

Introduction
Pulmonary cysts and cavities are commonly encountered on chest imaging. The differential diagnosis is broad because both congenital and acquired processes can cause such findings. The following chapter will review common diffuse cystic lung diseases and a systematic approach to diagnosis.
Definition
Cysts and cavities are foci of decreased lung density with discernible walls as assessed by high-resolution computed tomography (HRCT). In contrast, emphysematous airspaces typically lack a perceptible wall. A “cyst” is typically surrounded by a thin wall (≤ 2mm) of uniform thickness whereas a “lung cavity” is a gas-filled space with a relatively thick wall (>4mm), and may be surrounded by consolidation or mass.1 A cavity often develops from drainage of a necrotic lesion via the bronchial tree and may contain a fluid level. This distinction is useful since “cysts” are rarely malignant, but a cavitary lesion raises concern for malignancy, infection or vasculitis, especially in a high-risk patient.2
The distribution of cysts is classified as focal or multifocal vs diffuse (involving all lobes). The presence of lung cysts in a diffuse distribution limits the differential diagnosis to certain disorders and will be the focus of this chapter.
Lymphangioleiomyomatosis
Background
Lymphangioleiomyomatosis (LAM) is a rare, slowly progressive neoplastic and metastasizing disorder characterized by progressive cystic destruction of the lung with marked female predominance. Clinical manifestations include exertional dyspnea, recurrent pneumothoraces, chylous effusions in the chest and abdomen, and abdominal tumors such as renal angiomyolipomas (AMLs).
LAM can be associated with the tuberous sclerosis complex (TSC-LAM) or sporadic (S-LAM) in which patients do not have TSC gene mutations or its clinical manifestations. TSC is an autosomal dominant genetic disorder that in its most severe form is associated with seizures and cognitive impairment, and CNS, cutaneous, and systemic lesions. Diffuse cystic lung disease has been reported in 10% of men and 30% of women although studies indicate that symptomatic TSC-LAM is nearly completely limited to women.3 In S-LAM, lymphangiomas, AMLs and sclerotic bone lesions may accompany the cystic lung changes but CNS and skin lesions are lacking.4
Variable phenotypes in S-LAM in the face of negative TSC1/TSC2 genetic testing of peripheral blood leukocytes may be explained by genetic mosaicism, the occurrence of somatic cells in a single organism with different genetic compositions which occur from random errors in DNA replication after fertilization and during embryogenesis. For example, Han et al report the case of an otherwise normal man who presented with apparent S-LAM and mosaicism for a TSC2 mutation yet no other TSC manifestations including a normal brain MRI.5 Ogorek et al report a similar case of LAM with mosaicism for a pathogenic TSC2 mutation among 61 female patients with apparent sporadic LAM. The authors hypothesize whether bilateral AMLs and sclerotic bone lesions (present in their index case yet rare among the cohort) could predict mosaicism for TSC2 mutations in sporadic LAM.6
Pathogenesis
LAM is caused by mutations in either of the two known TSC genetic loci; TSC1 on chromosome 9q34 or TSC2 on chromosome 16p13.7,8 Dysregulation of the PI3/Akt signaling pathway enables activation of mechanistic target of rapamycin (mTOR) that promotes abnormal LAM cell proliferation and survival.9,10 Disease progression is aided by abnormal lymphangiogenesis, immune evasion mechanisms, and antiapoptotic effects of sex steroids such as estrogen.
A model for LAM suggests that LAM cells metastasize to the lung from a remote source hypothesized to be the lymphatic system11, the uterus12 or AMLs.9 Recently, single-cell transcriptomic analysis identified a unique population of LAM cells (LAMcore cells). The pulmonary and uterine LAMcore cells demonstrate similar gene expression profiles, and this observation provides support for the concept of a uterine source for pulmonary LAM cells.13 The novel LAMcore cell discovery may serve to develop biomarkers and future therapeutic targets.
Nearly 1/3 of LAM patients have abdominal or thoracic lymph node enlargement, usually due to lymphangiomyomatous tissue (Fig. 1).14,15 Clusters of LAM cells enter the venous circulation at the thoracic duct, disseminate through the pulmonary capillary bed, and can be found in chylous pleural fluid.11 Expression of lymphatic endothelial markers such as vascular endothelial growth factor receptors are critical to the process. Elevated levels of vascular endothelial growth factor D (VEGF-D > 800 pg/ml)) can be useful to distinguish LAM from other cystic lung diseases16 and predict response to therapy.17
Figure 1. Typical CT findings of LAM.
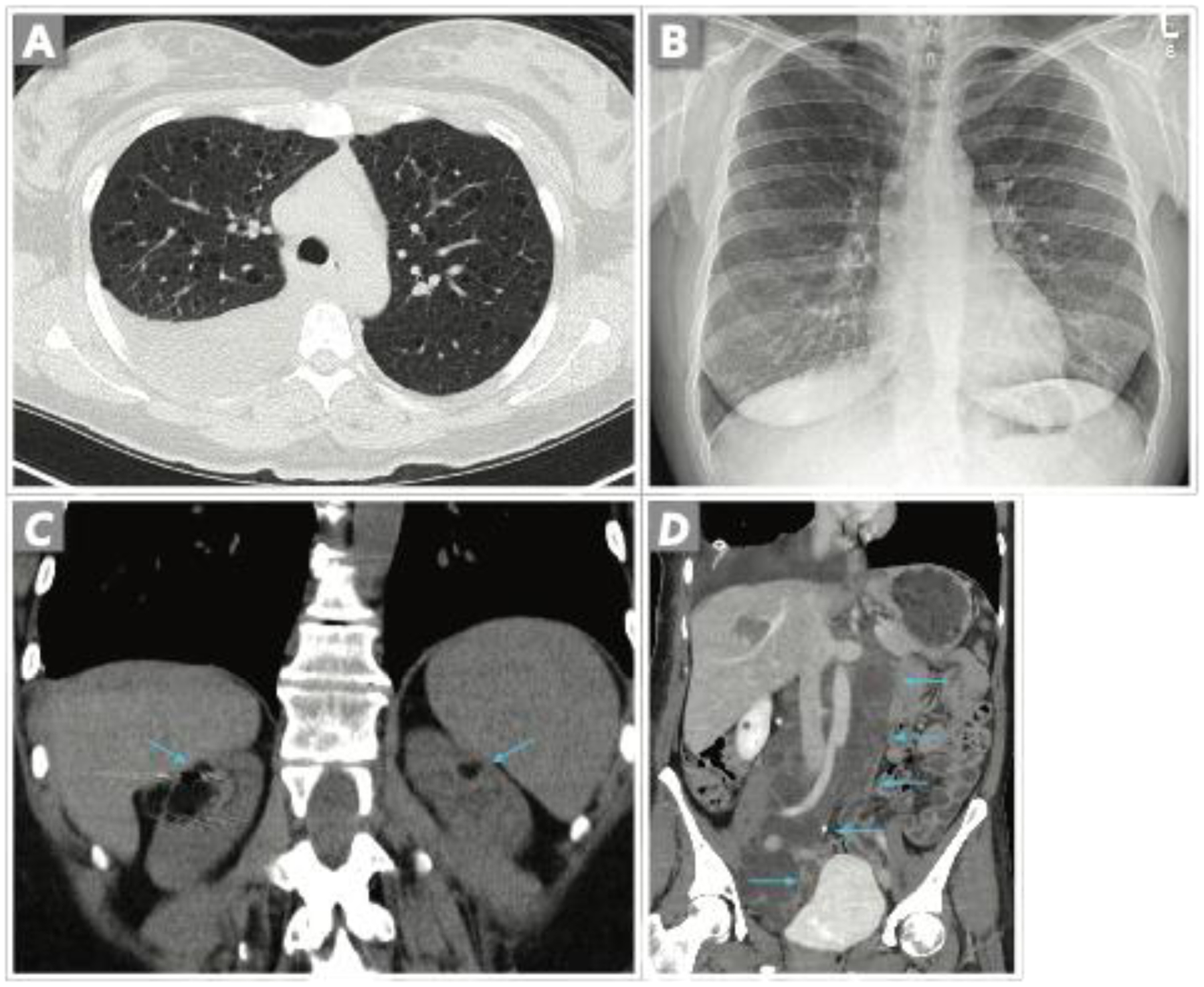
A. Axial CT image shows a moderate right chylous effusion and thin-walled cysts of variable sizes evenly distributed throughout the lungs. B. CXR shows a resolved effusion following 4 months of medical therapy. C. Coronal CT image in a different patient shows bilateral fat-containing renal angiomyolipomas (arrows). Metal artifact in the right kidney is from arterial embolization coils. D. Coronal CT in a different patient shows a large tubular and lobulated fluid attenuating structure in the retroperitoneum (arrows) compatible with retroperitoneal lymphangeioleiomyoma which resolved with prolonged therapy (not shown). A large right chylous effusion is also present.
Estrogen may play a critical role in LAM cell survival, proliferation, and destructive potential. Estrogen regulates gene transcription and may modulate signaling to activate mTOR. Animal studies have shown that estrogen may promote metastases and the survival of TSC-2 deficient cells18 whereas estrogen suppression (by ovariectomy or aromatase inhibition) decreases mTOR activity and inhibits myometrial proliferation.19,20 Targeting the estrogen-ERK pathway, along with the mTOR pathway, could be a potential therapeutic approach for LAM.
The pathogenesis of LAM has many similarities to the mechanisms of other human cancers such as immune evasion strategies and tissue destruction. For example, targeted monoclonal antibodies (e.g. anti-PD-L1) reduced tumor burden and prolonged survival in an animal model of TSC-LAM21,22 and suggests immunotherapy as a potential treatment strategy for human LAM.
Clinicoradiologic manifestations
LAM typically presents during the reproductive years although examples of LAM presenting well after menopause have been reported.23,24 Most patients are nonsmokers and LAM does not appear to be smoking-related.
The most common clinical presentations are exertional dyspnea from disease progression, spontaneous pneumothorax or incidental discovery of lung cysts. Pneumothorax is common (up to 2/3 of patients) with a high recurrence rate (70%),24–26 and may precede the diagnosis of LAM in the majority of patients. The accumulation of chyle in pleural and extrathoracic locations, such as the airway (chyloptysis) and genitourinary tract (chyluria) may occur. Fistulous communication with the gut can result in chyle in stool and retrograde chylous parenchymal congestion can present with ground glass or reticular change in the lung.27
AMLs are tumors composed of fat, smooth muscle and abnormal blood vessels, which most often affect the kidneys but can present anywhere in the chest and abdomen. AMLs are seen in 1/3 of S-LAM and nearly 90% of TSC-LAM patients.4 Renal cysts have been reported in patients with LAM, and concomitant polycystic kidney disease may develop from the genetic deletion of PKD1, which is adjacent to TSC2.28 Lymphangioleiomyomas are masses of LAM cell clusters within lymphatic vessels and lymph nodes that can mimic lymphomas, ovarian or renal cancers or other malignant tumors (Fig. 1D).29
Radiologic-pathologic correlation
On the chest radiograph, early signs of LAM include fine nodular, reticular or reticulonodular opacities. Over time, the reticular pattern may progress into a more irregular pattern, lung volumes may increase (50% of cases) and pulmonary cysts become visible.30 CT will invariably show diffuse cysts of varying size and profusion (Figure 1).31 The lung cysts in LAM are typically round, thin-walled (1–2 mm), and diffusely distributed throughout otherwise normal lung. The number of cysts is typically greater among those with symptomatic pulmonary impairment than in asymptomatic patients.30 The involvement of pulmonary cysts in the costophrenic sulci can distinguish LAM from pulmonary Langerhans Cell Histiocytosis (pLCH) that typically shows upper lung predominance with sparing of the sulci.32
The pulmonary cysts may develop from air-trapping by smooth muscle proliferation in the small airways.33 The LAM muscle cells stain with a monoclonal antibody, HMB-45 (human melanoma black-45), specific for LAM in this context (Fig. 2).34 There is profound lymphatic duct and lymph node involvement and obstruction, which accounts for the chylous accumulation in the pleura and peritoneum among patients with LAM yet unusual among TSC-LAM.35 Other lung findings described include septal thickening, presumed to be consequent to lymphatic obstruction36, centrilobular nodules that may reflect pneumocyte hyperplasia, and ground glass opacities or focal consolidations that may reflect hemosiderosis or hemorrhage.37
Figure 2. Histopathologic findings of LAM.

A. H&E stained section at low power shows a cystic structure with a nodule of LAM cells protruding into the cyst space in a polypoid manner. B. High power image of the LAM nodule demonstrating clusters of spindle shaped cells growing in a haphazard manner. C. HMB45 immunohistochemical stain of LAM cells shows patchy cytoplasmic staining. D. SMA (smooth muscle active) immunohistochemistry stain highlights the nodules of smooth muscle cells within the wall of a cyst in LAM. (Images courtesy of Dr. Carlyne D. Cool and Dr. Steve D. Groshong, Division of Pathology, National Jewish Health).
Renal angiomyolipomas are common (30–50% of patients) (Fig. 1). Other intrabdominal findings include hepatic angiomyolipomas, lymphangiomyomas, retroperitoneal lymph nodes and chylous peritoneum. The presence of characteristic lung cysts associated with either hepatic or renal AMLs and/or chylothorax supports a confident diagnosis of LAM.26
Management and Clinical Trails
Based on the landmark MILES trial (Multicenter International LAM Efficacy of Sirolimus),38 inhibition of mechanistic target of rapamycin (mTOR) is indicated for patients with abnormal lung function (FEV1 < 70% predicted), progressive lung disease or clinically significant chylous effusions. mTOR inhibitors are also effective for other presentations such as angiomyolipomas and lymphangeiomyomas and may decrease the frequency of pneumothoracies.39,40 The MILED (Multicenter Interventional LAM Early Disease) trial is investigating the safety and efficacy of low dose treatment to preserve lung function in earlier stages of disease (NCT03150914). Several clinical trials are investigating novel therapies involving mTOR inhibitors and other investigational treatments which can be found at The LAM Foundation website.41
Birt-Hogg-Dubé (BHD) Syndrome
Background
The BHD syndrome was first described in a case report of 2 siblings with unique skin lesions and a strong family history of similar skin lesions; one of the siblings later developed colon cancer.42 In 1977, Birt and colleagues described the autosomal dominance of the disorder among a large kindred with hereditary medullary carcinoma of the thyroid and skin lesions, which they diagnosed histologically as fibrofolliculomas.43 The majority of patients (~80%) currently present with diffuse pulmonary cysts.44
Pathogenesis
Numerous studies have defined the genetics and pathogenesis of BHDS. In 2001, the BHD gene locus was mapped to chromosome 17p45 and later narrowed to a 700kb region on chromosome 17p11.2.46 In 2005, Schmidt and colleagues reported germline mutations in 84% of affected families and more than 150 unique mutations in the folliculin (FLCN) gene have since been reported.47 Most result in loss-of-function mutations and support the role of FLCN as a tumor suppressor gene. The loss of FLCN leads to BHD-associated tumors such as kidney tumors, the most serious manifestation, occurring in up to 34% of patients by age 50.48 BHD syndrome has been associated with other neoplasms such as colorectal cancer,49 melanoma,50 thyroid and parathyroid tumors51 although data is limited to case reports and small series and the risk is uncertain.
Clinicoradiologic manifestations
The phenotypic expression of BHD syndrome is highly variable even among families sharing the same FLCN mutation. Patients may present with any combination of skin, pulmonary and renal findings. However, the absence of skin and renal manifestations does not exclude the diagnosis. Many BHD patients presenting with a pneumothorax are misdiagnosed as primary spontaneous pneumothorax or emphysema given the rarity of the syndrome.52
Skin manifestations include fibrofolliculomas, and acrochordons that are difficult to distinguish and may be spectrums of the same lesion.43 They are characterized by round, grayish-white papules 2–4mm in size and most often distributed along the face, trunk and neck, including the posterior ear (Fig. 3).53 Fibrofolliculomas are the most prevalent lesion and may be subtle so that patients may not seek medical care.
Figure 3. Characteristic findings of Birt-Hogg-Dube syndrome.

A. Axial CT shows elliptical thin-walled pulmonary cysts predominating in the paracardiac regions of the lower lungs. B. Subpleural cystic structure lined by bland alveolar cells without atypical morphology. C. White/gray papules on midface, forehead and post-auricular of a patient with Birt-Hogg-Dube syndrome characteristic of fibrofolliculomas or similar lesions (e.g., acrochordons). (Fig 3B, courtesy of Dr. Carlyne D. Cool, Division of Pathology, National Jewish Health).
Pulmonary cysts are the most common systemic manifestation and typically appear after the 4th and 5th decades but can appear during teenage years. They are elliptical or “floppy”, thin-walled and typically distributed along the basilar medial region of the lungs (Fig. 3). Studies suggest that the number and size of cysts typically remain stable.30 Cysts do not typically impact pulmonary function until the development of a pneumothorax. Histopathologic examination of cysts in BHD patients with recurrent pneumothorax reveal inner surfaces lined with type II pneumocyte-like cells suggesting slow-growing, hamartomatous cysts that may rupture.54
Escalon et al studied 47 subjects with isolated cystic lung disease in which thoracic radiologists were blinded to the final diagnoses, limited to BHD, LIP, or LAM.55 Lower lung-predominant cysts were significantly more likely among BHD or LIP compared with LAM, in which cysts were diffusely distributed. Furthermore, BHD patients were more likely to have elliptical, paramediastinal cysts. The authors propose an algorithm to reliably differentiate BHD from other cystic lung diseases (Fig. 4).55
Figure 4. Algorithm to reliably differentiate BHD from other cystic lung diseases (AJR 2019; 212: 1260–1264.

Management
Similar to LAM, pulmonary cysts predispose patients to develop pneumothoraces. Although the risk for pneumothorax is relatively low (~24%), the risk for recurrence is high (75%).56 For this reason, pleurodesis has been suggested with the first event to avoid the morbidity associated with repeat events.57 Certain activities, such as scuba diving, increase the risk of pneumothorax due to potential expansion of cysts from transthoracic pressure changes. Although air travel is considered safe among patients with diffuse cystic lung disease,58 subtle symptoms of pneumothorax may go unrecognized by patients.58 For this reason, patients with extensive cystic disease, prior pneumothoraces, or new symptoms of chest pain should seek evaluation prior to air travel. Tobacco smoking and of other substances is discouraged despite limited data that such exposure increased risk of pneumothorax.56
Screening for renal tumors should begin at age 20 (per expert opinion)59 or at the time of diagnosis preferably with magnetic resonance imaging, which is more sensitive and specific than ultrasonography, and avoids the cumulative radiation exposure with computed tomography. Surveillance should continue at least every 36 months until a mass is identified which will determine management.60 Most tumors have an indolent behavior. The risk of metastases increases with tumor size and kidney-sparing resection is recommended for tumors larger than 3cm in diameter, along with resection of all additional tumors detected during surgery.60
Genetic testing may detect germline mutations in the folliculin (FLCN) gene, which confirms the diagnosis. Some patients (~5%) may be FLCN mutation-negative by DNA sequencing yet carry intragenic deletions/duplications detectable by more advanced molecular diagnostic methods.61 The penetrance of FLCN mutations is high among affected families and genetic counseling should be encouraged for first-degree relatives of patients. Carriers of FLCN mutations should undergo regular (every 3 years) imaging surveillance for kidney tumors.60
Lymphoid interstitial pneumonia
Background
Lymphocytic Interstitial Pneumonia (LIP) is characterized by infiltration of the pulmonary interstitium by dense lymphoid tissue.62 The radiographic LIP pattern is most commonly seen as a pulmonary manifestation of systemic collagen vascular diseases.63 Other disease associations with LIP include dysproteinemias, infections (e.g., EBV or HTLV-1), and rarely drug reactions (e.g., phenytoin).64 The dense accrual of lymphoid tissue implies risk for lymphoproliferative disease, particularly small B cell lymphomas of extranodal marginal zone type, as well as polyclonal lymphoproliferative conditions associated with viral infections.63 Idiopathic LIP is rare and must be distinguished from systemic and lymphoproliferative conditions.
Pathogenesis
The histopathologic pattern involves a dense layer of lymphocytes, plasma cells and histiocytes with alveolar septal infiltration and along the bronchi and vasculature. Granulomatous and lymphocytic interstitial lung disease (GLILD) is considered a variant of LIP associated with common variable immunodeficiency (CVID), but cysts are rarely seen in GLILD.65 Germinal centers may become prominent along the airways and lymphatic channels in which lymphoproliferative conditions should be considered. In the idiopathic form of LIP, immunophenotyping would show absence of clonality.66 When the nodular lymphoid hyperplasia is prominent along the bronchioles then Sjogren’s syndrome should be strongly considered67 and co-existing amyloidosis may also be seen in this context.68 (Fig. 5B).
Figure 5.

A. Coronal CT in a patient with Sjogren’s syndrome shows multiple lower lung predominant thin-walled cysts with peribronchovascular predominance, compatible with LIP. B. Axial CT through the lower lungs in a patient with biopsy-proven LIP and amyloidosis in Sjogren’s syndrome shows a combination of irregular nodules, cysts and ground glass opacities. C. Low power image demonstrating diffuse interstitial infiltrate by mature lymphocytes and plasma cells. Cysts are seen adjacent to airways. D. Lymphoid follicles with germinal centers in a patient with Sjogren’s syndrome and cystic lung disease. E, F. CT scans two years apart in a patient with LIP and Sjogren syndrome show increased ground glass abnormality and septal thickening indicating progression of lymphocytic infiltrates. BAL demonstrated 60% lymphocytic predominance without monoclonality or infection. (Images C, D, images courtesy of Dr. Carlyne D. Cool, Division of Pathology, National Jewish Health).
Clinicoradiologic manifestations
The cystic airspaces associated with LIP range from 1 to 30mm, are typically peribronchovascular in distribution and appear to represent dilated small bronchi and bronchioles from partial obstruction by lymphocytic infiltration.69,70 Ground glass opacities, poorly defined centrolobular nodules, bronchovascular and septal thickening may also be seen.71 The ground glass opacities may improve with treatment but new cysts may develop in areas of centrilobular nodules and consolidations may evolve into honeycombing.72 Larger nodules (11–30 mm in diameter), consolidations, and pleural effusions are more common among LIP associated with lymphoma.67,73
Management considerations
The natural history of LIP varies according to the underlying disease process. When LIP is associated with autoimmune disease (e.g., Sjogren’s syndrome), management is based on the severity of pulmonary impairment and evidence of progression. Treatment is directed at the underlying condition and some regimens for extrapulmonary manifestations may also benefit the lung disease. It is important to exclude secondary conditions such as granulomatous and lymphocytic interstitial lung disease (GLILD) among patients with common variable immunodeficiency, HIV, and secondary infections from immune deficiency syndromes or secondary to immunosuppression. Lymphoma may develop in approximately 5% of patients with LIP with an increased risk in those with Sjogren’s syndrome.63 Malignant transformation may be suggested by larger nodules or those increasing in size, pleural effusions and alveolar consolidations67,73; polyclonality is key to differentiate LIP from lymphoma.74
Amyloidosis and light chain deposition
Background and pathogenesis
Amyloidosis is the abnormal deposition of low molecular weight proteins into highly structured fibrils which, in their native state, would otherwise circulate in plasma. The deposition of “amyloid deposits” result in a broad range of clinical manifestations depending on their type and location. The major types of systemic amyloidosis include the primary types (light chain [AL] and transthyretin [ATTR]), which account for the majority of systemic amyloid.75 Secondary amyloidosis (AA) is a rare systemic complication of chronic disease resulting in sustained production of serum amyloid A (SAA), an acute phase reactant, and most often affects the kidney leading to proteinuria.76 Light chain deposition disease (LCDD) reflects the presence of monoclonal deposits composed of light chains only and is typically associated with lymphoproliferative diseases with prominent kidney involvement.77
Clinicoradiologic manifestations
Pulmonary amyloidosis typically presents with multiple lung nodules. Cystic pulmonary amyloidosis is rare and may be associated with Sjogren syndrome or mucosa-associated lymphoid tissue (MALT) lymphoma (Fig. 6).78,79 The lung cysts are thought to arise from obstruction of distal airways from amyloid deposits.78 Pulmonary lung function tests may be normal, obstructive, or have an isolated reduction in diffusing capacity.78
Figure 6.

A. Axial CT image of thin-walled pulmonary cysts and nodules, some calcified, in a patient with biopsy-proved LIP and amyloidosis. B. Axial CT image of diffuse lung cysts in the mid and upper lung zones in a patient with advanced LCDD of the kidney secondary to multiple myeloma C. Nodular amyloidosis characterized by intraalveolar and interstitial deposits of amorphous eosinophilic material. D. Congo red stain highlights the amyloid material. (Image C, courtesy of Dr. Carlyne D. Cool, Division of Pathology, National Jewish Health. Image D, with permission, Rosane Duarte Achcar MD, FCAP, FASCP, Steve D. Groshong MD, PhD, Carlyne D. Cool MD, Differential Diagnoses in Surgical Pathology: Pulmonary Pathology, copyright 2017 Wolters Kluwer Health, Inc.
Management
The diagnosis of pulmonary amyloidosis requires lung biopsy to demonstrate amyloid deposits which stain with Congo red dye or exhibit apple-green birefringence under polarized light. Biopsy is usually considered for growth of a nodule to exclude other processes (e.g., lung cancer or lymphoma). LCDD can be progressive and lead to respiratory failure. Treatment is directed at the underlying lymphoproliferative disease, if present.80
Pulmonary Langerhans Cell Histiocytosis
Background
Pulmonary Langerhans Cell Histiocytosis (pLCH), previously called pulmonary eosinophilic granuloma or Histiocytosis X, is a smoking-related lung disease predominantly seen in young adults.81 By contrast, systemic Langerhans Cell Histiocytosis (LCH) is a rare histiocytic disorder characterized by single or multiple osteolytic lesions although histocytes can infiltrate any organ, particularly bones (although sparing the heart and kidneys). Systemic LCH may be diagnosed at any age but is more common in children (especially younger children) and has no apparent association with cigarette smoking.82 The two conditions appear to be unrelated yet are indistinguishable histologically when the systemic eosinophilic granuloma of LCH involves the lung.83
Pathogenesis
PLCH has two distinct histopathologic manifestations; a cellular and fibrotic phase. The natural history of pLCH includes the early cellular form with abundance of Langerhans cells and tissue eosinophilia.84 As the lesions age, Langerhans and other immune cells become progressively depleted and overshadowed by fibrosis. In some patients, only the residual stellate scar may be left with pulmonary function significantly compromised.85,86 In such cases, imaging may demonstrate diffuse disease yet biopsied tissue may exhibit only stellate fibrotic lesions centered on the terminal airways without any identifiable interstitial inflammatory disease. (Fig. 7).
Figure 7.

A,B Coronal CT images in a patient with pLCH show characteristic irregular cysts with mid to upper zone predominance, relative sparing of the costophrenic sulci, and scattered small nodules. C. Bronchiolocentric stellate (star-like) scars develop with aging of the lesions and depletion of the Langerhans cells. D. Immunohistochemical stain for CD1a highlights the Langerhans cells. (Images C, D courtesy of Dr. Steve D. Groshong, Division of Pathology, National Jewish Health).
Clinicoradiologic manifestations
pLCH typically presents in young adults (ages 20–40) who are cigarette smokers, with non-specific pulmonary symptoms (cough, dyspnea, chest pain), pneumothorax or may be incidentally diagnosed on imaging. Some may present with systemic symptoms (fatigue, weight, fever). Pulmonary cysts are seen in almost call cases; these may be thin- or thick-walled, often irregular in outline, and usually predominate in the mid and upper lungs (Fig. 7). Nodules are seen in many cases, and the combination of cysts and nodules in a cigarette smoker should always suggest pLCH. Extrapulmonary manifestations may involve the pituitary, bone, skin and lymph nodes and patients may present with bone pain or pathologic fractures, signs of diabetes insipidus or atypical skin lesions. Longitudinal study suggests that 5% of patients with pulmonary involvement alone at diagnosis may subsequently develop extrapulmonary manifestations.87
Management considerations
Evaluation should exclude other causes of cystic lung disease, including testing for alpha-1 antitrypsin deficiency particularly if there is airflow obstruction, and identify symptoms suggestive of extrapulmonary involvement. Lung function impairment, especially airflow obstruction, is predictive of adverse outcomes,84 and early diagnosis with lung function testing may facilitate early intervention. Once the diagnosis is confirmed, management includes (1) smoking cessation and avoidance of all second-hand smoking exposure, (2) consideration for pharmacotherapy (e.g., bronchodilators and chemotherapy), and (3) assessment for pulmonary-related complications such hypoxemic respiratory failure, pneumothoraces and secondary pulmonary hypertension.88
For systemic LCH and other histiocytic disorders, somatic mutations in the MAPK pathway, such as BRAF V600E, appear to correlate with more severe disease89 and treatment with vemurafenib, an inhibitor of BRAF V600 kinase, may decrease risk for disease progression.90 The role of targeted therapy for MAPK pathway mutations remains unclear, particularly for pLCH, however testing at the time of diagnosis should be considered whenever possible (ie., surgical lung biopsy) given the future potential for treatment in refractory cases.
Clinicoradiologic correlation
A systematic approach can narrow the differential diagnosis of diffuse cystic lung disease and starts with a detailed history and physical exam. For example, sicca symptoms may suggest autoimmune-related LIP, and smoking exposure is requisite for pLCH. A family history of recurrent pneumothoraces, skin lesions or kidney cancer may suggest BHD and physical exam may reveal BHD skin lesions or clues to TSC-LAM. Additional testing may provide diagnostic confirmation such as elevated serum VEGF-D level for LAM or pathogenic FLCN gene mutation for BHD. Other diseases such as amyloidosis and LCDD typically require tissue diagnosis. Gupta et al proposed an algorithmic approach to support the clinical evaluation for such diffuse cystic lung diseases. (Fig. 8)
Figure 8. An algorithmic approach to the diagnosis of diffuse cystic lung disease.

Modified from Gupta et al. 2015, Diffuse Cystic Lung Disease Part II, AJRCCM, Vol 192 p17–29, with permission of the American Thoracic Society. Copyright © 2022 American Thoracic Society. All rights reserved.
Conclusions
Lung cysts are commonly encountered with chest imaging and can be a diagnostic challenge. Expert review of chest HRCT is still the most valuable tool to narrow the diagnostic possibilities.91 An accurate diagnosis is required to facilitate treatment and, in some conditions, adequate surveillance of extrapulmonary manifestations for patients and affected family members.
Key Points:
Distinguish cysts from cavities and define anatomic distribution as focal vs diffuse
Identify tempo of disease progression and the clinical context
Review of high resolution computerized tomography (HRCT) scan with expert radiologists will help identify key features and refine diagnostic possibilities
Blood biomarkers and genetic testing may be required to establish diagnosis
Some conditions (ie. LAM and BHD) require surveillance of extrapulmonary manifestations such as renal and other abdominal tumors
Synopsis.
Cysts and cavities in the lung are commonly encountered on chest imaging. It is necessary to distinguish thin-walled lung cysts (≤ 2mm) from cavities and characterize their distribution as focal or multi-focal vs diffuse. Focal cavitary lesions are often caused by inflammatory, infectious, or neoplastic processes in contrast to diffuse cystic lung diseases. An algorithmic approach to diffuse cystic lung disease can help narrow the differential diagnosis and additional testing such as skin biopsy, serum biomarkers and genetic testing can be confirmatory. An accurate diagnosis is essential for management and disease surveillance of extrapulmonary complications.
Abbreviations:
- LAM
lymphangioleiomyomatosis
- BHD
Birt-Hogg-Dubé
- LIP
Lymphoid interstitial pneumonia
- pLCH
Pulmonary Langerhans Cell Histiocytosis
- AML
Angiomyolipoma
- HRCT
High-Resolution Computed Tomography
Footnotes
Disclosure statement: The authors have no disclosures related to the content of this work.
Contributor Information
Matthew Koslow, Assistant Professor of Medicine, Division of Pulmonary and Critical Care Medicine, Associate Co-Director of LAM and Rare Lung Disease Clinic, National Jewish Health.
David A. Lynch, Department of Radiology, National Jewish Health.
Gregory P. Downey, Executive Vice President for Academic Affairs, Professor, Departments of Medicine, Pediatrics and Immunology and Genomic Medicine, National Jewish Health.
References
- 1.Hansell DM, Bankier AA, MacMahon H, McLoud TC, Muller NL, Remy J. Fleischner Society: glossary of terms for thoracic imaging. Radiology. Mar 2008;246(3):697–722. doi: 10.1148/radiol.2462070712 [DOI] [PubMed] [Google Scholar]
- 2.Woodring JH, Fried AM. Significance of wall thickness in solitary cavities of the lung: a follow-up study. AJR Am J Roentgenol. Mar 1983;140(3):473–4. doi: 10.2214/ajr.140.3.473 [DOI] [PubMed] [Google Scholar]
- 3.Moss J, Avila NA, Barnes PM, et al. Prevalence and clinical characteristics of lymphangioleiomyomatosis (LAM) in patients with tuberous sclerosis complex. Am J Respir Crit Care Med. Aug 15 2001;164(4):669–71. doi: 10.1164/ajrccm.164.4.2101154 [DOI] [PubMed] [Google Scholar]
- 4.Ryu JH, Moss J, Beck GJ, et al. The NHLBI lymphangioleiomyomatosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med. Jan 1 2006;173(1):105–11. doi: 10.1164/rccm.200409-1298OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han MK, Tyburczy ME, Darling TN, et al. Apparent Sporadic Lymphangioleiomyomatosis in a Man as a Result of Extreme Mosaicism for a TSC2 Mutation. Ann Am Thorac Soc. Jul 2017;14(7):1227–1229. doi: 10.1513/AnnalsATS.201703-229LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogorek B, Hamieh L, Lasseter K, et al. Generalised mosaicism for TSC2 mutation in isolated lymphangioleiomyomatosis. Eur Respir J. Oct 2019;54(4)doi: 10.1183/13993003.00938-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.European Chromosome 16 Tuberous Sclerosis C. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. Dec 31 1993;75(7):1305–15. doi: 10.1016/0092-8674(93)90618-z [DOI] [PubMed] [Google Scholar]
- 8.van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. Aug 8 1997;277(5327):805–8. doi: 10.1126/science.277.5327.805 [DOI] [PubMed] [Google Scholar]
- 9.Henske EP, McCormack FX. Lymphangioleiomyomatosis - a wolf in sheep’s clothing. J Clin Invest. Nov 2012;122(11):3807–16. doi: 10.1172/JCI58709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krymskaya VP, McCormack FX. Lymphangioleiomyomatosis: A Monogenic Model of Malignancy. Annu Rev Med. Jan 14 2017;68:69–83. doi: 10.1146/annurev-med-050715-104245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumasaka T, Seyama K, Mitani K, et al. Lymphangiogenesis-mediated shedding of LAM cell clusters as a mechanism for dissemination in lymphangioleiomyomatosis. Am J Surg Pathol. Oct 2005;29(10):1356–66. doi: 10.1097/01.pas.0000172192.25295.45 [DOI] [PubMed] [Google Scholar]
- 12.Ando H, Ogawa M, Watanabe Y, et al. Lymphangioleiomyoma of the Uterus and Pelvic Lymph Nodes: A Report of 3 Cases, Including the Potentially Earliest Manifestation of Extrapulmonary Lymphangioleiomyomatosis. Int J Gynecol Pathol. May 2020;39(3):227–232. doi: 10.1097/PGP.0000000000000589 [DOI] [PubMed] [Google Scholar]
- 13.Guo M, Yu JJ, Perl AK, et al. Single-Cell Transcriptomic Analysis Identifies a Unique Pulmonary Lymphangioleiomyomatosis Cell. Am J Respir Crit Care Med. Nov 15 2020;202(10):1373–1387. doi: 10.1164/rccm.201912-2445OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tobino K, Johkoh T, Fujimoto K, et al. Computed tomographic features of lymphangioleiomyomatosis: evaluation in 138 patients. Eur J Radiol. Mar 2015;84(3):534–541. doi: 10.1016/j.ejrad.2014.12.008 [DOI] [PubMed] [Google Scholar]
- 15.Avila NA, Dwyer AJ, Rabel A, Moss J. Sporadic lymphangioleiomyomatosis and tuberous sclerosis complex with lymphangioleiomyomatosis: comparison of CT features. Radiology. Jan 2007;242(1):277–85. doi: 10.1148/radiol.2421051767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young LR, Inoue Y, McCormack FX. Diagnostic potential of serum VEGF-D for lymphangioleiomyomatosis. N Engl J Med. Jan 10 2008;358(2):199–200. doi: 10.1056/NEJMc0707517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young L, Lee HS, Inoue Y, et al. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med. Aug 2013;1(6):445–52. doi: 10.1016/S2213-2600(13)70090-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu JJ, Robb VA, Morrison TA, et al. Estrogen promotes the survival and pulmonary metastasis of tuberin-null cells. Proc Natl Acad Sci U S A. Feb 24 2009;106(8):2635–40. doi: 10.1073/pnas.0810790106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prizant H, Sen A, Light A, et al. Uterine-specific loss of Tsc2 leads to myometrial tumors in both the uterus and lungs. Mol Endocrinol. Sep 2013;27(9):1403–14. doi: 10.1210/me.2013-1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prizant H, Taya M, Lerman I, et al. Estrogen maintains myometrial tumors in a lymphangioleiomyomatosis model. Endocr Relat Cancer. Apr 2016;23(4):265–80. doi: 10.1530/ERC-15-0505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu HJ, Lizotte PH, Du H, et al. TSC2-deficient tumors have evidence of T cell exhaustion and respond to anti-PD-1/anti-CTLA-4 immunotherapy. JCI Insight. Apr 19 2018;3(8)doi: 10.1172/jci.insight.98674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maisel K, Merrilees MJ, Atochina-Vasserman EN, et al. Immune Checkpoint Ligand PD-L1 Is Upregulated in Pulmonary Lymphangioleiomyomatosis. Am J Respir Cell Mol Biol. Dec 2018;59(6):723–732. doi: 10.1165/rcmb.2018-0123OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta N, Lee HS, Ryu JH, et al. The NHLBI LAM Registry: Prognostic Physiologic and Radiologic Biomarkers Emerge From a 15-Year Prospective Longitudinal Analysis. Chest. Feb 2019;155(2):288–296. doi: 10.1016/j.chest.2018.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson SR, Tattersfield AE. Clinical experience of lymphangioleiomyomatosis in the UK. Thorax. Dec 2000;55(12):1052–7. doi: 10.1136/thorax.55.12.1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almoosa KF, Ryu JH, Mendez J, et al. Management of pneumothorax in lymphangioleiomyomatosis: effects on recurrence and lung transplantation complications. Chest. May 2006;129(5):1274–81. doi: 10.1378/chest.129.5.1274 [DOI] [PubMed] [Google Scholar]
- 26.Gupta N, Finlay GA, Kotloff RM, et al. Lymphangioleiomyomatosis Diagnosis and Management: High-Resolution Chest Computed Tomography, Transbronchial Lung Biopsy, and Pleural Disease Management. An Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guideline. Am J Respir Crit Care Med. Nov 15 2017;196(10):1337–1348. doi: 10.1164/rccm.201709-1965ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moua T, Olson EJ, Jean HC, Ryu JH. Resolution of chylous pulmonary congestion and respiratory failure in lymphangioleiomyomatosis with sirolimus therapy. Am J Respir Crit Care Med. Aug 15 2012;186(4):389–90. doi: 10.1164/ajrccm.186.4.389 [DOI] [PubMed] [Google Scholar]
- 28.Brook-Carter PT, Peral B, Ward CJ, et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease--a contiguous gene syndrome. Nat Genet. Dec 1994;8(4):328–32. doi: 10.1038/ng1294-328 [DOI] [PubMed] [Google Scholar]
- 29.Avila NA, Kelly JA, Chu SC, Dwyer AJ, Moss J. Lymphangioleiomyomatosis: abdominopelvic CT and US findings. Radiology. Jul 2000;216(1):147–53. doi: 10.1148/radiology.216.1.r00jl42147 [DOI] [PubMed] [Google Scholar]
- 30.Hansell David M DL H Page McAdams, Bankier Alexander A. Imaging of Diseases of the Chest. Fifth ed. MOSBY, Elsevier; 2010. [Google Scholar]
- 31.Carrington CB, Cugell DW, Gaensler EA, et al. Lymphangioleiomyomatosis. Physiologic-pathologic-radiologic correlations. Am Rev Respir Dis. Dec 1977;116(6):977–95. doi: 10.1164/arrd.1977.116.6.977 [DOI] [PubMed] [Google Scholar]
- 32.Raoof S, Bondalapati P, Vydyula R, et al. Cystic Lung Diseases: Algorithmic Approach. Chest. Oct 2016;150(4):945–965. doi: 10.1016/j.chest.2016.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.AL K. Katzenstein and Askin’s surgical pathology of non-neoplastic lung disease. 1997;(3rd Ed) [PubMed] [Google Scholar]
- 34.Bonetti F, Chiodera PL, Pea M, et al. Transbronchial biopsy in lymphangiomyomatosis of the lung. HMB45 for diagnosis. Am J Surg Pathol. Nov 1993;17(11):1092–102. doi: 10.1097/00000478-199311000-00002 [DOI] [PubMed] [Google Scholar]
- 35.Miller WT, Cornog JL Jr., Sullivan MA. Lymphangiomyomatosis. A clinical-roentgenologic-pathologic syndrome. Am J Roentgenol Radium Ther Nucl Med. Mar 1971;111(3):565–72. doi: 10.2214/ajr.111.3.565 [DOI] [PubMed] [Google Scholar]
- 36.Rappaport DC, Weisbrod GL, Herman SJ, Chamberlain DW. Pulmonary lymphangioleiomyomatosis: high-resolution CT findings in four cases. AJR Am J Roentgenol. May 1989;152(5):961–4. doi: 10.2214/ajr.152.5.961 [DOI] [PubMed] [Google Scholar]
- 37.Muller NL, Chiles C, Kullnig P. Pulmonary lymphangiomyomatosis: correlation of CT with radiographic and functional findings. Radiology. May 1990;175(2):335–9. doi: 10.1148/radiology.175.2.2326457 [DOI] [PubMed] [Google Scholar]
- 38.McCormack FX, Inoue Y, Moss J, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. Apr 28 2011;364(17):1595–606. doi: 10.1056/NEJMoa1100391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taveira-DaSilva AM, Hathaway O, Stylianou M, Moss J. Changes in lung function and chylous effusions in patients with lymphangioleiomyomatosis treated with sirolimus. Ann Intern Med. Jun 21 2011;154(12):797–805, W-292–3. doi: 10.7326/0003-4819-154-12-201106210-00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for renal angiomyolipoma in patients with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis: extension of a randomized controlled trial. Nephrol Dial Transplant. Jan 2016;31(1):111–9. doi: 10.1093/ndt/gfv249 [DOI] [PubMed] [Google Scholar]
- 41.The LAM Foundation. https://wwwthelamfoundationorg/.
- 42.Hornstein OP, Knickenberg M. Perifollicular fibromatosis cutis with polyps of the colon--a cutaneo-intestinal syndrome sui generis. Arch Dermatol Res (1975). Sep 12 1975;253(2):161–75. doi: 10.1007/BF00582068 [DOI] [PubMed] [Google Scholar]
- 43.Birt AR, Hogg GR, Dube WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. Dec 1977;113(12):1674–7. [PubMed] [Google Scholar]
- 44.Daccord C, Good JM, Morren MA, Bonny O, Hohl D, Lazor R. Birt-Hogg-Dube syndrome. Eur Respir Rev. Sep 30 2020;29(157) doi: 10.1183/16000617.0042-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khoo SK, Bradley M, Wong FK, Hedblad MA, Nordenskjold M, Teh BT. Birt-Hogg-Dube syndrome: mapping of a novel hereditary neoplasia gene to chromosome 17p12-q11.2. Oncogene. Aug 23 2001;20(37):5239–42. doi: 10.1038/sj.onc.1204703 [DOI] [PubMed] [Google Scholar]
- 46.Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. Aug 2002;2(2):157–64. doi: 10.1016/s1535-6108(02)00104-6 [DOI] [PubMed] [Google Scholar]
- 47.Schmidt LS, Nickerson ML, Warren MB, et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dube syndrome. Am J Hum Genet. Jun 2005;76(6):1023–33. doi: 10.1086/430842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pavlovich CP, Grubb RL 3rd, Hurley K, et al. Evaluation and management of renal tumors in the Birt-Hogg-Dube syndrome. J Urol. May 2005;173(5):1482–6. doi: 10.1097/01.ju.0000154629.45832.30 [DOI] [PubMed] [Google Scholar]
- 49.Nahorski MS, Lim DH, Martin L, et al. Investigation of the Birt-Hogg-Dube tumour suppressor gene (FLCN) in familial and sporadic colorectal cancer. J Med Genet. Jun 2010;47(6):385–90. doi: 10.1136/jmg.2009.073304 [DOI] [PubMed] [Google Scholar]
- 50.Mota-Burgos A, Acosta EH, Marquez FV, Mendiola M, Herrera-Ceballos E. Birt-Hogg-Dube syndrome in a patient with melanoma and a novel mutation in the FCLN gene. Int J Dermatol. Mar 2013;52(3):323–6. doi: 10.1111/j.1365-4632.2012.05742.x [DOI] [PubMed] [Google Scholar]
- 51.Lindor NM, Kasperbauer J, Lewis JE, Pittelkow M. Birt-Hogg-Dube syndrome presenting as multiple oncocytic parotid tumors. Hered Cancer Clin Pract. Oct 10 2012;10(1):13. doi: 10.1186/1897-4287-10-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gupta N, Sunwoo BY, Kotloff RM. Birt-Hogg-Dube Syndrome. Clin Chest Med. Sep 2016;37(3):475–86. doi: 10.1016/j.ccm.2016.04.010 [DOI] [PubMed] [Google Scholar]
- 53.Tong Y, Schneider JA, Coda AB, Hata TR, Cohen PR. Birt-Hogg-Dube Syndrome: A Review of Dermatological Manifestations and Other Symptoms. Am J Clin Dermatol. Feb 2018;19(1):87–101. doi: 10.1007/s40257-017-0307-8 [DOI] [PubMed] [Google Scholar]
- 54.Furuya M, Tanaka R, Koga S, et al. Pulmonary cysts of Birt-Hogg-Dube syndrome: a clinicopathologic and immunohistochemical study of 9 families. Am J Surg Pathol. Apr 2012;36(4):589–600. doi: 10.1097/PAS.0b013e3182475240 [DOI] [PubMed] [Google Scholar]
- 55.Escalon JG, Richards JC, Koelsch T, Downey GP, Lynch DA. Isolated Cystic Lung Disease: An Algorithmic Approach to Distinguishing Birt-Hogg-Dube Syndrome, Lymphangioleiomyomatosis, and Lymphocytic Interstitial Pneumonia. AJR Am J Roentgenol. Mar 19 2019:1–5. doi: 10.2214/AJR.18.20920 [DOI] [PubMed] [Google Scholar]
- 56.Toro JR, Pautler SE, Stewart L, et al. Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dube syndrome. Am J Respir Crit Care Med. May 15 2007;175(10):1044–53. doi: 10.1164/rccm.200610-1483OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gupta N, Seyama K, McCormack FX. Pulmonary manifestations of Birt-Hogg-Dube syndrome. Fam Cancer. Sep 2013;12(3):387–96. doi: 10.1007/s10689-013-9660-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu X, Cowl CT, Baqir M, Ryu JH. Air travel and pneumothorax. Chest. Apr 2014;145(4):688–694. doi: 10.1378/chest.13-2363 [DOI] [PubMed] [Google Scholar]
- 59.Gupta N, Vassallo R, Wikenheiser-Brokamp KA, McCormack FX. Diffuse Cystic Lung Disease. Part II. Am J Respir Crit Care Med. Jul 1 2015;192(1):17–29. doi: 10.1164/rccm.201411-2096CI [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stamatakis L, Metwalli AR, Middelton LA, Marston Linehan W. Diagnosis and management of BHD-associated kidney cancer. Fam Cancer. Sep 2013;12(3):397–402. doi: 10.1007/s10689-013-9657-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benhammou JN, Vocke CD, Santani A, et al. Identification of intragenic deletions and duplication in the FLCN gene in Birt-Hogg-Dube syndrome. Genes Chromosomes Cancer. Jun 2011;50(6):466–77. doi: 10.1002/gcc.20872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cha SI, Fessler MB, Cool CD, Schwarz MI, Brown KK. Lymphoid interstitial pneumonia: clinical features, associations and prognosis. Eur Respir J. Aug 2006;28(2):364–9. doi: 10.1183/09031936.06.00076705 [DOI] [PubMed] [Google Scholar]
- 63.Swigris JJ, Berry GJ, Raffin TA, Kuschner WG. Lymphoid interstitial pneumonia: a narrative review. Chest. Dec 2002;122(6):2150–64. doi: 10.1378/chest.122.6.2150 [DOI] [PubMed] [Google Scholar]
- 64.Cosgrove GP FM, Schwarz MI. Lymphoplasmacytic infiltrations of the lung. Interstitial Lung Diseases. 4th ed. Hamilton, Canada: BC Decker; 2003. [Google Scholar]
- 65.Wick La. Practical Pulmonary Pathology. 2011;
- 66.Julsrud PR, Brown LR, Li CY, Rosenow EC 3rd, Crowe JK. Pulmonary processes of mature-appearing lymphocytes: pseudolymphoma, well-differentiated lymphocytic lymphoma, and lymphocytic interstitial pneumonitis. Radiology. May 1978;127(2):289–96. doi: 10.1148/127.2.289 [DOI] [PubMed] [Google Scholar]
- 67.Strimlan CV, Rosenow EC 3rd, Divertie MB, Harrison EG Jr. Pulmonary manifestations of Sjogren’s syndrome. Chest. Sep 1976;70(03):354–61. doi: 10.1378/chest.70.3.354 [DOI] [PubMed] [Google Scholar]
- 68.Strimlan CV, Rosenow EC 3rd, Weiland LH, Brown LR. Lymphocytic interstitial pneumonitis. Review of 13 cases. Ann Intern Med. May 1978;88(5):616–21. doi: 10.7326/0003-4819-88-5-616 [DOI] [PubMed] [Google Scholar]
- 69.Ichikawa Y, Kinoshita M, Koga T, Oizumi K, Fujimoto K, Hayabuchi N. Lung cyst formation in lymphocytic interstitial pneumonia: CT features. J Comput Assist Tomogr. Sep-Oct 1994;18(5):745–8. doi: 10.1097/00004728-199409000-00012 [DOI] [PubMed] [Google Scholar]
- 70.Silva CI, Flint JD, Levy RD, Muller NL. Diffuse lung cysts in lymphoid interstitial pneumonia: high-resolution CT and pathologic findings. J Thorac Imaging. Aug 2006;21(3):241–4. doi: 10.1097/01.rti.0000213554.61752.73 [DOI] [PubMed] [Google Scholar]
- 71.Johkoh T, Muller NL, Pickford HA, et al. Lymphocytic interstitial pneumonia: thin-section CT findings in 22 patients. Radiology. Aug 1999;212(2):567–72. doi: 10.1148/radiology.212.2.r99au05567 [DOI] [PubMed] [Google Scholar]
- 72.Johkoh T, Ichikado K, Akira M, et al. Lymphocytic interstitial pneumonia: follow-up CT findings in 14 patients. J Thorac Imaging. Jul 2000;15(3):162–7. doi: 10.1097/00005382-200007000-00002 [DOI] [PubMed] [Google Scholar]
- 73.Zhu C, Hu J, Wu J, Cheng L. Transformation of lymphoid interstitial pneumonia (LIP) into malignant lymphoma in patients with Sjogren’s syndrome: a case report and literature review. J Cardiothorac Surg. Apr 15 2022;17(1):79. doi: 10.1186/s13019-022-01826-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Travis WD, Galvin JR. Non-neoplastic pulmonary lymphoid lesions. Thorax. Dec 2001;56(12):964–71. doi: 10.1136/thorax.56.12.964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ravichandran S, Lachmann HJ, Wechalekar AD. Epidemiologic and Survival Trends in Amyloidosis, 1987–2019. N Engl J Med. Apr 16 2020;382(16):1567–1568. doi: 10.1056/NEJMc1917321 [DOI] [PubMed] [Google Scholar]
- 76.Papa R, Lachmann HJ. Secondary, AA, Amyloidosis. Rheum Dis Clin North Am. Nov 2018;44(4):585–603. doi: 10.1016/j.rdc.2018.06.004 [DOI] [PubMed] [Google Scholar]
- 77.Pozzi C, D’Amico M, Fogazzi GB, et al. Light chain deposition disease with renal involvement: clinical characteristics and prognostic factors. Am J Kidney Dis. Dec 2003;42(6):1154–63. doi: 10.1053/j.ajkd.2003.08.040 [DOI] [PubMed] [Google Scholar]
- 78.Zamora AC, White DB, Sykes AM, et al. Amyloid-associated Cystic Lung Disease. Chest. May 2016;149(5):1223–33. doi: 10.1378/chest.15-1539 [DOI] [PubMed] [Google Scholar]
- 79.Lantuejoul S, Moulai N, Quetant S, et al. Unusual cystic presentation of pulmonary nodular amyloidosis associated with MALT-type lymphoma. Eur Respir J. Sep 2007;30(3):589–92. doi: 10.1183/09031936.00136605 [DOI] [PubMed] [Google Scholar]
- 80.Colombat M, Stern M, Groussard O, et al. Pulmonary cystic disorder related to light chain deposition disease. Am J Respir Crit Care Med. Apr 1 2006;173(7):777–80. doi: 10.1164/rccm.200510-1620CR [DOI] [PubMed] [Google Scholar]
- 81.Travis WD, Borok Z, Roum JH, et al. Pulmonary Langerhans cell granulomatosis (histiocytosis X). A clinicopathologic study of 48 cases. Am J Surg Pathol. Oct 1993;17(10):971–86. doi: 10.1097/00000478-199310000-00002 [DOI] [PubMed] [Google Scholar]
- 82.Howarth DM, Gilchrist GS, Mullan BP, Wiseman GA, Edmonson JH, Schomberg PJ. Langerhans cell histiocytosis: diagnosis, natural history, management, and outcome. Cancer. May 15 1999;85(10):2278–90. doi: [DOI] [PubMed] [Google Scholar]
- 83.Colby TV, Lombard C. Histiocytosis X in the lung. Hum Pathol. Oct 1983;14(10):847–56. doi: 10.1016/s0046-8177(83)80160-9 [DOI] [PubMed] [Google Scholar]
- 84.Vassallo R, Ryu JH, Colby TV, Hartman T, Limper AH. Pulmonary Langerhans’-cell histiocytosis. N Engl J Med. Jun 29 2000;342(26):1969–78. doi: 10.1056/NEJM200006293422607 [DOI] [PubMed] [Google Scholar]
- 85.Brauner MW, Grenier P, Tijani K, Battesti JP, Valeyre D. Pulmonary Langerhans cell histiocytosis: evolution of lesions on CT scans. Radiology. Aug 1997;204(2):497–502. doi: 10.1148/radiology.204.2.9240543 [DOI] [PubMed] [Google Scholar]
- 86.Wick KOLaMR. Practical Pulmonary Pathology: A Diagnostic Approach. 3rd Ed ed. Elsevier; 2018. [Google Scholar]
- 87.Benattia A, Bugnet E, Walter-Petrich A, et al. Long-term outcomes of adult pulmonary Langerhans cell histiocytosis: a prospective cohort. Eur Respir J. May 2022;59(5)doi: 10.1183/13993003.01017-2021 [DOI] [PubMed] [Google Scholar]
- 88.Gupta N, Vassallo R, Wikenheiser-Brokamp KA, McCormack FX. Diffuse Cystic Lung Disease. Part I. Am J Respir Crit Care Med. Jun 15 2015;191(12):1354–66. doi: 10.1164/rccm.201411-2094CI [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heritier S, Emile JF, Barkaoui MA, et al. BRAF Mutation Correlates With High-Risk Langerhans Cell Histiocytosis and Increased Resistance to First-Line Therapy. J Clin Oncol. Sep 1 2016;34(25):3023–30. doi: 10.1200/JCO.2015.65.9508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Diamond EL, Subbiah V, Lockhart AC, et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol. Mar 1 2018;4(3):384–388. doi: 10.1001/jamaoncol.2017.5029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gupta N, Meraj R, Tanase D, et al. Accuracy of chest high-resolution computed tomography in diagnosing diffuse cystic lung diseases. Eur Respir J. Oct 2015;46(4):1196–9. doi: 10.1183/13993003.00570-2015 [DOI] [PubMed] [Google Scholar]