [PubMed] [Google Scholar]
- 25. Noble CL, Abbas AR, Cornelius J, et al. . Regional variation in gene expression in the healthy colon is dysregulated in ulcerative colitis. Gut. 2008;57(10):1398–1405. [DOI] [PubMed] [Google Scholar]
- 26. Qin CX, Norling LV, Vecchio EA, et al. . Formylpeptide receptor 2: nomenclature, structure, signalling and translational perspectives: IUPHAR review 35. Br J Pharmacol. 2022;179(19):4617–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rabiet MJ, Macari L, Dahlgren C, Boulay F.. N-formyl peptide receptor 3 (FPR3) departs from the homologous FPR2/ALX receptor with regard to the major processes governing chemoattractant receptor regulation, expression at the cell surface, and phosphorylation. J Biol Chem. 2011;286(30):26718–26731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin JC, Chang C, Boschetti G, et al. . Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell. 2019;178(6):1493–1508.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Noble CL, Abbas AR, Lees CW, et al. . Characterization of intestinal gene expression profiles in Crohn’s disease by genome-wide microarray analysis. Inflamm Bowel Dis. 2010;16(10):1717–1728. [DOI] [PubMed] [Google Scholar]
- 30. Arijs I, De Hertogh G, Lemaire K, et al. . Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PLoS One. 2009;4(11):e7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Toedter G, Li K, Marano C, et al. . Gene expression profiling and response signatures associated with differential responses to infliximab treatment in ulcerative colitis. Am J Gastroenterol. 2011;106(7):1272–1280. [DOI] [PubMed] [Google Scholar]
- 32. Vanhove W, Peeters PM, Staelens D, et al. . Strong upregulation of AIM2 and IFI16 inflammasomes in the mucosa of patients with active inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(4):2673–2682. [DOI] [PubMed] [Google Scholar]
- 33. Arijs I, De Hertogh G, Lemmens B, et al. . Effect of vedolizumab (anti-alpha4beta7-integrin) therapy on histological healing and mucosal gene expression in patients with UC. Gut. 2018;67(1):43–52. [DOI] [PubMed] [Google Scholar]
- 34. Sandborn WJ, Feagan BG, Marano C, et al. ; PURSUIT-SC Study Group. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):85–95; quiz e14. [DOI] [PubMed] [Google Scholar]
- 35. Pavlidis P, Tsakmaki A, Pantazi E, et al. . Interleukin-22 regulates neutrophil recruitment in ulcerative colitis and is associated with resistance to ustekinumab therapy. Nat Commun. 2022;13(1):5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao JL, Lee EJ, Murphy PM.. Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J Exp Med. 1999;189(4):657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dorward DA, Lucas CD, Doherty MK, et al. . Novel role for endogenous mitochondrial formylated peptide-driven formyl peptide receptor 1 signalling in acute respiratory distress syndrome. Thorax. 2017;72(10):928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leslie J, Millar BJ, Del Carpio Pons A, et al. . FPR-1 is an important regulator of neutrophil recruitment and a tissue-specific driver of pulmonary fibrosis. JCI Insight. 2020;5(4):10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Manuc M, Ionescu EM, Milanesi E, et al. . Molecular signature of persistent histological inflammation in ulcerative colitis with mucosal healing. J Gastrointestin Liver Dis. 2020;29(2):159–166. [DOI] [PubMed] [Google Scholar]
- 40. Zhang J, Wu X, Wei S, Liu C, Wang X, Dong W.. Identified potential biomarkers may predict primary nonresponse to infliximab in patients with ulcerative colitis. Autoimmunity. 2022;55(8):538–548. [DOI] [PubMed] [Google Scholar]
- 41. Di Paola R, Fusco R, Gugliandolo E, et al. . Formyl peptide receptor 1 signalling promotes experimental colitis in mice. Pharmacol Res. 2019;141(2):591–601. [DOI] [PubMed] [Google Scholar]
- 42. Farooq SM, Stadnyk AW.. Neutrophil infiltration of the colon is independent of the FPR1 yet FPR1 deficient mice show differential susceptibilities to acute versus chronic induced colitis. Dig Dis Sci. 2012;57(7):1802–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cardini S, Dalli J, Fineschi S, Perretti M, Lungarella G, Lucattelli M.. Genetic ablation of the FPR1 gene confers protection from smoking-induced lung emphysema in mice. Am J Respir Cell Mol Biol. 2012;47(3):332–339. [DOI] [PubMed] [Google Scholar]
- 44. Liu M, Chen K, Yoshimura T, et al. . Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci Rep. 2012;2(1):786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wollam J, Riopel M, Xu YJ, et al. . Microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance. Diabetes. 2019;68(7):1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peters LA, Perrigoue J, Mortha A, et al. . A functional genomics predictive network model identifies regulators of inflammatory bowel disease. Nat Genet. 2017;49(10):1437–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wenzel-Seifert K, Grunbaum L, Seifert R.. Differential inhibition of human neutrophil activation by cyclosporins A, D, and H. Cyclosporin H is a potent and effective inhibitor of formyl peptide-induced superoxide formation. J Immunol. 1991;147(6):1940–1946. [PubMed] [Google Scholar]
- 48. Gschwendt M, Kittstein W, Marks F.. The weak immunosuppressant cyclosporine D as well as the immunologically inactive cyclosporine H are potent inhibitors in vivo of phorbol ester TPA-induced biological effects in mouse skin and of Ca2+/calmodulin dependent EF-2 phosphorylation in vitro. Biochem Biophys Res Commun. 1988;150(2):545–551. [DOI] [PubMed] [Google Scholar]
- 49. Marques PE, Amaral SS, Pires DA, et al. . Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology. 2012;56(5):1971–1982. [DOI] [PubMed] [Google Scholar]
- 50. Friedrich M, Pohin M, Jackson MA, et al. ; Oxford IBD Cohort Investigators. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med. 2021;27(11):1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data available via access to Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo) microarray/gene expression databases GSE11223, GSE20881, GSE16879, GSE23597, GSE59071, GSE73661, GSE92415, and GSE206285. Single-cell RNA sequencing data is available via Gene Expression Omnibus (GEO) GSE134809.