

Abstract
The borders between cell and developmental biology, always permeable, have largely dissolved. One manifestation is the blossoming of cilia biology, with cell and developmental approaches (increasingly complemented by human genetics, structural insights, and computational analysis) fruitfully advancing understanding of this fascinating, multifunctional organelle. The last eukaryotic common ancestor probably possessed a motile cilium, providing evolution with ample opportunity to adapt cilia to many jobs. Over the last decades, we have learned how nonmotile, primary cilia play important roles in intercellular communication. Reflecting their diverse motility and signaling functions, compromised cilia cause a diverse range of diseases collectively called ciliopathies. In this Review, we highlight how cilia signal, focusing on how second messengers generated in cilia convey distinct information, how cilia are a potential source of signals to other cells, how evolution may have shaped ciliary function, and how cilia research may address thorny outstanding questions.
Introduction
Eukaryotic cells are compartmentalized, tasking organelles with different functions. For example, aerobic respiration occurs in mitochondria and interpretation of genetic information occurs in nuclei. Like mitochondria and nuclei, cilia are organelles that are specialized, but for the tasks of motility and signal transduction. Here, we use the terms cilia and flagella interchangeably, underscoring that our discussion is limited to eukaryotes, with prokaryotic flagella being fundamentally different. Moreover, cilia can be divided into distinct subsets based on structure and function. Some cilia are motile and transport fluid or propel sperm. Other cilia are immotile interpret environmental information or intercellular cues (Figure 1A). These sensory immotile cilia were named primary cilia in reference to their appearance in lung development before (or primary to) motile cilia1. This nomenclature extended to encompass all immotile cilia2. However, as with most definitions in biology, the division of cilia into motile and sensory is not absolute, as motile cilia can have sensory functions.
Figure 1 -. Motile and immotile cilia differ structurally and LECA possessed a sophisticated cilium.
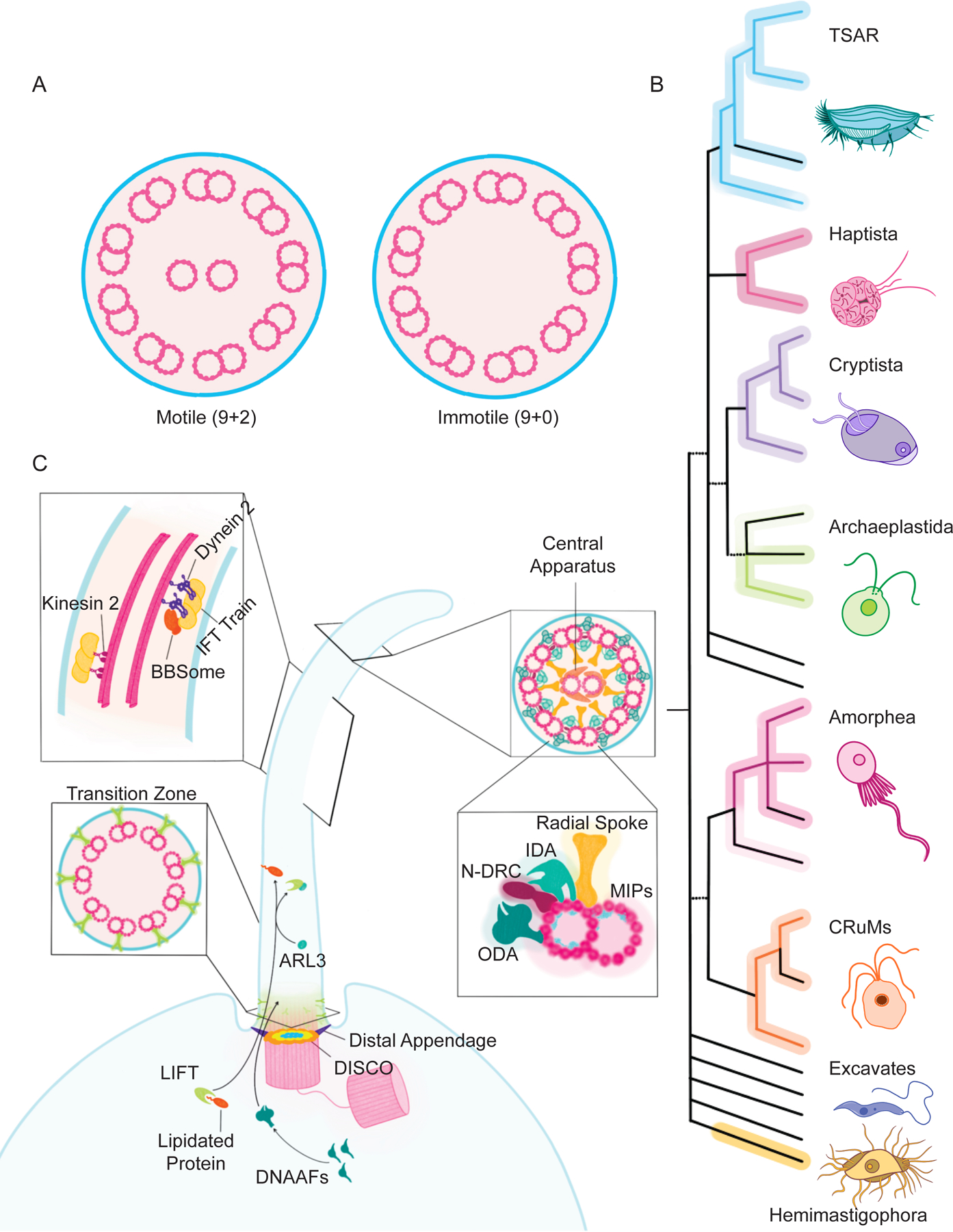
A) Most motile cilia (left) have an axoneme comprised of nine circumferential doublets and a central pair. Most primary cilia (right) have an axoneme lacking the central pair. B) A formulation of the eukaryotic tree of life, depicting major clades. Ciliated lineages are depicted with colored lines, unciliated groups are in black and lineages with both are in both. While some groups lack cilia (e.g., Rhodophyta), and some groups contain both ciliated and unciliated members (e.g., Chloroplastida), ciliated organisms exist in all extant clades. Thus, the last eukaryotic common ancestor was probably a ciliated protist. Figure modified from reference 164. C) Ciliated lineages share genes encoding ciliary proteins, including components of the LIFT system for transporting lipidated proteins. In the cilium, ARL3 triggers LIFT to deposit cargoes such as ARL13B and INPP5E into the ciliary membrane. Other evolutionarily ancient protein complexes involved in ciliary function include DISCO, a distal centriolar complex involved in forming distal appendages, DNAAFs involved in assembling dyneins, the transition zone controlling the protein composition of the ciliary membrane, IFT driving transport along the ciliary axoneme, the BBSome helping export select proteins from the cilium, the MIPs reinforcing the ciliary microtubules, and the central pair apparatus, radial spokes, and N-DRC that, together, coordinate the IDAs and ODAs that beat the cilia.
In this review, we highlight emerging understanding of how, like your radio antenna, primary cilia function in signal transduction. A complication to receiving information that all cells contend with is the lipid bilayer, the scant few nanometers of hydrophobicity that separate the highly regulated intracellular environment from the extracellular world. This delimiting membrane poses a challenge: to respond to the world, intracellular machinery must gain information from the other side of the membrane. Cells have solved this challenge in diverse ways, several of which use cilia. Moreover, cilia share a common structure but can be tuned to receive different types of information in different cell types.
Different modes of ciliary signaling share some requirements (e.g., diverse ciliary signaling pathways rely on a part of the ciliary base called the transition zone to control ciliary protein composition) and have some unique requirements (e.g., different ciliary signals are transduced via distinct ciliary receptors and effectors). Below, we detail how the cilium, an evolutionarily ancient organelle, acts as a dynamic signaling hub for the transduction of both intracellular and extracellular cues. We also reflect on the various roles of cilia in human physiology, and how evolution may have shaped this functional diversity.
Nothing in ciliary biology makes sense except in the light of evolution
Phylogenetics strongly suggests that the last common eukaryotic ancestor (LECA) was a ciliated organism3–5 (Figure 1B). Because of its ancestral origin, much ciliary biology is shared among currently existing species, and principles discovered in one organism can illuminate biology in others. For example, motor-driven intraflagellar transport (IFT) was discovered in the green alga Chlamydomonas6. IFT, critical for both ciliogenesis and ciliary maintenance, transports proteins both to the ciliary tip via Kinesin-2 and back to the ciliary base via Dynein-2 (Figure 1C). Identification of the Chlamydomonas genes encoding IFT components allowed homologs to be recognized and revealed that the IFT machinery is conserved across ciliated organisms.
Another evolutionarily ancient ciliary machine is the transition zone, a gate at the base of the cilium that helps to regulate ciliary protein composition. Because of the cilium’s deep evolutionary conservation, identification of transition zone components in one organism predicts transition zone components of other organisms. For example, CEP290 was discovered to be a component of the Chlamydomonas transition zone and subsequently recognized to be a component of the mammalian transition zone7,8. Many transition zone components are co-conserved with CEP290 among ciliated phyla, underscoring how, to paraphrase Theodosius Dobzhansky9, nothing makes sense except in the light of evolution. Humans are no exception to conservation of ciliary components. For example, human mutations in CEP290 can cause Joubert syndrome, which helped spur the realization that mutations in other transition zone components also cause Joubert syndrome10–12.
Consistent with the hypothesis that LECA possessed a cilium, many ciliary proteins beyond IFT and transition zone components are conserved across diverse extant ciliated organisms. Many of these evolutionarily ancient proteins are centriolar components required for ciliogenesis, such as the DISCO complex (including CEP90, OFD1, FOPNL)13.
Other ancient complexes are essential for ciliary motility. These include the axonemal dyneins, the outer dynein arm-docking complex (ODA-DC), the nexin-dynein regulatory complex (N-DRC) and the radial spokes. In addition, many microtubule inner proteins (MIPs) are widely conserved among existing diverse ciliated organisms. MIPs are required to strengthen the axonemal microtubules and regenerate ATP to fuel the dyneins14,15.
Additional ancient ciliary proteins include assemblies involved in trafficking proteins to or from cilia. These include lipidated protein intraflagellar targeting (LIFT) involved in trafficking of lipidated proteins to the ciliary membrane, and an octameric adaptor related to Bardet-Biedl syndrome called the BBSome16.
Not all proteins with conserved roles in ciliary function fulfill their role at the cilium proper. Despite not localizing to centrioles or cilia, cytoplasmic proteins such as the dynein axonemal assembly factors (DNAAFs) co-evolved with cilia and contribute to ciliary motility by assembling the axonemal dyneins before their import into cilia17.
Thus, extrapolation from extant organisms allows us to partially reconstruct the ancestral cilium of LECA. Most probably, this cilium contained a 9+2 axoneme possessing MIPs for structural reinforcement and generation of ATP from AMP and ADP, as well as radial spokes. These cilia were motile and used DNAAFs to generate axonemal dyneins, some of which docked onto the ODA-DC and were regulated by N-DRC. Within the cilium, IFT along the axonemal proteins was powered by Kinesin-2 and Dynein-2. Select lipidated proteins were transported to these cilia by LIFT, and proteins were transported out of the cilia via the BBSome, both across the transition zone.
While these reconstructions suggest that the LECA had a complex cilium, we should remain aware that living organisms may not fully represent the ancestral ciliary protein complement. For example, an evolutionarily ancient ciliary protein, EVC3, has been lost by extant vertebrates18.
LECA may have used its cilium both for motility and for sensing extracellular cues. Below, we describe our current understanding of how animal cilia use three distinct signaling systems: Hedgehog, Polycystin and G protein-coupled receptor (GPCR) signaling. These three pathways represent distinct modes of ciliary signaling and are associated with cilia biology in diverse organisms.
Cilia transduce vertebrate Hedgehog signals
Patterning of tissues during embryonic development depends on a surprisingly small number of signaling pathways. One critical means of intercellular communication in developing animals is Hedgehog signaling19. Last century witnessed the discovery of Hedgehog proteins as secreted lipoproteins sensed by the receptor Patched (PTCH) to regulate the central transducer, Smoothened (SMO), thereby controlling the activity of the downstream GLI transcription factor effectors20,21. This century has seen a burgeoning understanding of how many animals use primary cilia for Hedgehog signal transduction.
How do cilia transduce Hedgehog signals? Central to many signal transduction pathways are post-translational modifications (e.g., phosphorylation by receptor tyrosine kinases) or changes in protein-protein interactions (e.g., steroid hormone receptor separation from HSP90). Certainly, Hedgehog signaling involves post-translational modifications and changes in protein-protein interactions. Many of these modifications and changes occur in one subcellular locale, the primary cilium, and Hedgehog signaling depends critically on regulated changes in the composition of the primary cilium.
Not only are cilia required for vertebrate Hedgehog signaling, but all of the central components of the Hedgehog signal transduction pathway (PTCH, SMO, PKA, KIF7, SUFU and GLI) spend at least some of their time at the cilium22–25. In the absence of Hedgehog stimuli, the cilium possesses PTCH and the GLI proteins are bound to SUFU to prevent transcriptional activity (Figure 2). Some GLI proteins are cleaved to generate a truncated repressor form (GliR) that keeps the Hedgehog transcriptional program off. Hedgehog stimuli induces PTCH ciliary exit and SMO ciliary accumulation, resulting in the formation of GLI activators (GliA). When activated, GLI cleavage is prevented, GLI dissociates from SUFU26, and GLI turns the Hedgehog transcriptional program on in the nucleus.
Figure 2 – Ciliary Hedgehog signaling translates different ciliary protein composition into different transcriptional outputs.
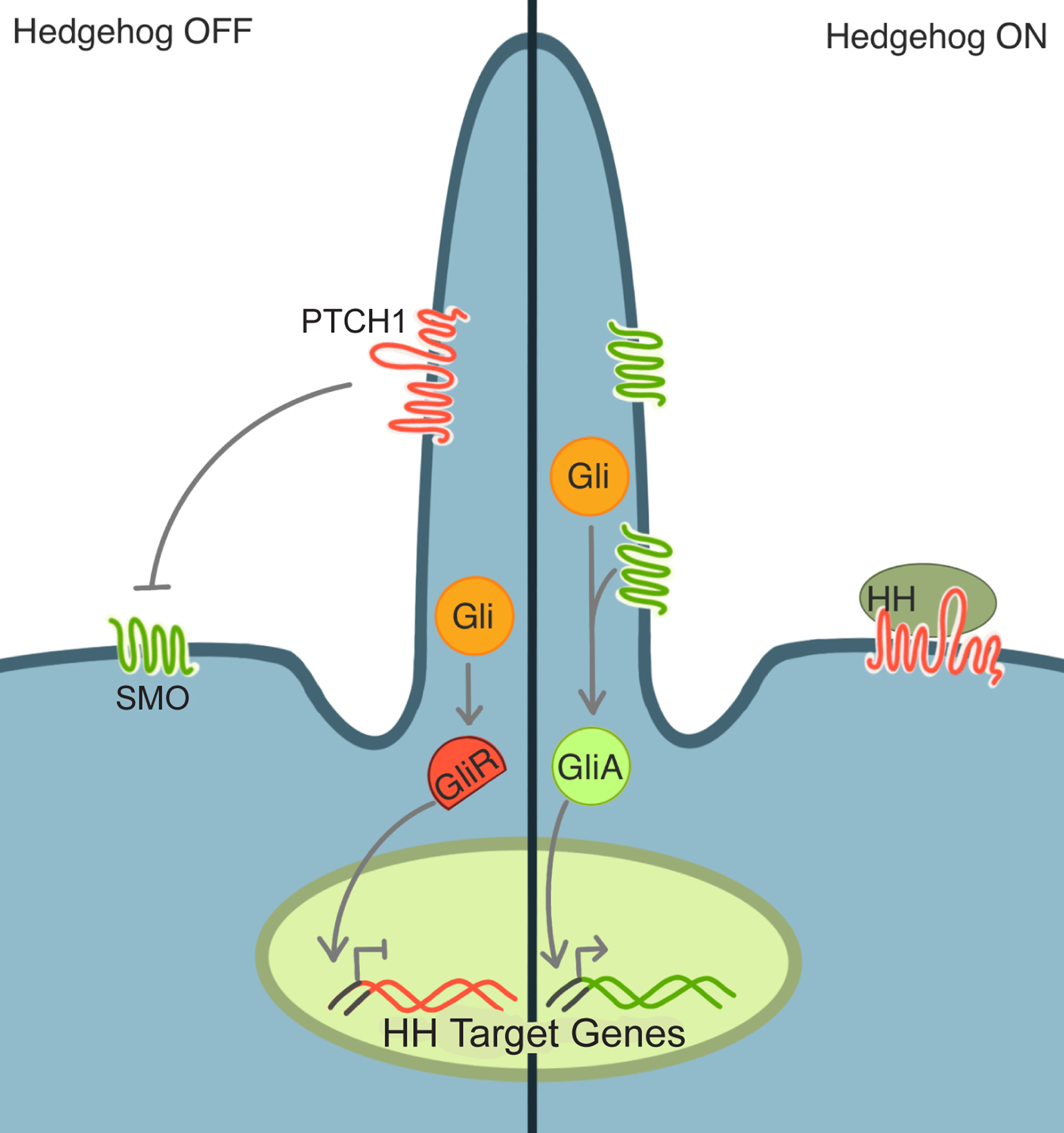
In the absence of Hedgehog (HH) signals (left), PTCH is present in the cilium and keeps SMO out and inactive. Without active SMO in the cilium, GLI transcription factors are converted into a repressive form (GliR), translocate to the nucleus and keep the Hedgehog transcription program off. In the presence of HH signals (right), SMO accumulates in the cilium and converts GLI into an activator form (GliA) which induces the Hedgehog transcriptional program.
How SMO activation leads to GLI activation remains a major gap in understanding. In Drosophila, the SMO C-terminus can interact directly with a GLI-containing protein complex27. The Drosophila and vertebrate SMO C-termini are highly divergent, indicating that vertebrates are likely to activate GLI through a distinct mechanism.
A key intermediary between SMO and GLI is protein kinase A (PKA), which promotes GLI conversion into GliR28–30. Vertebrate SMO can bind and inhibit PKA31,32. Thus, vertebrate SMO may regulate GLI by blocking PKA-mediated phosphorylation. As Hedgehog signals are morphogens capable of specifying multiple different fates depending on concentration and duration, GLI proteins may be in more states than on and off, with intermediate states transducing the mid-range of the morphogen gradient. How the ciliary machinery parses characteristics of the morphogen gradient to direct differentiation along multiple trajectories remains unclear.
In addition to its distinct protein composition, cilia possess a distinct lipid composition33. Ciliary lipids may also be critical to Hedgehog signal transduction. One subclass of lipids are sterols. A diverse group of oxidized sterols called oxysterols bind and activate SMO34–36. Several SMO-activating oxysterols are enriched in cilia, suggesting that the distinct lipid composition of cilia may be one reason why SMO requires the cilium to activate the downstream pathway signaling37. The upstream regulator of SMO, PTCH, can transport sterols across the cell membrane38–40. Hedgehog binding to PTCH1 could block this transport, potentially increasing sterol concentration and activating SMO. It will be interesting to ascertain whether endogenous SMO is regulated by oxysterols and/or cholesterol, and whether the regulation of SMO varies by cell type or organism.
Despite the aforementioned conservation of ciliary machinery, exploration of cilia function in diverse organisms has pointed out how evolution has tinkered with ciliary Hedgehog signaling. Whereas vertebrates and sea urchins require cilia for Hedgehog signaling, cilia are dispensable for Hedgehog signaling in many Drosophila tissues and in planaria41–44. Did the animal ancestor use cilia for Hedgehog signaling? Proteomic profiling of cilia from sea urchins (an early-branching non-chordate deuterostome), sea anemones (non-bilaterians) and choanoflagellates (protists thought to be the closest known relative of animals) revealed that Hedgehog signaling components are associated with cilia in sea anemones, suggesting that the ancestor of bilateria may have employed cilia for Hedgehog signaling18.
In addition to most known ciliary components, comparing the ciliary proteomes of diverse species identified ciliary proteins of unknown function18. For example, this comparison indicated that DYRK2 could be a ciliary kinase involved in Hedgehog signaling, subsequently validated by mammalian genetics45.
A parsimonious model is that evolution has played with a basal ciliary Hedgehog pathway, either by removing its dependence on cilia in one ecdysozoan (Drosophila) or by dispensing with it altogether in another (C. elegans). Evolution is most probably continuing to shape ciliary Hedgehog signaling in vertebrates, as alterations in ciliary Hedgehog signaling may underlie relatively recent changes in the eyes of cavefish, the heads of wolves and dogs, and the limbs of flightless birds46–48.
Future comparisons between cilium-dependent and -independent Hedgehog signaling may help illuminate which attributes are conveyed by the cilium itself. For example, comparing how ciliary and nonciliary Hedgehog signaling differ in kinetics or fidelity may reveal the design principles optimized by both forms of information transmission. One possibility is that relying on protein trafficking to and from cilia may increase signaling fidelity but sacrifice signal transduction speed.
Cilia transduce Polycystin signaling
In addition to their essential role in vertebrate embryonic development, cilia can be important in the homeostasis of adult tissues. PKD1 and PKD2 (encoding Polycystin-1 and 2) are justifiably most famous for being the two genes in which mutations cause autosomal dominant polycystic kidney disease (PKD)49,50. PKD is a human disease characterized by progressive formation of cysts, leading to kidney failure. Polycystin-1 and 2 are founding members of a subfamily of transient receptor potential (TRP) channels.
The TRP superfamily is comprised of a diverse cohort of tetrameric channel-forming integral membrane proteins. Compared to other members of the TRP superfamily, the Polycystin channel proteins are noncanonical. For example, Polycystin channels are heterotetramers comprised of one Polycystin-1 subunit and three Polycystin-2 subunits51 (Figure 3A).
Figure 3 – Polycystin channels protect the kidney from cystogenesis.
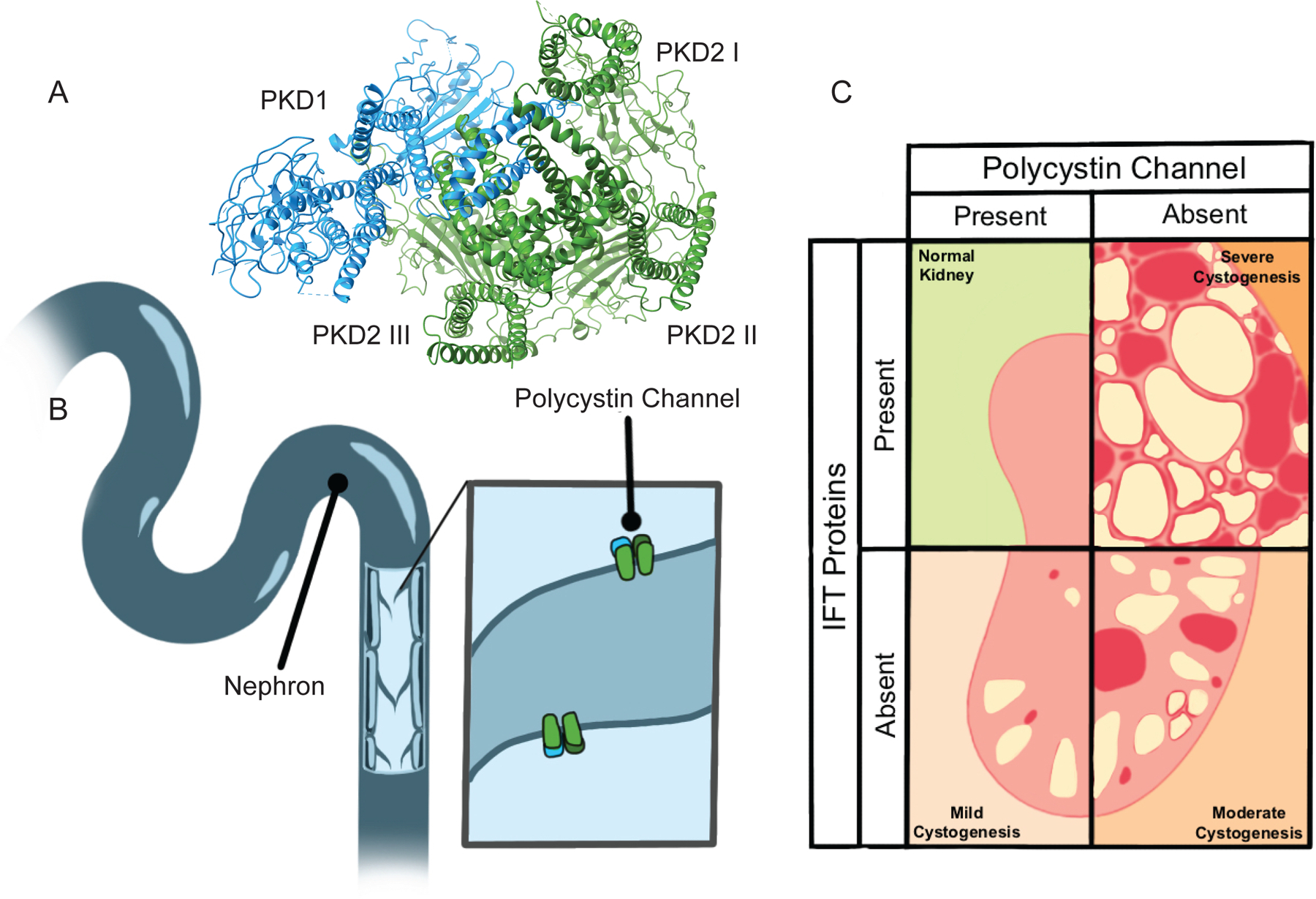
A) Polycystin channels are heterotetramers each comprised of one PKD1 subunit and three PKD2 subunits. Image of PDB 6A70 human PKD1/PKD2 complex (Su et al., 2018) created with UCSF ChimeraX. B) Almost all nephron epithelial cells project a Polycystin channel-bearing cilium into the lumen through which filtrate flows. C) Removing cilia from nephron epithelial cells causes mild cystogenesis. Removing Polycystin function by abrogating either PKD1 or PKD2 causes severe cystogenesis. Removing both cilia and Polycystin function causes moderate cystogenesis, revealing cilia-dependent cyst activation (CDCA) (Ma et al., 2013).
In the kidney, most cells of the nephron project a primary cilium into the nephron lumen (Figure 3B). Although Polycystin-1 and 2 localize to several subcellular locations (e.g., sites of membrane protein biosynthesis such as the ER), they prominently localize to cilia52–54. Deletion of cilia in the kidney epithelium is sufficient to induce cystogenesis, suggesting that cilia, like Polycystin-1/2 channels, restrain renal epithelial proliferation55–57.
However, cilia play a complex role in kidney cystogenesis. Cysts resulting from loss of cilia are smaller than the dramatic cysts caused by loss of Polycystin-1/2 function (with cilia present). Moreover, loss of both Polycystin-1/2 function and cilia also results in more mild cystogenesis58. One interpretation of these genetic interactions is that cilia act epistatically to PKD1/2 channels and that cilia both restrain and promote renal epithelial proliferation (Figure 3C).
How should we conceptualize these dual functions of cilia in cystogenesis? A perspective comes from understanding of ciliary Hedgehog signaling. Hedgehog signals pattern many tissues, including the neural tube and the limb bud. In the neural tube, Hedgehog signals induce ventral fates. Loss of Hedgehog signaling causes a dorsalization of the neural tube, and gain of Hedgehog signaling causes ventralization of the neural tube. In the limb bud, Hedgehog signals specify the number and identity of the digits. Loss of Hedgehog signaling causes a reduction in digit number and gain of Hedgehog signaling causes a gain in digit number. Unintuitively, loss of cilia causes dorsalization of the neural tube (an apparent loss of Hedgehog signaling) and a gain in digit number (an apparent gain of Hedgehog signaling)42,59. This apparent paradox is resolved by recognizing that cilia are essential for both generating the GLI activator and repressor. In the neural tube, Hedgehog patterning is effected by generating GLI activator, whereas in the limb bud, Hedgehog patterning is effected by inhibiting GLI repressor. Thus, loss of cilia removes both GLI activator and repressor in both tissues, but the phenotypic consequences are distinct because of differential reliance on GLI activator or repressor.
As kidney cilia either potentiate or restrain cystogenesis in different genetic contexts, they may function similarly to Hedgehog-transducing cilia: instead of generating both GLI activators and repressors, kidney cilia may generate cyst activators and repressors. In this model, loss of kidney cilia may compromise the formation of a cyst repressor, leading to cystogenesis. In addition, cilia may also generate cyst activators, potentially explaining the aggressive cystogenesis when Polycystin-1/2 channels are absent but cilia are present.
Fascinatingly, restoration of Polycystin-1/2 function in mouse cysts shrinks kidney cysts and reverses associated histological changes such as fibrosis60. Thus, if we could discover how Polycystin-1/2 transduces information to control cell proliferation, we might gain insight into pharmacologically tractable approaches to reversing polycystic kidney disease.
A different flavor of Polycystin channel is deployed in the left-right organizer early in development to specify the vertebrate left-right axis. While Polycystin-2 works with Polycystin-1 to limit kidney epithelial cell proliferation, Polycystin-2 collaborates with a paralog of Polycystin-1 called Polycystin-1-like-1 to help the embryo tell left from right61–63.
Even more than with Hedgehog signaling, deep mysteries about Polycystin remain to be explored. What are the molecular identities of these hypothesized Polycystin-regulated and cilia-dependent cyst activators and repressors? What cations do Polycystin-1/2 flux to influence kidney epithelial growth and morphology? What cues regulate Polycystin-1/2 activity? In addition to Polycystin-1, Polycystin-1-like-1 and Polycystin-2, we possess three more paralogs of Polycystin-1 and two paralogs of Polycystin-2. If Polycystin-2 paralogs can form functional homomeric channels and productively heterodimerize with all Polycystin-1 paralogs, we may possess 18 different flavors of Polycystin channels. Are each of these possible combinations formed? If so, what do they sense and what do they do? If these other forms of Polycystin channels function in development and physiology, do they all function at cilia and do they function through shared or distinct effectors?
It is likely that answers to these questions will depend on insights into Polycystin signaling derived from studies in diverse organisms. Homologs of Polycystin-2 are present in diverse ciliated organisms, including the green alga Chlamydomonas. In Chlamydomonas, Polycystin-2 localizes to two rows along the flagella and organizes a ciliary structure required for efficient swimming64. Interestingly, Chlamydomonas lacks a clear Polycystin-1 homolog, suggesting that Polycystin-2-type proteins can function independently of Polycystin-1 proteins. In Drosophila, a homolog of Polycystin-2 localizes to the sperm flagellum and is important for the ability of sperm to navigate the female reproductive tract65,66. In C. elegans, Polycystin-2 and an ortholog of Polycystin-1 localize to select neuronal cilia and function in male mating52,67. Together, these results reveal that Polycystins may have ancient roles in cilia, perhaps dating to the Neoproterozoic Era over 500 million years ago, and that evolution has subsequently deployed Polycystin channels for diverse functions in extant descendants.
Cilia transduce signaling by select GPCRs
Beyond Hedgehog and Polycystin signaling, we have known for decades that select GPCRs sense environmental cues at specialized cilia: the photoreceptor outer segment is a highly modified primary cilium at which Opsins sense light, and olfactory neurons possess atypical cilia adorned by odorant-sensing GPCRs that sense environmental chemicals. The sensitivity of these sensory cilia can be near perfect: amazingly, photoreceptor cells can detect single photons and olfactory neurons can detect single molecules of pheromone68,69. Ciliary signaling can also be blazingly fast with photoreceptor peak dim flash response happening at 140ms70.
A more recent surprise of cilia biology has been that select GPCRs also localize to the primary cilia outside of sensory organs. For example, SSTR3, 5HT6, MCHR1, GPR161, GPR88, FFAR4 and MC4R are all GPCRs from different subfamilies that localize to cilia71–75.
Beyond vision and olfaction, what types of information do ciliary GPCRs communicate? Clues from both mouse and human genetics point to critical roles for primary cilia in energy homeostasis. In mice, deletion of cilia from adult neurons causes obesity55. Likewise, in humans, mutations that disrupt ciliary function cause ciliopathies characterized by obesity, including Bardet-Biedl syndrome, Carpenter syndrome, Alström syndrome and MORM76. These human diseases are called ciliopathies to denote their common origin in compromised ciliary function77.
The most common monogenic cause of human obesity are mutations in MC4R, encoding the ciliary localized GPCR Melanocortin receptor 475,78,79. Thus, an appealing model is that ciliopathies cause obesity because cilia are fundamentally required for MC4R-mediated control of long-term energy homeostasis (Figure 4A).
Figure 4 – Different ciliary environments facilitate specific signaling cascades.
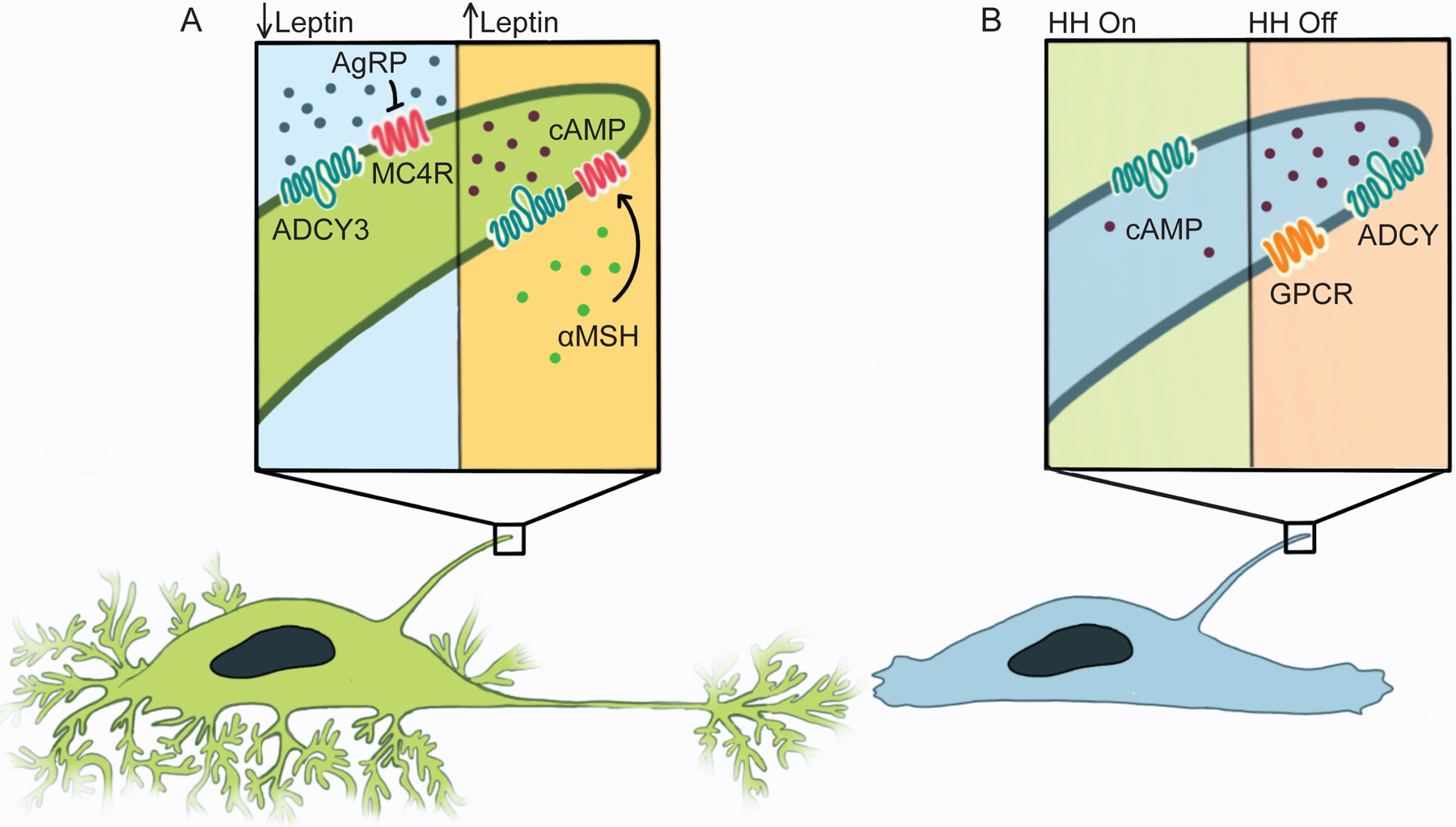
A) Left: When peripheral energy stores are depleted, Leptin levels fall, leading Leptin-responsive neurons in the hypothalamus to make AgRP, an inverse agonist of MC4R. In the absence of MC4R activity, ADCY3 in neuronal cilia does not generate cAMP, spurring feeding. Right: When peripheral energy stores are repleted, Leptin triggers hypothalamic neurons to make αMSH, an MC4R agonist (green). Stimulating MC4R induces ADCY3 to generate ciliary cAMP, suppressing feeding. B) cAMP generated in the cilium activates a ciliary pool of PKA to turn off Hedgehog signal transduction.
Many GPCRs signal through cAMP, and GPCRs can be Gαs-coupled to induce cAMP formation (e.g., MC4R) or Gαi-coupled to inhibit cAMP formation (e.g., SSTR3) via regulation of adenylyl cyclases. Adenylyl cyclase 3 (ADCY3) exquisitely localizes to neuronal cilia and mutations in ADCY3 cause obesity in both mice and humans, suggesting that cAMP generation within cilia is key for energy homeostasis80–82. Attenuating cAMP generation specifically in mouse hypothalamic cilia causes hyperphagia and obesity83, further suggesting that not only does MC4R localize to neuronal cilia, but MC4R regulation of ciliary cAMP controls feeding behavior. Thus, it is likely that feeding behavior is regulated by MC4R activation of ADCY3 to generate cAMP specifically within cilia. If so, MC4R regulation of long-term energy homeostasis can serve as one example of how neuronal cilia control mammalian behavior.
The finding that MC4R requires cilia to signal raises the interesting question of whether other ciliary GPCRs, such as FFAR4, also require cilia to signal. In preadipocytes, FFAR4 localizes to cilia and promotes differentiation84. During differentiation, the nascent adipocyte loses its cilium85. After differentiation, FFAR4 localizes to the plasma membrane and sensitizes the adipocyte to Insulin86–88. These findings raise the intriguing possibility that the same GPCR and ligand may, in one cell state, signal at the cilium with one output and, in another cell state, signal at the plasma membrane with a different output. Thus, by placing a receptor either at the ciliary membrane or at the plasma membrane, cells may respond to the same ligand in two distinct ways, and this choice of placement may be dependent on factors such as stage of differentiation.
Interesting questions about the function of GPCRs at cilia include how ligands are delivered to neuronal cilia and how cells distinguish closely related GPCRs (e.g., SSTR3 and SSTR1) to send one to the cilium and the other to the plasma membrane. For example, if a single cilium contains multiple receptor types, can the cell distinguish their outputs or are they summed? And, importantly, how do cells distinguish second messengers produced at cilia from those produced elsewhere?
The cilium and the cell body can differentially interpret second messengers
The discovery in the 1960s by Sutherland and colleagues that many hormones communicate via the second messenger cAMP was critiqued because it was deemed improbable that different cues could trigger distinct responses through a single chemical entity89,90. In the 1980s, Brunton and colleagues proposed that cAMP is compartmentalized within the cell such that different hormones produce distinct pools of cAMP that are interpreted in distinct ways91,92.
How different cAMP pools communicate different information remains an area of active research. As described above, FFAR4 is an example of a GPCR that may signal sequentially at the ciliary membrane and the plasma membrane. What happens when distinct GPCRs signal at the ciliary membrane and the plasma membrane simultaneously? One possibility is that physically sequestering signaling pathways to different parts of the cell surface allows parallel information processing to avoid, in the terminology of information theory, intersignal interference. While cells are likely to use many strategies to compartmentalize signaling so as not to confuse the information content, one possible strategy would be to take advantage of the physical differences of the cilium and cell body to distinguish cAMP in each subcellular domain.
To investigate whether the cilium represents a functionally distinct cAMP compartment, we and others have developed tools to control the subcellular production of cAMP. One optogenetic system to exert temporal control of cAMP production uses bacterial photoactivatable adenylyl cyclase (bPAC), which generates cAMP in response to blue light93. To distinguish the functions of cilium-generated and cell body-generated cAMP, we generated transgenic zebrafish that express bPAC targeted to cilia (Cilia-bPAC) or cytoplasm (Cyto-bPAC)94.
Because cAMP and its effector, PKA, negatively regulate the Hedgehog pathway, Hedgehog signal transduction is one potential interpreter of ciliary and cytoplasmic cAMP. Among the many tissues patterned by Hedgehog signals are somites. Surprisingly, generating moderate amounts of cytoplasmic cAMP did not attenuate Hedgehog patterning of somites, but generating the same amount of cAMP within cilia did94. Similarly, a ciliary-targeted GPCR that generates cAMP affected Hedgehog signaling, but the same GPCR outside the cilium did not94. Perhaps these artificial systems mimic the function of GPR161, a ciliary Gαs-coupled GPCR that inhibits Hedgehog signaling during embryonic development74. Thus, cAMP generated in different subcellular sites imparts different information, and the cilium and cell body are functionally distinct cAMP compartments (Figure 4B).
A corollary suggested by the finding that ciliary cAMP inhibits Hedgehog signaling is that a ciliary Gαi-coupled GPCR might be able to lower ciliary cAMP and activate Hedgehog signaling. Indeed, stimulation of the ciliary Gαi-coupled GPCR, SSTR3, with its ligand, somatostatin, can activate the Hedgehog pathway in NIH-3T3 cells94. Given the capacity of this ciliary GPCR to activate the Hedgehog signaling pathway when expressed heterologously, it will be interesting to assess whether ciliary GPCRs beyond GPR161 function via GLI modulation.
How might the cilium act as a distinct cAMP compartment? Could there be a barrier to prevent the mixing of ciliary and nonciliary cAMP? Unlike mitochondria and most other organelles, cilia are not entirely membrane bound and are thus contiguous with the cytoplasm. cAMP biosensors revealed that cAMP freely diffuses between the cilium and cytoplasm94,95, indicating that there was no barrier preventing the mixing of the two pools of cAMP. Similarly, examination of calcium diffusion between the cytoplasm and the cilium revealed no barrier between the two96. Thus, two second messengers, cAMP and calcium, freely diffuse between the two compartments, raising the vexing question of how cells distinguish ciliary and cytoplasmic signals.
Computational modeling suggested that the narrow entrance and elongated shape of the cilium imposes different cAMP dynamics in the cytoplasm and cilium: because of the cilium’s low volume and high surface-to-volume ratio relative to the cell body, small absolute changes in cAMP within the cilium translate into large increases in ciliary cAMP concentration and negligible changes in cytoplasmic cAMP concentration94. Similarly, small absolute increases in ciliary calcium can cause large increases in ciliary calcium concentration without perturbing cytoplasmic calcium concentration96.
In the sea urchin sperm flagellum, the effectors of cAMP and calcium are probably orders of magnitude more abundant than the second messengers themselves97. Thus, as long as effectors have sufficient affinity for their second messengers, activation of effectors within the flagellum happens before the second messenger has a chance to diffuse into the cytoplasm, be exported, or get degraded, a phenomenon termed kinetic compartmentalization97. Similar principles may operate within primary cilia to contribute to different effects of ciliary and cytosolic second messengers. Additional plausible parameters may contribute to functional separation of ciliary and cell body signaling (e.g., degradation of cAMP by phosphodiesterases, differential localization of cAMP effectors), but geometry alone may be sufficient to explain how cells distinguish ciliary and cytoplasmic second messengers.
Distinguishing pools of second messengers is likely to be essential for cellular functioning. For example, sperm make use of cAMP and calcium both in the flagellum to control swimming direction and in the head to control acrosomal exocytosis98. Mixing the second messengers between the two compartments would disrupt fertilization. If the unique geometry of the cilium is used to distinguish ciliary and plasma membrane signaling, then one might expect that flooding the system with cAMP would erase the ability of the cell to make that distinction. Indeed, the production of super-physiological levels of cAMP using bPAC erases the distinction and allows cytoplasmically produced cAMP to affect Hedgehog signaling94.
Such experiments raise interesting questions about how the functional compartmentalization between ciliary and cell body signaling is maintained over different signaling regimes. For example, do negative regulators of second messengers (e.g., cAMP phosphodiesterases or calcium buffers) help prevent crosstalk between the two compartments? Additionally, if the second messengers remain restricted to their compartment of origin, the effectors can be in common. However, if signaling within the ciliary compartment must be communicated elsewhere within the cell (as exemplified by Hedgehog signaling, which must ultimately affect nuclear transcription), second messengers must work via effectors specific to their compartment of origin. An ambassador’s effectiveness depends on being identified with their country.
The cilium is a source of extracellular signals
Initially observed in Chlamydomonas99,100, a growing body of evidence supports a role for the cilium not just as a receiver and interpreter of extracellular information, but also as a source. In Chlamydomonas, flagella are the source of proteolytic enzyme-bearing extracellular vesicles that degrade the cell wall to liberate daughter cells following mitosis99. Activated Chlamydomonas gametes shed vesicles that specifically bind the flagellae of other gametes, hinting at signaling functions101.
Similarly, in C. elegans, ciliated sensory neurons release extracellular vesicles containing Polycystins, and purified vesicles trigger mating behaviors in male worms52,67,102. In male C. elegans, select neurons release ciliary vesicles into the luminal space that surrounds their cilia and then through a pore into the external environment of the animal. Interestingly, bbs-8 and rab-28 mutants accumulate extracellular vesicles in the lumen surrounding the cilium, but do not show any change in the release of vesicles into the external environment (Figure 5). Therefore, there may be two functionally distinct steps for ciliary vesicle release in C. elegans: ectocytosis from the neuron and then subsequent expulsion into the environment for extracellular communication103. In mammals, RAB28 promotes the shedding of the photoreceptor outer segment and RAB28 mutations cause a form of retinal degeneration, suggesting that RAB28 may have different roles in the regulation of ciliary ectocytosis in nematodes and vertebrates66,104.
Figure 5 -. Some C. elegans cephalic male neuronal cilia release extracellular vesicles.
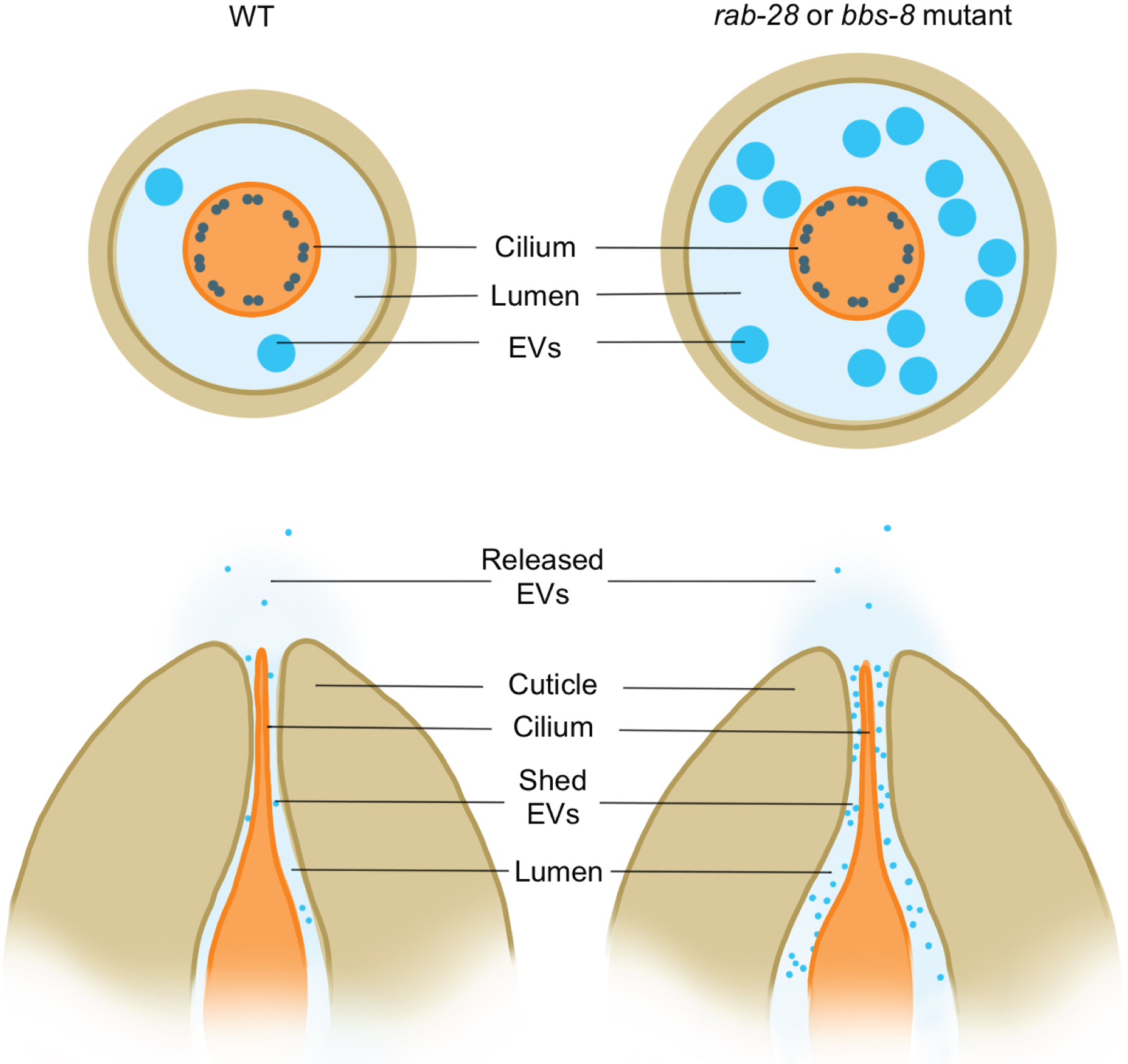
Mutations in rab-28 or bbs-8 increase ectocytosis of extracellular vesicles into the lumen surrounding the cilia.
The photoreceptor outer segment is a specialized primary cilium containing stacks of membrane disks concentrated with Opsins, key to phototransduction. Opsins contain 11-cis-retinal which absorbs light to undergo isomerization to all-trans-retinal. In addition to sensing light, these Opsins are damaged by light. Disks containing photooxidized Opsins and all-trans-retinal are shed from the distal outer segment and taken up by the adjoining retinal pigmented epithelium, which rejuvenates the 11-cis-retinal and sends it back to the photoreceptors105. Defects in outer segment turnover cause forms of retinal degeneration, highlighting the importance of this specialized form of ciliary ectocytosis for vision maintenance.
Formation of the disks requires branched actin polymerization and a transmembrane protein called Peripherin106,107. In the absence of Peripherin, the photoreceptor fails to form internalized disks and, instead, sheds them as extracellular vesicles108. These findings suggest that photoreceptor disk formation may be a modification of ciliary ectocytosis, a variant that accumulates the Opsin-bearing vesicles internally, not externally, to enhance light capture. Although photoreceptors are highly specialized, these results raise the question of whether other cell types alter ectocytosis to control ciliary membrane topology and signaling sensitivity.
Ectocytotic control of ciliary composition may occur outside of photoreceptors. In primary ciliated cells, upon SSTR3 stimulation and inhibition of BBSome-mediated retrieval, ciliary ectocytosis increases109,110. Similar to photoreceptor disk shedding, ectocytosis requires Actin109. The ability of the BBSome to promote GPCR retrieval into the cell body and to attenuate ectocytosis suggests that there are at least two mechanisms for controlling ciliary GPCR content.
Which proteins are shed in ciliary vesicles? Curiously, in the absence of the BBSome, a subset of GPCRs are lost from neuronal cilia111–113. Perhaps only certain neuronal cell types activate ectocytosis in the absence of the BBSome, or perhaps only some types of GPCRs exit cilia via ectocytosis.
Ciliation and deciliation -- the cilium as a transient organelle
With very few exceptions (e.g., metazoan oocytes lack centrosomes, red blood cells lack nuclei), cellular organelles are present across cell types. But unlike the nucleus, ER and Golgi, not all cells have a cilium114. For example, blood cells, both red and white, lack cilia.
Ciliation can vary not just by cell type, but also within a cell type. For example, in the skin, many hair follicle cells are ciliated, but few cells in the interfollicular epidermis are ciliated115 (Figure 6). The dispensability of the cilium for many cellular functions may allow tissues to tune individual cells, even neighboring cells of the same type, to different extracellular cues. For example, the few ciliated cells that line the stomach may respond to different cues than the rest of the gastric epithelium116.
Figure 6 – Not all animal cells possess cilia.
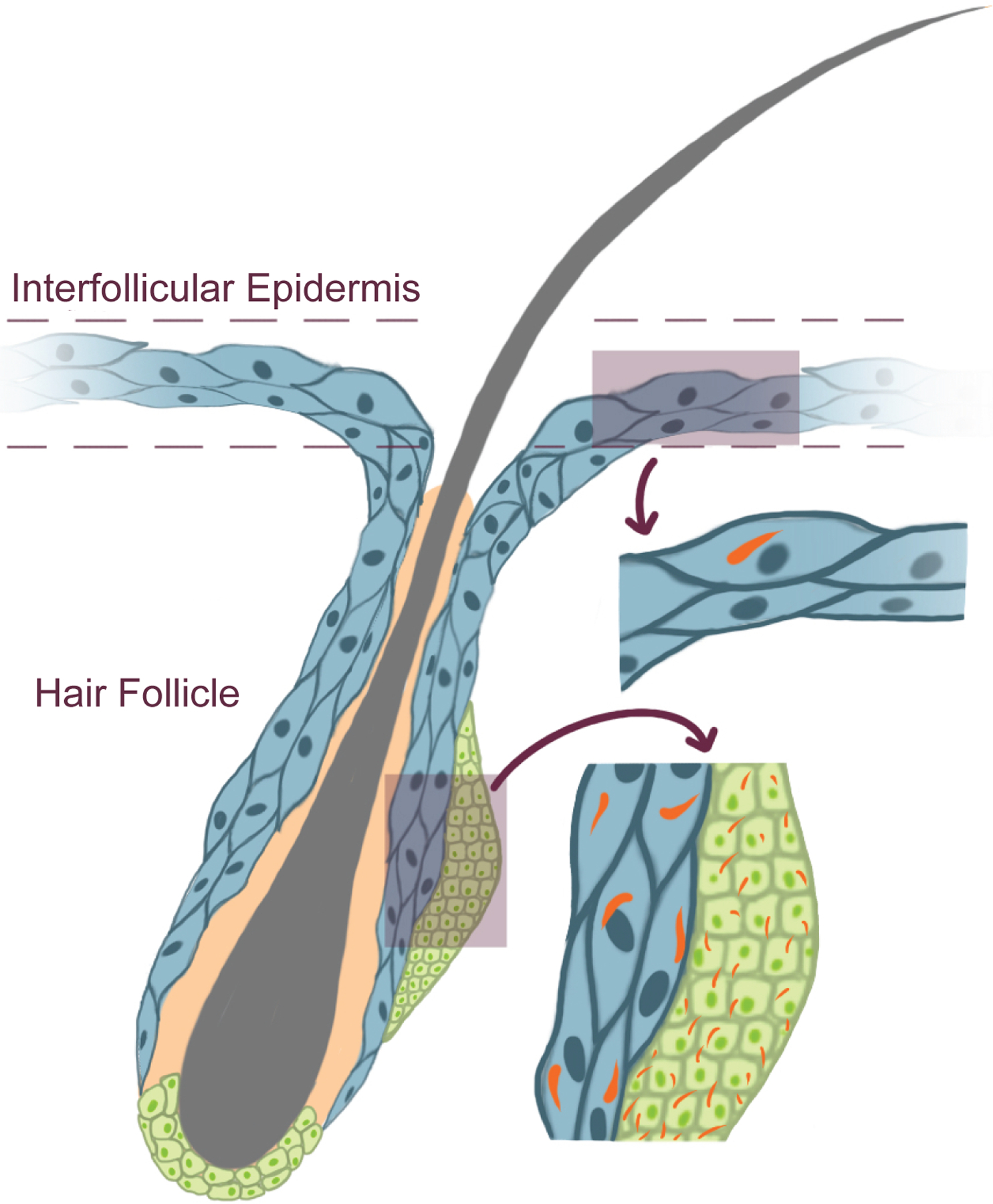
For example, the skin is comprised of hair follicles and the interfollicular epidermis. Most cells making up the interfollicular epidermis are unciliated. In contrast, most cells of the hair follicle are ciliated. What controls which cells are ciliated and which are not remains unknown.
In addition, some types of cells have cilia only during certain stages of their differentiation. For example, during early stages of neural tube development, the neural epithelium lacks cilia. Soon thereafter, the neural epithelium acquires cilia and uses them for dorsoventral patterning. Conversely, the endodermal epithelium is ciliated early in development but loses cilia as it differentiates into the intestinal epithelium117.
Control of the timing of ciliation may participate in the timing of responsiveness to extracellular information. A longstanding conundrum has been how cells accurately interpret a morphogen gradient when that morphogen gradient must be generated over time. One possible solution to this conundrum would be to separate generation of the gradient and interpretation of the gradient into different time periods. To pattern the dorsoventral axis of the neural tube, SHH and IHH are expressed at the midline from gastrulation stages. As the neural tube ciliates after gastrulation, temporal control of ciliation may allow neural epithelial cells to ignore Hedgehog cues while the gradient is being created, then, upon ciliating, interpret that mature gradient to accurately read out their dorsoventral position within the neural tube.
Thus, regulation of ciliation may help regulate cellular responses to cues, both spatially and temporally. We do not understand the mechanisms by which cells are ciliated. Some insight may come from the fact that almost all cells retract their cilia before entering mitosis. Cilia extend from a centriole, so deciliation prior to mitosis releases one of the centrioles so that it can become part of the spindle pole. How ciliary disassembly occurs remains poorly understood, although Aurora A kinase is part of the process118.
Presumably, ciliary disassembly before mitosis frees up the mother centriole to participate in spindle pole formation. Ciliate protists solve the problem of dual roles of centrioles in ciliation and spindle function in a different way: they assemble their mitotic spindles independently of centrioles and thus the basal bodies remain attached to the cell surface throughout the entire lifecycle119,120.
Do mammalian cells take advantage of the retractability of cilia to shape their responses to non-developmental cues? Multiciliated cells that line the upper airway and clear mucus out of the lung avoid deciliation altogether by being terminally differentiated. However, upon infection by Respiratory syncytial virus (RSV), a serious respiratory pathogen often affecting young children, multiciliated cells in the airway lose their cilia121,122. Loss of motile cilia compromises airway clearing, promotes the formation of mucus plugs and predisposes children to opportunistic infections by other respiratory pathogens123. While it is likely that the loss of cilia is caused by virally mediated damage, it is possible that aspects of this loss of cilia are adaptive. As respiratory viruses may bud from the ciliary membrane124, it is possible that retraction of cilia could attenuate viral production. Are there inhibitors of ciliogenesis that cells turn on to negatively regulate ciliogenesis under stressful conditions such as RSV infection? Transcriptomic or proteomic analysis of RSV infected airway epithelial cells may help identify negative regulators of ciliogenesis.
Deciliation is also employed by the devastating frog-killing chytrid Batrachochytrium dendrobatidis125. Batrachochytrium zoospores can swim using a motile cilium126. Batrachochytrium sense the mucins secreted by frog skin, triggering a transition called encystment during which the cilium is quickly resorbed and the centriole is repurposed for the mitotic spindle during nuclear divisions127. Efforts to make chytrid species genetically tractable may shed light on how they resorb their cilia126,128.
Other unicellular organisms also are flagellated only part time. Naegleria gruberi is a free-living protist that can generate two distinct cellular architectures: its feeding, reproducing, crawling amoeba-like form can persist indefinitely without flagella and even without centrioles129. However, when stressed, it transforms into a flagellated swimmer to search out better conditions. Astonishingly, this transformation involves de novo assembly of basal bodies and construction of flagella in about an hour. A similar phenomenon occurs in Tetramitus, a related free-living amoeba that not only constructs four flagella via de novo basal body assembly but can reproduce in both its flagellated and amoeboid states130,131.
Not to be outdone by its distant cousin is Plasmodium, the protist parasite that causes malaria. Plasmodium lacks flagella for most of its lifecycle132. In the blood, Plasmodium can become a gametocyte which, if swallowed by a feeding mosquito, undergoes three lightning-fast rounds of mitosis and flagellogenesis, resulting in eight flagellated gametes in just 10–15 minutes. These flagella power Plasmodium mating in the mosquito’s gut. As flagella are essential only for this one stage in its lifecycle, it is possible that a Plasmodium mutant lacking, for example, an outer arm dynein (e.g., PF3D7_1213600, PF3D7_1020100 or PF3D7_1114000) could be maintained in its asexual stage in the lab but fail to propagate outside the lab133.
Similarly, choanoflagellates can retract their flagella to adopt amoeboid forms134. Thus, it is possible that the unicellular ancestor of multicellular animals had evolved the ability to adopt either ciliated or unciliated forms, and animals differentially deployed these programs for cell specialization. For example, in sponges, ciliated choanocytes may represent deployment of the ancestral flagellated program and unciliated archeocytes may represent deployment of the ancestral unflagellated program. As noted above, we do not understand how vertebrate cells decide whether to generate a cilium or not. Part of the decision is probably made at the level of transcriptional control. Two transcriptional regulators of ciliogenesis, RFX and FOXJ1, are providing clues to the evolutionary history of ciliogenesis programs.
In C. elegans, an RFX transcription factor is essential for ciliogenesis135. Homologs of RFX are required for ciliogenesis in specific cell types in mice: RFX3 is required for motile ciliogenesis, RFX4 is required for ciliogenesis in parts of the brain, and RFX2 is required for sperm flagellogenesis136–138. In a choanoflagellate, an RFX transcription factor promotes ciliogenesis and is necessary for expression of another transcription factor, FOXJ1139. In mammals, FOXJ1 regulates genes involved in ciliary motility140,141. Thus, RFX and FOXJ1 are likely part of an ancient transcriptional network employed by animal ancestors to build cilia and subsequently modified in animal lineages142,143.
A more even sampling of the eukaryotic tree will yield a more complete understanding of how cilia are built by extant descendants of LECA. For example, are there additional conserved nontranscriptional regulators of ciliation? And what additional factors does RFX need to induce ciliogenesis in unciliated cells?
Human genetics and disease inform ciliary biology
Among the diverse organisms with conserved ciliary biology are humans. Consequently, ciliary dysfunction in people causes disease. Mutations in more than 200 genes cause 35 distinct ciliopathies, each with partially overlapping phenotypic manifestations. Perhaps because clinicians can get to know their patients extremely well and follow them for years, clinicians provide perceptive insights into ciliary functions.
Clinicians have often collaborated closely with biologists to uncover how cilia participate in human physiology. Perhaps the most famous of these interactions were those that led to the first recognized ciliopathy, Primary Ciliary Dyskinesia (PCD). In the early 1970s, Bjorn Afzelius was working at the University of Stockholm as a PhD electron microscopist. A physician, Rune Eliasson, solicited Dr. Afzelius’s help in analyzing the ultrastructure of sperm of infertile men144.
Electron microscopy of these sperm revealed the absence of dynein arms145. In addition to infertility, these men had the hallmarks of PCD (then called Kartagener syndrome): bronchiectasis, chronic sinusitis and, sometimes, situs inversus. Association of these ciliary defects with low or no mucociliary transport in the nose led to the realization that PCD is caused by defective ciliary motility146. Thus, cutting-edge experimental approaches and clinical insights were complementary in discovering that ciliary motility is essential for lung health and left-right axis patterning.
Since the discovery of ciliary motility defects as the cause of PCD, mutations resulting in ciliary dysfunction have been uncovered as causes of other human ciliopathies. One example is Bardet-Biedl syndrome (BBS), characterized by retinal degeneration, polydactyly, kidney cysts, anosmia, cognitive impairment, and obesity. Many of the genes mutated in BBS affect the BBSome, involved in exporting membrane-associated proteins from the cilium147,148.
The ongoing revolution in the cell biological understanding of disease allows for a shift in classification, from causative tissue to causative subcellular process. The reclassification of diverse inherited diseases as ciliopathies highlights the commonalities between them and may serve to point out shared biology and shared therapeutic approaches going forward. Similar reclassification of other types of non-ciliary diseases by which subcellular process underlies them may help other fields recognize therapeutically exploitable commonalities for diseases previously regarded as unrelated. One example may be emerging recognition of the role of the integrated stress response (ISR) in a range of neurological diseases for which ISR inhibitor ISRIB may be useful149.
Beyond inherited ciliopathies, Hedgehog-associated cancers such as basal cell carcinoma and medulloblastoma highlight how cilia can participate in acquired diseases. These cancer cells are ciliated and require their cilia to transduce oncogenic Smoothened signaling115,150. Another example of how cilia may participate in acquired disease may be some forms of Parkinson’s disease, characterized by loss of dopaminergic neurons in the brain. Gain-of-function mutations in Leucine Rich Repeat Kinase 2 (LRRK2) are a rare cause of Parkinson’s disease151. Increased LRRK2 activity inhibits ciliogenesis152–155. Correspondingly, mice bearing Parkinson’s disease-causing mutations in LRRK2 or lacking the counteracting PPM1H phosphatase exhibit a disruption of cilia in about half of striatal cholinergic neurons and astrocytes153,156. Normally, these cholinergic neurons respond to Hedgehog signals to support the survival of dopaminergic neurons157. Thus, an intriguing model of how mutations in LRRK2 cause Parkinson’s disease is that they attenuate striatal ciliogenesis, limiting Hedgehog signaling, thereby leading to loss of dopaminergic neurons (Figure 7). Perhaps because altered ciliary function is limited to a fraction of striatal cholinergic neurons and astrocytes, mutant LRRK2 does not cause widespread ciliary dysfunction. It will be interesting to discover whether cilia play a role in more common forms of Parkinson’s disease, how germline mutations result in cell type-specific ciliary disruptions, and what other acquired diseases alter ciliary function.
Figure 7 -. A model of ciliary involvement in neurological disease.
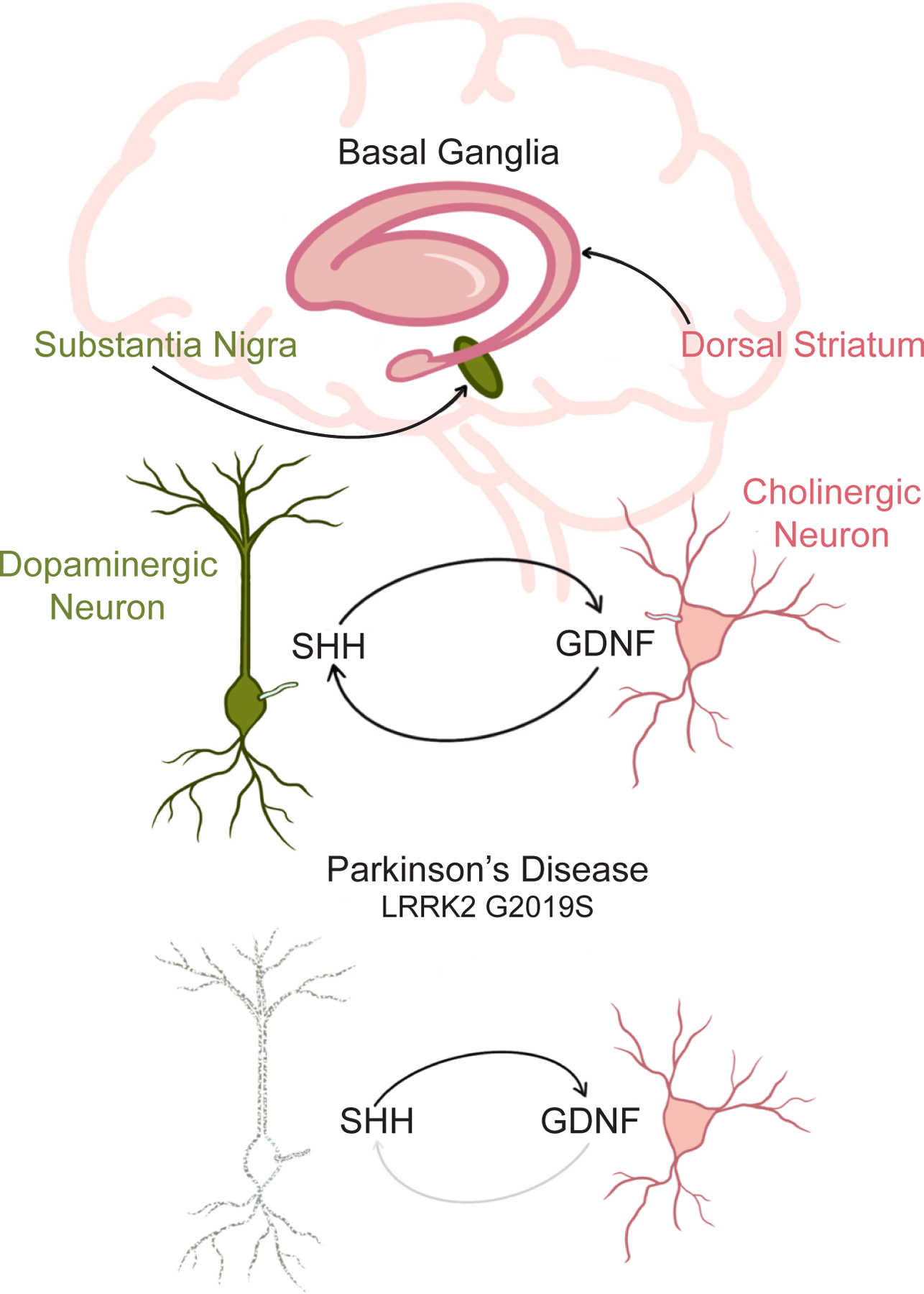
In Parkinson’s disease, dopaminergic neurons in the substantial nigra of the basal ganglia are lost. These neurons are supported by GDNF produced by cholinergic neurons in the striatum. A rare form of Parkinson’s disease is caused by gain-of-function mutations (e.g., G2019S) in a kinase, LRRK2, that disrupts cilia in the striatum. With compromised cilia, the cholinergic neurons do not respond to the Hedgehog signals produced by the dopaminergic neurons and, consequently, fail to produce GDNF, perhaps leading to the death of the dopaminergic neurons.
This discovery of an organelle at the origin of disparate diseases and the coining of the term ciliopathy indicates how nosology, the classification of disease, may be changing. Nosology reflects progress in our understanding of biology: for example, classical Greek physicians recognizing that diabetics produce excess urine, focused their attention on the kidney. Not until the eighteenth century did a more sensitive test, tasting urine, allow for subclassification of diabetes into two forms: mellitus (“from honey”) and insipidus (“without taste”). This modest diagnostic advance helped to move physicians’ focus from the kidney to the causative organs, reclassifying diabetes as a pancreatic disease (mellitus) and a pituitary disease (insipidus). Similarly, a diverse list of diseases grouped based on their affected tissues are now being reclassified based on which organelle underlies their etiology, with ciliopathies being one key example.
Conclusion
Cilia are ancient, complex machines comprised of several hundred different components that have been evolving for hundreds of millions of years to move and interpret diverse environmental and intercellular signals. Investigating cilia will continue to illuminate general principles of subcellular organization and information transmission. For example, understanding how cilia are compartmentalized will elucidate how transduction machinery is organized within a subcellular space to interpret extracellular cues. Understanding how ciliary lipids control signaling will reveal how organelle-specific lipids regulate protein function. Understanding how cells distinguish ciliary and plasma membrane signals will uncover how the cell holds multiple conversations simultaneously without confusing the information. Thus, investigating cilia will help reveal fundamental ways in which cells use specialized subcellular compartments to interpret their environments. Studying how cilia are constructed and signal will continue to identify human mutations that cause ciliopathies, how those mutations cause disease and, perhaps someday, how we can cure those diseases.
Beyond the fascinating field of cilia biology, there has been concern lately about the current and future status of the field of developmental biology158–160. Part of the concern stems from a perceived reduction in the importance of developmental biology research. Perhaps this perception stems from a constricted definition of what developmental biology entails. While the traditional domain of “good old God-fearing developmental biology”161 contains many important, unanswered questions, a more expansive definition of developmental biology might be the study of all biological processes requiring more than one cell and relevant to embryogenesis. Under such a definition, stem cell biology, organoids, embryoids, regeneration and computational approaches applied to cell populations (e.g., pseudotime analysis) are all subdomains or tools within developmental biology. When considered from this perspective, developmental biology is experiencing explosive growth.
The recent exciting discoveries in cilia biology described above underscore how cell biological tools can help drive discovery of important and general principles in developmental biology. Of course, apart from cilia, there are many instances of how cell biological approaches have led to fascinating developmental biology discoveries. For example, investigating how the actin cytoskeleton is spatially regulated has helped reveal how convergent extension drives neural tube closure162 . And investigating how cadherins control cell-cell adhesion has shed light on how mechanical forces sculpt cell shape163.
Just as biochemistry and genetics are simultaneously fields in their own rights and the source of experimental approaches used by developmental biologists, it is likely that cell biology will both remain its own domain and continue to provide insights, approaches and tools that inform developmental biology. Blurring of the borders between previously distinct fiefdoms of biology can be disorienting as it disrupts long-standing definitions of fields. Still, the borders between biochemistry, genetics, cell biology and developmental biology, always imprecise, are only becoming more so, with cilia biology being a prime example of how synergistic cell and developmental biological perspectives drive discovery164.
The advances detailed in this Review underscore the importance of cilia in evolutionary, developmental, cell, and human biology. Evolutionarily conserved machinery hints at ancestral ciliary function and continues to inspire avenues of inquiry into ciliary functions in animals living today. Hedgehog, Polycystin and GPCR signaling are examples of how ciliary signaling shapes embryonic development, maintains adult tissue populations, and provides a platform for reception of extracellular sensory cues. The cilium, despite being incompletely membrane bound, also functions as a distinct compartment that responds to local signaling molecule concentrations. Beyond its role as a signaling receiver, there is evidence that cilia can produce signals to be received by other cells. Additionally, there is great diversity in the spatiotemporal dynamics of ciliation amongst species and even amongst tissue types within one organism. This variety and complexity can not only be appreciated at the cellular level but also at the level of human disease, as ciliary defects cause a wide variety of diseases depending on the tissue location and type of the ciliary dysfunction. Ciliary biology lies at the boundary between biological disciplines, lending itself beautifully to a diverse range of tools, analyses, and inquiries that can be rigorously applied to understanding this complex organelle.
Footnotes
Declaration of Interests
J. Reiter is a founder of Renasant Bio and Sen Therapeutics and an advisor to Maze Therapeutics.
References
- 1.Sorokin SP (1968). Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J. Cell Sci 3, 207–230. 10.1242/jcs.3.2.207. [DOI] [PubMed] [Google Scholar]
- 2.Bloodgood RA (2009). From central to rudimentary to primary: the history of an underappreciated organelle whose time has come. The primary cilium. Methods Cell Biol. 94, 3–52. 10.1016/s0091-679x(08)94001-2. [DOI] [PubMed] [Google Scholar]
- 3.Carvalho-Santos Z, Azimzadeh J, Pereira-Leal José.B., and Bettencourt-Dias M (2011). Tracing the origins of centrioles, cilia, and flagella. J. Cell Biol 194, 165–175. 10.1083/jcb.201011152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavalier-Smith T (2022). Ciliary transition zone evolution and the root of the eukaryote tree: implications for opisthokont origin and classification of kingdoms Protozoa, Plantae, and Fungi. Protoplasma 259, 487–593. 10.1007/s00709-021-01665-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metchnikoff É, and Metchnikoff É (1886). Embryologische Studien an Medusen : Ein Beitrag zur Genealogie der Primitiv-organe (A. Hölder) 10.5962/bhl.title.5982. [DOI] [Google Scholar]
- 6.Kozminski KG, Johnson KA, Forscher P, and Rosenbaum JL (1993). A motility in the eukaryotic flagellum unrelated to flagellar beating. PNAS USA 90, 5519–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Craige B, Tsao C-C, Diener DR, Hou Y, Lechtreck K-F, Rosenbaum JL, and Witman GB (2010). CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol 190, 927–940. 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, García-Verdugo JM, Katsanis N, Hildebrandt F, Reiter JF (2011). A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet 43, 776–784. 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dobzhansky T (1964). Biology, Molecular and Organismic. Am. Zool 4, 443–452. 10.1093/icb/4.4.443. [DOI] [PubMed] [Google Scholar]
- 10.Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, Fazzi E, Signorini S, Louie CM, Bellacchio E (2006). Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet 38, 623–625. 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 11.Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, et al. (2006). The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet 38, 674–681. 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 12.Van De Weghe JC, Gomez A, and Doherty D (2022). The Joubert–Meckel–Nephronophthisis Spectrum of Ciliopathies. Annu. Rev. Genomics Hum. Genet. 23, 301–329. 10.1146/annurev-genom-121321-093528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar D, Rains A, Herranz-Pérez V, Lu Q, Shi X, Swaney DL, Stevenson E, Krogan NJ, Huang B, Westlake C, et al. (2021). A ciliopathy complex builds distal appendages to initiate ciliogenesis. J. Cell Biol 220, e202011133. 10.1083/jcb.202011133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma M, Stoyanova M, Rademacher G, Dutcher SK, Brown A, and Zhang R (2019). Structure of the Decorated Ciliary Doublet Microtubule. Cell 179, 909–922.e12. 10.1016/j.cell.2019.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dougherty GW, Mizuno K, Nöthe-Menchen T, Ikawa Y, Boldt K, Ta-Shma A, Aprea I, Minegishi K, Pang Y-P, Pennekamp P, et al. (2020). CFAP45 deficiency causes situs abnormalities and asthenospermia by disrupting an axonemal adenine nucleotide homeostasis module. Nat. Commun 11, 5520. 10.1038/s41467-020-19113-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reiter JF, and Leroux MR (2017). Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol 18, 533–547. 10.1038/nrm.2017.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Legendre M, Zaragosi L-E, and Mitchison HM (2021). Motile cilia and airway disease. Semin. Cell Dev. Biol 110, 19–33. 10.1016/j.semcdb.2020.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Sigg MA, Menchen T, Lee C, Johnson J, Jungnickel MK, Choksi SP, Garcia G, Busengdal H, Dougherty GW, Pennekamp P, et al. (2017). Evolutionary Proteomics Uncovers Ancient Associations of Cilia with Signaling Pathways. Dev. Cell 43, 744–762.e11. 10.1016/j.devcel.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ingham PW, and McMahon AP (2001). Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087. 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 20.Nüsslein-Volhard C, and Wieschaus E (1980). Mutations affecting segment number and polarity in Drosophila. Nature 287, 795–801. 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 21.Nüsslein-Volhard C, Wieschaus E, and Kluding H (1984). Mutations affecting the pattern of the larval cuticle inDrosophila melanogaster : I. Zygotic loci on the second chromosome. Wilehm Roux Arch. Dev. Biol 193, 267–282. 10.1007/BF00848156. [DOI] [PubMed] [Google Scholar]
- 22.Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DYR, and Reiter JF (2005). Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021. 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 23.Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, and Yoder BK (2005). Gli2 and Gli3 Localize to Cilia and Require the Intraflagellar Transport Protein Polaris for Processing and Function. PLoS Genet. 1, e53. 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He M, Subramanian R, Bangs F, Omelchenko T, Liem KF, Kapoor TM, and Anderson KV (2014). The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat. Cell Biol 16, 663–672. 10.1038/ncb2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rohatgi R, Milenkovic L, and Scott MP (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372–376. 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 26.Niewiadomski P, Niedziółka SM, Markiewicz Ł, Uśpieński T, Baran B, and Chojnowska K (2019). Gli Proteins: Regulation in Development and Cancer. Cells 8, 147. 10.3390/cells8020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robbins DJ, Nybakken KE, Kobayashi R, Sisson JC, Bishop JM, and Thérond PP (1997). Hedgehog elicits signal transduction by means of a large complex containing the kinesin-related protein costal2. Cell 90, 225–234. 10.1016/s0092-8674(00)80331-1. [DOI] [PubMed] [Google Scholar]
- 28.Jiang J, and Struhl G (1995). Protein kinase A and hedgehog signaling in drosophila limb development. Cell 80, 563–572. 10.1016/0092-8674(95)90510-3. [DOI] [PubMed] [Google Scholar]
- 29.Tuson M, He M, and Anderson KV (2011). Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development 138, 4921–4930. 10.1242/dev.070805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niewiadomski P, Kong JH, Ahrends R, Ma Y, Humke EW, Khan S, Teruel MN, Novitch BG, and Rohatgi R (2014). Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 6, 168–181. 10.1016/j.celrep.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arveseth CD, Happ JT, Hedeen DS, Zhu J-F, Capener JL, Klatt Shaw D, Deshpande I, Liang J, Xu J, Stubben SL, et al. (2021). Smoothened transduces Hedgehog signals via activity-dependent sequestration of PKA catalytic subunits. PLoS Biol. 19, e3001191. 10.1371/journal.pbio.3001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Happ JT, Arveseth CD, Bruystens J, Bertinetti D, Nelson IB, Olivieri C, Zhang J, Hedeen DS, Zhu J-F, Capener JL, et al. (2022). A PKA inhibitor motif within SMOOTHENED controls Hedgehog signal transduction. Nat. Struct. Mol. Biol 29, 990–999. 10.1038/s41594-022-00838-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen TD, Truong ME, and Reiter JF (2022). The Intimate Connection Between Lipids and Hedgehog Signaling. Front. Cell Dev. Biol 10, 876815. 10.3389/fcell.2022.876815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nachtergaele S, Mydock LK, Krishnan K, Rammohan J, Schlesinger PH, Covey DF, and Rohatgi R (2012). Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol 8, 211–220. 10.1038/nchembio.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qi X, Liu H, Thompson B, McDonald J, Zhang C, and Li X (2019). Cryo-EM structure of oxysterol-bound human Smoothened coupled to a heterotrimeric Gi. Nature 571, 279–283. 10.1038/s41586-019-1286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deshpande I, Liang J, Hedeen D, Roberts KJ, Zhang Y, Ha B, Latorraca NR, Faust B, Dror RO, Beachy PA, et al. (2019). Smoothened stimulation by membrane sterols drives Hedgehog pathway activity. Nature 571, 284–288. 10.1038/s41586-019-1355-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raleigh DR, Sever N, Choksi PK, Sigg MA, Hines KM, Thompson BM, Elnatan D, Jaishankar P, Bisignano P, Garcia-Gonzalo FR, et al. (2018). Cilia-Associated Oxysterols Activate Smoothened. Mol. Cell 72, 316–327.e5. 10.1016/j.molcel.2018.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Bulkley DP, Xin Y, Roberts KJ, Asarnow DE, Sharma A, Myers BR, Cho W, Cheng Y, and Beachy PA (2018). Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 175, 1352–1364.e14. 10.1016/j.cell.2018.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qi C, Di Minin G, Vercellino I, Wutz A, and Korkhov VM (2019). Structural basis of sterol recognition by human hedgehog receptor PTCH1. Sci. Adv 5, eaaw6490. 10.1126/sciadv.aaw6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinnebrew M, Woolley RE, Ansell TB, Byrne EFX, Frigui S, Luchetti G, Sircar R, Nachtergaele S, Mydock-McGrane L, Krishnan K, Newstead S, Sansom MSP, Douglas CF, Diebold C, Rohatgi R (2022). Patched 1 regulates Smoothened by controlling sterol binding to its extracellular cysteine-rich domain. Sci. Adv 8, eabm5563. 10.1126/sciadv.abm5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, and Anderson KV (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87. 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 42.Warner JF, McCarthy AM, Morris RL, and McClay DR (2014). Hedgehog signaling requires motile cilia in the sea urchin. Mol. Biol. Evol 31, 18–22. 10.1093/molbev/mst176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yazawa S, Umesono Y, Hayashi T, Tarui H, and Agata K (2009). Planarian Hedgehog/Patched establishes anterior–posterior polarity by regulating Wnt signaling. Proceedings of the National Academy of Sciences 106, 22329–22334. 10.1073/pnas.0907464106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, and Anderson KV (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87. 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 45.Yoshida S, Aoki K, Fujiwara K, Nakakura T, Kawamura A, Yamada K, Ono M, Yogosawa S, and Yoshida K (2020). The novel ciliogenesis regulator DYRK2 governs Hedgehog signaling during mouse embryogenesis. eLife 9, e57381. 10.7554/eLife.57381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bergström A, Stanton DWG, Taron UH, Frantz L, Sinding M-HS, Ersmark E, Pfrengle S, Cassatt-Johnstone M, Lebrasseur O, Girdland-Flink L, et al. (2022). Grey wolf genomic history reveals a dual ancestry of dogs. Nature 607, 313–320. 10.1038/s41586-022-04824-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burga A, Wang W, Ben-David E, Wolf PC, Ramey AM, Verdugo C, Lyons K, Parker PG, and Kruglyak L (2017). A genetic signature of the evolution of loss of flight in the Galapagos cormorant. Science 356, eaal3345. 10.1126/science.aal3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamamoto Y, Stock DW, and Jeffery WR (2004). Hedgehog signalling controls eye degeneration in blind cavefish. Nature 431, 844–847. 10.1038/nature02864. [DOI] [PubMed] [Google Scholar]
- 49.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millán JL, Gamble V, and Harris PC (1995). The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet 10, 151–160. 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 50.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, et al. (1996). PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272, 1339–1342. 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 51.Su Q, Hu F, Ge X, Lei J, Yu S, Wang T, Zhou Q, Mei C, and Shi Y (2018). Structure of the human PKD1-PKD2 complex. Science 361, eaat9819. 10.1126/science.aat9819. [DOI] [PubMed] [Google Scholar]
- 52.Barr MM, DeModena J, Braun D, Nguyen CQ, Hall DH, and Sternberg PW (2001). The Caenorhabditis elegans autosomal dominant polycystic kidney disease gene homologs lov-1 and pkd-2 act in the same pathway. Curr. Biol 11, 1341–1346. 10.1016/s0960-9822(01)00423-7. [DOI] [PubMed] [Google Scholar]
- 53.Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, and Witman GB (2002). Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr. Biol 12, R378–380. 10.1016/s0960-9822(02)00877-1. [DOI] [PubMed] [Google Scholar]
- 54.Yoder BK, Hou X, and Guay-Woodford LM (2002). The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol 13, 2508–2516. 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 55.Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, and Yoder BK (2007). Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr. Biol 17, 1586–1594. 10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LSB, Somlo S, and Igarashi P (2003). Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. PNAS USA 100, 5286–5291. 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, and Cole DG (2000). Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J. Cell. Biol 151, 709–718. 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma M, Tian X, Igarashi P, Pazour GJ, and Somlo S (2013). Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet 45, 1004–1012. 10.1038/ng.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu A, Wang B, and Niswander LA (2005). Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development 132, 3103–3111. 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- 60.Dong K, Zhang C, Tian X, Coman D, Hyder F, Ma M, and Somlo S (2021). Renal plasticity revealed through reversal of polycystic kidney disease in mice. Nat. Genet 53, 1649–1663. 10.1038/s41588-021-00946-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Field S, Riley K-L, Grimes DT, Hilton H, Simon M, Powles-Glover N, Siggers P, Bogani D, Greenfield A, and Norris DP (2011). Pkd1l1 establishes left-right asymmetry and physically interacts with Pkd2. Development 138, 1131–1142. 10.1242/dev.058149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pennekamp P, Karcher C, Fischer A, Schweickert A, Skryabin B, Horst J, Blum M, and Dworniczak B (2002). The ion channel polycystin-2 is required for left-right axis determination in mice. Curr. Biol 12, 938–943. 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]
- 63.Yoshiba S, Shiratori H, Kuo IY, Kawasumi A, Shinohara K, Nonaka S, Asai Y, Sasaki G, Belo JA, Sasaki H, et al. (2012). Cilia at the node of mouse embryos sense fluid flow for left-right determination via Pkd2. Science 338, 226–231. 10.1126/science.1222538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu P, Lou X, Wingfield JL, Lin J, Nicastro D, and Lechtreck K (2020). Chlamydomonas PKD2 organizes mastigonemes, hair-like glycoprotein polymers on cilia. J. Cell Biol 219, e202001122. 10.1083/jcb.202001122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao Z, Ruden DM, and Lu X (2003). PKD2 cation channel is required for directional sperm movement and male fertility. Curr. Biol 13, 2175–2178. 10.1016/j.cub.2003.11.053. [DOI] [PubMed] [Google Scholar]
- 66.Watnick TJ, Jin Y, Matunis E, Kernan MJ, and Montell C (2003). A flagellar polycystin-2 homolog required for male fertility in Drosophila. Curr. Biol 13, 2179–2184. 10.1016/j.cub.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 67.Barr MM, and Sternberg PW (1999). A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature 401, 386–389. 10.1038/43913. [DOI] [PubMed] [Google Scholar]
- 68.Baylor DA, Lamb TD, and Yau KW (1979). Responses of retinal rods to single photons. J. Physiol 288, 613–634. [PMC free article] [PubMed] [Google Scholar]
- 69.Leinders-Zufall T, Lane AP, Puche AC, Ma W, Novotny MV, Shipley MT, and Zufall F (2000). Ultrasensitive pheromone detection by mammalian vomeronasal neurons. Nature 405, 792–796. 10.1038/35015572. [DOI] [PubMed] [Google Scholar]
- 70.Calvert PD, Govardovskii VI, Krasnoperova N, Anderson RE, Lem J, and Makino CL (2001). Membrane protein diffusion sets the speed of rod phototransduction. Nature 411, 90–95. [DOI] [PubMed] [Google Scholar]
- 71.Berbari NF, Johnson AD, Lewis JS, Askwith CC, and Mykytyn K (2008). Identification of Ciliary Localization Sequences within the Third Intracellular Loop of G Protein-coupled Receptors. MBoC 19, 1540–1547. 10.1091/mbc.e07-09-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brailov I, Bancila M, Brisorgueil MJ, Miquel MC, Hamon M, and Vergé D (2000). Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res. 872, 271–275. 10.1016/s0006-8993(00)02519-1. [DOI] [PubMed] [Google Scholar]
- 73.Händel M, Schulz S, Stanarius A, Schreff M, Erdtmann-Vourliotis M, Schmidt H, Wolf G, and Höllt V (1999). Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience 89, 909–926. 10.1016/s0306-4522(98)00354-6. [DOI] [PubMed] [Google Scholar]
- 74.Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, and Jackson PK (2013). The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 152, 210–223. 10.1016/j.cell.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 75.Siljee JE, Wang Y, Bernard AA, Ersoy BA, Zhang S, Marley A, Von Zastrow M, Reiter JF, and Vaisse C (2018). Subcellular localization of MC4R with ADCY3 at neuronal primary cilia underlies a common pathway for genetic predisposition to obesity. Nat. Genet 50, 180–185. 10.1038/s41588-017-0020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vaisse C, Reiter JF, and Berbari NF (2017). Cilia and Obesity. Cold Spring Harb. Perspect. Biol 9, a028217. 10.1101/cshperspect.a028217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stoetzel C, Laurier V, Davis EE, Muller J, Rix S, Badano JL, Leitch CC, Salem N, Chouery E, Corbani S, et al. (2006). BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet 38, 521–524. 10.1038/ng1771. [DOI] [PubMed] [Google Scholar]
- 78.Vaisse C, Clement K, Guy-Grand B, and Froguel P (1998). A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat. Genet 20, 113–114. 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- 79.Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, and O’Rahilly S (1998). A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat. Genet 20, 111–112. 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- 80.Grarup N, Moltke I, Andersen MK, Dalby M, Vitting-Seerup K, Kern T, Mahendran Y, Jørsboe E, Larsen CVL, Dahl-Petersen IK, et al. (2018). Loss-of-function variants in ADCY3 increase risk of obesity and type 2 diabetes. Nat. Genet 50, 172–174. 10.1038/s41588-017-0022-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saeed S, Bonnefond A, Tamanini F, Mirza MU, Manzoor J, Janjua QM, Din SM, Gaitan J, Milochau A, Durand E, et al. (2018). Loss-of-function mutations in ADCY3 cause monogenic severe obesity. Nat. Genet 50, 175–179. 10.1038/s41588-017-0023-6. [DOI] [PubMed] [Google Scholar]
- 82.Wang Z, Li V, Chan GCK, Phan T, Nudelman AS, Xia Z, and Storm DR (2009). Adult Type 3 Adenylyl Cyclase–Deficient Mice Are Obese. PLoS One 4, e6979. 10.1371/journal.pone.0006979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y, Bernard A, Comblain F, Yue X, Paillart C, Zhang S, Reiter JF, and Vaisse C (2021). Melanocortin 4 receptor signals at the neuronal primary cilium to control food intake and body weight. J. Clin. Invest 131, e142064, 142064. 10.1172/JCI142064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hilgendorf KI, Johnson CT, Mezger A, Rice SL, Norris AM, Demeter J, Greenleaf WJ, Reiter JF, Kopinke D, and Jackson PK (2019). Omega-3 Fatty Acids Activate Ciliary FFAR4 to Control Adipogenesis. Cell 179, 1289–1305.e21. 10.1016/j.cell.2019.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kopinke D, Roberson EC, and Reiter JF (2017). Ciliary Hedgehog Signaling Restricts Injury-Induced Adipogenesis. Cell 170, 340–351.e12. 10.1016/j.cell.2017.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gotoh C, Hong Y-H, Iga T, Hishikawa D, Suzuki Y, Song S-H, Choi K-C, Adachi T, Hirasawa A, Tsujimoto G, et al. (2007). The regulation of adipogenesis through GPR120. Biochem. Biophys. Res. Commun 354, 591–597. 10.1016/j.bbrc.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 87.Miyauchi S, Hirasawa A, Iga T, Liu N, Itsubo C, Sadakane K, Hara T, and Tsujimoto G (2009). Distribution and regulation of protein expression of the free fatty acid receptor GPR120. Naunyn Schmiedebergs Arch. Pharmacol 379, 427–434. 10.1007/s00210-008-0390-8. [DOI] [PubMed] [Google Scholar]
- 88.Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, and Olefsky JM (2010). GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698. 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sutherland EW, and Robison GA (1969). The role of cyclic AMP in the control of carbohydrate metabolism. Diabetes 18, 797–819. 10.2337/diab.18.12.797. [DOI] [PubMed] [Google Scholar]
- 90.Sutherland EW, Robison GA, and Butcher RW (1968). Some Aspects of the Biological Role of Adenosine 3’,5’-monophosphate (Cyclic AMP). Circulation 37, 279–306. 10.1161/01.CIR.37.2.279. [DOI] [Google Scholar]
- 91.Brunton LL, Hayes JS, and Mayer SE (1981). Functional compartmentation of cyclic AMP and protein kinase in heart. Adv. Cyclic Nucleotide Res 14, 391–397. [PubMed] [Google Scholar]
- 92.Buxton IL, and Brunton LL (1983). Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem 258, 10233–10239. [PubMed] [Google Scholar]
- 93.Stierl M, Stumpf P, Udwari D, Gueta R, Hagedorn R, Losi A, Gärtner W, Petereit L, Efetova M, Schwarzel M, et al. (2011). Light modulation of cellular cAMP by a small bacterial photoactivated adenylyl cyclase, bPAC, of the soil bacterium Beggiatoa. J. Biol. Chem 286, 1181–1188. 10.1074/jbc.M110.185496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Truong ME, Bilekova S, Choksi SP, Li W, Bugaj LJ, Xu K, and Reiter JF (2021). Vertebrate cells differentially interpret ciliary and extraciliary cAMP. Cell 184, 2911–2926.e18. 10.1016/j.cell.2021.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Marley A, Choy RW-Y, and Zastrow M. von (2013). GPR88 Reveals a Discrete Function of Primary Cilia as Selective Insulators of GPCR Cross-Talk. PLoS One 8, e70857. 10.1371/journal.pone.0070857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Delling M, DeCaen PG, Doerner JF, Febvay S, and Clapham DE (2013). Primary cilia are specialized calcium signaling organelles. Nature 504, 311–314. 10.1038/nature12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Trötschel C, Hamzeh H, Alvarez L, Pascal R, Lavryk F, Bönigk W, Körschen HG, Müller A, Poetsch A, Rennhack A, et al. (2020). Absolute proteomic quantification reveals design principles of sperm flagellar chemosensation. EMBO J 39, e102723. 10.15252/embj.2019102723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Buffone MG ed. (2016). Sperm Acrosome Biogenesis and Function During Fertilization (Springer International Publishing; ) 10.1007/978-3-319-30567-7. [DOI] [Google Scholar]
- 99.Wood CR, Huang K, Diener DR, and Rosenbaum JL (2013). The Cilium Secretes Bioactive Ectosomes. Curr. Biol 23, 906–911. 10.1016/j.cub.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dentler W (2013). A Role for the Membrane in Regulating Chlamydomonas Flagellar Length. PLS One 8, e53366. 10.1371/journal.pone.0053366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cao M, Ning J, Hernandez-Lara CI, Belzile O, Wang Q, Dutcher SK, Liu Y, and Snell WJ (2015). Uni-directional ciliary membrane protein trafficking by a cytoplasmic retrograde IFT motor and ciliary ectosome shedding. eLife 4, e05242. 10.7554/eLife.05242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang J, Silva M, Haas LA, Morsci NS, Nguyen KCQ, Hall DH, and Barr MM (2014). C. elegans ciliated sensory neurons release extracellular vesicles that function in animal communication. Curr. Biol 24, 519–525. 10.1016/j.cub.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Akella JS, Carter SP, Nguyen K, Tsiropoulou S, Moran AL, Silva M, Rizvi F, Kennedy BN, Hall DH, Barr MM, Blacque OE (2020). Ciliary Rab28 and the BBSome negatively regulate extracellular vesicle shedding. eLife 9, e50580. 10.7554/eLife.50580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jespersgaard C, Hey AB, Ilginis T, Hjortshøj TD, Fang M, Bertelsen M, Bech N, Jensen H, Larsen LJ, Tümer Z, Rosenberg T, Brøndum-Nielsen K, Møller LB, Grønskov K (2020). A Missense Mutation in RAB28 in a Family with Cone-Rod Dystrophy and Postaxial Polydactyly Prevents Localization of RAB28 to the Primary Cilium. Invest. Ophthalmol. Vis. Sci 61, 29. 10.1167/iovs.61.2.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baehr W, Wu SM, Bird AC, and Palczewski K (2003). The retinoid cycle and retina disease. Vision Res. 43, 2957–2958. 10.1016/j.visres.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 106.Spencer WJ, Lewis TR, Phan S, Cady MA, Serebrovskaya EO, Schneider NF, Kim K-Y, Cameron LA, Skiba NP, Ellisman MH, Arshavsky VY (2019). Photoreceptor disc membranes are formed through an Arp2/3-dependent lamellipodium-like mechanism. PNAS USA 116, 27043–27052. 10.1073/pnas.1913518117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Salinas RY, Pearring JN, Ding J-D, Spencer WJ, Hao Y, and Arshavsky VY (2017). Photoreceptor discs form through peripherin-dependent suppression of ciliary ectosome release. J. Cell Biol 216, 1489–1499. 10.1083/jcb.201608081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Spencer WJ, Lewis TR, Pearring JN, and Arshavsky VY (2020). Photoreceptor Discs: Built Like Ectosomes. Trends Cell Biol. 30, 904–915. 10.1016/j.tcb.2020.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nager AR, Goldstein JS, Herranz-Pérez V, Portran D, Ye F, Garcia-Verdugo JM, and Nachury MV (2017). An Actin Network Dispatches Ciliary GPCRs into Extracellular Vesicles to Modulate Signaling. Cell 168, 252–263.e14. 10.1016/j.cell.2016.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ojeda Naharros I, and Nachury MV (2022). Shedding of ciliary vesicles at a glance. J. Cell Sci 135, jcs246553. 10.1242/jcs.246553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Berbari NF, Lewis JS, Bishop GA, Askwith CC, and Mykytyn K (2008). Bardet–Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. PNAS 105, 4242–4246. 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Domire JS, Green JA, Lee KG, Johnson AD, Askwith CC, and Mykytyn K (2011). Dopamine receptor 1 localizes to neuronal cilia in a dynamic process that requires the Bardet-Biedl syndrome proteins. Cell Mol. Life Sci 68, 2951–2960. 10.1007/s00018-010-0603-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stubbs T, Koemeter-Cox A, Bingman JI, Zhao F, Kalyanasundaram A, Rowland LA, Periasamy M, Carter CS, Sheffield VC, Askwith CC, et al. (2022). Disruption of dopamine receptor 1 localization to primary cilia impairs signaling in striatal neurons. J. Neurosci 42, 6692–6705. 10.1523/JNEUROSCI.0497-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bangs FK, Schrode N, Hadjantonakis A-K, and Anderson KV (2015). Lineage specificity of primary cilia in the mouse embryo. Nat. Cell Biol 17, 113–122. 10.1038/ncb3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wong SY, Seol AD, So P-L, Ermilov AN, Bichakjian CK, Epstein EH, Dlugosz AA, and Reiter JF (2009). Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat. Med 15, 1055–1061. 10.1038/nm.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Saqui-Salces M, Dowdle WE, Reiter JF, and Merchant JL (2012). A high-fat diet regulates gastrin and acid secretion through primary cilia. FASEB J 26, 3127–3139. 10.1096/fj.11-197426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xie C, Abrams SR, Herranz-Pérez V, García-Verdugo JM, and Reiter JF (2021). Endoderm development requires centrioles to restrain p53-mediated apoptosis in the absence of ERK activity. Dev. Cell 56, 3334–3348.e6. 10.1016/j.devcel.2021.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, and Golemis EA (2007). HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129, 1351–1363. 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cohen J, Adoutte A, Grandchamp S, Houdebine LM, Beisson J (1982). Immunocytochemical study of microtubular structures throughout the cell cycle of paramecium. Biol. Cell 44, 35–44. [Google Scholar]
- 120.Debec A, Sullivan W, and Bettencourt-Dias M (2010). Centrioles: active players or passengers during mitosis? Cell Mol. Life Sci 67, 2173–2194. 10.1007/s00018-010-0323-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jumat MR, Yan Y, Ravi LI, Wong P, Huong TN, Li C, Tan BH, Wang DY, and Sugrue RJ (2015). Morphogenesis of respiratory syncytial virus in human primary nasal ciliated epithelial cells occurs at surface membrane microdomains that are distinct from cilia. Virology 484, 395–411. 10.1016/j.virol.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 122.Smith CM, Kulkarni H, Radhakrishnan P, Rutman A, Bankart MJ, Williams G, Hirst RA, Easton AJ, Andrew PW, and O’Callaghan C (2014). Ciliary dyskinesia is an early feature of respiratory syncytial virus infection. Eur. Respir. J 43, 485–496. 10.1183/09031936.00205312. [DOI] [PubMed] [Google Scholar]
- 123.Ampofo K, Bender J, Sheng X, Korgenski K, Daly J, Pavia AT, and Byington CL (2008). Seasonal invasive pneumococcal disease in children: role of preceding respiratory viral infection. Pediatrics 122, 229–237. 10.1542/peds.2007-3192. [DOI] [PubMed] [Google Scholar]
- 124.Zhang L, Bukreyev A, Thompson CI, Watson B, Peeples ME, Collins PL, and Pickles RJ (2005). Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J. Virol 79, 1113–1124. 10.1128/JVI.79.2.1113-1124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rosenblum EB, Voyles J, Poorten TJ, and Stajich JE (2010). The Deadly Chytrid Fungus: A Story of an Emerging Pathogen. PLoS Pathog. 6, e1000550. 10.1371/journal.ppat.1000550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Medina EM, Robinson KA, Bellingham-Johnstun K, Ianiri G, Laplante C, Fritz-Laylin LK, and Buchler NE (2020). Genetic transformation of Spizellomyces punctatus, a resource for studying chytrid biology and evolutionary cell biology. eLife 9, e52741. 10.7554/eLife.52741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Robinson KA, Prostak SM, Campbell Grant EH, and Fritz-Laylin LK (2022). Amphibian mucus triggers a developmental transition in the frog-killing chytrid fungus. Curr. Biol 32, 2765–2771.e4. 10.1016/j.cub.2022.04.006. [DOI] [PubMed] [Google Scholar]
- 128.Swafford AJM, Hussey SP, and Fritz-Laylin LK (2020). High-efficiency electroporation of chytrid fungi. Sci. Rep 10, 15145. 10.1038/s41598-020-71618-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fritz-Laylin LK, Ginger ML, Walsh C, Dawson SC, and Fulton C (2011). The Naegleria genome: a free-living microbial eukaryote lends unique insights into core eukaryotic cell biology. Res. Microbiol 162, 607–618. 10.1016/j.resmic.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fulton C (1970). Transformation of Tetramitus Amebae into Flagellates. Science 167, 1269–1270. 10.1126/science.167.3922.1269. [DOI] [PubMed] [Google Scholar]
- 131.Bunting M (1922). A Preliminary Note on Tetramitus, a Stage in the Life Cycle of a Coprozoic Amoeba. PNAS 8, 294–300. 10.1073/pnas.8.10.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sinden R, Talman A, Marques S, Wass M, and Sternberg M (2010). The flagellum in malarial parasites. COMICR 13, 491–500. 10.1016/j.mib.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 133.Githui EK, De Villiers EP, and McArthur AG (2009). Plasmodium possesses dynein light chain classes that are unique and conserved across species. Infect. Genet. Evol 9, 337–343. 10.1016/j.meegid.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 134.Brunet T, Albert M, Roman W, Coyle MC, Spitzer DC, and King N (2021). A flagellate-to-amoeboid switch in the closest living relatives of animals. eLife 10, e61037. 10.7554/eLife.61037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Swoboda P, Adler HT, and Thomas JH (2000). The RFX-type transcription factor DAF-19 regulates sensory neuron cilium formation in C. elegans. Mol. Cell 5, 411–421. 10.1016/s1097-2765(00)80436-0. [DOI] [PubMed] [Google Scholar]
- 136.Ashique AM, Choe Y, Karlen M, May SR, Phamluong K, Solloway MJ, Ericson J, and Peterson AS (2009). The Rfx4 transcription factor modulates Shh signaling by regional control of ciliogenesis. Sci. Signal 2, ra70. 10.1126/scisignal.2000602. [DOI] [PubMed] [Google Scholar]
- 137.Bonnafe E, Touka M, AitLounis A, Baas D, Barras E, Ucla C, Moreau A, Flamant F, Dubruille R, Couble P, et al. (2004). The transcription factor RFX3 directs nodal cilium development and left-right asymmetry specification. Mol. Cell Biol 24, 4417–4427. 10.1128/MCB.24.10.4417-4427.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kistler WS, Baas D, Lemeille S, Paschaki M, Seguin-Estevez Q, Barras E, Ma W, Duteyrat J-L, Morlé L, Durand B, et al. (2015). RFX2 Is a Major Transcriptional Regulator of Spermiogenesis. PLoS Genet. 11, e1005368. 10.1371/journal.pgen.1005368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Coyle MC, Tajima AM, Leon F, Choksi SP, Yang A, Espinoza S, Hughes TR, Reiter JF, Booth DS, and King N (2022). An RFX transcription factor regulated ciliogenesis in the progenitors of choanoflagellates and animals. bioRxiv. 10.1101/2022.11.11.515474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yu X, Ng CP, Habacher H, and Roy S (2008). Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat. Genet 40, 1445–1453. 10.1038/ng.263. [DOI] [PubMed] [Google Scholar]
- 141.Choksi SP, Babu D, Lau D, Yu X, and Roy S (2014). Systematic discovery of novel ciliary genes through functional genomics in the zebrafish. Development 141, 3410–3419. 10.1242/dev.108209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Vij S, Rink JC, Ho HK, Babu D, Eitel M, Narasimhan V, Tiku V, Westbrook J, Schierwater B, and Roy S (2012). Evolutionarily Ancient Association of the FoxJ1 Transcription Factor with the Motile Ciliogenic Program. PLoS Genet. 8, e1003019. 10.1371/journal.pgen.1003019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Piasecki BP, Burghoorn J, and Swoboda P (2010). Regulatory Factor X (RFX)-mediated transcriptional rewiring of ciliary genes in animals. PNAS 107, 12969–12974. 10.1073/pnas.0914241107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Berdon WE, and Willi U (2004). Situs inversus, bronchiectasis, and sinusitis and its relation to immotile cilia: history of the diseases and their discoverers-Manes Kartagener and Bjorn Afzelius. Pediatr. Radiol 34, 38–42. 10.1007/s00247-003-1072-9. [DOI] [PubMed] [Google Scholar]
- 145.Afzelius BA (1976). A human syndrome caused by immotile cilia. Science 193, 317–319. 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 146.Eliasson R, Mossberg B, Camner P, and Afzelius BA (1977). The immotile-cilia syndrome. A congenital ciliary abnormality as an etiologic factor in chronic airway infections and male sterility. N. Engl. J. Med 297, 1–6. 10.1056/NEJM197707072970101. [DOI] [PubMed] [Google Scholar]
- 147.Liu P, and Lechtreck KF (2018). The Bardet–Biedl syndrome protein complex is an adapter expanding the cargo range of intraflagellar transport trains for ciliary export. PNAS 115, E934–E943. 10.1073/pnas.1713226115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ye F, Nager AR, and Nachury MV (2018). BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J. Cell Biol 217, 1847–1868. 10.1083/jcb.201709041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368, eaat5314. 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Han Y-G, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, and Alvarez-Buylla A (2009). Dual and opposing roles of primary cilia in medulloblastoma development. Nat. Med 15, 1062–1065. 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Alessi DR, and Sammler E (2018). LRRK2 kinase in Parkinson’s disease. Science 360, 36–37. 10.1126/science.aar5683. [DOI] [PubMed] [Google Scholar]
- 152.Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, Tonelli F, Martinez TN, Lorentzen E, Pfeffer SR, et al. (2017). Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife 6, e31012. 10.7554/eLife.31012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dhekne HS, Yanatori I, Gomez RC, Tonelli F, Diez F, Schüle B, Steger M, Alessi DR, and Pfeffer SR (2018). A pathway for Parkinson’s Disease LRRK2 kinase to block primary cilia and Sonic hedgehog signaling in the brain. eLife 7, e40202. 10.7554/eLife.40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Purlyte E, Dhekne HS, Sarhan AR, Gomez R, Lis P, Wightman M, Martinez TN, Tonelli F, Pfeffer SR, and Alessi DR (2018). Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J 37, 1–18. 10.15252/embj.201798099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sobu Y, Wawro PS, Dhekne HS, Yeshaw WM, & Pfeffer SR (2021). Pathogenic LRRK2 regulates ciliation probability upstream of tau tubulin kinase 2 via Rab10 and RILPL1 proteins. Proceedings of the National Academy of Sciences of the United States of America, 118(10), e2005894118. 10.1073/pnas.2005894118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Khan SS, Sobu Y, Dhekne HS, Tonelli F, Berndsen K, Alessi DR, and Pfeffer SR (2021). Pathogenic LRRK2 control of primary cilia and Hedgehog signaling in neurons and astrocytes of mouse brain. eLife 10, e67900. 10.7554/eLife.67900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gonzalez-Reyes LE, Verbitsky M, Blesa J, Jackson-Lewis V, Paredes D, Tillack K, Phani S, Kramer ER, Przedborski S, and Kottmann AH (2012). Sonic Hedgehog Maintains Cellular and Neurochemical Homeostasis in the Adult Nigrostriatal Circuit. Neuron 75, 306–319. 10.1016/j.neuron.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Johnston DS (2015). The Renaissance of Developmental Biology. PLOS Biology 13, e1002149. 10.1371/journal.pbio.1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Stern CD (2022). Reflections on the past, present and future of developmental biology. Developmental Biology 488, 30–34. 10.1016/j.ydbio.2022.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zon L (2019). Improving the visibility of developmental biology: time for induction and specification. Development 146, dev174631. 10.1242/dev.174631. [DOI] [PubMed] [Google Scholar]
- 161.Bilder D (2016). The Maturation of Development. Dev. Cell 38, 569–570. 10.1016/j.devcel.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Walck-Shannon E, and Hardin J (2014). Cell intercalation from top to bottom. Nat Rev Mol. Cell Biol 15, 34–48. 10.1038/nrm3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hayashi T, and Carthew RW (2004). Surface mechanics mediate pattern formation in the developing retina. Nature 431, 647–652. 10.1038/nature02952. [DOI] [PubMed] [Google Scholar]
- 164.Burki F, Roger AJ, Brown MW, and Simpson AGB (2020). The New Tree of Eukaryotes. Trends Ecol. Evol 35, 43–55. 10.1016/j.tree.2019.08.008. [DOI] [PubMed] [Google Scholar]