

Abstract
Objective
To identify gut microbiome features associated with MRI lesion burden in persons with pediatric‐onset multiple sclerosis (symptom onset <18 years).
Methods
A cross‐sectional study involving the Canadian Paediatric Demyelinating Disease Network study participants. Gut microbiome features (alpha diversity, phylum‐ and genus‐level taxa) were derived using 16S rRNA sequencing from stool samples. T1‐ and T2‐weighted lesion volumes were measured on brain MRI obtained within 6 months of stool sample procurement. Associations between the gut microbiota and MRI metrics (cube‐root‐transformed) were assessed using standard and Lasso regression models.
Results
Thirty‐four participants were included; mean ages at symptom onset and MRI were 15.1 and 19.0 years, respectively, and 79% were female. The T1‐ and T2‐weighted lesion volumes were not significantly associated with alpha diversity (age and sex‐adjusted p > 0.08). At the phylum level, high Tenericutes (relative abundance) was associated with higher T1 and T2 volumes (β coefficient = 0.25, 0.37) and high Firmicutes, Patescibacteria or Actinobacteria with lower lesion volumes (β coefficient = −0.30 to −0.07). At the genus level, high Ruminiclostridium, whereas low Coprococcus 3 and low Erysipelatoclostridium were associated with higher lesion volumes.
Interpretation
Our study characterized the gut microbiota features associated with MRI lesion burden in pediatric‐onset MS, shedding light onto possible pathophysiological mechanisms.
Introduction
Multiple sclerosis (MS) is a chronic autoimmune disease characterized by inflammatory demyelination of the central nervous system. MS typically manifests in adulthood, but pediatric‐onset (<18 years of age) affects up to 5% of the MS population. 1 Pediatric‐onset MS is predominantly relapsing–remitting and is associated with a highly active course. 2 The involvement of white or gray matter related to inflammatory activity can be clinically silent but evident on MRI. Persons with pediatric‐onset MS have comparable, if not greater, total MRI lesion burden relative to adult‐onset individuals with similar clinical disease durations. 3 , 4 , 5 Although the relationship between early MRI and long‐term outcome in individuals with pediatric‐onset MS is not well understood, the lesion burden seen on MRI in the early stages of adult‐onset MS predicts long‐term disability. 6
The growing understanding of the role of microbiota in the development of the immune system suggests that the gut microbiota may affect MS pathogenesis, 7 , 8 including inflammatory activity in the brain. 9 The total T2 lesion volume on brain MRI is a marker for focal inflammatory lesion burden, 10 , 11 and chronic T1‐hypointense lesion volume represents irreversible focal lesional tissue degeneration. 11 , 12 , 13 This raises the possibility that a person's gut resident microbes might influence MS disease burden, as measured by MRI lesion burden.
Compared to studies of adults with MS, studies of individuals with pediatric‐onset MS likely provide an earlier window into understanding the initiation of the disease and related disease activity. Previous studies comparing individuals with pediatric‐onset MS and unaffected controls revealed differences in the taxonomic composition, connectivity, and functional potential of the gut microbiota. 14 , 15 , 16 , 17 Alpha diversity, measuring the richness and evenness within a microbial community, is an important overall indicator of gut health 18 ; shifts in the relative abundance of individual taxa, on the other hand, have the potential to reveal the dysbiosis of gut microbiota that is associated with the MS disease activity. To date, relatively little is known of the relationship between these gut microbiota features and MRI‐related disease activity in the pediatric‐onset MS population. 19
A better understanding of the connections between gut microbiota and MRI lesion activities could guide future novel biomarkers of disease activity or suggest novel drug or intervention‐related targets. In this cross‐sectional study, we investigated the relationship between the gut microbiota composition and brain MRI lesion burden in persons with pediatric‐onset MS (symptom onset <18 years of age). The MRI metrics included total T2‐hyperintense lesion volume and chronic T1‐hypointense lesion volume. The gut microbiota composition metrics included alpha diversity (richness and evenness) and taxa abundance at the phylum and genus levels.
Methods
Enrollment, data collection, procurement of stool samples, and related sequencing have been described previously in detail. 15 Briefly, participants were enrolled through the Canadian Paediatric Demyelinating Disease Study (23 sites in Canada and 1 in the United States). Informed consent or assent was obtained from participants and their parents/legal guardians. Ethical approval was obtained from each institution's research ethics board (the Hospital for Sick Children in Toronto, the Children's hospital of Philadelphia, the Universities of: British Columbia, Calgary, Manitoba, McGill, Memorial, and Western). All methods were performed in accordance with the relevant guidelines and regulations. All participants were <18 years of age at MS symptom onset, met the 2017 McDonald diagnostic criteria for MS and were negative for serum myelin oligodendrocyte glycoprotein (MOG) antibodies, if a test result was available. The MOG antibodies were tested using live, MOG IgG1‐specific cell‐based assay (Oxford, UK) with full‐ or short‐length human MOG. 20 , 21 Demographic and clinical characteristics were collected via standardized forms facilitated by the site coordinator, including date of birth, MS symptom onset, sex assigned at birth, and country of residence (Canada/USA). Information on body mass index (BMI), categorized into normal or obese (>30, the 85th percentile of the cohort), current or prior use of a disease‐modifying drug (DMD) for the treatment of MS, and the Expanded Disability Status Scale score (EDSS) was also collected around the time of stool sample procurement. The 7‐point Bristol Stool Scale 22 was also completed by the participant or parent/caregiver at the time of stool sample procurement and was categorized into hard (types 1–2), medium (3–5), or loose (6, 7).
Stool samples were procured between 2015 and 2019, shipped on ice and stored at −80°C in a central location. Participants were required to have no exposure to systemic antibiotics or corticosteroids in the 30 days before stool sample procurement. After all samples were procured, Zymo Quick‐DNA™ Fecal/Soil Microbe Miniprep Kit (D6010) was used to extract DNA from each stool sample. The 16S rRNA gene (V4 region) of all samples included in this study was amplified and sequenced via Illumina MiSeq‐600 V3 platform in one batch at the National Microbiology Laboratory (Winnipeg, Manitoba, Canada). The resulting sequences were then denoised and clustered into amplicon sequence variants (ASVs) using Deblur (v.1.1.0) and QIIME2 (Quantitative Insights Into Microbial Ecology; v.2019.4) with taxonomy assigned using the SILVA database (v.132).
Research MRI scans were obtained using standardized 1.5 or 3 Tesla brain MRI protocols, and all images were processed centrally at the McConnell Brain Imaging Centre, McGill University. T1 and T2 lesion were segmented automatically and manually corrected, as previously described. 5 , 23 T2‐weighted hyperintense lesions had to be at least 3 voxels in size. T1‐weighted hypointense lesions were segmented on postcontrast T1‐weighted images with contrast enhanced regions excluded from the segmentation. For inclusion in the present study, participants were required to have a research brain MRI scan performed within 180 days of a stool sample procurement. T2 lesion volume was the primary MRI metric of interest with the T1 lesion volume being the secondary metrics. If more than one research MRI scan was available within the time frame, the closest one to the stool sample collection was selected.
Cohort characteristics are presented descriptively. The T1 and T2 lesion volumes were transformed by the cube root to achieve an approximately normal distribution. Hereafter, the lesion volume refers to cube‐root‐transformed lesion volume unless stated otherwise. The relationships between the T1 and T2 lesion volumes and participant characteristics were assessed by the Kruskal–Wallis rank sum test for categorical variables and Pearson's product–moment correlation for continuous variables.
Alpha diversity was assessed as species evenness and richness (Shannon index, Margalef's index and Chao1 index; see Table S1) using amplicon sequence variants (ASVs) rarefied to 16,181 sequences. Linear regression was used to examine the association between these alpha diversity indices and T1 and T2 lesion volumes with and without adjusted for age at MRI and sex. Separately, at the phylum and genus levels, a Least Absolute Shrinkage and Selection Operator (Lasso) regression was used to determine the most prominent individual ASVs associated with T1 and T2 lesion volumes. The Lasso regression is a type of linear regression that uses shrinkage to avoid over fitting of the data and is well suited for variable selection with high dimensional data. 24 , 25 All individual ASVs with more than 20% of nonzero counts were included in the Lasso model after normalization, using the median‐of‐ratios method (R‐package DESeq2 [Differential Expression of Sequencing Data]). In total, 9 phyla (8 bacteria and 1 archaea) and 169 genera (168 bacteria and 1 archaea) were included in the Lasso analyses as binary variables after being categorized as high relative abundance (above the median of the normalized relative abundance) and low relative abundance (below or equal to the median). Since taxonomic relative abundance was usually highly skewed, it was categorized to avoid unduly large influence from a few extreme values. The optimal tuning parameter λ of the Lasso regression was determined by implementing a repeated grid search cross‐validation 26 with 10,000 runs of K‐fold cross‐validation (K = 10 for T2, K = 9 for T1 as ≥3 individuals per fold is required). The mean cross‐validation error for each λ was stored from each run, and the λ with the smallest average of the mean cross‐validation errors over all runs was selected for the final model. The linear and Lasso regression results were expressed as β coefficients and 95% confidence intervals (CIs). The 95% CIs of the β coefficients from the Lasso regressions were calculated using 10,000 bootstrap resamples. The value of β represents the mean difference in cube‐root‐transformed T1 and T2 lesion volumes between those with high relative abundance and with low relative abundance after adjusting for the other ASVs selected by the Lasso model; a positive (negative) β value indicates that high relative abundance was associated with a higher (lower) lesion volume. For descriptive purpose, the boxplots of lesion volumes were generated by the relative abundance level of the selected phyla and genera. No patient characteristics were included in our main Lasso regression analyses. To assess whether the results may be affected by the other factors, a sensitivity analysis was carried out in which the following potential confounders were included: age at MRI (continuous), sex, DMD exposure (ever vs never), country of residence (Canada vs USA), and Bristol Stool Scale (medium [types 3–5] vs hard or loose [types 1–2, 6–7]). The Bristol Stool Scale was grouped into two levels in this sensitivity analysis as only two participants' samples were categorized as loose (types 6–7) (see Supplementary Material—Sensitivity Analysis). The analyses in this paper are exploratory in nature, and no power calculation was conducted a priori. Due to limited knowledge regarding the relationship between the MRI outcomes and the gut microbiome composition, a meaningful power calculation was not feasible. Statistical analyses were performed using R (v. 4.0.2).
Results
Descriptive summaries
The cohort selection flow chart is shown in Fig. S1. The descriptive summaries of the cohort characteristics are presented in Table 1. A total of 34 individuals with pediatric‐onset MS were included in this study. Nearly 80% were female and 76% were residents of Canada, with the rest living in the United States. Approximately three‐quarters of participants had a medium stool consistency, and five participants (15%) were considered overweight or obese (BMI > 30).
Table 1.
Characteristics of the pediatric‐onset multiple sclerosis cases.
| Characteristic measured at stool sample collection, unless stated otherwise | Multiple sclerosis cases (total n = 34) |
|---|---|
| Female: n (%) | 27 (79%) |
| Country of residence: Canada/USA a , n | 26/8 |
| Age at symptom onset, years: mean (SD; range) | 15.1 (2.4; 6.3–17.8) |
| Age at MRI, years: mean (SD; range) | 19.0 (4.3; 7.3–27.4) |
| Clinical disease duration at MRI, years: mean (SD; range) | 4.0 (3.9; 0.1–11.1) |
| Expanded Disability Status Scale b : median (IQR) | 1.5 (1, 1.5) |
| Time since last relapse to MRI, years: median (IQR) | 1.0 (0.4, 5.1) |
| Presence of comorbidities c : n (%) | 13 (38%) |
| Ever exposed to disease‐modifying drug d : n (%) | 27 (79%) |
| Body Mass Index: crude median (IQR) | 23.2 (20.4, 26.2) |
| Cigarette smoking (passive or active) ever prestool sample: n (%) | 5 (15%) |
| Bristol Stool Scale: median (IQR) | 3.5 (3, 4) |
| Hard (types 1–2)/Medium (types 3–5)/Loose (types 6–7) | 7/25/2 |
| Block Kids Screener: dietary intake per day, range | |
| % protein caloric intake | 15%–18% |
| % fat caloric intake | 31%–38% |
| % carbohydrate caloric intake | 47%–58% |
| Fiber, grams | 8.8–13.7 |
| Number of participants with available/valid diet data | 22 |
| Alpha diversity of the gut microbiota, mean (SD; IQR) | |
| Shannon index (evenness and richness) | 0.68 (0.10; 0.66, 0.72) |
| Margalef's index (richness) | 21.1 (6.4; 17.2, 23.2) |
| Chao 1 index (richness) | 241.4 (74.4; 197.4, 279.3) |
| MRI metrics, median (IQR; range) | |
| T2 lesion volume, cm3 | 3.911 (1.389, 9.916; 0.033–27.135) |
| T1 lesion volume e , cm3 | 0.642 (0.075, 1.959; 0–5.907) |
Percentage denominator reflects those without missing data for the corresponding variable.
IQR, interquartile range (first and third quartiles); range, minimum–maximum; SD, standard deviation.
Canada: 19 from Ontario, 4 from Quebec, 2 from Manitoba, 1 from Alberta; USA: all 8 from Pennsylvania area.
Assessed at the clinical visit closest to the stool sample procurement, which had a median time to stool sample procurement of 11 days (IQR: 5–31 days).
Reported comorbidities included asthma, psoriasis, atopic dermatitis, moderate depression, anxiety, milk allergy, attention‐deficit/hyperactivity disorder, gastroesophageal reflux, and migraine.
All “ever exposed to disease‐modifying drug” participants had been exposed within 3 months of stool sample procurement; 12 ever received beta‐interferon, 6 glatiramer acetate, 4, dimethyl fumarate, 3 natalizumab, 2 fingolimod, 2 rituximab, and 1 teriflunomide.
T1 lesion results were available in 29 participants and were missing in 5 participants due to unavailable postcontrast T1‐weighted scan.
The average (standard deviation [SD]) age at MS symptom onset and at MRI measurement were 15.1 (2.4) and 19.0 (4.3) years, respectively. The mean clinical disease duration at the time of MRI was 4.0 (3.9) years. The median time (interquartile range [IQR]) since the most recent relapse (pre‐MRI) was 1.0 (0.4–5.1) year. The median (IQR) time between the MRI measurement and stool sample collection was 12 (5–34) days and ranged from 0 to 168 days. No study participant had a relapse during this time period.
Most participants had a relatively low disability burden as measured by EDSS; 32 of 34 participants scored 2.5 or below, considered “minimal disability.” Most participants (79%) were exposed to at least one DMD before stool sample collection. Thirteen (38%) of the participants had at least one comorbidity prior to or at the time of MRI (the most common being asthma and atopic dermatitis).
The median T2 lesion volume was 3.911 cm3 (IQR: 1.389, 9.916 cm3). The median (IQR) after cube root transformation was 1.6 (1.1, 2.1). T1 lesion volume was missing in five participants since a postcontrast image was unavailable. The median T1 lesion volume for the remaining 29 participants was 0.642 cm3 (0.075, 1.959 cm3) and 0.9 (0.4, 1.3) after transformation.
Age at MS onset, age at MRI, and disease duration at MRI were not highly correlated with either of the T2 or T1 lesion volume (all Pearson's correlations < 0.34, p‐values > 0.05; see Table S2). The T2 or T1 lesion volume also did not differ significantly by sex, country of residence, Bristol Stool Scale (hard, normal, loose), DMD exposure, or BMI (normal vs obese) (Kruskal–Wallis rank sum test, p‐values > 0.10; see Table S3). Therefore, these participant characteristics were not considered as potential confounders in the main Lasso regressions.
Gut microbiota alpha diversity
Two alpha diversity metrics (Margalef's index and Chao 1 index) were initially associated with the T2 lesion volume in univariate analyses, but none remained significant after age at MRI and sex adjustments (all p‐values > 0.08, Table 2). None of the alpha diversity metrics (Shannon index, Margalef's index and Chao 1 index) were associated with T1 lesion volume (univariate and age at MRI and sex adjusted p > 0.13, Table 2).
Table 2.
The association between the gut microbiota alpha diversity indices and brain MRI lesion volumes in the pediatric‐onset MS cohort.
| Alpha diversity | Unadjusted | Adjusted | ||
|---|---|---|---|---|
| β (95% CI) | p‐value | β (95% CI) | p‐value | |
| T2 lesion volume (cube‐root‐transformed) | ||||
| Shannon index (evenness and richness) | −0.12 (−0.36, 0.12) | 0.34 | −0.12 (−0.34, 0.11) | 0.33 |
| Margalef's index (richness) | 0.24 (0.01, 0.46) | 0.047 | 0.18 (−0.06, 0.42) | 0.14 |
| Chao 1 index (richness) | 0.25 (0.03, 0.48) | 0.036 | 0.21 (−0.02, 0.44) | 0.086 |
| T1 lesion volume (cube‐root‐transformed) | ||||
| Shannon index (evenness and richness) | −0.20 (−0.62, 0.22) | 0.35 | −0.36 (−0.78, 0.06) | 0.11 |
| Margalef's index (richness) | 0.12 (−0.07, 0.31) | 0.22 | 0.07 (−0.13, 0.26) | 0.53 |
| Chao 1 index (richness) | 0.11 (−0.08, 0.31) | 0.27 | 0.07 (−0.13, 0.27) | 0.48 |
Interpretation: The β values and their 95% confidence intervals (CIs) were assessed using linear regression. The β value represents the mean change in lesion volume (cube root transformed) associated per 1 standard deviation (SD) increase in the alpha diversity index. The SDs are as follows: 0.1 (Shannon), 6.4 (Margalef's), and 74.4 (Chao 1) (see Tables 1 and S1). A positive β value indicates that a higher index value was associated with a higher brain T2 or T1 lesion volume.
Phylum‐level gut microbiota
The Lasso regression identified five phyla associated with T2 lesion volume and four phyla associated with T1 lesion volume (Fig. 1); all belong to Bacteria. The only phylum within Archaea included in the analysis, Euryarchaeota, was not selected by the Lasso regression. The two most influential phyla on T2 lesion volume were Firmicutes and Tenericutes. The two most influential phyla on T1 lesion volume were Actinobacteria and Tenericutes. A high relative abundance of Tenericutes was associated with a higher T2 or T1 lesion volume (β coefficient [95% CI] = 0.37 [0.00–0.85] for T2, 0.25 [0.00–0.60] for T1). Meanwhile, a high relative abundance of Firmicutes, Patescibacteria, and Actinobacteria was associated with a lower T2 or T1 lesion volume (β coefficient of Firmicutes = −0.30 [−0.79, 0.00] for T2, −0.07 [−0.48, 0.00] for T1; β coefficient of Patescibacteria = −0.21 [−0.63, 0.00] for T2, −0.18 [−0.49, 0.00] for T1; β coefficients of Actinobacteria = −0.18 [−0.54, 0.00] for T2, −0.22 [−0.55, 0.00] for T1). A summary of the relative abundance of the phyla selected by the Lasso regression can be found in Table S4. The distributions of the lesion volumes by the relative abundance level of these phyla are shown in Fig. S2. Although, in the sensitivity analysis when the other covariates were included, Actinobacteria and Verrucomicrobia were no longer selected for T2 lesion volume, and one additional phylum Proteobacteria was selected for T1 lesion volume albeit with a relatively weak association (see Table S5).
Figure 1.
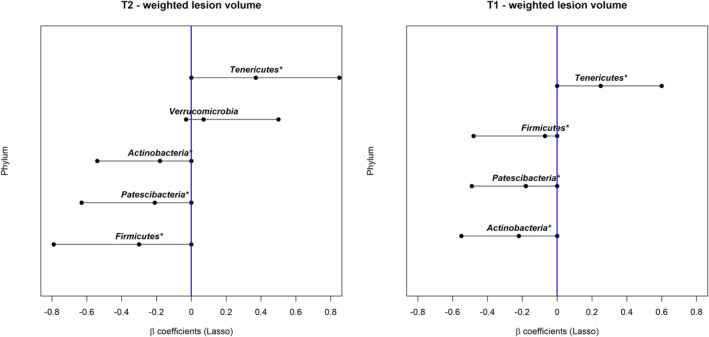
The association between the gut microbiota (phylum level) and brain MRI lesion volumes (cube root transformed) in the pediatric‐onset MS cohort. “*” indicates that the taxon was identified for both T1 and T2 lesion volumes. Interpretation: The β values and their 95% confidence intervals were assessed using Lasso regression. The β value represents the mean difference in lesion volume (cube root transformed) between the participants with normalized gut microbiota relative abundance above the median vs those below or equal to the median after adjusting for the other ASVs in the model; a positive β value indicates that a high relative abundance was associated with a higher lesion volume. A difference of 0.33 and 0.18 corresponds to 20% above or below the overall sample mean for the (cube root transformed) T2 and T1 lesion volumes (mean = 1.6 and 0.9, respectively).
Genus level gut microbiota
The Lasso regression selected 10 dominant genera associated with T2 lesion volume, a high relative abundance of five genera was associated with a higher T2 lesion volume and five associated with a lower T2 lesion volume. The only genus within Archaea included in the analysis, Methanobrevibacter, was not selected by the Lasso regression. All but one were classified within the phylum Firmicutes. One belonged to the phylum Actinobacteria, which was uncultured at the genus level but classified under the family Atopobiaceae (Fig. 2). Seven genera, all but one being from the phylum Firmicutes, were associated with T1 lesion volume; a high relative abundance of two genera was associated with a higher lesion volume and five were associated with a lower lesion volume. The genus whose relative abundance associated with the greatest increase in the lesion volumes was Ruminiclostridium (β coefficients = 0.10 [0.00, 0.42] for T2, 0.18 [0.00, 0.34] for T1) and the one associated with the largest decrease was Coprococcus 3 (β coefficient = −0.31 [−0.54, 0.00] for T2, −0.28 [−0.52, 0.00] for T1), followed by Erysipelatoclostridium (β coefficient = −0.24 [−0.50, 0.00] for T2, −0.14 [−0.34, 0.00] for T1). A summary of the relative abundance and the complete taxonomic level information of the genera selected by the Lasso regression can be found in Table S4 and Fig. S3, respectively. The distributions of the lesion volumes by the relative abundance level of these genera are shown in Fig. S4. In the sensitivity analysis with the other covariates, the findings remain largely unchanged except for two taxa Candidatus Stoquefichus and Adlercreutzia, that were weakly associated with T1 lesion volume (close to null β coefficient values) in the main analysis, were no longer selected (see Table S6).
Figure 2.
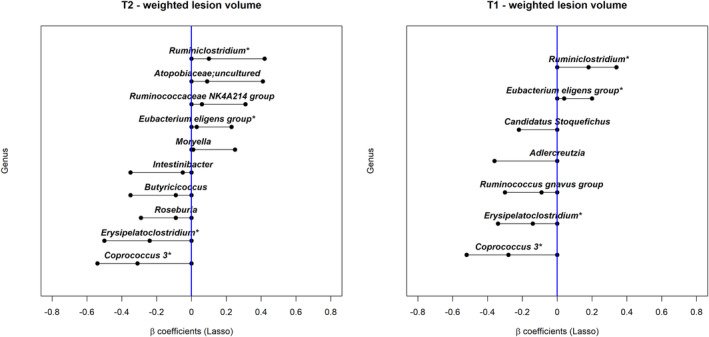
The association between the gut microbiota (genus level) and brain MRI lesion volumes (cube root transformed) in the pediatric‐onset MS cohort. “*” indicates that the taxon was identified for both T1 and T2 lesion volumes. Interpretation: The β values and their 95% confidence intervals were assessed using Lasso regression. The β value represents the mean difference in lesion volume (cube root transformed) between the participants with normalized gut microbiota relative abundance above the median vs those below or equal to the median after adjusting for the other ASVs in the model; a positive β value indicates that a high relative abundance was associated with a higher lesion volume. A difference of 0.33 and 0.18 corresponds to 20% above or below the overall sample mean for the (cube root transformed) T2 and T1 lesion volumes (mean = 1.6 and 0.9, respectively).
Discussion
In this cross‐sectional study of persons with pediatric‐onset MS, we did not detect a clear association between the alpha diversity (evenness and richness) of the gut microbiota and MRI lesion burden. However, at the taxonomic level, we identified several phylum‐ and genus‐level microbes associated with both T1 and T2 lesion volumes. These findings, combined with others, 19 suggest that the gut microbiota has potential to play various roles in modifying MS activity, which should be examined in future studies. Ultimately, this may offer novel opportunity to alter the disease course in MS.
Of the five phyla identified, four were associated with both T1 and T2 lesion volumes in the same direction: Tenericutes, Firmicutes, Patesibacteria, and Actinobacteria. One, Verrucomicrobia, was only associated with T2 lesion volume, although the association was relatively weak (see Fig. 1 and Table S5). At the genus level, four genera were associated with both T1 and T2 lesion volumes, and were in the same direction. Six genera were exclusively associated with T2 lesion volume and three were exclusively associated with T1 lesion volume (see Fig. 2 and Table S6). We observed relatively fewer taxa associated with T1 lesion volume. This may be due to the smaller sample size and lower variability in T1 lesion volume.
The presence of phylum Tenericutes was associated with higher T1 and T2 lesion burden. Tenericutes is typically reported as being of relatively low abundance in the human gut microbiota, and little appears known of its role or functionality in a human health or disease context. This phylum was only present in eight participants (24%) in our cohort, and most clades were from the order RF39, class Mollicutes, and were typically unnamed at the genus level. Although Tenericutes has not been previously correlated with MS activity, several studies have found other links with MS 14 , 27 , 28 as well as other conditions, such as obesity 29 and inflammatory bowel disease. 30 One study found that members of the phylum Tenericutes were enriched in the relapsing–remitting form of experimental autoimmune encephalitis (EAE) relative to disease naive mice. 27 A previous case–control study, reported higher Tenericutes abundance in those with pediatric‐onset MS, especially among the DMD naïve group, compared to healthy controls. 14 Similarly, a recent Italian study comparing adults with MS (n = 15) and their unaffected family members (n = 15) 28 also found a higher relative abundance of Tenericutes in those with MS.
Of the other phyla identified, most were associated with lower brain MRI lesion volume. For example, a high relative abundance of Firmicutes was associated with lower T1 and T2 lesion volumes. Many members within Firmicutes are thought to exert anti‐inflammatory effects. 31 , 32 , 33 Our findings broadly agree with a previous study in which depletion of Firmicutes was found in persons with pediatric‐onset MS compared to controls. 19 However, in our study, the direction of the association varied across different genera classified within the phylum Firmicutes. This observation is consistent with the diverse functionality of its member taxa. For example, a high relative abundance of the genera Ruminiclostridium and Ruminococcaceae NK4A214 group from the family Ruminococcaceae was associated with a higher lesion burden. In contrast, the genus Butyricicoccus from the same family was associated with a lower lesion burden. Also within the Firmicutes, a high relative abundance of the genus Erysipelatoclostridium from the family Erysipelotrichaceae, and the genera Coprococcus 3 and Roseburia from the butyrate‐producing family Lachnospiraceae, was all associated with a lower lesion burden. Butyrate is a short‐chain fatty acid (SCFA) and a key metabolite produced by some gut microbes, which has been shown to have neuroprotective effects. 34 Decreased butyrate producers in MS have been reported. 32 , 35 Interestingly, the presence of Roseburia has been associated with better cognitive function in other neurodegenerative diseases, 36 presumed to be mediated via SCFA production. Our study also found a high abundance of the phylum Actinobacteria associated with a lower T2 volume. Presuming that a higher abundance of this phylum infers a beneficial effect, our findings agree with observations seen in another study from our group, which also reported a relative depletion of Actinobacteria in persons with pediatric‐onset MS (who were also participants from the Canadian Paediatric Demyelinating Disease Study) versus unaffected controls. 15 However, this finding remains to be verified as Actinobacteria was not selected in our sensitivity analysis that included other covariates. Finally, in our study, the phylum Patescibacteria was also associated with a lower T1 and T2 volume, although little is known about its role in health and disease.
Few studies have reported a relationship between the gut microbiome and MRI features in persons with MS. To our knowledge, none has studied the association of the gut microbiota and cumulative lesion burden on MRI in the pediatric‐onset MS population. A study of 55 pediatric‐onset MS patients in the United States 19 examined the gut microbiome and new lesion activity on brain MRI. They found higher abundance of butyrate‐producing microbes associated with low risk of subsequent disease activity. Another study from the United States 37 reported that, based on analysis of univariate correlations adjusted for age, the abundances of several species in the genus Clostridium were associated with a higher T2 lesion volume and a lower whole brain volume in adult individuals with RRMS (n = 116). Although Clostridium was not identified in our analysis, the study identified species from Roseburia and Coprococcus as associated with a higher brain volume in the RRMS participants. Caution should be taken when comparing findings between studies due to differences in the methodology, including study design, stool sample processing, sequencing and bioinformatics pipeline, analytical (statistical) approaches, and covariate adjustment. A well‐designed study is needed to compare the microbiome and its relationship to MRI lesion burden between adult‐onset and pediatric‐onset MS.
We examined two main MRI metrics, the total T1 and T2 lesion volumes, routinely used for monitoring the cumulative disease burden in MS. T2‐weighted hyperintense lesions could represent a wide range of pathologic processes and are not specific for lesion severity.
Chronic hypointense areas on T1‐weighted MRIs are thought to be more pathologically specific, representing permanent tissue destruction. 13 As expected these two metrics were highly correlated in our cohort, hence, it was not surprising that several identified taxa were associated with both lesion volumes. Not all pathological features of MS can be studied using conventional MRI scans and the association between lesion volume and clinical presentation or change is typically modest. Other MRI metrics, such as brain volume, particularly thalamic volume loss, and lesion volumes at specific brain regions, spinal cord lesions, injuries in the gray matter, such as gray matter volume loss or cortical lesions, would also be of interest for future studies.
A strength of this study is that we adopted the Lasso regression model, which allowed us to consider all candidate ASVs simultaneously. The relationships between different gut microbes are complex, and their relative abundances are interrelated. Instead of assessing the association between each ASV and the MRI metrics individually and adjusting for multiple comparisons, our approach captures the joint effect of the ASVs. We chose to dichotomize the ASV relative abundance into low and high categories to avoid the issues with highly skewed ASV counts, making the results more robust, although there was some loss of data granularity. Another study strength was that our cohort consists of young individuals relatively close to their disease initiation (median disease duration was 4 years), thus reducing (although not eliminating entirely) many confounders experienced when studying adults with long‐term MS.
Our study has several limitations, including being cross‐sectional such that our observed associations do not imply causality. In addition, MS is a highly heterogeneous chronic disease; both lesion volumes and microbial composition evolve over time and are subject to short‐term fluctuation. 38 Therefore, the potentially complex relationship between MRI lesion burden and gut microbiota features might not be reliably captured by cross‐sectional data. Without repeated MRIs and stool samples, we were not able to evaluate the stability of our findings. However, an earlier study of a group of individuals with at least two stool samples 39 suggested that the gut microbiota composition in our population exhibited relative stability over a short interval (2–25 months). Our study focused on gut microbiome composition and did not investigate the relationship between the lesion volumes and biochemical pathway features (functionality) of the gut microbiome. Our sample size is modest since pediatric‐onset MS remains relatively rare, and resulted in wide confidence intervals and limited our ability to detect mild associations and adjust for potential confounders. It is also possible that some of the identified associations may only be present in our cohort. Verifying the results in future studies with larger cohorts would be valuable. Our study was not design to examine the mediation effect of SCFA production or the impact of past DMD exposure; both of these aspects warrant future investigations.
Our study provides valuable insight into the design of such future studies. Our findings could guide the determination of sample size that will allow for adequate power to detect meaningful differences as well as controlling for potential confounders. Careful considerations should be given to identifying and controlling for potential confounding factors through study design and analysis. To reduce the impact of the random variability in gut microbiota composition on the study findings, it would be desirable to collect multiple stool samples when feasible. 38 As advanced sequencing technologies, such as shotgun metagenomic sequencing with deep sequencing, become more accessible, it will be possible to obtain data with better taxonomic coverage and resolution, or allow for analysis of function potential, and biochemical pathway. Which MRI metrics to be included also requires consideration depending on the pathological features of interest and the sensitivity of the metrics. It would be valuable to measure the SCFA levels in stool or serum samples. This would enable the study to examine the correlation between the abundance of SCFA‐producing bacteria and the levels of SCFA, as well as their correlation with MRI features. Another aspect to investigate is whether DMD alters the association between the microbiome and MRI features. Ideally, stool samples and MRI scans should be collected both before and after the initiation of DMD.
In summary, our findings add to the understanding of the association of the gut microbiota with the MRI lesion burden in pediatric‐onset MS. At the taxonomic level, several microbes were found to be associated with either a higher or lower lesion burden and were also found to be either enriched or depleted in persons with MS compared to healthy controls in previous studies. This raises the possibility that they could play a role in both the development of MS and the disease course, both of which are worthy of future investigation. Future work could include gaining an in‐depth understanding of the functionality of our identified microbes and their interaction with the central nervous system, as well as examining whether these microbes predict changes in lesion volume and future disease progression, or if modification of these microbes could prevent MS disease activity or progression.
Author Contributions
FZ, YZ, and HT contributed to conception and design of the study, acquisition, and interpretation of data, and drafted the first version of the manuscript, tables and figures. FZ conducted statistical analysis, and YZ provided input on statistical analysis. YZ, JF, GVD, CB, MG, JH, EW, and HT facilitated obtaining funding (PI: Tremlett, The Multiple Sclerosis Scientific and Research Foundation, EGID: 2636). DA, AB‐O, RAM, JOM, EAY, and BB were part of the original Canadian Pediatric Demyelinating Disease Network study and facilitated collection of the Canada‐USA cohort characteristics and MRI data. JH facilitated training of study coordinators the collection of stool samples. CBe oversaw the biobanking. MG, NK, and GVD oversaw the 16S rRNA sequencing and bioinformatics; Cbo, performed the stool extractions and 16S rRNA sequencing; JF, AM, NK, and FZ performed the bioinformatics. JH and EW facilitated data access to the US Network of Pediatric MS Centers study cohort. All authors contributed to the interpretation of the data and provided a critical review of the manuscript.
Conflict of Interest Statement
Feng Zhu and Yinshan Zhao were funded through research grants held by HT, including The Multiple Sclerosis Scientific and Research Foundation (PI: Tremlett, EGID: 2636). Douglas L. Arnold is funded by the Canadian MS Society, the International Progressive MS Alliance, the Canadian Institutes of Health Research, and the US Department of Defense. He has received personal compensation for serving as a Consultant for Alexion, Biogen, Celgene, Frequency Therapeutics, GENeuro, Genentech, Merck/EMD Serono, Novartis, Roche, and Sanofi. Dr. Arnold has an ownership interest in NeuroRx. Amit Bar‐Or is funded by the National Institutes of Health, ITN, National Mulitple Sclerosis Society, and Multiple Sclerosis Society of Canada. ABO has participated as a speaker in meetings sponsored by and received consulting fees and/or grant support from: Janssen/Actelion; Atara Biotherapeutics, Biogen Idec, Celgene/Receptos, Roche/Genentech, Medimmune, Merck/EMD Serono, Novartis, Sanofi‐Genzyme. Charles Bernstein is supported in part by the Bingham Chair in Gastroenterology. He has consulted to or served on advisory boards for Abbvie Canada, Amgen Canada, Bristol Myers Squibb Canada, JAMP Pharmaceuticals, Janssen Canada, Pfizer Canada, Sandoz Canada, Takeda, and has received unrestricted educational grants from Abbvie Canada, Janssen Canada, Pfizer Canada, Bristol Myers Squibb Canada, and Takeda Canada. He has been on the speaker's bureau of Abbvie Canada, Janssen Canada, Pfizer Canada, and Takeda Canada. He has received research grants from Abbvie Canada, Amgen Canada, Pfizer Canada, and Sandoz Canada and contract grants from Janssen. Ruth Ann Marrie receives research funding from: CIHR, Multiple Sclerosis Society of Canada, Multiple Sclerosis Scientific Foundation, National Multiple Sclerosis Society, the Consortium of Multiple Sclerosis Centers, the Arthritis Society and US Department of Defense. She is a co‐investigator on a study funded in part by Biogen Idec and Roche Canada. Ali Mirza is funded through the MS Society of Canada endMS Doctoral Studentship (EGID: 3246) and the Multiple Sclerosis Scientific and Research Foundation (PI: Tremlett, EGID: 2636). Julia O'Mahony receives research funding from: Multiple Sclerosis Society of Canada, Multiple Sclerosis Scientific Foundation, and Consortium of Multiple Sclerosis Centers. Gary Van Domselaar is the Chief Bioinformatics Scientist with the National Microbiology Laboratory—Public Health Agency of Canada and has received research support in the last 3 years from the National Multiple Sclerosis Society, the Canadian Institute of Health Research, and Genome Canada. E. Ann Yeh has received research support in the last 3 years from the National Multiple Sclerosis Society, Canadian Institutes of Health Research, National Institutes of Health, Ontario Institute of Regenerative Medicine, Stem Cell Network, SickKids Foundation, Peterson Foundation, Multiple Sclerosis Society of Canada, and the Multiple Sclerosis Scientific Research Foundation. She has received funding for investigator‐initiated research from Biogen and has served on scientific advisory boards for Biogen, Alexion, and Hoffman‐LaRoche. Brenda Banwell serves as a consultant to Novartis, UCB, and Roche. Dr Banwell provides nonremunerated advice on clinical trial design to Novartis, Biogen, and Teva Neuroscience. BB is funded by the National Multiple Sclerosis Society, National Institutes of Health, and Mutliple Sclerosis Society of Canada. Emmanuelle Waubant is funded by the National Mutliple Sclerosis Society, the National Institues of Health, PCORI and the Race to Erase MS. EW has received consulting honoraria from Jazz Pharma, Emerald, and DBV. She volunteers on a clinical trial committee for Novartis. Helen Tremlett has, in the last 5 years, received research support from the Canada Research Chair Program, the National Multiple Sclerosis Society, the Canadian Institutes of Health Research, the Multiple Sclerosis Society of Canada, the Multiple Sclerosis Scientific Research Foundation, and the EDMUS Foundation (“Fondation EDMUS contre la sclérose en plaques”). In addition, in the last 5 years, has had travel expenses or registration fees prepaid or reimbursed to present at CME conferences from the Consortium of Multiple Sclerosis Centres (2018), National Multiple Sclerosis Society (2018), ECTRIMS/ACTRIMS (2017–2022), and American Academy of Neurology (2019). Speaker honoraria are either declined or donated to an MS charity or to an unrestricted grant for use by HT's research group. Christine Bonner, Janace Hart, Natalie Knox, and Morag Graham declare no potential conflict of interest.
Supporting information
Data S1 Supporting information.
Acknowledgements
This study was supported by the Multiple Sclerosis Scientific and Research Foundation (#EGID: 2636; PI:Tremlett). The funding source was not involved in the study design, the collection, analysis, and interpretation of the data, or in the decision to submit this article for publication. We are grateful for all the participants' involvement, especially children and teenagers with MS and their parents. We are also grateful to all the investigators and study teams at each site involved in the Canadian Paediatric Demyelinating Disease Network study, without whom this study would not have been possible. We acknowledge the important contributions of the Tremlett team (University of British Columbia); Thomas Duggan in facilitating study set‐up, coordination and data collection; Bonnie Leung for study coordination; Michael Sargent (Department of Internal Medicine, and the University of Manitoba Inflammatory Bowel Disease Clinical and Research Centre laboratory, Winnipeg, Canada) for managing the stool biobank, and Jessica D. Forbes (University of Toronto, Toronto, Canada) for assisting with the original grant application.
Feng Zhu and Yinshan Zhao contributed equally to the manuscript.
Funding Statement
This work was funded by Multiple Sclerosis Scientific and Research Foundation grant #EGID: 2636.
Data Availability Statement
The authors can be contacted for data access; requests will be assessed on a case‐by‐case basis, based on the scientific rigor of the proposed research question.
References
- 1. Yeh EA, Chitnis T, Krupp L, et al. Pediatric multiple sclerosis. Nat Rev Neurol. 2009;5(11):621‐631. [DOI] [PubMed] [Google Scholar]
- 2. Waubant E, Banwell B, Wassmer E, et al. Clinical trials of disease‐modifying agents in pediatric MS: opportunities, challenges, and recommendations from the IPMSSG. Neurology. 2019;92(22):e2538‐e2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waubant E, Chabas D, Okuda DT, et al. Difference in disease burden and activity in pediatric patients on brain magnetic resonance imaging at time of multiple sclerosis onset vs adults. Arch Neurol. 2009;66(8):967‐971. [DOI] [PubMed] [Google Scholar]
- 4. Yeh EA, Weinstock‐Guttman B, Ramanathan M, et al. Magnetic resonance imaging characteristics of children and adults with paediatric‐onset multiple sclerosis. Brain. 2009;132(Pt 12):3392‐3400. [DOI] [PubMed] [Google Scholar]
- 5. Ghassemi R, Narayanan S, Banwell B, et al. Quantitative determination of regional lesion volume and distribution in children and adults with relapsing‐remitting multiple sclerosis. PLoS One. 2014;9(2):e85741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brownlee WJ, Altmann DR, Prados F, et al. Early imaging predictors of long‐term outcomes in relapse‐onset multiple sclerosis. Brain. 2019;142(8):2276‐2287. [DOI] [PubMed] [Google Scholar]
- 7. Cryan JF, O'Riordan KJ, Sandhu K, Peterson V, Dinan TG. The gut microbiome in neurological disorders. Lancet Neurol. 2020;19(2):179‐194. [DOI] [PubMed] [Google Scholar]
- 8. Forbes JD, Bernstein CN, Tremlett H, van Domselaar G, Knox NC. A fungal world: could the gut mycobiome be involved in neurological disease? Front Microbiol. 2018;9:3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rothhammer V, Mascanfroni ID, Bunse L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med. 2016;22(6):586‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bruck W, Bitsch A, Kolenda H, et al. Inflammatory central nervous system demyelination: correlation of magnetic resonance imaging findings with lesion pathology. Ann Neurol. 1997;42(5):783‐793. [DOI] [PubMed] [Google Scholar]
- 11. Filippi M, Agosta F. Imaging biomarkers in multiple sclerosis. J Magn Reson Imaging. 2010;31(4):770‐788. [DOI] [PubMed] [Google Scholar]
- 12. van Walderveen MA, Kamphorst W, Scheltens P, et al. Histopathologic correlate of hypointense lesions on T1‐weighted spin‐echo MRI in multiple sclerosis. Neurology. 1998;50(5):1282‐1288. [DOI] [PubMed] [Google Scholar]
- 13. Hemond CC, Bakshi R. Magnetic resonance imaging in multiple sclerosis. Cold Spring Harb Perspect Med. 2018;8(5):a028969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tremlett H, Fadrosh DW, Faruqi AA, et al. Gut microbiota in early pediatric multiple sclerosis: a case‐control study. Eur J Neurol. 2016;23(8):1308‐1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tremlett H, Zhu F, Arnold D, et al. The gut microbiota in pediatric multiple sclerosis and demyelinating syndromes. Ann Clin Transl Neurol. 2021;8(12):2252‐2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mirza AI, Zhu F, Knox N, et al. Metagenomic analysis of the pediatric‐onset multiple sclerosis gut microbiome. Neurology. 2022;98(10):e1050‐e1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mirza AI, Zhu F, Knox N, et al. The metabolic potential of the paediatric‐onset multiple sclerosis gut microbiome. Mult Scler Relat Disord. 2022;63:103829. [DOI] [PubMed] [Google Scholar]
- 18. Magurran AE. Measuring Biological Diversity. Blackwell Pub.; 2004. [DOI] [PubMed] [Google Scholar]
- 19. Horton MK, McCauley K, Fadrosh D, et al. Gut microbiome is associated with multiple sclerosis activity in children. Ann Clin Transl Neurol. 2021;8(9):1867‐1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waters P, Woodhall M, O'Connor KC, et al. MOG cell‐based assay detects non‐MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Waters P, Fadda G, Woodhall M, et al. Serial anti‐myelin oligodendrocyte glycoprotein antibody analyses and outcomes in children with demyelinating syndromes. JAMA Neurol. 2020;77(1):82‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blake MR, Raker JM, Whelan K. Validity and reliability of the Bristol stool form scale in healthy adults and patients with diarrhoea‐predominant irritable bowel syndrome. Aliment Pharmacol Ther. 2016;44(7):693‐703. [DOI] [PubMed] [Google Scholar]
- 23. Francis SJ. Automatic lesion identification in MRI of multiple sclerosis patients. M.Sc. McGill University; 2004. [Google Scholar]
- 24. Tibshirani R. Regression shrinkage and selection via the lasso. J R Stat Soc Series B Stat Methodol. 1996;58(1):267‐288. [Google Scholar]
- 25. Friedman JHT, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1‐22. [PMC free article] [PubMed] [Google Scholar]
- 26. Krstajic D, Buturovic LJ, Leahy DE, Thomas S. Cross‐validation pitfalls when selecting and assessing regression and classification models. J Chem. 2014;6(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gandy KAO, Zhang J, Nagarkatti P, Nagarkatti M. The role of gut microbiota in shaping the relapse‐remitting and chronic‐progressive forms of multiple sclerosis in mouse models. Sci Rep. 2019;9(1):6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Galluzzo P, Capri FC, Vecchioni L, et al. Comparison of the intestinal microbiome of Italian patients with multiple sclerosis and their household relatives. Life (Basel). 2021;11(7):620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Santos VM, Brito AKP, Amorim AT, et al. Evaluation of fecal microbiota and its correlation with inflammatory, hormonal, and nutritional profiles in women. Braz J Microbiol. 2022;53(2):1001‐1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Imhann F, Vich Vila A, Bonder MJ, et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. 2018;67(1):108‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tremlett H, Fadrosh DW, Faruqi AA, et al. Gut microbiota composition and relapse risk in pediatric MS: a pilot study. J Neurol Sci. 2016;363:153‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mirza A, Forbes JD, Zhu F, et al. The multiple sclerosis gut microbiota: a systematic review. Mult Scler Relat Disord. 2020;37:101427. [DOI] [PubMed] [Google Scholar]
- 33. Cao Y, Shen J, Ran ZH. Association between Faecalibacterium prausnitzii reduction and inflammatory bowel disease: a meta‐analysis and systematic review of the literature. Gastroenterol Res Pract. 2014;2014:872725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dalile B, Van Oudenhove L, Vervliet B, et al. The role of short‐chain fatty acids in microbiota‐gut‐brain communication. Nat Rev Gastroenterol Hepatol. 2019;16(8):461‐478. [DOI] [PubMed] [Google Scholar]
- 35. Noto D, Miyake S. Gut dysbiosis and multiple sclerosis. Clin Immunol. 2022;235:108380. [DOI] [PubMed] [Google Scholar]
- 36. Toh TS, Chong CW, Lim SY, et al. Gut microbiome in Parkinson's disease: new insights from meta‐analysis. Parkinsonism Relat Disord. 2022;94:1‐9. [DOI] [PubMed] [Google Scholar]
- 37. Cox LM, Maghzi AH, Liu S, et al. Gut microbiome in progressive multiple sclerosis. Ann Neurol. 2021;89(6):1195‐1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vandeputte D, De Commer L, Tito RY, et al. Temporal variability in quantitative human gut microbiome profiles and implications for clinical research. Nat Commun. 2021;12(1):6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liang G, Zhu F, Mirza AI, et al. Stability of the gut microbiota in persons with paediatric‐onset multiple sclerosis and related demyelinating diseases. Mult Scler. 2022;28(11):1819‐1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting information.
Data Availability Statement
The authors can be contacted for data access; requests will be assessed on a case‐by‐case basis, based on the scientific rigor of the proposed research question.