

Summary
Exercise mediates tissue metabolic function through direct and indirect adaptations to acylcarnitine (AC) metabolism, but the exact mechanisms are unclear. We found that circulating medium-chain acylcarnitines (AC) (C12-C16) are lower in active/endurance trained human subjects compared to sedentary controls, and this is correlated with elevated cardiorespiratory fitness and reduced adiposity. In mice, exercise reduced serum AC and increased liver AC, and this was accompanied by a marked increase in expression of genes involved in hepatic AC metabolism and mitochondrial β-oxidation. Primary hepatocytes from high-fat fed, exercise trained mice had increased basal respiration compared to hepatocytes from high-fat fed sedentary mice, which may be attributed to increased Ca2+ cycling and lipid uptake into mitochondria. The addition of specific medium- and long-chain AC to sedentary hepatocytes increased mitochondrial respiration, mirroring the exercise phenotype. These data indicate that AC redistribution is an exercise-induced mechanism to improve hepatic function and metabolism.
Subject areas: Health sciences, Physiology, Cell biology
Graphical abstract
Highlights
-
•
Chronic exercise training reduces serum acylcarnitines (AC) in mice and humans
-
•
Hepatic AC facilitates mitochondrial respiration
-
•
Exercise enhances liver AC metabolism by increasing fatty acid uptake and Ca2+ cycling
Health sciences; Physiology; Cell biology
Introduction
A growing aging population has led to an increased prevalence of various metabolic disorders including obesity and type 2 diabetes. An impaired ability to adapt to different substrate utilization,1,2,3 including impaired fatty acid oxidation and metabolism, contributes to altered body fat distribution and is tightly linked to insulin resistance.4 Thus, strategies that improve substrate utilization and enhance metabolic flexibility are required to combat obesity, insulin resistance, and type 2 diabetes; one such strategy is exercise. Exercise-induced benefits on human metabolite profiles include increases in oxidation of fatty acids and energy expenditure.5,6,7,8,9,10,11
Exercise training is associated with numerous improvements in metabolite profiles,5,6,7,8,9,10 studies have been predominantly in plasma,10 but few studies have investigated tissue-specific metabolite changes induced by exercise. Metabolomic studies in exercise-trained individuals have identified circulating metabolites that are associated with increased gene expression of mitochondrial fatty acid oxidation.8,10,12,13 Studies have also indicated that exercise mediates tissue metabolic function through direct and indirect adaptations to acylcarnitine (AC) metabolism.7,14 AC are byproducts of fatty acid oxidation that fuel mitochondrial respiration and have emerged as important indicators of fuel selection during exercise.7,15,16 AC may also have bioactive properties that are independent from energy production, acting to mediate membrane dynamics, membrane-bound ion transporters, or intracellular signaling.17
Exercise is also an important modulator of hepatic metabolism.18,19 In humans, exercise is associated with a reduction in liver triglycerides in obese patients with non-alcoholic fatty liver disease (NAFLD) or type 2 diab,etes,20,21 indicating enhanced utilization of fat within the liver. Improvements in hepatic metabolism are promoted by increased muscle-liver substrate flux, which might be necessary to mediate the metabolic benefits of exercise training.22 AC can be readily utilized as fuel sources by the liver,23 and in turn, the liver acts as an important hub for fuel redistribution to other tissues.14,23,24,25 Although the effect of exercise on hepatic metabolism has been explored, a role for exercise training to modulate liver AC and mediate hepatic metabolic adaptations has not been thoroughly investigated.
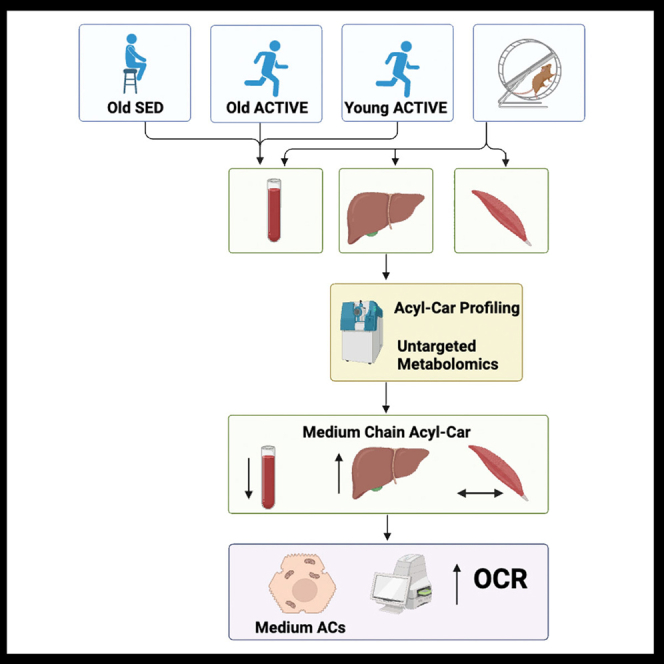
In the current study, we investigated the role of exercise to regulate AC in both humans and rodents and determined that exercise reduces circulating short-, medium-, and long-chain ACs. In humans, AC were reduced in physically active/trained individuals, and this was associated with lower adiposity and greater cardiorespiratory fitness. In mice, AC analysis of the muscle, serum, and liver showed that exercise training shifted short-, medium-, and long-chain ACs within and across tissues and identified the liver as a highly dynamic metabolic hub in response to exercise training. Notably, exercise training in mice resulted in dramatic reduction in medium and long-chain ACs in serum and improved medium- and long-chain AC handling and metabolism in the liver. Lastly, we define a role for medium- and long-chain ACs as activators of hepatic mitochondrial respiration in response to exercise through a mechanism involving enhanced hepatic lipid uptake and Ca2+ cycling.
Results
Chronic exercise training differentially regulates circulating acylcarnitine levels in human subjects
We first performed an exploratory analysis to determine whether chronic physical activity (i.e., endurance exercise training) and age alter the systemic levels of acylcarnitines (AC) in three groups of male and female human subjects. Young active/endurance trained adults (YA, n = 13; 10 Males, 3 Females; Age = 32.3 ± 5.3 years; VO2peak = 56.1 ± 8.3 mL/kg/min), older active/endurance trained adults (OA; n = 13; 11 Males, 2 Females; Age = 68.3 ± 3.5 years; VO2peak = 34.5 ± 4.2 mL/kg/min), and older sedentary adults (OS; n = 16; 10 Males, 6 Females; Age = 70.7 ± 5.0 years; VO2peak = 19.1 ± 3.9 mL/kg/min) participated in the study. (Table S1). Lipidomics analysis examined a panel of 87 acylcarnitines in serum samples obtained at rest following an overnight fast. There were 8 total acylcarnitines that were significantly different (-log10(p value)>1.3) between sedentary (OS) and active/endurance trained (YA + OA) groups when controlled for age and sex, with 3 acylcarnitines lower in the active/endurance trained group and 5 acylcarnitines higher (Figure 1A). We next applied targeted, quantitative MS/MS analysis to measure the abundance of a subset of acylcarnitines among the three separate groups. Figure 1B provides a heatmap of the acylcarnitines that were present in at least 75% of the participants from each of the groups (n = 30 acylcarnitines). The majority of circulating acylcarnitines were lowest in the young active/endurance trained group in comparison to the older sedentary group. Several acylcarnitines (C10:3, C14:3, C16:2, C18:3, C18:1, C18:0, C20:3, C20:2, and C22:0) showed age-related differences, where acylcarnitine levels for both older sedentary and older active/endurance trained individuals were higher than young active counterparts. This is in line with previous work showing age increases serum acylcarnitine levels in humans.26,27 Interestingly, several acylcarnitines showed differences related to physical activity/exercise training. In particular, medium-chain acylcarnitines (C10:0, C14:0, and C16:0) (Figures 1C–1E) were lowest in the young active group and highest in the older sedentary group, while older active/endurance trained adults presented an intermediate profile that closely resembled the young active/endurance trained group. These results are similar to previous exercise training intervention studies that showed a reduction in serum medium- and long-chain acylcarnitines in obese adults.7,28 Further, long-chain acylcarnitine C20:1 showed an opposite trend, with older sedentary individuals presenting lower levels than active/endurance trained counterparts (Figure 1F). An interaction effect between group and sex was not evident, suggesting sex was not a driving factor for group differences in circulating acylcarnitine levels (Tables S2 and S3).
Figure 1.

Circulating acylcarnitines are differentially regulated with chronic endurance exercise training in humans
(A) Volcano plot of 87 acylcarnitines comparing the fold difference between active/endurance trained (young and old) and sedentary adults (old only). p values were derived from ANCOVA analysis where age and biological sex were taken as co-variants. Dashed line indicates level of significance (-log10(p value)>1.3; p value<0.05). Blue dots indicate acylcarnitines that were more abundant in the sedentary group (log2(Fold Change) < 0) while the gold dots indicate acylcarnitines that were more abundant in the active group (log2(Fold Change) > 0).
(B) Heatmap of acylcarnitines that were present in the majority (>75%) of young active/endurance trained (YA), older active/endurance trained (OA), and older sedentary (OS) adults. Data are presented as group averages and row normalized values, with blue indicating lowest abundance (0%) and red indicating highest abundance (100%).
(C‒F) Circulating concentrations of C10:0 (C), C14:0 (D), C16:0 (E), and C20:1 (F) acylcarnitines in young active/endurance trained (n = 11–13; orange circles), older active/endurance trained (n = 13; green circles), and older sedentary (n = 15–16; light blue circles). For C:10 acylcarnitines, two data points (1 young active/endurance trained, 1 older sedentary) were not included in the analysis as these points were statistical outliers (Grubb’s test). For C20:1, two data points were excluded from the young active/endurance trained group due to values being below the level of detection. ∗, p value<0.05; ∗∗, p value<0.01 (one-way ANOVA and Tukey’s multiple comparisons test).
(G‒V) Pearson correlations of resting circulating acylcarnitines (C10:0 (G, K, O, and S), C14:0 (H, L, P, and T), C16:0 (I, M, Q, and U), and C20:1 (J, N, R, and V) with VO2peak (G–J; n = 40–42), daily energy expenditure attributed to activity (K‒N; n = 39–41), total fat mass (O‒R; n = 40–42), and visceral adipose tissue (VAT) mass (S–V; n = 40–42). Correlations include data from young active/endurance trained adults (orange circles; n = 12–13), older active/endurance trained adults (green circles; n = 13), and older sedentary adults (light blue circles; n = 14–16). Data are mean ± SD.
Circulating acylcarnitines are associated with cardiorespiratory fitness and adiposity in humans
To further evaluate the role of physical activity on circulating acylcarnitines, we performed Pearson correlation analysis with clinical variables. First, we determined the relationship between circulating acylcarnitines with markers of cardiorespiratory fitness (i.e., VO2peak). In line with the findings in Figures 1C–1F, lower levels of medium-chain acylcarnitines (C10:0, C14:0, and C16:0) were associated with elevated cardiorespiratory fitness (Figures 1G–1I), while levels of C20:1 acylcarnitines were not associated with VO2peak (Figure 1J). We also determined whether acylcarnitines were related to daily physical activity levels. Specifically, lower levels of medium-chain acylcarnitines (C10:0, C14:0, and C16:0) were associated with higher daily energy expenditure that was attributed to physical activity (Figures 1K‒1M). In contrast, elevated serum levels of long-chain acylcarnitines (C20:1) were associated with higher daily energy expenditure attributed to physical activity (Figure 1N).
As serum acylcarnitines are associated with metabolic disorders,29,30 we next evaluated the relationship between circulating acylcarnitines and adiposity. Using dual energy x-ray absorptiometry (DEXA) scans, we evaluated total fat mass and visceral adipose tissue (VAT) mass in humans. In contrast to markers of cardiorespiratory fitness and daily physical activity energy expenditure (Figures 1G and 1N), higher levels of C10:0, C14:0, and C16:0 acylcarnitines were associated with greater adiposity, including elevated total fat (Figures 1O‒1Q) and VAT mass (Figures 1S‒1U), while higher C20:1 acylcarnitine levels were negatively associated with adiposity (Figures 1R and 1V). Collectively, these data indicate that lower serum concentration of medium-chain acylcarnitines are associated with elevated cardiorespiratory fitness and lower adiposity in active/endurance trained adults.
Exercise training shifts the metabolite profile in mouse liver and serum
Accumulation of ACs in circulation is associated with metabolic disorders, and diversion of ACs to specific tissues for oxidation may be a means to reduce disease risk. Our human data (Figure 1) indicated that circulating ACs are reduced in exercise-trained individuals, but due to the limitations in human studies, it is uncertain where the ACs may be metabolized in response to exercise training. To determine the effects of exercise on AC tissue accumulation, we utilized a rodent model and investigated the AC profile of the serum, liver, and skeletal muscle (triceps) in high-fat fed male mice. Chronic exercise (3 weeks) did not result in any difference in body weight among sedentary (inactive or SED) or exercise-trained (active or EX) mice (Figures S1A and S1B). EX mice ran ∼4.6 km/day (Figure S1C). There was no change in total fat mass but % fat mass was reduced in EX mice (Figures S1D and S1E). Total lean mass and % lean mass were increased with exercise (Figures S1F and S1G), and fat-to-lean ratio was decreased in EX mice (Figure S1H).
To determine the effects of exercise training on the AC profile in mice, we performed targeted and untargeted metabolomics in the serum, liver, and skeletal muscle (triceps) to survey 15 amino acids (AA) and 67 acylcarnitines (ACs) in the serum, liver, and muscle. Two-way hierarchical clustering of auto-scaled metabolites showed that EX significantly altered ACs (yellow) in the serum (Figure 2A; Table 1), liver (Figure 2B; Table 2), and muscle (Figure 2C; Table S4).
Figure 2.

Chronic exercise training shift the hepatic metabolomic profile in mice fed high-fat diet and alters serum ACs and hydroxylated ACs
Heatmap showing a two-way hierarchical clustering of (A) serum, (B) liver, or (C) triceps, highlighting that ACs account for major shifts following exercise training. Total AC and AC-OH in (D) serum, (E) liver, and (F) muscle, (G) Short AC, (H) medium AC, and (I) long AC in serum, liver, and muscle.
(J) Chord visualization of significant, positive (chords represent r ≥ 0.5, p < 0.05) short ACs correlations demonstrate greater inter-tissue connectivity between muscle-liver (blue-to-red chords) and liver-serum (yellow-to-red chords), inter-tissue correlations. Conversely, exercise training reduced inter-tissue positive correlations (r ≥ 0.5, p < 0.05). of short AC.
(K) Chord representation (r ≥ 0.5, p < 0.05) of medium ACs correlations demonstrate greater inter-tissue connectivity between muscle-liver (blue-to-red chords) and many muscle intra-tissue correlations (more chords), while exercise training robustly reduced intra- and inter-tissue correlations (r ≥ 0.5, p < 0.05).
(L) Chord visualization (r ≥ 0.5, p < 0.05) shows a great degree of coordination between muscle-liver (blue-to-red chords) and intra-tissue liver and muscle correlations (more chords) while exercise training drastically reduced the intra- and inter-tissue correlations (r ≥ 0.5, p < 0.05).
Table 1.
Summary of significant exercise effects in serum metabolites in mice
| Metabolite class | Metabolite | SED | EX |
|---|---|---|---|
| Short AC | C2a | 4.76 ± 1.28 | 2.88 ± 0.30 |
| C3a | 0.1276 ± 0.0134 | 0.0791 ± 0.0064 | |
| C4/Ci4a | 0.1198 ± 0.0206 | 0.0555 ± 0.0032 | |
| C5:1a | 0.0893 ± 0.0031 | 0.0778 ± 0.0048 | |
| C5a | 0.0559 ± 0.0070 | 0.0395 ± 0.0029 | |
| C4-OHa | 0.0738 ± 0.0083 | 0.0487 ± 0.0039 | |
| C5-OH/C3-DCa | 0.0666 ± 0.0052 | 0.0523 ± 0.0032 | |
| C8a | 0.0101 ± 0.0015 | 0.0067 ± 0.0006 | |
| C10:1a | 0.0062 ± 0.0008 | 0.0043 ± 0.0005 | |
| C8:1-DC | 0.0036 ± 0.0003 | 0.0047 ± 0.0004 | |
| C10-OH/C8-DCa | 0.0124 ± 0.0010 | 0.0089 ± 0.0009 | |
| Medium AC | C12a | 0.0121 ± 0.0018 | 0.0073 ± 0.0006 |
| C14a | 0.0348 ± 0.0030 | 0.0252 ± 0.0017 | |
| C16a | 0.1268 ± 0.0087 | 0.1014 ± 0.0078 | |
| Long AC | C18a | 0.0753 ± 0.0029 | 0.0622 ± 0.0040 |
| C20:1-OH/C18:1-DCa | 0.0079 ± 0.0004 | 0.0046 ± 0.0005 |
Metabolite concentrations are expressed as μM. Data are presented as means ± SEM. Values are statistically different assessed with One-way ANOVA Student test at p < 0.05.
Table 2.
Summary of significant exercise effects in liver metabolites in mice
| Metabolite class | Metabolite | SED | EX |
|---|---|---|---|
| Short AC | C3a | 0.10 ± 0.01 | 0.07 ± 0.01 |
| C4/Ci4a | 0.12 ± 0.01 | 0.05 ± 0.01 | |
| C5:1a | 0.0436 ± 0.0041 | 0.0597 ± 0.0043 | |
| C8:1-OH/C6:1-DCa | 0.0135 ± 0.0011 | 0.0168 ± 0.0011 | |
| C8:1-DCa | 0.0107 ± 0.0016 | 0.0162 ± 0.0017 | |
| C10-OH/C8-DCa | 0.0124 ± 0.0016 | 0.0172 ± 0.0017 | |
| Medium AC | C12a | 0.0129 ± 0.0012 | 0.0161 ± 0.0013 |
| C14-OH/C12-DCa | 0.0032 ± 0.0005 | 0.0054 ± 0.0005 | |
| C16:2a | 0.0066 ± 0.0011 | 0.0112 ± 0.0012 | |
| C16:2-OH/C14:2-DCa | 0.0028 ± 0.0007 | 0.0054 ± 0.0007 | |
| C16:1-OH/C14:1-DCa | 0.0000 ± 0.0000 | 0.1019 ± 0.0245 | |
| C16-OH/C14-DCa | 0.0051 ± 0.0008 | 0.0010 ± 0.0009 | |
| Long AC | C18:3-OH/C16:3-DCa | 0.0017 ± 0.0002 | 0.0027 ± 0.0002 |
| C18:2-OH/C16:2-DCa | 0.0041 ± 0.0007 | 0.0095 ± 0.0007 | |
| C18:1-OH/C16:1-DCa | 0.0077 ± 0.0014 | 0.01361 ± 0.0015 | |
| C18-OH/C16-DCa | 0.0041 ± 0.0005 | 0.0064 ± 0.0005 | |
| C20:3-OH/C18:3-DC | 0.0011 ± 0.0002 | 0.0023 ± 0.0002 | |
| C22:4a | 0.0015 ± 0.0002 | 0.0006 ± 0.0002 |
Data are presented as means ± SEM. Values are statistically different assessed with One-way ANOVA Student test at p < 0.05.
Metabolite concentrations are expressed as μM.
Given that hierarchical clustering showed that ACs were regulated by exercise training, we investigated exercise-induced adaptations to AC profiles and metabolism. Similar to human data (Figure 1), EX reduced both total ACs (predominantly short and medium ACs) and incompletely oxidized hydroxylated ACs (AC-OH) in serum (Figure 2D; Table 1). Principal component analysis (PCA) indicated little separation of serum ACs between SED and EX (Figure S2A), but circulating AC-OH profiles showed separation between groups (Figure S2B).
In the liver, EX did not affect total abundance of AC but increased medium and long ACs and also AC-OH (Figure 2E; Table 2; Figures S2C and S2D). In muscle, exercise did not alter abundance of AC and AC-OH (Figure 2F) but shifted AC and AC-OH profiles (Table S4; Figures S2E and S2F). Thus, EX shifts ACs and AC-OH primarily in the liver and serum.
Exercise mediates the coordination of short, medium, and long-chain acylcarnitines across tissues
Aberrant accumulation and metabolism of short-, medium-, and long-chain ACs have been implicated in the pathogenesis of insulin resistance.23,31,32 Acyl-CoAs are formed within the cytosol by attaching L-carnitine to activate long-chain fatty acids, which are then transported into the peroxisomes and mitochondria as substrates for fatty acid oxidation.23 Failure to oxidize short-, medium-, and long-chain ACs within the mitochondria is an indicator of incomplete fatty acid (FA) utilization or an overload of mitochondrial oxidative capacity16,29 which can worsen insulin resistance in the context of obesity and type 2 diabetes.29,33 Given that exercise altered the total abundance of ACs, we further stratified the effects of exercise on ACs of varying lengths (Figures 2G–2I).
EX reduced short-chain ACs in serum but did not alter liver or muscle levels (Figure 2G). Medium ACs were decreased in serum and increased in the liver but were not altered in muscle (Figure 2H). EX did not affect total abundance of long-chain ACs (length C18-C22) in serum or muscle but increased long-chain ACs in the liver (Figure 2I). Together these data indicate that EX training decreases short and medium ACs in the serum, but increases medium and long ACs in the liver. Because accumulation of ACs in serum is an indicator of poor metabolic health, these data provide a potential role for exercise to regulate whole-body metabolism by reducing serum ACs and shifting AC accumulation to the liver.
To determine the relative coordination of ACs among tissues, we used chord graphical representation which allows us to trace arches for highly connected metabolites (Pearson r ≥ 0.5 or 0.8, p < 0.05). Lines that begin and end within the same tissue (color) denote intra-tissue correlations, but if the tissues (color) are different, they indicate inter-tissue correlations between individual metabolites (lines). Chord diagram representation of highly correlated short-chain AC demonstrates that EX reduces short-chain AC in the serum and reduces inter-tissue coordination of short-chain ACs (Figure 2J). Regarding medium chain AC, EX produces robust alterations in medium-chain AC compared to SED by reducing positive coordination within muscle and liver (fewer lines), and increasing inter-tissue correlations across the muscle, liver, and serum (Figure 2K), indicating that the liver may contribute to AC clearance from circulation. Chord representation for long ACs demonstrated that EX reduced positive correlations (fewer lines) across the muscle, liver, and serum compared to SED (Figure 2L). Together these data indicate that EX training alters medium and long-chain ACs in the liver and supports the idea that one mechanism for exercise is to regulate whole-body metabolism by shifting AC accumulation to the liver.
Exercise increases acylcarnitine metabolism and β-oxidation across tissues
We next investigated the molecular regulation of tissue AC metabolism provoked by EX by measuring expression of genes and proteins involved in mitochondrial function, AC metabolism, and mitochondrial β-oxidation. In the mouse liver, EX increased expression of genes involved in mitochondrial function (Nrf1), AC metabolism (Hnf4a, Octn2, Cpt1a, and Crat), and mitochondrial β-oxidation (Cpt2, Hadha, Hadhb, and Acadvl) (Figure 3A). EX also increased hepatic protein expression of Complex-IV, CPT1A, and CACT in the liver (Figures 3B and 3C). These data indicate that EX increases expression of genes and proteins involved in fatty acid metabolism and mitochondrial function in the liver of HFD mice.
Figure 3.

Exercise-trained mice have greater acylcarnitine metabolism, β-oxidation, respiration, and fatty acid uptake in the liver
Expression of genes (A) and proteins (B and C) involved in mitochondrial and AC metabolism and β-oxidation in the liver. Data are mean ± SEM (n = 6 per group), ∗p ≤ 0.05 One-way ANOVA. Red box on CACT indicates a well that was left blank and did not factor into the analysis.
(D) Bioenergetic profile of isolated hepatocytes from sedentary and exercise-trained mice and (E) basal OCR, ATP turnover, and maximal respiration in isolated hepatocytes. Data are mean ± SEM (n = 6, 3 replicates per group); ∗p ≤ 0.05 One-way ANOVA.
(F) Incubation of primary hepatocytes from sedentary or exercise-trained mice with BODIPY (fluorophore pentanoic acid) and within punctae (mitochondria), and etomoxir incubation.
(G) Quantification of fluorescent signal in primary hepatocytes/area mm2. Data are mean ± SEM (n = 12 replicates per group); ∗p ≤ 0.05 vs. SED One-way ANOVA.
(H) Expression of genes in liver of sedentary and exercise-trained HFD-fed mice. Data are mean ± SEM (n = 6 per group), fold-change vs. SED; ∗p ≤ 0.05 One-way ANOVA.
(I) Model for exercise-training to increase pathways related to mitochondrial AC translocation, oxidation, and electron transport chain (ETC), resulting in enhanced hepatic mitochondrial function in high-fat fed mice.
In tibialis anterior (TA) muscle, EX increased expression of genes involved in mitochondrial function (Csand Tfam), AC metabolism (Cpt1b and Cact), and mitochondrial β-oxidation (Cpt2, Hadha, Hadhb, and Acadvl) (Figure S3A). EX also increased protein levels of CS and CPT1B in TA (Figures S3B and S3C), and together with elevated gene expression, this indicates increased mitochondrial function in muscle with EX, as has been established.34,35,36
Exercise training increases respiration and fatty acid uptake in isolated hepatocytes
Due to the changes in mitochondrial transcripts and proteins, we examined whether EX training induces cell-autonomous improvements in hepatocyte mitochondrial bioenergetics. Hepatocytes were isolated from mice that were fed a high-fat diet for 3 weeks and concomitantly SED or EX trained. EX significantly increased mitochondrial oxygen consumption rate (OCR) in isolated hepatocytes, enhancing basal, maximal, and non-mitochondrial respiration (Figures 3D and 3E). Thus, EX training enhances the cell-autonomous improvements in hepatocyte mitochondrial bioenergetics.
EX reduces serum ACs in humans and in mice, and EX mice have elevated hepatic AC oxidation and cell-autonomous increases in hepatic mitochondrial respiration. To determine how exercise increases AC metabolism, we investigated the effect of exercise on hepatic fatty acid uptake using BODIPY (pentanoic acid) in isolated hepatocytes. Hepatocytes from EX mice have increased lipid-dye uptake (Figures 3F and 3G) compared to SED hepatocytes. Inhibition of fatty acid oxidation via etomoxir in hepatocytes negates the effects of EX on BODIPY uptake (Figures 3F and 3G). This enhanced fatty acid uptake in EX hepatocytes (in the absence of etomoxir) is accompanied by the increased expression of hepatic AC transporters Slc22a1 and Slc16a1 (Figure 3H). Together, these data indicate that EX in HFD-fed mice increases expression of genes and proteins involved in AC metabolism and β-oxidation, changes the AC profile across tissues, and increases to markers of increased respiration and fatty acid uptake. In particular, EX promotes gene and protein adaptations in the liver that may be involved with the changes in the metabolomic and AC profiles (Figure 3I).
Exercise training improves hepatocyte calcium cycling and transport to sustain elevated AC metabolism
Given the cell-autonomous effects of EX on hepatic mitochondrial respiration, we further explored EX-activated mechanisms that promote mitochondrial function in the liver. In addition to fuel supply (ACs), the transport of ions across the mitochondria, in particular calcium (Ca2+), fosters mitochondrial energy production. Previous studies have shown that Ca2+ flux across the inner mitochondrial membrane mediates cellular bioenergetics.37 However, the extent to which EX enhances hepatic Ca2+ handling is unclear.
To determine the role of EX on Ca2+ handling, we measured expression of genes involved in Ca2+ handling in the liver and determined that EX increased expression of genes involved in store-operated Ca2+ regulation including endoplasmic reticulum-resident Serca (Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase 1) channel, and the mitochondrial Ca2+ gatekeepers Micu-1 and -2 (Mitochondrial calcium uptake), but not Mcu (Mitochondrial calcium uniporter) (Figure 4A). Moreover, EX increases protein levels of SERCA and MICU-1 in the liver but does not affect MCU (Figures 4B–4E). These data show that in addition to increasing mitochondrial respiration (Figures 3D and 3E), EX increases mitochondrial Ca2+ channels that support ATP production in the liver.
Figure 4.

Exercise training enhances Ca2+ cycling
(A) Expression of genes and (B‒E) proteins in liver of sedentary and exercise-trained HFD-fed mice. Data are mean ± SEM (n = 6 per group), fold-change vs. SED; ∗p ≤ 0.05 One-way ANOVA. Ca2+ at (F) basal (F0), and after (G) insulin and (H) thapsagarin stimulation in primary hepatocytes. Ca2+ is measured with selective fluorescent indicator Fura-2 AM. Data are mean ± SEM (n = 35 cells, 2 replicates per group); ∗p ≤ 0.05 vs. SED One-way ANOVA.
In order to define the effect of EX on Ca2+ cycling, we measured steady state, intracellular free Ca2+ levels (basal), following stimulation of extracellular Ca2+ uptake (insulin), and after blocking the intracellular storage of free Ca2+ within the ER (thapsigargin) in hepatocytes isolated from SED and EX mice fed HFD. EX hepatocytes had increased Ca2+ stores at baseline (Figure 4F), and following incubation with both insulin (Figure 4G) and thapsigargin (Figure 4H), indicating that hepatocytes from EX trained mice have greater Ca2+ storage at baseline and have enhanced Ca2+ cycling.38
Accumulation of ACs drives increase in hepatic mitochondrial respiration
To delineate the physiological impact of ACs on hepatic mitochondrial energetics, we incubated unstimulated primary hepatocytes with short-chain (C3), medium-chain (C10, C14, and C16), and long-chain ACs (C18) that were impacted by exercise training in either physically active subjects (Figure 1) or mice (Table 1). While no changes were observed in hepatocytes treated with short-chain ACs (Figure S4A), incubation of isolated hepatocytes with medium-chain and long-chain ACs increased mitochondrial oxygen consumption rate (OCR), including basal, maximal, ATP production, and non-mitochondrial respiration (Figures S4B–S4E). We next determined whether ACs alone could recapitulate the EX mitochondrial phenotype in SED hepatocytes. Incubation of SED hepatocytes with C10 increased basal OCR to the level of EX hepatocytes but did not alter maximal or non-mitochondrial OCR (Figures 5A and 5B). Similarly, incubation with the long-chain C18 AC increased basal OCR to the level of EX hepatocytes (Figures 5C and 5D). Although EX increases Cpt1a and Cact in the liver (Figures 3A–3C), the increase in mitochondrial respiration in SED hepatocytes incubation with medium-chain and long-chain ACs was not accompanied by increased transcription of Cpt1a or Cact (Figures S5A and S5B).
Figure 5.

Incubation with medium and long ACs rescues respiration in isolated hepatocytes
(A) Bioenergetic profile and (B) basal OCR and maximal respiration in isolated hepatocytes from sedentary or exercise-trained mice and after incubation of C10 (10μM). Data are mean ± SEM (n = 6, 3 replicates per group); ∗p ≤ 0.05; ∗∗∗p < 0.001 vs. SED One-way ANOVA.
(C) Bioenergetic profile and (D) basal OCR and maximal respiration in isolated hepatocytes from sedentary or exercise-trained mice and after incubation of C18 (10μM). Data are mean ± SEM (n = 6, 3 replicates per group); ∗p ≤ 0.05; ∗∗∗p < 0.001 vs. SED One-way ANOVA.
(E) Bioenergetic profile and (F) basal OCR and maximal respiration in isolated hepatocytes from sedentary mice and after incubation of C10 (10μM) with the addition of etomoxir. Data are mean ± SEM (n = 6, 3 replicates per group); ∗p ≤ 0.05; ∗∗∗p < 0.001 vs. SED One-way ANOVA.
(G) Bioenergetic profile and (H) basal OCR and maximal in isolated hepatocytes from sedentary mice and after incubation of C18 (10μM) with the addition of etomoxir. Data are mean ± SEM (n = 6, 3 replicates per group); ∗p ≤ 0.05; ∗∗∗p < 0.001 vs. SED One-way ANOVA.
(I) Bioenergetic profile and (J) maximal respiration in isolated hepatocytes from sedentary mice and after incubation of C10 (10μM) with the addition of CACT inhibitor. Data are mean ± SEM (n = 6, 3 replicates per group); ∗p ≤ 0.05; ∗∗∗p < 0.001 vs. SED One-way ANOVA.
(K) Bioenergetic profile and (L) maximal respiration in isolated hepatocytes from sedentary mice and after incubation of C18 (10μM) with the addition of CACT inhibitor. Data are mean ± SEM (n = 3, 2 replicates per group); ∗p ≤ 0.05; ∗∗∗p < 0.001 vs. SED One-way ANOVA.
(M) Gene expression in primary hepatocytes from HFD-fed mice after incubation with PBS, C3, C10, and C18. Data are mean ± SEM (n = 3 replicates per group); ∗p ≤ 0.05 vs. PBS One-way ANOVA.
To determine if the increased amount of ACs in the liver is due to an overproduction of ACs within the liver or an increased uptake from circulation, hepatocytes from sedentary or exercise-trained mice were isolated and incubated with C18 for 4 h, and then metabolomics were performed on both the media and the hepatocytes. Interestingly, C18 was not different in the media among groups, but the C18 was significantly reduced in the EX hepatocytes compared to SED.
While not conclusive, since there is no difference in C18 in the media but reduced C18 in the EX hepatocytes, these data suggest that the increase in ACs in the liver could be due to increased AC metabolism (Figures S6A and S6B).
We further investigated the mechanism through which C10 and C18 activate mitochondrial respiration by incubating hepatocytes using etomoxir which inhibits CPT1A, a rate-limiting step in fatty acid oxidation. Addition of etomoxir did not affect basal or maximal OCR (Figures 5E and 5F). This is likely because short- and medium-ACs bypass CPT1a and diffuse to the inter-mitochondrial membrane space, and then are transported by inner mitochondrial membrane-bound CACT.39 C18 increased respiration in SED hepatocytes, and treatment with etomoxir blunted the effect of C18 in SED hepatocytes (Figures 5G and 5H). We next treated isolated hepatocytes from SED mice with a carnitine-acylcarnitine translocase (CACT) inhibitor in the presence of exogenous C10 and C18. Incubation with C10 or C18 increased respiration in hepatocytes, but treatment with the CACT inhibitor prevented the effect of exogenous C10 or C18 to fuel or mimic the exercise-induced increase in mitochondrial energetics or maximal respiration (Figures 5I‒5L). Together, these data reveal that augmenting SED mitochondrial respiration with medium and long ACs is dependent on mitochondrial oxidation and that EX enhances cell autonomous mitochondrial respiration that is fueled by fatty acid metabolism.
Since high concentrations of ACs can also have detergent properties, we investigated if the effect of the ACs to increase the bioenergetic profile in isolated hepatocytes. Hepatocytes isolated from sedentary mice were incubated with PBS, C10, C18, Triton X-100, Tween 80, and SDS. Only incubation with C10 or C18 increased the bioenergetic profile of isolated hepatocytes suggesting that the effect of ACs to increase respiration is independent from their non-specific detergent properties (Figures S6C–S6E).
Incubation of primary hepatocytes with medium and long ACs increased the expression of genes related to AC uptake (Slc22a1and Slc16a1), AC metabolism (Pgc1a, Pgc1b, Cpt1a, Cact, and Cpt2), hepatic β-oxidation (Hadha/b, Acadvl, Acadm, and Acads), and Ca2+ handling (Micu-1 and Mcu) (Figure 5M), but incubation with short-chain AC C3 decreased expression of these genes (Figure 5M), indicating a potential for ACs to alter the molecular machinery that leads to changes in Ca2+ dynamics observed with exercise training. These data suggest that exercise increases the molecular machinery to import fatty acids and increase AC metabolism, and this is demonstrated by increased fatty acid uptake and mitochondrial respiration. The increased energetics demand provoked by exercise involves an increase in Ca2+ cycling in livers of mice, and the exercise-induced increase in respiration and Ca2+ cycling is specific to AC accumulation, as treatment of hepatocytes with ACs induce a similar molecular profile and increase in respiration as observed with exercise training.
Discussion
Obesity and type 2 diabetes are often characterized by a reduced capacity to regulate lipid mobilization and oxidize lipids. Exercise is an important therapeutic tool to restore metabolic flexibility and increase insulin sensitivity; however, the mechanism by which exercise training improves lipid trafficking and oxidation—particularly in the liver—has not been exclusively defined. In the current study, we demonstrate that physically active (endurance trained) human subjects have lower circulating serum medium-chain ACs. Lower serum ACs are associated with increased physical activity, improved cardiorespiratory fitness, and reduced adiposity. Likewise, exercise training in mice reduced circulating short- and medium-chain ACs but increased medium- and long-chain hepatic ACs while enhancing hepatic AC metabolism and fatty acid β-oxidation. In mice, exercise training amplifies mitochondrial respiration in isolated hepatocytes, and supplementation of medium- and long-chain AC (C10 and C18, respectively) to SED hepatocytes increases mitochondrial respiration to the level of EX hepatocytes. Furthermore, EX training increases hepatic uptake of fatty acids and thus contributes to serum AC clearance, and together with enhanced hepatocyte Ca2+ cycling, EX supports hepatic mitochondrial function. These data indicate that EX training induces metabolic adaptations that facilitate hepatic AC metabolism and drive mitochondrial bioenergetic adaptations in the liver.
Fatty acid trafficking, defined by the ability to regulate lipid mobilization to be used by tissues to fuel fatty acid oxidation,1,2,40 is largely impaired in patients who are obese or have type 2 diabetes. As a consequence, the fatty acid byproducts, including ACs, are elevated in plasma of patients who are obese or have type 2 diabetes.41 Exercise is an important environmental stimulus that synchronizes nutrient availability to tissue energy production.4,42 Compared to sedentary subjects, increased physical activity produces a distinct serum metabolomic signature,5,6,7,8,9,10 and this is mainly driven by reductions in circulating ACs.7,14 Our data demonstrate that physical activity reduces medium-chain ACs and this is associated with lower adiposity and greater aerobic capacity in human participants. Thus, serum ACs and AC utilization are likely important biomarkers of exercise training and could be utilized as biological sensors of improved metabolic flexibility.
Mitochondrial AC oxidation is a tightly regulated process that is activated to meet energy requirements. Exercise training accelerates fuel flux within and across tissues and impacts hepatic metabolism that utilizes amino acids, fatty acids (FA), and lactate to facilitate fuel delivery to other tissues in the form of ketones, urea, and glucose.18,20,21,43,44,45,46 Intracellular fatty acids are activated by acyl-CoA synthetase and conjugated with carnitine by CPT1A, the rate-limiting enzyme in β-oxidation.47 CPT1A generates ACs of varying lengths, which are sequentially transported into the mitochondrial matrix by CACT to be further broken down by the mitochondrial fatty acid-oxidation pathway (CPT2, ACADVL, ACADL, ACADM, ACADS, and HADHA/B). Oxidation of ACs produces the necessary reducing power in the form of NADH/FADH2 to produce ATP. Although ACs are important for energy production, incomplete oxidation and accumulation of AC overload the mitochondrial capacity that leads to impaired metabolism.23,30,48 Our current findings show that exercise training reduces serum AC and increases hepatic AC metabolism and β-oxidation in rodents. This indicates that exercise may coordinate the restructuring of AC metabolism across tissues, improving clearance of ACs from circulation, which could contribute to improved metabolic function.
An important remaining question is whether improved AC uptake and metabolism by the liver increases mitochondrial AC oxidation. This is relevant, given that greater AC funneling to hepatocytes will lead to increased mitochondrial energy production. We show that incubation with medium-chain ACs like C10 and myristoyl-carnitine (C14), as well as palmitoyl-carnitine (C16), or long-chain ACs like stearoyl-carnitine (C18), increase mitochondrial oxygen consumption in primary hepatocytes. Our data demonstrates that in addition to increasing hepatic lipid uptake, exercise training increases hepatic mitochondrial respiration, indicating that the increased AC uptake could be intricately coupled to mitochondrial bioenergetics. Given that AC transporters are reduced in hepatocytes from sedentary vs. EX mice, we hypothesized that the addition of ACs to hepatocytes isolated from sedentary mice would rescue mitochondrial respiration to the level of their EX-trained hepatocytes. Inhibition of CPT1A or CACT minimally altered the effects of C10 or C18 on mitochondrial respiration. These data must be interpreted with caution as the CPT1A inhibitor etomoxir may have several off-target effects and can sequester free CoA, which could inhibit oxidation of ACs of varying chain lengths,49,50,51 but together suggest that hepatic AC uptake and mitochondrial AC oxidation are enhanced by exercise training to improve liver bioenergetic function.
Although the effects of exercise on liver metabolism have been well documented,52,53,54,55,56 the mechanisms by which exercise training mediates hepatic mitochondrial function remains elusive. Studies have shown that exercise promotes the clearance of ACs in mice5 and humans,14 and in the current study we show that exercise increases liver mitochondrial energetics and AC metabolism. To pinpoint the mechanism by which exercise increases hepatic mitochondrial respiration, we focused on a major driver of mitochondrial ATP production, hepatocyte Ca2+ cycling. Studies have shown that uptake of Ca2+ into the mitochondria directly activates components of the TCA cycle to increase NADH/FADH2 production and power the electron transport chain, thus consuming more oxygen and producing more energy.37,57 Hepatocytes from obese mice have impaired Ca2+ cycling,58,59 but a single bout of exercise (swimming) can activate mitochondrial function and enhance mitochondrial Ca2+ uptake.60 Our data show that in addition to increasing basal and maximal mitochondrial capacity, exercise training increases expression of mitochondrial Ca2+ gatekeepers, MICU-1 and MICU-2, resulting in increased Ca2+ storage and cycling which can fuel mitochondria bioenergetics.38,57 Future studies that target key steps of hepatic AC metabolism through gain- and loss-of-function experiments are needed to identify the AC-dependent and independent mechanisms provoked by exercise training that leads to the positive effects on lipid trafficking and to fully elucidate the interaction of AC and Ca2+ handling.
Together, these data indicate that physical activity in humans and mice reduces serum AC concentrations, enhances hepatic AC metabolism, and increases hepatic mitochondrial respiration. In addition to increasing cell-autonomous hepatic mitochondrial respiration, availability of ACs with exercise training drives the elevated Ca2+ responses, contributing to improved metabolic function. These data demonstrate the important adaptations in hepatic AC metabolism with exercise training and the potential of ACs as biomarkers of metabolic flexibility that dynamically respond to dietary and lifestyle inputs.
Limitations of the study
There are several limitations to the study. In our animal study, only male mice were fed a high-fat diet, at one age, after a fixed period and type of exercise-training program were investigated. It is possible that female mice, mice fed a chow diet, or mice of a different age, would respond differently to exercise. In our human study, we did not include a young, sedentary cohort and it is possible that this cohort would have responded differently to exercise than the older cohort. In addition, we acknowledge that to unequivocally demonstrate where the AC products are metabolized would require the use of (13C)AC into mice or isolated hepatocytes, which we did not measure in this study. Future work will focus on addressing these limitations.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CS | Thermo Fisher Scientific | Cat # PA5-22126 |
| CPT1A | Sigma-Aldrich | Cat # ABS65 |
| CPT1B | Sigma-Aldrich | Cat # SAB2108822 |
| SLC25A20 | Sigma-Aldrich | Cat # SAB2102185 |
| CACT | Sigma-Aldrich | Cat # SAB2102185 |
| OXPHOS | Invitrogen | Cat # 45-8099 |
| MCU (D2Z3B) | Cell Signaling Technology | Cat # 14997S |
| CBARA1/MICU1 (D4P8Q) | Cell Signaling Technology | Cat # 12524S |
| Anti-Rabbit | Sigma-Aldrich | Cat # NA934V |
| Anti-Mouse | Sigma-Aldrich | Cat # NA921V |
| Chemicals, peptides, and recombinant proteins | ||
| High-fat diet (60% kcal from Fat) | Research Diets Inc. | Cat #D12492i |
| Qiazol | Qiagen | Cat # 79306 |
| Type I Collagenase | Worthington | Cat # LS004197 |
| Propionyl-L-carnitine (C3) | Cayman chemicals | Cat # 9001873 |
| Valeryl-L-carnitine (C5) | Cayman chemicals | Cat # 27871 |
| Decanoyl-L-carnitine (C10) | Cayman chemicals | Cat # 26549 |
| Myristoyl-L-carnitine (C14) | Cayman chemicals | Cat # 26559 |
| Palmitoyl-D-carnitine (C16) | Cayman chemicals | Cat # 26552 |
| Stearoyl-L-carnitine (C18) | Cayman chemicals | Cat # 26556 |
| Arachidonoyl-L-carnitine (C20:4) | Cayman chemicals | Cat # 900939 |
| Propanesulfonate (CACT inhibitor) | Sigma-Aldrich | Cat #H6883-5G |
| Fluo-4 | Thermo Fisher Scientific | Cat #F14201 |
| Insulin, Cell culture | Roche | Cat # 11376497001 |
| Thapsigarin | Sigma-Aldrich | Cat # T-650 |
| BODIPY | Thermo Fisher Scientific | Cat #D3834 |
| SYBR green | QuantaBio | Cat # 95047-500 |
| Critical commercial assay | ||
| Seahorse XFe24 | Agilent Technologies | Cat # 102340-100 |
| Experimental models: Organisms/strains | ||
| Wild-type mice | Charles River laboratories | C57BL/6J |
| Software and algorithms | ||
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com |
| ImageJ | NIH | Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.net/ij/, 1997–2018. |
| Analyst TF 1.7 | Sciex | https://sciex.com/products/software/analyst-tf-software |
| Metaboanalyst | Metaboanalyst | https://www.metaboanalyst.ca/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Kristin Stanford (kristin.stanford@osumc.edu).
Materials availability
This study does not generate unique reagents.
Data and code availability
-
•
Original western blot images are available in the supplemental file. This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Human subject recruitment
All participants provided written informed consent and the study protocol was approved by the Institutional Review Board at AdventHealth Orlando. Inclusion criteria for participation in the study included stable weight, non-smoker, BMI <35 kg/m2, resting blood pressure ≤150 mmHg syst olic and ≤95 mmHg diastolic. Exclusion criteria included inability or unwillingness to comply with the protocol, clinically significant CVD including MI within the past year, the presence of peripheral vascular disease, hepatic disease, renal disease, muscular or neuromuscular disease, hematologic/oncologic disease, peripheral neuropathy, orthopedic limitations, history of pulmonary emboli, history of alcohol or substance abuse, current use of blood thinners or any medication that can alter glucose homeostasis.
Male and female participants were recruited from the greater Orlando, FL area and classified into one of three groups: Young active/endurance trained adults (YA, n = 13; 10 Males, 3 Females; Age = 32.3 ± 5.3 years; VO2peak = 56.1 ± 8.3 mL/kg/min), older active/endurance trained adults (OA; n = 13; 11 Males, 2 Females; Age = 68.3 ± 3.5 years; VO2peak = 34.5 ± 4.2 mL/kg/min), and older sedentary adults (OS; n = 16; 10 Males, 6 Females; Age = 70.7 ± 5.0 years; VO2peak = 19.1 ± 3.9 mL/kg/min) participated in the study. (Table S1). Participants in the active/endurance trained groups (both young and older) were required to be engaged in endurance exercise training (running, cycling, swimming) at least 3 days/wk without extensive lay off over the previous 6 months, while sedentary performed one or less structured exercise session/wk.61 Along with self-reported assessments, participants wore a triaxial accelerometer (SenseWear Pro Armband, BodyMedia Inc., Pittsburgh, PA) consistently for at least 7 days on their upper left arm, except while showering or bathing, to objectively assess physical activity levels.62,63 Days with wear time of at least 85% were used for analysis.
Method details
Clinical assessments in human subjects
Cardiorespiratory fitness (VO2peak) was examined by a graded exercise protocol on a cycle ergometer. Heart rate, blood pressure, and ECG were constantly monitored throughout the test. Tests were terminated when volitional exhaustion was reached or one of three criteria were met, which included: 1) plateau or decline in VO2, 2) respiratory exchange ratio (RER) increased to 1.10 or higher, and/or 3) participant’s heart rate increased to within 10 beats of the age-predicted maximum (208 – [0.7 x age]).64 Body composition was assessed by dual energy X-ray absorptiometry (DEXA) using a GE lunar iDXA whole-body scanner.61 Lean and fat mass was analyzed with Encore software. Following an overnight fast, a blood sample was obtained at rest and used for lipidomics analysis.
Mice and training paradigm
Seven-week-old male C57BL/6 mice were placed on a high-fat diet (60% kcal from fat; Research Diets Inc.) for 3 weeks and further subdivided into two groups: sedentary (placed in a static cage; inactive, or SED) or exercise-trained (housed with running wheels for 3 weeks; normal activity, or EX). Body weight was measured using an OHAUS NV212 scale. Body fat and lean mass were measured using an EchoMRI instrument (EchoMRI LLC) with canola oil calibration. After 3 weeks, mice were sacrificed following an overnight fast and removal of running wheels to eliminate any acute effects of exercise. At the time of sacrifice, tissues (liver, testes, and muscle) were flash frozen and stored at −80°C until processing. All procedures were followed as approved by the IACUC at The Ohio State University and the Joslin Diabetes Center.
Structural lipidomics in human serum samples
Structural lipidomic analysis was performed using a modified Bligh and Dyer extraction with a customized, automated liquid-liquid extraction sequence on a Hamilton Robotics StarLET Robot System. Four mL of chloroform:methanol (1:1, by vol) was added to the samples followed by addition of 25 μL of a cocktail mixture of deuterium-labeled, odd chain, and extremely low naturally abundant internal standards. A front and back extraction were performed as previously described.65,66
Electrospray ionization (ESI) MS was performed on a Sciex TripleTOF 5600+ coupled with a direct injection loop on an Eksigent Ekspert MicroLC 200 system as previously described.67 The MS/MSALL data were acquired by Analyst TF 1.7 software (Sciex) and processed with MultiQuant 1.2.2.5 (Sciex) using an in-house database of lipid species for identification. Concentrations of lipid species were quantified by comparison of peak area of a diagnostic fragment(s) for each lipid species to the peak area of the internal standard.
Metabolite analysis in rodent tissues
In mice, panels of amino acids (AA), organic acids (OA), acylcarnitines (AC) and nucleotides (NUC) were measured by targeted MS/MS, GC/MS, and LC-MS/MS, respectively, as described previously.68,69,70,71,72 Absolute levels of metabolites are reported, calculated relative to stable isotope-labeled internal standards added to serum, media, or extracts of liver, testes, and triceps skeletal muscle samples.
Primary hepatocyte isolation and acylcarnitine treatment
Mouse hepatocytes were isolated as previously described.73 Briefly, mice were anesthetized with isofluorane and perfused via cardiac puncture with 20mL of Kreb’s Ringer containing 1mM EDTA (37C, 7.5 mL/min), followed by perfusion of 30mL Kreb’s Ringer containing 0.15mM CaCl2 and 0.5 mg/mL type I Collagenase (Worthington). The liver was removed, minced into pieces, and chilled to 4°C in 40mL of Krebs Ringer. The tissue was dispersed by pipetting and filtered through a 100-μm nylon cell strainer. The filtrate was centrifuged for 3 min at 50G. The supernatant was removed and the cell pellet was resuspended with 40mL of freshly-prepared, cold Krebs Ringer. This was repeated 2x. Cells were then resuspended in William’s Media supplemented with 10% FBS, 10 μg/mL epidermal growth factor (Sigma), 0.1 μM dexamethasone, 4.4 μM nicotinamide and 100U/mL penicillin and 100 μg/mL streptomycin. Cells (30 k/well) were seeded on laminin-treated plates. All experiments with primary hepatocytes were completed within 48 h of isolation.
For AC experiments, primary liver hepatocytes were isolated and plated at 30 K/well and incubated overnight. The next day, cells were incubated with incremental concentrations of short-chain ACs (1.0, 5.0, 10.0 mM), medium-chain ACs (10.0, 50.0, 100.0 μM), or long-chain ACs (10.0, 50.0, 100.0 μM).74,75 Selected ACs species for in vitro incubation were short-chain ACs propionyl-L-carnitine (C3) and valeryl-L-carnitine (C5), medium-chain ACs decanoyl-L-carnitine (C10), myristoyl-L-carnitine (C14), and palmitoyl-DL-carnitine (C16), and long-chain ACs stearoyl-L-carnitine (C18) and arachidonyl-L-carnitine (C20:4) (Cayman chemicals). Hepatocytes were exposed to ACs for 4hrs prior to mitochondrial respiration assays. A separate cohort of primary hepatocytes were incubated with either 10uM Sodium Dodecyl Sulfate (SDS), 0.3% Triton X-100 or 0.3% Tween 20. Hepatocytes were exposed to these detergents for 4hrs prior to mitochondrial respiration assays.
Hepatocyte mitochondrial respiration
Isolated hepatocytes from exercise or sedentary mice were seeded onto laminin-coated 24 well Seahorse Plates at 30K cells/well according to standard protocols. Cells were treated for 4 h with respective ACs or were untreated. The oxygen-consumption rates (OCR; to assess mitochondrial respiration) were measured in a Seahorse XFe24 instrument using the standard protocol of 3-min mix, 2-min wait and 3-min measure.76 Basal rates are determined by subtracting the non-mitochondrial respiration from the baseline values. Carbonyl cyanide-p-trifluoromethoxy-phenyl-hydrazon (FCCP; 2 μM) was used to determine the cells’ maximal respiratory capacity by allowing the electron transport chain to function at its maximal rate (maximal respiratory capacity is derived by subtracting non-mitochondrial respiration from the FCCP rate). Oligomycin (a complex V inhibitor; 2 μM) was used to derive ATP-linked respiration (by subtracting the oligomycin rate from baseline cellular OCR) and proton leak respiration (by subtracting non-mitochondrial respiration from the oligomycin rate). AntimycinA/Rotenone (mitochondrial inhibitors; 0.5 μM) was used to determine non-mitochondrial respiration. In a subset of experiments, Etomoxir was added for 4 h and used to block CPT1a-mediated import of fatty acids into the mitochondria (10 μM). Carnitine acylcarnitine translocase (CACT) is responsible for passive transport of carnitine and carnitine-fatty acid complexes across the inner mitochondrial membrane. The CACT inhibitor, 3-(N,N-Dimethylpalmitylammonio) propanesulfonate (Sigma #H6883-5g), was added 15 min before the start of the respiration assay (400μM). Data from wells of the same treatment group (n = 8–10 wells per timepoint; 3 plates per condition) were averaged together and analyzed directly using Wave software. For the normalization of respiration to protein content, cells were lysed in EBT buffer and protein concentration was measured by Bradford assay.
Hepatocyte calcium imaging and lipid uptake
Hepatocytes were plated on laminin-coated glass slides and placed on the stage of an IonOptix Automated Cell Finder microscope system and superfused (∼1 mL/min at 30°C) with contractile buffer containing (in mM, pH 7.4) 4 KCl, 131 NaCl, 1 MgCl2, 10 HEPES, 1 CaCl2, and 10 glucose. The cells were visualized using a 40X objective. The cells were then loaded with (0.5 μmol/L) Fluo-4 (Cat.#F14201; ThermoFisher), a single-wavelength green fluorescent calcium indicator (excitation at 480 nm, emission at 525 nm) to measure free calcium in the cells. Basal calcium levels were recorded (under no stimulation), the same cells were then treated with 200nM insulin (Cat.# 11376497001; Roche), then 2uM thapsigarin (Cat.# T-650; Sigma) with calcium levels recorded after each treatment. All fluo-4 data were normalized to the plate background. At least 10 cells per mouse/treatment group were analyzed. For the AC experiments, the plated cells were incubated with 10 μM of C3, C10, C18 or PBS during the 1-h fluo-4 incubation/de-esterification time prior to the calcium recordings.
For lipid uptake experiments, isolated hepatocytes from EX and SED HFD-fed mice (n = 12 replicates) were serum starved with Williams media for 1 h. Cells were washed with PBS 1X, then supplemented with BODIPY (C5-BODIPY; ThermoFisher) to a final concentration of 2.5uM, and incubated in a CO2 incubator in the dark for 30 min. Cells were washed twice with PBS and then imaged while in PBS. Imaging was done using FITC imaging filter at 10X and 20x and quantified as (Mean/Area)∗10−6 using ImageJ.
Biochemical methods
mRNA levels in frozen tissues or in hepatocytes following 4 h incubation with ACs were measured by quantitative Real-Time PCR (qRT-PCR) (Roche LightCycler 480II) using SYBR Green detection (QuantaBio). The primers used are included in Table S5.77 Tissue processing and immunoblotting were performed as previously described.78 Briefly, liver, tibialis anterior muscle, and testes lysates were solubilized in Laemmli buffer, separated by SDS-PAGE, and transferred to nitrocellulose membranes. The membranes were incubated with antibodies specific for citrate synthase (CS) (Cat# PA5-22126 1:1000; ThermoFisher), carnitine palmitoyl-transferase 1a (CPT1A) (Cat# ABS65 1:1000; Sigma), carnitine/acylcarnitine translocase (CACT) (Cat# SAB2102185 1:1000; Sigma), oxidative phosphorylation cocktail (OXPHOS) (Cat# 45–8099 1:1000; Invitrogen), mitochondrial calcium uniporter (MCU) (Cat# 149975 1:1000; Cell Signaling Technology), mitochondrial calcium uptake 1 (MICU1) (Cat# 12524S 1:1000; Cell Signaling Technology) (Table S6). The immunoreactive proteins were detected with enhanced chemiluminescence and quantified by densitometry.
Quantification and statistical analysis
Statistical analysis
Human data are presented as mean ± standard deviation (SD) and significance defined as p < 0.05. One-way ANOVA and Tukey’s post hoc analysis were used to determine group differences, while Pearson correlation coefficients were used to examine the relationship between circulating acylcarnitines and clinical markers of physical activity and body composition. To determine the influence of physical activity/endurance exercise training on circulating acylcarnitines, the young and older active groups were combined and compared to the older sedentary group. ANCOVA was used where age and biological sex were taken as co-variants. The number of human subjects used per experiment are found in the figure legends.
For mouse data, statistical significance was defined as p < 0.05 and determined by Student t-tests (n = 6–10 per group). For seahorse data, all plots represent 8–10 wells per time point per treatment group done in triplicate, and error bars are SEM. Heatmap analysis were performed using a two-way hierarchical clustering using Euclidean measures to obtain distance matrix and complete agglomeration method for clustering with R package R package “ggplots” and Metaboanalyst.79 Principal component analysis (PCA) was performed using Metaboanalyst79 and plotting the PC1 and 2 with 95% confidence ellipses. Chord diagrams were created by obtaining the adjacency matrix with Pearson correlation coefficient r ≥ 0.5 for amino acids, organic acids, and nucleotides and r ≥ 0.8 for short and long acylcarnitines both at p ≤ 0.05 then visualized using Circos table viewer v0.63-9.80 Throughout the text, ∗, p value<0.05; ∗∗, p value<0.01, and ∗∗∗, p value<0.001, and comparisons are specifically indicated in the figure legends.
Acknowledgments
This work was supported by NIH grant R01HL138738 to KIS and R01AG060542 to KIS and PMC, K01AG044437 to PMC, R01-DK-101043 and R01-DK101043-S1 to LJG, P30-DK124723 to CBN, and the Joslin Diabetes Center DRC (P30 DK36836). DHS was supported by AHAPOST831250. KMP was supported by F31-HL152648-01A1 and T32-HL134616. PV was supported by AHAPRE903654. Correspondence for the human data should be directed to paul.coen@adventhealth.com. The authors gratefully appreciate the contributions of our study participants and acknowledge the excellent technical assistance of the core staff at the AdventHealth Translational Research Institute.
Author contributions
This study was conceived and designed by D.H.S., J.M.H., P.M.C., and K.I.S. P.M.C. and B.H.G. were responsible for the human study concept and design at AdventHealth Orlando. Human data were analyzed by J.M.H., A.M.B., B.H.G., and P.M.C. Mouse in vivo experiments were carried out by D.H.S., L.A.B., K.M.P., and P.V. In vitro experiments were performed by D.H.S., L.A.B., and K.M.P. Human metabolomics was performed by E.Y.C., N.R.N., V.B., V.V.T., and M.A.K. Mouse metabolomics was performed by C.M. and O.I. C.B.N. provided oversight for all metabolomics experiments. C.B.N., L.J.G., and K.I.S. designed the mouse metabolomics experiment. D.H.S., J.M.H., S.N., P.V., A.M.B., P.M.C., and K.I.S. analyzed and interpreted the data, prepared figures, and completed statistical analysis. Manuscript was written by D.H.S., J.M.H., P.M.C., and K.I.S. with input from other authors. P.M.C. and K.I.S. are the guarantors of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors gave final approval for publication.
Declaration of interests
P.M.C. is a consultant for Astellas/Mitobridge, Incorporate. K.I.S. is a consultant for Lygenesis.
Published: February 1, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109083.
Contributor Information
Paul M. Coen, Email: paul.coen@adventhealth.com.
Kristin I. Stanford, Email: kristin.stanford@osumc.edu.
Supplemental information
References
- 1.Kelley D.E., Simoneau J.A. Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. J. Clin. Invest. 1994;94:2349–2356. doi: 10.1172/JCI117600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelley D.E., Mandarino L.J. Hyperglycemia normalizes insulin-stimulated skeletal muscle glucose oxidation and storage in noninsulin-dependent diabetes mellitus. J. Clin. Invest. 1990;86:1999–2007. doi: 10.1172/JCI114935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodpaster B.H., Sparks L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017;25:1027–1036. doi: 10.1016/j.cmet.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fritzen A.M., Lundsgaard A.M., Kiens B. Tuning fatty acid oxidation in skeletal muscle with dietary fat and exercise. Nat. Rev. Endocrinol. 2020;16:683–696. doi: 10.1038/s41574-020-0405-1. [DOI] [PubMed] [Google Scholar]
- 5.Xiang L., Zhang H., Wei J., Tian X.Y., Luan H., Li S., Zhao H., Cao G., Chung A.C.K., Yang C., et al. Metabolomics studies on db/db diabetic mice in skeletal muscle reveal effective clearance of overloaded intermediates by exercise. Anal. Chim. Acta. 2018;1037:130–139. doi: 10.1016/j.aca.2017.11.082. [DOI] [PubMed] [Google Scholar]
- 6.Schranner D., Kastenmüller G., Schönfelder M., Römisch-Margl W., Wackerhage H. Metabolite Concentration Changes in Humans After a Bout of Exercise: a Systematic Review of Exercise Metabolomics Studies. Sports Med. Open. 2020;6:11. doi: 10.1186/s40798-020-0238-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J., Light A.R., Hoppel C.L., Campbell C., Chandler C.J., Burnett D.J., Souza E.C., Casazza G.A., Hughen R.W., Keim N.L., et al. Acylcarnitines as markers of exercise-associated fuel partitioning, xenometabolism, and potential signals to muscle afferent neurons. Exp. Physiol. 2017;102:48–69. doi: 10.1113/ep086019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huffman K.M., Koves T.R., Hubal M.J., Abouassi H., Beri N., Bateman L.A., Stevens R.D., Ilkayeva O.R., Hoffman E.P., Muoio D.M., Kraus W.E. Metabolite signatures of exercise training in human skeletal muscle relate to mitochondrial remodelling and cardiometabolic fitness. Diabetologia. 2014;57:2282–2295. doi: 10.1007/s00125-014-3343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis G.D., Farrell L., Wood M.J., Martinovic M., Arany Z., Rowe G.C., Souza A., Cheng S., McCabe E.L., Yang E., et al. Metabolic signatures of exercise in human plasma. Sci. Transl. Med. 2010;2:33ra37. doi: 10.1126/scitranslmed.3001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morville T., Sahl R.E., Moritz T., Helge J.W., Clemmensen C. Plasma Metabolome Profiling of Resistance Exercise and Endurance Exercise in Humans. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108554. [DOI] [PubMed] [Google Scholar]
- 11.van Loon L.J., Jeukendrup A.E., Saris W.H., Wagenmakers A.J. Effect of training status on fuel selection during submaximal exercise with glucose ingestion. J. Appl. Physiol. 1999;87:1413–1420. doi: 10.1152/jappl.1999.87.4.1413. [DOI] [PubMed] [Google Scholar]
- 12.Bain J.R., Stevens R.D., Wenner B.R., Ilkayeva O., Muoio D.M., Newgard C.B. Metabolomics applied to diabetes research: moving from information to knowledge. Diabetes. 2009;58:2429–2443. doi: 10.2337/db09-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neufer P.D., Bamman M.M., Muoio D.M., Bouchard C., Cooper D.M., Goodpaster B.H., Booth F.W., Kohrt W.M., Gerszten R.E., Mattson M.P., et al. Understanding the Cellular and Molecular Mechanisms of Physical Activity-Induced Health Benefits. Cell Metab. 2015;22:4–11. doi: 10.1016/j.cmet.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Xu G., Hansen J.S., Zhao X.J., Chen S., Hoene M., Wang X.L., Clemmesen J.O., Secher N.H., Häring H.U., Pedersen B.K., et al. Liver and Muscle Contribute Differently to the Plasma Acylcarnitine Pool During Fasting and Exercise in Humans. J. Clin. Endocrinol. Metab. 2016;101:5044–5052. doi: 10.1210/jc.2016-1859. [DOI] [PubMed] [Google Scholar]
- 15.Seifert E.L., Fiehn O., Bezaire V., Bickel D.R., Wohlgemuth G., Adams S.H., Harper M.E. Long-chain fatty acid combustion rate is associated with unique metabolite profiles in skeletal muscle mitochondria. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams S.H., Hoppel C.L., Lok K.H., Zhao L., Wong S.W., Minkler P.E., Hwang D.H., Newman J.W., Garvey W.T. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J. Nutr. 2009;139:1073–1081. doi: 10.3945/jn.108.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCoin C.S., Knotts T.A., Adams S.H. Acylcarnitines--old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 2015;11:617–625. doi: 10.1038/nrendo.2015.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trefts E., Williams A.S., Wasserman D.H. Exercise and the Regulation of Hepatic Metabolism. Prog. Mol. Biol. Transl. Sci. 2015;135:203–225. doi: 10.1016/bs.pmbts.2015.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thyfault J.P., Bergouignan A. Exercise and metabolic health: beyond skeletal muscle. Diabetologia. 2020;63:1464–1474. doi: 10.1007/s00125-020-05177-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura Y., Tanaka Y., Sato F., Choi J.B., Watada H., Niwa M., Kinoshita J., Ooka A., Kumashiro N., Igarashi Y., et al. Effects of diet and exercise on muscle and liver intracellular lipid contents and insulin sensitivity in type 2 diabetic patients. J. Clin. Endocrinol. Metab. 2005;90:3191–3196. doi: 10.1210/jc.2004-1959. [DOI] [PubMed] [Google Scholar]
- 21.Sullivan S., Kirk E.P., Mittendorfer B., Patterson B.W., Klein S. Randomized trial of exercise effect on intrahepatic triglyceride content and lipid kinetics in nonalcoholic fatty liver disease. Hepatology. 2012;55:1738–1745. doi: 10.1002/hep.25548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu C., Hoene M., Plomgaard P., Hansen J.S., Zhao X., Li J., Wang X., Clemmesen J.O., Secher N.H., Häring H.U., et al. Muscle-Liver Substrate Fluxes in Exercising Humans and Potential Effects on Hepatic Metabolism. J. Clin. Endocrinol. Metab. 2020;105:1196–1209. doi: 10.1210/clinem/dgz266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schooneman M.G., Vaz F.M., Houten S.M., Soeters M.R. Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes. 2013;62:1–8. doi: 10.2337/db12-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simcox J., Geoghegan G., Maschek J.A., Bensard C.L., Pasquali M., Miao R., Lee S., Jiang L., Huck I., Kershaw E.E., et al. Global Analysis of Plasma Lipids Identifies Liver-Derived Acylcarnitines as a Fuel Source for Brown Fat Thermogenesis. Cell Metab. 2017;26:509–522.e6. doi: 10.1016/j.cmet.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mashek D.G. Hepatic fatty acid trafficking: multiple forks in the road. Adv. Nutr. 2013;4:697–710. doi: 10.3945/an.113.004648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jarrell Z.R., Smith M.R., Hu X., Orr M., Liu K.H., Quyyumi A.A., Jones D.P., Go Y.M. Plasma acylcarnitine levels increase with healthy aging. Aging (Albany NY) 2020;12:13555–13570. doi: 10.18632/aging.103462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu Z., Zhai G., Singmann P., He Y., Xu T., Prehn C., Römisch-Margl W., Lattka E., Gieger C., Soranzo N., et al. Human serum metabolic profiles are age dependent. Aging Cell. 2012;11:960–967. doi: 10.1111/j.1474-9726.2012.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodríguez-Gutiérrez R., Lavalle-González F.J., Martínez-Garza L.E., Landeros-Olvera E., López-Alvarenga J.C., Torres-Sepúlveda M.R., González-González J.G., Mancillas-Adame L.G., Salazar-Gonzalez B., Villarreal-Pérez J.Z. Impact of an exercise program on acylcarnitines in obesity: a prospective controlled study. J. Int. Soc. Sports Nutr. 2012;9:22. doi: 10.1186/1550-2783-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koves T.R., Ussher J.R., Noland R.C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J.R.B., Newgard C.B., et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 30.Mihalik S.J., Goodpaster B.H., Kelley D.E., Chace D.H., Vockley J., Toledo F.G.S., DeLany J.P. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity. 2010;18:1695–1700. doi: 10.1038/oby.2009.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koves T.R., Li P., An J., Akimoto T., Slentz D., Ilkayeva O., Dohm G.L., Yan Z., Newgard C.B., Muoio D.M. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J. Biol. Chem. 2005;280:33588–33598. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- 32.Holland W.L., Knotts T.A., Chavez J.A., Wang L.P., Hoehn K.L., Summers S.A. Lipid mediators of insulin resistance. Nutr. Rev. 2007;65:S39–S46. doi: 10.1111/j.1753-4887.2007.tb00327.x. [DOI] [PubMed] [Google Scholar]
- 33.Muoio D.M. Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell. 2014;159:1253–1262. doi: 10.1016/j.cell.2014.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parry H.A., Roberts M.D., Kavazis A.N. Human Skeletal Muscle Mitochondrial Adaptations Following Resistance Exercise Training. Int. J. Sports Med. 2020;41:349–359. doi: 10.1055/a-1121-7851. [DOI] [PubMed] [Google Scholar]
- 35.Fitts R.H., Booth F.W., Winder W.W., Holloszy J.O. Skeletal muscle respiratory capacity, endurance, and glycogen utilization. Am. J. Physiol. 1975;228:1029–1033. doi: 10.1152/ajplegacy.1975.228.4.1029. [DOI] [PubMed] [Google Scholar]
- 36.Baldwin K.M., Cooke D.A., Cheadle W.G. Time course adaptations in cardiac and skeletal muscle to different running programs. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1977;42:267–272. doi: 10.1152/jappl.1977.42.2.267. [DOI] [PubMed] [Google Scholar]
- 37.Arruda A.P., Hotamisligil G.S. Calcium Homeostasis and Organelle Function in the Pathogenesis of Obesity and Diabetes. Cell Metab. 2015;22:381–397. doi: 10.1016/j.cmet.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shanmughapriya S., Rajan S., Hoffman N.E., Zhang X., Guo S., Kolesar J.E., Hines K.J., Ragheb J., Jog N.R., Caricchio R., et al. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci. Signal. 2015;8:ra23. doi: 10.1126/scisignal.2005673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Houten S.M., Wanders R.J.A., Ranea-Robles P. Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim. Biophys. Acta, Mol. Basis Dis. 2020;1866 doi: 10.1016/j.bbadis.2020.165720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelley D.E., Mokan M., Simoneau J.A., Mandarino L.J. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J. Clin. Invest. 1993;92:91–98. doi: 10.1172/JCI116603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.I S Sobczak A., A Blindauer C., J Stewart A. Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes. Nutrients. 2019;11 doi: 10.3390/nu11092022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy R.M., Watt M.J., Febbraio M.A. Metabolic communication during exercise. Nat. Metab. 2020;2:805–816. doi: 10.1038/s42255-020-0258-x. [DOI] [PubMed] [Google Scholar]
- 43.Hallsworth K., Fattakhova G., Hollingsworth K.G., Thoma C., Moore S., Taylor R., Day C.P., Trenell M.I. Resistance exercise reduces liver fat and its mediators in non-alcoholic fatty liver disease independent of weight loss. Gut. 2011;60:1278–1283. doi: 10.1136/gut.2011.242073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schultz A., Mendonca L.S., Aguila M.B., Mandarim-de-Lacerda C.A. Swimming training beneficial effects in a mice model of nonalcoholic fatty liver disease. Exp. Toxicol. Pathol. 2012;64:273–282. doi: 10.1016/j.etp.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 45.Rector R.S., Uptergrove G.M., Morris E.M., Borengasser S.J., Laughlin M.H., Booth F.W., Thyfault J.P., Ibdah J.A. Daily exercise vs. caloric restriction for prevention of nonalcoholic fatty liver disease in the OLETF rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2011;300:G874–G883. doi: 10.1152/ajpgi.00510.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harrison B.C., Bell M.L., Allen D.L., Byrnes W.C., Leinwand L.A. Skeletal muscle adaptations in response to voluntary wheel running in myosin heavy chain null mice. J. Appl. Physiol. 2002;92:313–322. doi: 10.1152/japplphysiol.00832.2001. [DOI] [PubMed] [Google Scholar]
- 47.Esser V., Britton C.H., Weis B.C., Foster D.W., McGarry J.D. Cloning, sequencing, and expression of a cDNA encoding rat liver carnitine palmitoyltransferase I. Direct evidence that a single polypeptide is involved in inhibitor interaction and catalytic function. J. Biol. Chem. 1993;268:5817–5822. [PubMed] [Google Scholar]
- 48.Aguer C., McCoin C.S., Knotts T.A., Thrush A.B., Ono-Moore K., McPherson R., Dent R., Hwang D.H., Adams S.H., Harper M.E. Acylcarnitines: potential implications for skeletal muscle insulin resistance. FASEB J. 2015;29:336–345. doi: 10.1096/fj.14-255901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Divakaruni A.S., Hsieh W.Y., Minarrieta L., Duong T.N., Kim K.K.O., Desousa B.R., Andreyev A.Y., Bowman C.E., Caradonna K., Dranka B.P., et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab. 2018;28:490–503.e7. doi: 10.1016/j.cmet.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yao C.H., Liu G.Y., Wang R., Moon S.H., Gross R.W., Patti G.J. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of β-oxidation. PLoS Biol. 2018;16 doi: 10.1371/journal.pbio.2003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma Y., Wang W., Devarakonda T., Zhou H., Wang X.Y., Salloum F.N., Spiegel S., Fang X. Functional analysis of molecular and pharmacological modulators of mitochondrial fatty acid oxidation. Sci. Rep. 2020;10:1450. doi: 10.1038/s41598-020-58334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith R.L., Soeters M.R., Wüst R.C.I., Houtkooper R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018;39:489–517. doi: 10.1210/er.2017-00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.E L., Lu J., Burns J.M., Swerdlow R.H. Effect of exercise on mouse liver and brain bioenergetic infrastructures. Exp. Physiol. 2013;98:207–219. doi: 10.1113/expphysiol.2012.066688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Santos-Alves E., Marques-Aleixo I., Rizo-Roca D., Torrella J.R., Oliveira P.J., Magalhães J., Ascensão A. Exercise modulates liver cellular and mitochondrial proteins related to quality control signaling. Life Sci. 2015;135:124–130. doi: 10.1016/j.lfs.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 55.Fletcher J.A., Meers G.M., Linden M.A., Kearney M.L., Morris E.M., Thyfault J.P., Rector R.S. Impact of various exercise modalities on hepatic mitochondrial function. Med. Sci. Sports Exerc. 2014;46:1089–1097. doi: 10.1249/MSS.0000000000000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCoin C.S., Von Schulze A., Allen J., Fuller K.N.Z., Xia Q., Koestler D.C., Houchen C.J., Maurer A., Dorn G.W., 2nd, Shankar K., et al. Sex modulates hepatic mitochondrial adaptations to high-fat diet and physical activity. Am. J. Physiol. Endocrinol. Metab. 2019;317:E298–E311. doi: 10.1152/ajpendo.00098.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nemani N., Dong Z., Daw C.C., Madaris T.R., Ramachandran K., Enslow B.T., Rubannelsonkumar C.S., Shanmughapriya S., Mallireddigari V., Maity S., et al. Mitochondrial pyruvate and fatty acid flux modulate MICU1-dependent control of MCU activity. Sci. Signal. 2020;13 doi: 10.1126/scisignal.aaz6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ali E.S., Rychkov G.Y., Barritt G.J. Metabolic Disorders and Cancer: Hepatocyte Store-Operated Ca(2+) Channels in Nonalcoholic Fatty Liver Disease. Adv. Exp. Med. Biol. 2017;993:595–621. doi: 10.1007/978-3-319-57732-6_30. [DOI] [PubMed] [Google Scholar]
- 59.Wilson C.H., Ali E.S., Scrimgeour N., Martin A.M., Hua J., Tallis G.A., Rychkov G.Y., Barritt G.J. Steatosis inhibits liver cell store-operated Ca(2)(+) entry and reduces ER Ca(2)(+) through a protein kinase C-dependent mechanism. Biochem. J. 2015;466:379–390. doi: 10.1042/BJ20140881. [DOI] [PubMed] [Google Scholar]
- 60.Tate C.A., Wolkowicz P.E., McMillin-Wood J. Exercise-induced alterations of hepatic mitochondrial function. Biochem. J. 1982;208:695–701. doi: 10.1042/bj2080695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Distefano G., Standley R.A., Zhang X., Carnero E.A., Yi F., Cornnell H.H., Coen P.M. Physical activity unveils the relationship between mitochondrial energetics, muscle quality, and physical function in older adults. J. Cachexia Sarcopenia Muscle. 2018;9:279–294. doi: 10.1002/jcsm.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berntsen S., Hageberg R., Aandstad A., Mowinckel P., Anderssen S.A., Carlsen K.H., Andersen L.B. Validity of physical activity monitors in adults participating in free-living activities. Br. J. Sports Med. 2010;44:657–664. doi: 10.1136/bjsm.2008.048868. [DOI] [PubMed] [Google Scholar]
- 63.Wefers J.F., Woodlief T.L., Carnero E.A., Helbling N.L., Anthony S.J., Dubis G.S., Jakicic J.M., Houmard J.A., Goodpaster B.H., Coen P.M. Relationship among physical activity, sedentary behaviors, and cardiometabolic risk factors during gastric bypass surgery-induced weight loss. Surg. Obes. Relat. Dis. 2017;13:210–219. doi: 10.1016/j.soard.2016.08.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.American College of Sports Medicine. Whaley M.H., Brubaker P.H., Otto R.M., Armstrong L.E. Lippincott Williams & Wilkins; 2006. ACSM's Guidelines for Exercise Testing and Prescription, 7th Edition. [Google Scholar]
- 65.Han X., Gross R.W. Shotgun lipidomics: electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom. Rev. 2005;24:367–412. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- 66.Gao F., McDaniel J., Chen E.Y., Rockwell H., Lynes M.D., Tseng Y.H., Sarangarajan R., Narain N.R., Kiebish M.A. Monoacylglycerol Analysis Using MS/MS(ALL) Quadruple Time of Flight Mass Spectrometry. Metabolites. 2016;6 doi: 10.3390/metabo6030025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao F., McDaniel J., Chen E.Y., Rockwell H.E., Drolet J., Vishnudas V.K., Tolstikov V., Sarangarajan R., Narain N.R., Kiebish M.A. Dynamic and temporal assessment of human dried blood spot MS/MS(ALL) shotgun lipidomics analysis. Nutr. Metab. 2017;14:28. doi: 10.1186/s12986-017-0182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.White P.J., McGarrah R.W., Grimsrud P.A., Tso S.C., Yang W.H., Haldeman J.M., Grenier-Larouche T., An J., Lapworth A.L., Astapova I., et al. The BCKDH Kinase and Phosphatase Integrate BCAA and Lipid Metabolism via Regulation of ATP-Citrate Lyase. Cell Metab. 2018;27:1281–1293.e7. doi: 10.1016/j.cmet.2018.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Walejko J.M., Christopher B.A., Crown S.B., Zhang G.F., Pickar-Oliver A., Yoneshiro T., Foster M.W., Page S., van Vliet S., Ilkayeva O., et al. Branched-chain alpha-ketoacids are preferentially reaminated and activate protein synthesis in the heart. Nat. Commun. 2021;12:1680. doi: 10.1038/s41467-021-21962-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Newgard C.B., An J., Bain J.R., Muehlbauer M.J., Stevens R.D., Lien L.F., Haqq A.M., Shah S.H., Arlotto M., Slentz C.A., et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gooding J.R., Jensen M.V., Dai X., Wenner B.R., Lu D., Arumugam R., Ferdaoussi M., MacDonald P.E., Newgard C.B. Adenylosuccinate Is an Insulin Secretagogue Derived from Glucose-Induced Purine Metabolism. Cell Rep. 2015;13:157–167. doi: 10.1016/j.celrep.2015.08.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cordell R.L., Hill S.J., Ortori C.A., Barrett D.A. Quantitative profiling of nucleotides and related phosphate-containing metabolites in cultured mammalian cells by liquid chromatography tandem electrospray mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008;871:115–124. doi: 10.1016/j.jchromb.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 73.Mooij H.L., Bernelot Moens S.J., Gordts P.S.M., Stanford K., Foley E., van den Boogert M.W., Witjes J., Hassing H.C., Tanck M., van de Sande M.J., et al. Ext1 heterozygosity causes a modest effect on postprandial lipid clearance in humans. J. Lipid Res. 2015;56:665–673. doi: 10.1194/jlr.M053504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seiler S.E., Koves T.R., Gooding J.R., Wong K.E., Stevens R.D., Ilkayeva O.R., Wittmann A.H., DeBalsi K.L., Davies M.N., Lindeboom L., et al. Carnitine Acetyltransferase Mitigates Metabolic Inertia and Muscle Fatigue during Exercise. Cell Metab. 2015;22:65–76. doi: 10.1016/j.cmet.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lehmann R., Zhao X., Weigert C., Simon P., Fehrenbach E., Fritsche J., Machann J., Schick F., Wang J., Hoene M., et al. Medium chain acylcarnitines dominate the metabolite pattern in humans under moderate intensity exercise and support lipid oxidation. PLoS One. 2010;5 doi: 10.1371/journal.pone.0011519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu M., Neilson A., Swift A.L., Moran R., Tamagnine J., Parslow D., Armistead S., Lemire K., Orrell J., Teich J., et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 77.Stanford K.I., Takahashi H., So K., Alves-Wagner A.B., Prince N.B., Lehnig A.C., Getchell K.M., Lee M.Y., Hirshman M.F., Goodyear L.J. Maternal exercise improves glucose tolerance in female offspring. Diabetes. 2017;66:2124–2136. doi: 10.2337/db17-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lessard S.J., Rivas D.A., Alves-Wagner A.B., Hirshman M.F., Gallagher I.J., Constantin-Teodosiu D., Atkins R., Greenhaff P.L., Qi N.R., Gustafsson T., et al. Resistance to aerobic exercise training causes metabolic dysfunction and reveals novel exercise-regulated signaling networks. Diabetes. 2013;62:2717–2727. doi: 10.2337/db13-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pang Z., Chong J., Zhou G., de Lima Morais D.A., Chang L., Barrette M., Gauthier C., Jacques P.É., Li S., Xia J. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021;49:W388–W396. doi: 10.1093/nar/gkab382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Original western blot images are available in the supplemental file. This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.