

Abstract
Background:
Clinical, molecular, and genetic epidemiology studies displayed remarkable differences between ever- and never-smoking lung cancer.
Methods:
We conducted a stratified multi-population (European, East Asian, and African descent) association study on 44,823 ever-smokers and 20,074 never-smokers to identify novel variants that were missed in the non-stratified analysis. Functional analysis including expression quantitative trait loci (eQTL) colocalization and DNA damage assays, and annotation studies were conducted to evaluate the functional roles of the variants. We further evaluated the impact of smoking quantity on lung cancer risk for the variants associated with ever-smoking lung cancer.
Results:
Five novel independent loci, GABRA4, intergenic region 12q24.33, LRRC4C, LINC01088, and LCNL1 were identified with the association at two or three populations (P < 5 × 10−8). Further functional analysis provided multiple lines of evidence suggesting the variants affect lung cancer risk through excessive DNA damage (GABRA4) or cis-regulation of gene expression (LCNL1). The risk of variants from 12 independent regions, including the well-known CHRNA5, associated with ever-smoking lung cancer was evaluated for never-smokers, light-smokers (packyear ≤ 20), and moderate-to-heavy-smokers (packyear > 20). Different risk patterns were observed for the variants among the different groups by smoking behavior.
Conclusions:
We identified novel variants associated with lung cancer in only ever- or never-smoking groups that were missed by prior main-effect association studies.
Impact:
Our study highlights the genetic heterogeneity between ever- and never-smoking lung cancer and provides etiologic insights into the complicated genetic architecture of this deadly cancer.
Introduction
Genome-wide association studies (GWAS) have been fruitful in the past two decades and more than 50 susceptibility loci have been identified in lung cancer (1). However, previously identified loci only account for a limited proportion of heritability, implying additional susceptibility loci that are not yet revealed. The missing variants may include low allele frequency variants [minor allele frequency (MAF) < 0.01] and those that affect lung cancer risk through genetic/environmental interactions that cannot be disclosed by regular main-effect association studies (2, 3). Smoking is the leading environmental risk factor contributing to lung cancer and >80% of patients with lung cancer have a history of tobacco smoking (4). Lung cancer in never-smokers, although much less common compared with lung cancer in ever-smokers, is still estimated to be the seventh leading cause of cancer-related deaths (5). Remarkable differences have been identified in both clinical and molecular epidemiology studies between ever- and never-smoking lung cancer (6). Quite a few genetic variants have been reported in ever-smoking lung cancer such as the well-known CHRNA5/A3/B4 gene region, TP63, TERT, and CYP2A6 genes (7, 8). However, fewer studies have been focused on identifying genetic loci within smoking behavior subgroups. Some susceptibility loci have also been identified in never-smoking lung cancer. For example, VTI1A and ACVR1B were found to be associated with lung cancer in Chinese and European never-smoking women (9, 10). Variants affecting the expression of hTERT and TP63 have also been associated with lung cancer in never-smokers (11). These findings suggest the heterogeneity in genetic architecture between ever- and never-smoking lung cancer.
To date, the majority of GWASs have been conducted in European (EUR) and East Asian populations (EAS), while African descent (AFR) populations have been under-represented. A multi-population GWAS including AFR populations will help clarify the varying effects of smoking on the risk for lung cancer among the major ancestral populations, identify novel variants with effects across multiple populations, and evaluate the heterogeneity in lung cancer risk across ancestral groups.
One challenge in GWAS is to delineate the relationship between the genetic variants and the biological mechanisms underlying the statistical findings. Various functional annotation tools have been developed to infer the functional role of genetic findings such as CADD and RegulomeDB (12–14). Expression quantitative trait loci (eQTL) analysis has also been commonly used in GWAS to infer the cis-regulation of nearby gene expression for the variants (15). Recently, DNA damage assays have also been applied in lung cancer GWAS to characterize candidate genes as lung cancer risk genes are enriched in the DNA damageome, proteins that can result in high DNA damage when overproduced (16, 17). For example, significantly increased DNA damage levels were observed in CHEK2, ATM, POMC, MLNR, MME, and PPIL6, genes that were found to be associated with lung cancer, in DNA damage assay, suggesting that genetic variants may promote lung cancer through DNA damage regulation (16, 17). An integrative functional analysis has the potential to provide multi-layered evidence for a more comprehensive understanding of the GWAS findings.
In 2022, we performed a multi-population GWAS, including EUR, EAS, and AFR populations, and identified five novel susceptibility loci associated with lung cancer (16). Leveraging this rich resource, we performed a comprehensive study of genetic variants associated with ever- and never-smoking lung cancer aiming to: (i) identify novel variants involved in only ever- or never-smoking groups that were missed by prior regular GWAS studies; (ii) explore the functional roles of the identified variants; (iii) investigate the impact of tobacco smoking on risk effect of the genetic variants associated with ever-smoking lung cancer.
Materials and Methods
Genotype data
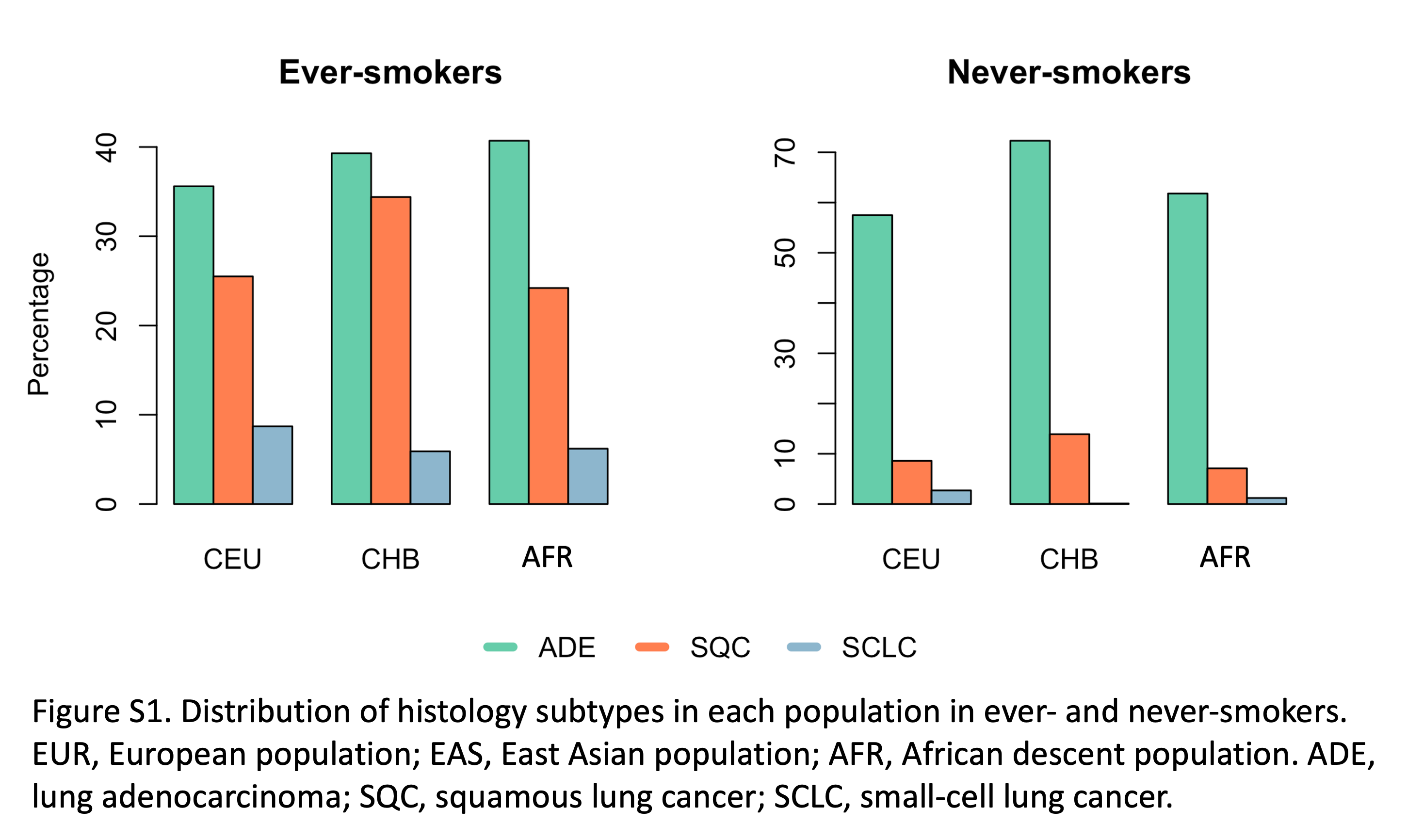
The imputed genotypes from the INTEGRAL (Integrative Analysis of Lung Cancer Etiology and Risk)-ILCCO (International Lung Cancer Consortium) lung cancer consortium were applied in this study [reference panel HRC (r1.1)]. Detailed information about genotype imputation and data quality control can be found in our previous publication in 2022 (16). About 9,000,000 high-quality imputed SNPs (information score ≥ 0.8) from a total of 64,897 individuals, including 44,823 ever-smokers and 20,074 never-smokers were analyzed in the study. The individuals came from 10 studies with diverse ancestry populations including EUR, EAS, and AFR (Table 1; Supplementary Table S1), and about 2,000 ancestry-informative markers were used to infer the ancestry information of the individuals. A total of 72.1% of the individuals are inferred with European ancestry (EUR, N = 46,786), compared with 19.1% with Asian ancestry (EAS, N = 12,423) and 8.8% with African ancestry (AFR, N = 5,688; ref. 16). About 35%–40% of the patients with ever-smoking lung cancer were diagnosed with lung adenocarcinoma (ADE) across the populations, and 25%–34% of the patients were diagnosed with squamous carcinoma (SQC; Supplementary Fig. S1). ADE is the predominant subtype in never-smoking patients and accounts for >57% of patients in all the populations. Small cell lung cancer (SCLC) is much less common compared with ADE and SQC in ever-smokers (9.79%) and very few cases occur in never-smokers (0.54%).
Table 1.
Sample size distribution from each population in the study.
| EUR | EAS | AFR | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Strata | CONTROL | CASE | Total | CONTROL | CASE | Total | CONTROL | CASE | Total |
| Ever-smokers | |||||||||
| Overall | 16,165 | 22,018 | 38,183 | 1,032 | 1,495 | 2,527 | 2,309 | 1,804 | 4,113 |
| ADE | 16,165 | 7,838 | 24,003 | 1,032 | 586 | 1,618 | 2,309 | 734 | 3,043 |
| SQC | 16,165 | 5,619 | 21,784 | 1,032 | 514 | 1,546 | 2,309 | 436 | 2,745 |
| SCLC | 16,165 | 1,919 | 18,084 | 1,032 | 88 | 1,120 | 2,309 | 111 | 2,420 |
| Never-smokers | |||||||||
| Overall | 6,396 | 2,207 | 8,603 | 4,335 | 5,561 | 9,896 | 1,405 | 170 | 1,575 |
| ADE | 6,396 | 1,268 | 7,664 | 4,335 | 4,019 | 8,354 | 1,405 | 105 | 1,510 |
| SQC | 6,396 | 189 | 6,585 | 4,335 | 771 | 5,106 | 1,405 | 12 | 1,417 |
| SCLC | 6,396 | 60 | 6,456 | 4,335 | 4 | 4,339 | 1,405 | 2 | 1,407 |
Note: Sample size of each strata is displayed in the table.
Abbreviations: ADE, lung adenocarcinoma; AFR, African population; EAS, East Asian population; EUR, European population; Overall, overall lung cancer; SCLC, small cell lung cancer; SQC, squamous lung cancer.
Association analysis of lung cancer in ever- and never-smokers
Smoking status was self-reported and was categorized into never-smokers and ever-smokers (including both current smokers and former smokers). We conducted separate GWAS in the ever- and never-smoking groups for EUR, EAS, and AFR populations and then performed a meta-analysis to combine information from each population separately according to the ever- and never-smoking strata. In addition, we adjusted for study sites in the analysis by including a categorical variable for each site along with conducting a principal components analysis to allow for residual effects of population structure, finding through univariate χ2 tests that the first three principal components were significantly associated with disease status. Therefore, we also adjusted for these principal components in the analysis. Significant SNPs were selected on the basis of two criteria: (i) with the same direction of risk effect and P value < 0.1 in two or three populations (so the association evidence comes from at least two populations); (ii) and with a joint P value < 5 × 10−8 in meta-analysis. For the significant variants with low allele frequency (MAF < 0.01), we further validated the signals with Firth logistic regression, a method designed for rare variants association test to reduce small-sample bias in regular logistic regression (18). The variants that were not significant in the Firth test were removed from the final report. The stratified GWAS analysis was conducted in overall lung cancer as well as ADE, SQC, and SCLC subtypes. The genomic inflation factor (the lambda value) was calculated to examine whether there was an inflated type I error rate in association analysis. The lambda value adjusted by sample size was also calculated using the formula:
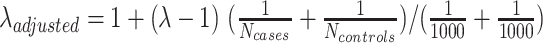 . PLINK 1.07 was used for GWAS and meta-analysis. R-4.0.2 and R package logistic 1.2 were applied for Firth logistic regression analysis.
. PLINK 1.07 was used for GWAS and meta-analysis. R-4.0.2 and R package logistic 1.2 were applied for Firth logistic regression analysis.
For the variants/regions that were significantly associated with ever-smoking lung cancer, including the novel variants identified in this study and the variants identified from prior GWAS studies, we selected the most significant variant from each region and further examined their risk effect in never-smokers, light-smokers [packyear (packyr) ≤ 20], and moderate-to-heavy-smokers (MtoH-smokers; packyr > 20) trying to explore whether there are different risk patterns among the variants across different smoking subgroups. We adjusted for the first three principal components and study sites in the analysis.
Functional annotation analysis
The web-based tool RegulomeDB was used to infer the regulatory potential of significant variants by integrating high-throughput, experimental datasets from ENCODE and other sources (13). For each variant, it calculates a probability score indicating their likelihood of being a regulatory element or a sequence motif. Another web server, RBPmap, was used to identify potential RNA-binding protein (RBP) binding motifs in all transcripts overlapping with alternative and reference alleles (14). A sequence of 61 bp, including 30 bp upstream/downstream of the candidate SNP was provided as the input for motif search. Transcription factor binding motifs or RBP binding motifs with P value <0.05 for either the reference or the alternative allele were identified as putative binding sites.
GWAS-eQTL colocalization analysis
Genotype and gene expression rpkm (reads per kilobase million) data from 377 lung tissue samples with EUR ancestry were downloaded from GTEx (phs000424.GTEx. v7.p2). The average rpkm for the gene was used if there were duplicated samples and individuals with rpkm < 0.25 were removed from the analysis. The SNPs from within ± 250 kb of each candidate variant were retrieved from both GTEx and GWAS data. The z-score from the association between genotype and gene expression data (GTEx) was plotted against those from the GWAS analysis for each retrieved SNP to examine the correlation between eQTL and GWAS studies. The eQTL analysis was conducted using program R-4.0.2.
Human cell line, reagents, and DNA damage assays
The MRC5-SV40 human lung fibroblast cell line (male, SV40-immortalized, source: Dr. Stephen P. Jackson Lab via Dr. Kyle Miller) was maintained in DMEM, high glucose medium (Gibco, #11965118) containing 10% FBS (Gibco, #10438034), 2 mmol/L l-glutamine, 100 μg/mL streptomycin, and 100 μg/mL penicillin (Gibco, #10378016). The cell line was authenticated via short tandem repeat analysis (ATCC, July 2018) immediately before freezing in liquid nitrogen and was routinely checked for Mycoplasma contamination (ABM, G238). The passage number was limited to a maximum of 30. Gating entry clones for each of the candidate genes, such as GABRA4 (IOH27675) and NF2F1 (IOH3781), were acquired from the Kenneth Scott cDNA library at Baylor College of Medicine. They were then further subcloned into an N-terminal EmGFP tagged vector (pcDNA6.2/N-EmGFP-DEST, Invitrogen), using Gateway LR Clonase II Enzyme Mix (Invitrogen, #11791020). The previously cloned EmGFP-Tubulin was used as a control (PMID: 30633903).
Plasmid transfections were performed using GenJet In Vitro DNA Transfection Reagent Ver. II (SignaGen, #SL100489). To further characterize the candidate genes, flow-cytometric DNA damage assays were performed as previously described in the MRC5-SV40 cell line with transient candidate gene overexpression (19, 20). Briefly, MRC5-SV40 human lung fibroblasts cells were fixed, permeabilized, and, stained with γH2AX antibody (#05-636, Sigma), then samples were measured by a BD LSRFortessa flow cytometer and analyzed using the FlowJo software. For overproduction experiments, cells with mock transfection were used to set the threshold gating to determine the percentage of GFP− and γH2AX− cells, with 0.5% of control cells gated as the damage threshold as validated previously. The DNA damage ratio caused by protein overproduction is defined by (Q2/Q3)/(Q1/Q4), where Q2 is the number of transfected damage-positive cells; Q3 is the number of transfected damage-negative cells; Q1 is the number of untransfected damage positive cells, and Q4 is the number of untransfected damage-negative cells.
DNA damage assays with benzo[a]pyrene (Bap; #48564, Sigma) were carried out under similar conditions that do not involve exogenous agent exposure. Briefly, BaP (8 μmol/L) was added when cells were transfected with plasmids, and incubated for 72 hours, followed by flow-cytometric DNA damage assays as described above.
Data availability
The following publicly available datasets were used in this work: Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial, phs000093.v2.p2; FLCCA study, phs000716.v1.p1; EAGLE study, phs000336.v1.p1; NCI study of African-Americans, phs001210.v1.p1; German, SLRI, IARC, and MD Anderson Cancer Center studies, phs000876.v2.p1; Oncoarray study, phs001273.v3.p2; imputed Oncoarray study using HRC reference panel, phs001273.v4.p2; Affymetrix study, phs001681. v1.p1. The eQTL data from GTEx was obtained from https://gtexportal. org/home/datasets (phs000424.GTEx.v7. p2; ref. 16).
Materials and correspondence
Correspondence should be addressed to Y. Li or C.I Amos. Material requests should be addressed to C.I Amos.
Results
Genetic variants associated with ever- or never-smoking lung cancer
Genome-wide association analyses were conducted in ever- and never-smokers in overall lung cancer as well as other lung cancer subtypes. Figure 1A displays the Manhattan plots of the signals from the stratified analysis. QQ-plots of the P values from the association analysis and adjusted genomic inflation values (lambda values) by sample size displayed no inflated type I error rate in the analysis (Fig. 1A right). We identified a few significant variants in ever- and never-smoking lung cancer, including the significant variants from known genes, such as AK5, TP63, TERT, etc., which are summarized in Supplementary Table S2 (labeled in black in Fig. 1A). Table 2 lists the risk variants with association evidence from only ever- or never-smoking individuals (not from both groups) including the well-known 15q25.1 region, which only shows associations in ever-smokers. Six candidate variants were identified in the study, but one of the variants, rs7985487, was removed from the final report due to not reaching genome-wide significance in the Firth test check despite being associated with lung cancer in the EUR and AFR population (P_firth = 4.00 × 10−7; Supplementary Table S3). In the end, five variants, including two variants associated with ever-smoking lung cancer, rs62303696 from GABRA4 and rs58778970 from intergenic region 12q24.33; and three variants from never-smoking lung cancer, rs4756620 from LRRC4C, rs1383429 from LINC01088 and rs968516 from LCNL1, were reported as novel findings (labeled in red at Fig. 1A). Multiple supporting variants in strong linkage disequilibrium (LD) (r2 ≥ 0.8) surrounding the five SNPs were identified indicating the reliability of the signals except for SNP rs4756620, for which only one supporting variant with r2 of 0.6 was detected in the region (Fig. 1B). To check the authenticity of the signal at rs4756620, we further checked the imputation quality of this SNP and found that this SNP was genotyped in four of the 10 studies (Supplementary Table S4). We examined the association using only genotyped data from these four studies and rs4756620 had P values of 9.79 × 10−7 (OR = 0.61, N = 7132) EAS and 6.49 × 10−2 (OR = 0.70, N = 1,387) in AFR population. We believe the association at rs4756620 was reliable and we reported it as a novel susceptibility locus associated with never-smoking lung adenocarcinoma.
Figure 1.

Signals from genome-wide association analysis in ever- and never-smoking lung cancer. A, Manhattan plot (left) and QQ-plot (right) of signals from the analysis. The y-axis was truncated at 17 in the plots denoted by red *. The known lung cancer variants were labeled in black and novel variants identified in this study were labeled in red. Some variants with joint P values < 5 × 10−8 and with association evidence from only one population were not labeled in the plot. x-axis was truncated at 5 in QQ-plots. Lambda value was calculated for each analysis. Both normal lambda and lambda adjusted by sample size (indicated by #) were calculated for each stratum except for never-smoking SCLC where the number of cases was < 1,000. No inflated type I error rate was detected. B, regional plots of signals identified in each population and meta-analysis. The color intensity reflected the extent of linkage disequilibrium index (r2) with the target SNP denoted in purple.
Table 2.
Variants associated with lung cancer in only ever- or never-smokers.
| EAF | OR _P | |||||||
|---|---|---|---|---|---|---|---|---|
| Strata | SNP | Position | Gene | EUR|EAS|AFR | Weighted score | EUR|EAS|AFR | Joint effect size (P value) | Q |
| Ever-smokers | ||||||||
| LUNG | rs62303696* | 4p12 | GABRA4 | 0.074|0.275|0.028 | 0.94 | 1.17 (2.71 × 10−7)|1.22 (4.81 × 10−3)| 1.33 (6.08 × 10−2) | 1.18 (1.22 × 10−9) | 0.62 |
| LUNG | rs55781567 | 15q25.1 | CHRNA5 | 0.414|0.039|0.299 | 0.99 | 1.31 (5.67 × 10−69)|0.99 (9.65 × 10−1)| 1.32 (8.51 × 10−8) | 1.31 (1.66 × 10−74) | 0.65 |
| SQUAM | rs17879961# | 22q12.1 | CHEK2 | 0.002|0.000|0.000 | 0.89 | 0.25 (2.93 × 10−11)| NA|NA | 0.25 (2.93 × 10−11) | NA |
| SCLC | rs58778970* | 12q24.33 | Intergenic | 0.134|0.007|0.190 | 0.92 | 1.33 (1.50 × 10−7)|0.77 (8.05 × 10−1)| 1.53 (2.40 × 10−2) | 1.34 (1.58 × 10−8) | 0.67 |
| Never-smokers | ||||||||
| ADE | rs4756620* | 11p12 | LRRC4C | 0.998|0.977|0.810 | 0.91 | 0.76 (5.62 × 10−1)|0.57 (1.37 × 10−8)| 0.64 (1.28 × 10−2) | 0.59 (6.51 × 10−10) | 0.74 |
| SQC | rs6757055# | 2q34 | IKZF2 | 0.962|0.909|0.917 | 0.96 | 1.44 (1.94 × 10−1)|0.56 (1.51 × 10−11)| 0.71 (6.49 × 10−1) | 0.61 (1.11 × 10−9) | 0.01 |
| SQC | rs1383429* | 4q21.21 | LINC01088 | 0.909|0.878|0.492 | 0.97 | 0.73 (8.74 × 10−2)|0.64 (5.57 × 10−9)| 1.56 (3.13 × 10−1) | 0.67 (6.44 × 10−9) | 0.12 |
| SQC | rs968516* | 9q34.3 | LCNL1 | 0.947|0.966|0.923 | 0.86 | 0.62 (4.10 × 10−2)|0.36(8.07 × 10−10)| 0.92 (9.47 × 10−1) | 0.34 (8.19 × 10−10) | 0.12 |
| Never-smoking women | ||||||||
| Overall | rs12265047 | 10q25.2 | VTI1A | 0.949|0.701|0.626 | 0.93 | 0.63 (4.64 × 10−5)|0.77 (4.53 × 10−13)| 0.63 (3.29 × 10−3) | 0.75 (1.10 × 10−17) | 0.68 |
| ADE | rs7962469 | 12q13.13 | ACVR1B | 0.684|0.674|0.443 | 0.90 | 1.12 (5.61 × 10−2)|1.18 (1.63 × 10−6)| 1.74 (3.14 × 10−3) | 1.18 (3.73 × 10−8) | 0.03 |
Note: The risk variants with association evidence from only ever- or never-smoking individuals (not from both groups).
Abbreviations: EAF, effective allele frequency. Q indicates the heterogeneity p value. EUR: European population; EAS, East Asian population; AFR, African population. Weighted score indicated the imputation quality score weighted by sample size from the studies. #, known variants identified from previous studies but shown to be related to lung cancer in only ever- or never-smoking group. *, novel variants identified in this study.
Table 2 displays detailed information for the variants associated with ever- or never-smoking lung cancer. rs62303696, located at 3′ UTR (untranslated region) of GABRA4, was identified in ever-smoking overall lung cancer with a joint P value of 1.22 × 10−9 and OR of 1.18. The evidence of association was detected in all three continental populations with P values of 2.71 × 10−7, 4.81 × 10−3, and 6.08 × 10−2 from the EUR, EAS, and AFR populations, respectively. The SNP rs58778970 was identified in ever-smoking small cell lung cancer (P = 1.58 × 10−8, OR = 1.34). The association evidence came from both European (P = 1.50 × 10−7, OR = 1.33) and AFR populations (P = 2.40 × 10−2, OR = 1.53). Three SNPs, rs4756620 (P = 6.51 × 10−10, OR = 0.59), rs1383429 (P = 6.44 × 10−9, OR = 0.67) and rs968516 (P = 8.19 × 10−10, OR = 0.34) were identified in never-smoking lung cancer. It was noted that all these three variants achieved genome-wide significance in the EAS population (P < 5 × 10−8) and were replicated in either the EUR or AFR population. We compared the risk effect between ever- and never-smoking groups for the newly identified variants, finding that all five of these novel variants were significant in either the ever- or never-smoking group and not significant in non-stratified analysis which explains why these variants were not discovered in prior GWAS studies (Fig. 2A).
Figure 2.

Heterogeneous effect of genetic variants in lung cancer. A, forest plot of association in ever-, never-smoking, and non-stratified analysis for new variant identified in the study. B, Bar chart of lung cancer risk in never-, light- (packyr ≤ 20), and MtoH-smokers (packyr > 20) for variants with association in ever-smoking lung cancer in EUR. *, novel variants identified in this study. y-axis was truncated at 4. 95% error bars were plotted for each measurement. The variants are divided into four groups according to the risk pattern in different smoking groups.
Some known variants were associated with lung cancer in only ever- or never-smoking population
Aside from the novel findings, the stratified analysis also found that some of the previously identified susceptibility loci were associated with lung cancer in only the ever- or never-smoking group. Our previous study found evidence for an association between rs6757055 at IKZF2 and squamous lung cancer in the East Asian population (OR = 0.23, P = 8.39×10−11; Fig. 2A; ref. 16). Furthermore, stratified analysis displayed this variant was more significant in the never-smoking squamous lung cancer in the EAS population (OR = 0.19, P = 1.51 × 10−11) and not significant in the ever-smoking group (OR = 1.05, P = 0.37).
rs17879961, a rare variant located in the exon of the CHEK2 gene, has been reported to be negatively associated with squamous lung cancer (16, 21). The results from our study showed that it was non-significant in the non-smoking group (OR = 0.59, P = 0.56); and it had an OR of 0.25 and P value of 2.93 × 10−11 in the ever-smoking group (Fig. 2A). However, this variant had a less significant risk effect (OR = 0.26, P = 5.86 × 10−11) when combining ever- and never-smoking groups together. The sample size in the never-smoking squamous lung cancer cohort is relatively small (N = 6,865) and further study is required before it can be determined whether rs17879961 is associated with lung cancer in only ever-smoking individuals.
Validation of lung cancer susceptibility loci in never-smoking women using data from African-descent populations
VTI1A and ACVR1B were previously reported to be associated with never-smoking lung cancer in both Asian and European women (10, 11). However, there is no report about the association in AFR population due to the under-represented AFR participants in previous lung cancer GWAS studies. In our analysis, rs12265047, from VTI1A, had an OR of 0.63 (P = 4.64 × 10−5), 0.77 (P = 4.53 × 10−13), and 0.63 (P = 3.29 × 10−3) in never-smoking women from the EUR, EAS, and AFR population, respectively (Table 2). The rs7962469, located in ACVR1B, was associated with elevated risk for lung adenocarcinoma in both EUR (OR = 1.12, P = 5.61 × 10−2) and EAS (OR = 1.18, P = 1.63 × 10−6) never-smoking women in our study, and a stronger risk effect in the never-smoking female in AFR population (OR = 1.74, P = 3.14 × 10−3).
Evaluation of the impact of smoking on lung cancer risk
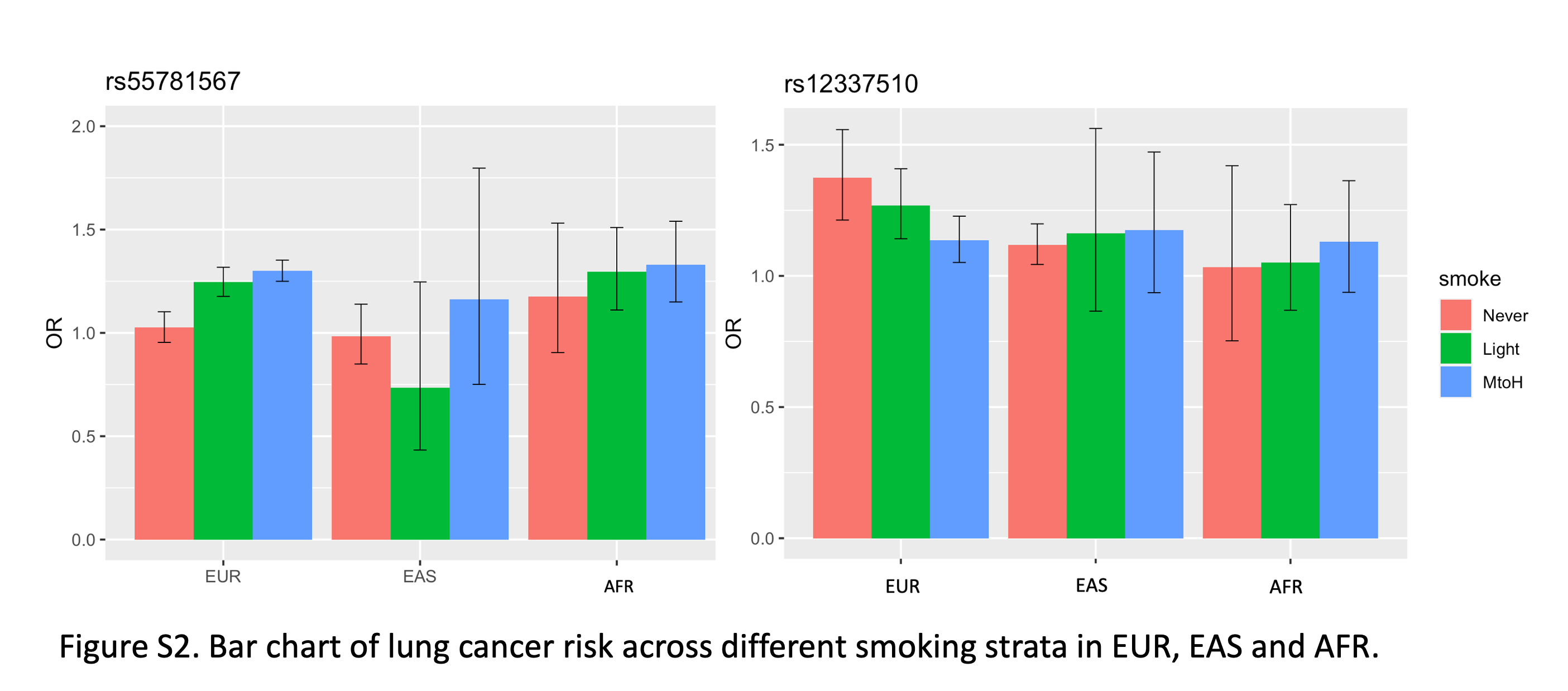
For the variants with association evidence in ever-smoking lung cancer, including the known variants identified from previous GWAS studies, we compared their lung cancer risk in never-, light- (packyr ≤ 20), and MtoH-smokers (packyr >20) in EUR, EAS, and AFR population, respectively. Because of the smaller sample size in the EAS and AFR population, there was limited power for most of the variants from these two populations, so we focused on the analysis in EUR population (Supplementary Table S5). The bar chart in Fig. 2B displayed the ORs in different smoking groups for variants from 12 independent regions. Most of the known variants, such as TERT, TP63, and ROS1, had association evidence from both ever- and never-smoking group and we observed similar risk effects across different types of smokers, so they were identified in prior non-stratified GWAS studies. rs55781567, located in CHRNA5, had association evidence from only ever-smokers and we observe similar lung cancer risk in MtoH-smokers (OR = 1.30, P = 6.17 × 10−39) compared with light-smokers (OR = 1.25, P = 3.19 × 10−14). A similar pattern was observed in AFR population, OR = 1.29 and P = 9.68 × 10−4 in light-smokers versus OR = 1.33 and P = 1.28 × 10−4 in MtoH-smokers (Supplementary Fig. S2 left). Some variants displayed slightly elevated risk in MtoH-smokers. For example, rs17879961 at CHEK2 had an OR of 0.10 and P value of 5.18 × 10−3 in light-smokers versus OR of 0.27 and a P value of 5.68 × 10−9 in MtoH-smokers; rs2523593 at HLA region had an OR of 1.16 and P value of 5.37 × 10−3 in light-smokers versus OR of 1.30 and P value of 1.12 × 10−14 in MtoH-smokers. rs12337510 at MTAP showed higher OR in never-smokers1.37 (P = 6.28 × 10−7) compared with an OR of 1.14 (P = 1.28 × 10−3) in MtoH-smokers. However, we did not see a similar pattern in either EAS or AFR population although it was significant in the other two populations (Supplementary Fig. S2 right).
Functional analysis of identified novel variants
We first conducted functional annotation analysis using RegulomeDB to evaluate how these identified variants affect lung cancer risk. All five new variants are located in non-coding regions such as 3′ or 5′ UTR, intronic, and intergenetic regions. The query from the RegulomeDB database showed that all five variants were located within peaks from more than one chromatin immunoprecipitation sequencing (CHIP-seq), DNase I hypersensitive sites sequencing (DNase-seq), or Formaldehyde-assisted isolation of regulatory elements sequencing (FAIRE-seq); experiment suggesting that they were located within regulatory DNA regions (Supplementary Table S6). Two SNPs, rs62303696 located at the 3′ UTR in the GABRA4 gene, and rs1383429 located in the intronic region in LINC01088, are predicted to be regulatory variants with probability > 0.6. CHIP-seq peaks are also detected at both of these two SNPs suggesting they were located in binding sites for regulatory proteins such as transcription factors, histone modifications, etc. (Fig. 3A). Position weight matrix analysis predicted that rs1383429 was a highly conserved SNP in sequence motifs (Fig. 3B).
Figure 3.

Functional analysis of the novel variants identified in ever- and never-smoking lung cancer. A, CHIP-seq peaks were identified at rs62303696 and rs1383429 by query from RegulomeDB. 0.61 and 0.69 in the brackets indicated the calculated probability of being a regulatory variant. B, the predicted sequence motif including the highly conservative allele at rs1383429. C, annotation results from RBPmap. The RBPs with significant sequence motifs (P < 0.05) between reference (yellow) and alternative (green) alleles were displayed for each variant located within coding gene. The size of circle indicates the significance of the sequence motif. D, eQTL analysis for rs968516 and LCNL1 gene. The x axis denotes the Z-score from association analysis between genotype and LCNL1 gene expression from GTEx data from lung tissues. y axis denotes the Z-score from GWAS analysis. The color intensity reflected the extent of linkage disequilibrium index (r2) with SNP rs968516 denoted by arrow.
We also evaluated and compared the RBPs with significant sequence motifs between reference and alternative alleles. Figure 3C displays the RBPs with significant motifs (P < 0.05) for novel variants located within coding genes. rs58778970 was located in an intergenic region and thus was removed from the analysis. We noticed different RBPs with significant motifs between reference and alternative alleles for the variants. For example, there were 13 RBPs for the reference allele of rs1383429 while only two were for the alternative allele. rs4756620 had two RBPs for the alternative allele but three additional RBPs for the reference allele. These findings, combined with the results from RegulomeDB, suggest that the two variants might regulate lung cancer risk by interacting with different regulatory proteins such as transcription factors and RBPs.
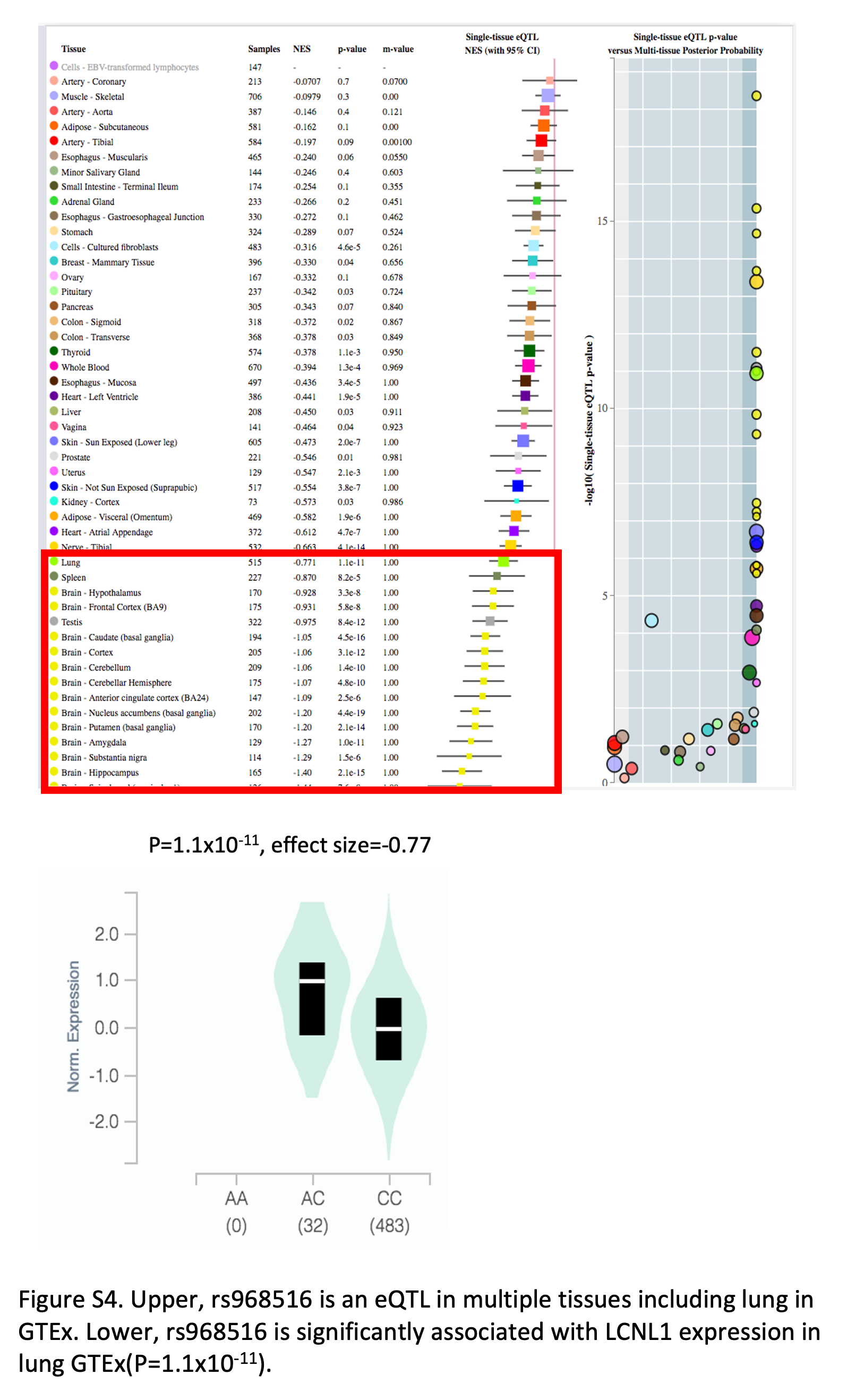
eQTL analysis was conducted to evaluate the association between lung cancer risk and nearby gene expression for each of the five novel variants. The z-score from the association between genotype and nearby GTEx was plotted against the z-score from GWAS analysis showing a strong association between lung cancer risk and LCNL1 gene expression for rs968516 and approximately 2,200 surrounding SNPs that were in strong LD with it (r2 > 0.8). These results suggested rs968516 could affect lung cancer risk in never-smokers through regulation of LCNL1 gene expression (Fig. 3D).
We performed DNA damage assays on each candidate gene following the procedures as displayed in Fig. 4A. We found that overproduced EmGFP fusions of GABRA4 and NR2F1 promoted DNA damage, measured by sensitive flow cytometric assays (Fig. 4B–F). BaP is one of the cigarette smoke carcinogens involved in lung tumorigenesis. Because GABRA4 was nominated from the lung cancer smoking analysis, we hypothesized that BaP exposure might enhance GABRA4-induced DNA damage. BaP exposure for 72 hours significantly increased GABRA4-induced DNA double-strand breaks, but not in tubulin overproducing cells (Fig. 4G and H). This observation supports the hypothesis that low-dose environmental mutagens can further titrate out DNA repair and cause amplified DNA damage in cells that have elevated endogenous DNA damage (Fig. 4I).
Figure 4.
![Figure 4. DNA damage assay at GABRA4 and NR2F1. A–F, GABRA4 and NR2F1 are lung cancer–associated DNA damageome proteins. A, Endogenous DNA damage assay scheme. B, GABRA4 and NR2F1 overproduction promotes DNA damage, respectively, quantified by H2AX levels using flow cytometry. DNA damage levels are compared and normalized to tubulin overproducing cells. Bar: median. n ≥ 7. Two-sample two-sided t test assuming equal variances. *, P = 0.0273; ***, P = 0.0003. 3–5, gating strategy and representative flow cytometric density plots. C, mock transfection. D, Tubulin overproduction. E, NF2F1 overproduction. F, Histograms showing that NF2F1 overproduced cells increase high-DNA damage subpopulations compared with Tubulin in GFP+ cells. G–I, Benzo[a]pyrene (BaP) potentiates GABRA4-induced DNA damage. G, Endogenous and exogenous agent DNA damage assay scheme. H, BaP exposure sensitizes GABRA4-induced DNA damage. A total of 8 μmol/L BaP exposure for 72 hours induces additional DNA damage in GABRA4 overproduced but not tubulin overproduced cells. n ≥ 7. Two-sample two-sided t test assuming equal variances. ns, not significant; P = 0.7047; *, P = 0.0137; **, P = 0.0034. I, Model: GABRA4 overproduction increases endogenous DNA damage and then potentially overloads DNA repair pathways. The addition of an exogenous agent (BaP, e.g.) causes more DNA damage that cannot be repaired and lead to DNA damage catastrophe.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10905670_389fig4.jpg)
DNA damage assay at GABRA4 and NR2F1. A–F, GABRA4 and NR2F1 are lung cancer–associated DNA damageome proteins. A, Endogenous DNA damage assay scheme. B, GABRA4 and NR2F1 overproduction promotes DNA damage, respectively, quantified by H2AX levels using flow cytometry. DNA damage levels are compared and normalized to tubulin overproducing cells. Bar: median. n ≥ 7. Two-sample two-sided t test assuming equal variances. *, P = 0.0273; ***, P = 0.0003. 3–5, gating strategy and representative flow cytometric density plots. C, mock transfection. D, Tubulin overproduction. E, NF2F1 overproduction. F, Histograms showing that NF2F1 overproduced cells increase high-DNA damage subpopulations compared with Tubulin in GFP+ cells. G–I, Benzo[a]pyrene (BaP) potentiates GABRA4-induced DNA damage. G, Endogenous and exogenous agent DNA damage assay scheme. H, BaP exposure sensitizes GABRA4-induced DNA damage. A total of 8 μmol/L BaP exposure for 72 hours induces additional DNA damage in GABRA4 overproduced but not tubulin overproduced cells. n ≥ 7. Two-sample two-sided t test assuming equal variances. ns, not significant; P = 0.7047; *, P = 0.0137; **, P = 0.0034. I, Model: GABRA4 overproduction increases endogenous DNA damage and then potentially overloads DNA repair pathways. The addition of an exogenous agent (BaP, e.g.) causes more DNA damage that cannot be repaired and lead to DNA damage catastrophe.
Discussion
Differences in genomic features have been identified in lung cancer between ever- and never-smokers such as genetic variants, gene mutation, gene expression, and DNA methylation profiles, etc (6). For example, the well-known CHRNA5/A3/B4 gene region was associated with nicotine dependence and lung cancer in ever-smokers, both in prior studies and more definitively in this study (7, 15, 21, 22). Leveraging the genotype from three continental populations, we identified five novel susceptibility loci associated with lung cancer, including GABRA4 and intergenic region 12q24.33 from ever-smokers; LRRC4C, LINC01088, and LCNL1 from never-smokers. All five variants have significant association in one smoking group and no effect in the other. These findings display heterogeneity in genetic predisposition to lung cancer between different smoking groups and highlight the complicated genetic architecture of this deadly disease. Gene–environment interaction analysis is another approach commonly used to identify variants with differential risk effects between groups. For the five novel variants, we further examined their interaction effect with smoking status in lung cancer risk using genotype data from CEU in the Oncoarray study, the study with the largest sample size of European individuals (N = 29,905), and none of them were significant (P < 0.05; Supplementary Table S7). These results illustrated that stratified GWAS was imperative for the identification of novel variants with effect only in subgroups that cannot be revealed by regular GWAS or genome-wide interaction studies and for prioritizing likely causal mechanisms as well.
IKZF2 was identified as a novel variant in lung cancer in our prior non-stratified GWAS study (16). The re-evaluation of variants in IKZF2 showed it was involved in only never-smoking lung cancer. rs6757055, located at IKZF2, is an uncommon variant with a MAF of 0.091 in EAS population. Our collaborator at Nanjing, P.R. China further validated this signal using data from six independent study sites in China, including a total of 8,407 never-smokers, and the final joint analysis showed an OR of 0.56 and a P value of 7.77 × 10−12 in never-smoking squamous lung cancer (Supplementary Fig. S3; Supplementary Table S8; ref. 23). Five of the study sites have MAF varying from 0.003 to 0.006 and one study site with MAF of 0.012.
One challenge in GWAS studies is that the variants identified in one population have often failed to be replicated in other populations. VTI1A was first discovered to be associated with lung cancer in Asian never-smoking women and then validated with nominal significance in European never-smoking women; ACVR1B was first reported in lung adenocarcinoma in European never-smokers and then reported in Asian women never-smokers (9–11, 24, 25). Little is known about their association with lung cancer in the AFR population. We successfully validated their association in people with AFR ancestry for the first time as far as we know. These two variants, together with the novel variant at GABRA4 (rs62303696), are the only three susceptibility loci associated with ever- or never-smoking lung cancer in all three continental populations (Table 2). These findings demonstrate that the inclusion of AFRs in the multi-population GWAS is crucial for a better understanding of genomic and environmental variations underpinning lung cancer. However, the AFR sample size is still limited (N = 5,688) in our study which limits our ability to identify novel variants in this population.
For the variants with association evidence in ever-smoking lung cancer, we evaluated their risk effect in never-, light- and MtoH-smokers with European ancestry. Among the 12 tested variants selected from independently associated regions, some variants displayed consistent risk effects across the different smoking groups; some displayed risk effects in only ever-smokers but not never-smokers; and some displayed slightly increased lung cancer risk in MtoH-smokers compared with light-smokers such as rs17879961 at CHEK2 and rs2523593 from HLA region (Fig. 2B). These observations suggested both tobacco smoking and genetic factors contribute to lung cancer risk and the heterogeneous disease mechanisms behind those susceptibility loci involved in smoking lung cancer.
As we step into the post-GWAS era, the ultimate goal is to understand the biological consequences of the statistical associations. We adopted multiple approaches for functional inference and obtained multiple layers of evidence supporting the regulatory role of the identified novel variants in ever- and never-smoking lung cancer. For example, rs968516, identified in never-smoking squamous lung cancer, was shown to affect lung cancer risk through regulation of nearby LCNL1 gene expression. It is also an eQTL in multiple tissues including the lung (Supplementary Fig. S4). rs62303696, identified in ever-smoking lung cancer, is located in the 3′ UTR region of GABRA4, a gene that has been reported to be related to alcohol use disorder in the European population (26). A systematic study showed that approximately 3% of GWAS hits were located within the 3′ UTR region (27). Genetic variations in 3′ UTR may change the binding sites for RBPs and miRNAs and lead to differential gene expression. DNase-seq and CHIP-seq experiments showed that rs62303696 was located within regions sensitive to cleavage by DNase I and DNA binding sites for transcription factors NR2F1 and JUNB (Fig. 3A). Further RBP analysis showed that the reference allele of rs62303696 enabled a binding motif for RBM6 while the alternative allele didn't (Fig. 3C). Aside from being reported as an alternative splicing factor and a putative tumor suppressor gene, RBM6 has been identified as a regulator involved in the repair of DNA double-strand breaks in a recent study (28–31). We further discovered GABRA4-induced DNA damage in lung fibroblast cell line which offered one mechanistic explanation for lung cancer: increased DNA damage and mutagenesis caused by upregulation of GABRA4 may underlie tumorigenesis and poor clinical prognosis. These integrated results suggest that rs62303696 could affect lung cancer risk in smokers through increased DNA damage and genome instability (Fig. 4).
In summary, we performed a multi-population GWAS stratified by smoking status in lung cancer, and we identified five novel variants associated with ever- or never-smoking lung cancer. The extensive functional analysis provided evidence for the functional roles of the identified variants and provided insights into the molecular mechanism underlying lung carcinogenesis. Our study highlighted the genetic heterogeneity between ever- and never-smoking lung cancer and provided helpful etiologic insights into the complicated genetic architecture of this deadly disease.
Supplementary Material
Supplementary Table S1 displays the number of ever- and never-smokers from each of the ten studies.
Supplementary Table S2 shows the signals at known susceptibility loci from ever- and never-smoking lung cancer.
Supplementary Table S3 shows the signals at rs7985487 in Firth test validation.
Supplementary Table S4 shows the imputation quality score of identified variants in different studies.
Supplementary Table S5 shows the signals for the variants in stratified analysis by smoking behavior
Supplementary Table S6 displays epigenomic evidence for the variants being located within genomic regions associated with regulatory activity.
Supplementary Table S7 shows the signals from interaction with smoking status for the identified variants in lung cancer.
Supplementary Table S8 displays the sample size from independent Asian studies.
Supplementary Figure S1 displays the distribution of histology subtypes in each population in ever- and never-smokers.
{kind=link}
Supplementary Figure S2 shows the bar chart of lung cancer risk across different smoking strata in EUR, EAS and AFR.
{kind=link}
Supplementary Figure S3 displays the Validation of the association effect of rs6757077 at IKZF2 using data from six independent study sites in China.
{kind=link}
Supplementary Figure S4 shows the results of eQTL analysis of rs968516 in GTEx.
{kind=link}
Acknowledgments
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the NIH, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from dbGaP accession number phs000424.v7. p2. Thanks to Kathryn Edwards and Zachary Xiao for proofreading the article.
The research in this study is supported by the NCI of the NIH under award numbers U19CA203654, U01CA243483, R21CA235464; by Cancer Prevention Research Institute of Texas (CPRIT) under award numbers RR170048, RR160097T, RR180061; by the Department of Health and Human Services contracts under award numbers HHSN26820100007C, HHSN268201700012C, 75N92020C00001; by NCI of the NIH under award number X01HG007491 under contract number HHSN268201200008I.
The CARET study is funded by the NCI of the NIH under award numbers U01CA063673, UM1CA167462, and U01CA167462; the Harvard Lung Cancer Study is funded by the NCI of the NIH under award number U01CA209414; the Asian validation study is funded by the National Natural Science Foundation of China under award number 81820108028; the Liverpool Lung Project is supported by MPAD Roy Castle Lung Foundation (UK); the EAGLE study is supported by the intramural program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute of the NIH; the ReSoLuCENT study is funded by the Weston Park Hospital Cancer Charity. Acknowledgment to the George IsAFRc Family Fund for Cancer Research for grant support. J.E. Bailey-Wilson was supported by the Intramural Research Program of the National Human Genome Research Institute at NIH. J. Xia was supported by the National Institute of Environmental Health Sciences of the NIH under award number KK99ES033259, the Nebraska Health Care Funding Act LB692, and Cancer and Smoking Disease Research Program LB595. S.M. Rosenberg was supported by NCI of the NIH under award number R01CA250905 and by the National Institute on Aging of NIH under award number DP1AG072751. This project was also supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the CPRIT Core Facility Support under award number RP180672, the NIH under award number P30CA125123 and S10RR024574, and the assistance of Joel M Sederstrom. C.I Amos and C. Cheng are Research Scholars at the Cancer Prevention Institute of Texas.
Footnotes
Note: Supplementary data for this article are available at Cancer Epidemiology, Biomarkers & Prevention Online (http://cebp.aacrjournals.org/).
Authors' Disclosures
S.M. Rosenberg reports part of this work was funded by U.S. NIH grant R01-CA250905. A. Risch reports grants from Deutsche Krebshilfe and NIH during the conduct of the study. S.M. Arnold reports other support from Merck Sharp & Dohme, AstraZeneca Pharma, Ellipses Pharma Limited, AbbVie Incorporated, LabCorp, Exelixis, Incyte Corp, Eli Lilly, Beigene, and Kinnate Biopharma Inc outside the submitted work. M.P.A. Davies reports grants from Roy Castle Lung Cancer Foundation during the conduct of the study. L. Le Marchand reports grants from NCI during the conduct of the study. M.C. Aldrich reports grants from NIH/NCI during the conduct of the study; personal fees from Guardant Health outside the submitted work. A.G. Schwartz reports grants from NIH during the conduct of the study. K.S. Purrington reports grants from NCI during the conduct of the study. S.M. Pinney reports grants from NCI and National Institute of Environmental Health Sciences during the conduct of the study. C.I. Amos reports grants from NCI during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
Y. Li: Conceptualization, methodology, writing–original draft. X. Xiao: Formal analysis. J. Li: Formal analysis. Y. Han: Formal analysis. C. Cheng: Supervision. G.F. Fernandes: Resources. S.E. Slewitzke: Resources. S.M. Rosenberg: Resources. M. Zhu: Validation. J. Byun: Formal analysis. Y. Bossé: Resources. J.D. McKay: Data curation. D. Albanes: Data curation. S. Lam: Data curation. A. Tardon: Data curation. C. Chen: Data curation. S.E. Bojesen: Data curation. M. Landi: Data curation. M. Johansson: Data curation. A. Risch: Data curation. H. Bickeböller: Data curation. H.-E. Wichmann: Data curation. D.C. Christiani: Data curation. G. Rennert: Data curation. S.M. Arnold: Data curation. G.E. Goodman: Data curation. J.K. Field: Data curation. M.P.A. Davies: Data curation. S. Shete: Data curation. L. Le Marchand: Data curation. G. Liu: Data curation. R.J. Hung: Resources. A.S. Andrew: Data curation. L.A. Kiemeney: Data curation. R. Sun: Resources. S. Zienolddiny: Data curation. K. Grankvist: Data curation. M. Johansson: Data curation. N.E. Caporaso: Data curation. A. Cox: Resources. Y.-C. Hong: Data curation. P. Lazarus: Data curation. M.B. Schabath: Data curation. M.C. Aldrich: Data curation. A.G. Schwartz: Data curation. I. Gorlov: Resources. K.S. Purrington: Data curation. P. Yang: Data curation. Y. Liu: Resources. J.E. Bailey-Wilson: Data curation. S.M. Pinney: Data curation. D. Mandal: Data curation. J.C. Willey: Resources. C. Gaba: Data curation. P. Brennan: Data curation. J. Xia: Formal analysis. H. Shen: Validation. C.I. Amos: Supervision, funding acquisition, writing–review and editing.
References
- 1. Bosse Y, Amos C. A decade of GWAS results in lung cancer. Cancer Epidemiol Biomarkers Prev 2018;27:363–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Momozawa Y, Mizukami K. Unique roles of rare variants in the genetics of complex diseases in humans. J Hum Genet 2021;66:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Y, Shen S, Wei Y, Zhu Y, Li Y, Chen J, et al. A large-scale genome-wide gene-gene interaction study of lung cancer susceptibility in Europeans with a trans-ethnic validation in Asians. J Thorac Oncol 2022;17:974–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walser T, Cui X, Yanagawa J, Lee JM, Heinrich E, Lee G, et al. Smoking and lung cancer–the role of inflammation. Proc Am Thorac Soc 2008;5:811–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rivera GA, Wakelee H. Lung cancer in never smokers. Adv Ep Med Biol 2016;893:43–57. [DOI] [PubMed] [Google Scholar]
- 6. Sun S, Schiller JH, Gazdar AF. Lung cancer in never-smokers- a different disease. Nat Rev Can 2007;7:778–90. [DOI] [PubMed] [Google Scholar]
- 7. Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet 2008;40:616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patel YM, Park SL, Han Y, Wilkens LR, Bickeböller H, Rosenberger A, et al. Novel association of genetic markers affecting CYP2A6 activity and lung cancer risk. Cancer Res 2016;76:5768–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lan Q, Hsiung CA, Keitaro Matsuo K, Hong YC, Seow A, Wang Z, et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat Genet 2012;44:1330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Spitz MR, Gorlov IP, Amos CI, Dong Q, Chen W, Etzel CJ, et al. Variants in inflammation genes are implicated in risk of lung cancer in never smokers exposed to second-hand smoke. Cancer Discov 2011;1:420–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hung RJ, Spitz MR, Houlston RS, Schwartz AG, Field JK, Ying J, et al. Lung cancer risk in never-smokers of European descent is associated with genetic variation in the 5p15.33 TERT-CLPTM1Ll region. J Thorac Oncol 2019;14:1360–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47:D886–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012;22:1790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paz I, Kosti I, Ares M Jr, Cline M, Mandel-Gutfreund Y. RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res 2014;42:W361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet 2017;49:1126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Byun J, Han Y, Li Y, Xia J, Long E, Choi J, et al. Cross-ancestry genome-wide meta-analysis of 61,047 cases and 947,237 controls identifies new susceptibility loci contributing to lung cancer. Nat Genet 2022;54:1167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Y, Xia J, McKay J, Tsavachidis S, Xiao X, Spitz MR, et al. Rare deleterious germline variants and risk of lung cancer. NPJ Precis Oncol 2021;5;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang X. Firth logistic regression for rare variant association tests. Front Genet 2014;5:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xia J, Chiu L-Y, Nehring RB, Núñez MAB, Mei A, Perez M, et al. Bacteria-to-human protein networks reveal origins of endogenous DNA damage. Cell 2019;176:127–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bossé Y, Li Z, Xia J, Manem V, Carreras-Torres R, Gabriel A, et al. Transcriptome-wide association study reveals candidate causal genes for lung cancer. Int J Cancer 2020;146:1862–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, McKay JD, Rafnar T, Wang Z, Timofeeva MN, Broderick P, et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat Genet 2014;46:736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet 2007;16:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dai J, Lv J, Zhu M, Wang Y, Qin N, Ma H, et al. Identification of risk loci and a polygenic risk score for lung cancer: a large-scale prospective cohort study in Chinese populations. Lancet Respir Med 2019;7:881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen LS, Saccone NL, Culverhouse RC, Bracci PM, Chen CH, Dueker Net al. Smoking and genetic risk variation across populations of European, Asian, and African American ancestry—a meta-analysis of chromosome 15q25. Genet Epidemiol 2012;36:340–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Z, Seow W, Shiraishi K, Hsiung CA, Matsuo K, Liu J, et al. Meta-analysis of genome-wide association studies identifies multiple lung cancer susceptibility loci in never-smoking Asian women. Hum Mol Genet 2016;25:620–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou H, Sealock JM, Sanchez-Roige S, Clarke TK, Levey DF, Cheng Z, et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat Neurosci 2020;23:809–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Steri M, Idda ML, Whalen MB, Orrù V. Genetic variants in mRNA untranslated regions. Wiley Interdiscip Rev RNA 2018;9:e1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heath E, Sablitzky F, Morgan GT. Subnuclear targeting of the RNA-binding motif protein RBM6 to splicing speckles and nascent transcripts. Chromosome Res 2010;18:851–72. [DOI] [PubMed] [Google Scholar]
- 29. Bechara EG, Sebestyen E, Bernardis I, Eyras E, Valcarcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell 2013;52:720–33. [DOI] [PubMed] [Google Scholar]
- 30. Wang Q, Wang F, Zhong W, Ling H, Wang J, Cui J, et al. RNA-binding protein RBM6 as a tumor suppressor gene represses the growth and progression in laryngocarcinoma. Gene 2019;697:26–34. [DOI] [PubMed] [Google Scholar]
- 31. Wistuba II, Behrens C, Virmani AK, Mele G, Milchgrub S, Girard L, et al. High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res 2000;60:1949–60. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1 displays the number of ever- and never-smokers from each of the ten studies.
Supplementary Table S2 shows the signals at known susceptibility loci from ever- and never-smoking lung cancer.
Supplementary Table S3 shows the signals at rs7985487 in Firth test validation.
Supplementary Table S4 shows the imputation quality score of identified variants in different studies.
Supplementary Table S5 shows the signals for the variants in stratified analysis by smoking behavior
Supplementary Table S6 displays epigenomic evidence for the variants being located within genomic regions associated with regulatory activity.
Supplementary Table S7 shows the signals from interaction with smoking status for the identified variants in lung cancer.
Supplementary Table S8 displays the sample size from independent Asian studies.
Supplementary Figure S1 displays the distribution of histology subtypes in each population in ever- and never-smokers.
Supplementary Figure S2 shows the bar chart of lung cancer risk across different smoking strata in EUR, EAS and AFR.
Supplementary Figure S3 displays the Validation of the association effect of rs6757077 at IKZF2 using data from six independent study sites in China.
Supplementary Figure S4 shows the results of eQTL analysis of rs968516 in GTEx.
Data Availability Statement
The following publicly available datasets were used in this work: Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial, phs000093.v2.p2; FLCCA study, phs000716.v1.p1; EAGLE study, phs000336.v1.p1; NCI study of African-Americans, phs001210.v1.p1; German, SLRI, IARC, and MD Anderson Cancer Center studies, phs000876.v2.p1; Oncoarray study, phs001273.v3.p2; imputed Oncoarray study using HRC reference panel, phs001273.v4.p2; Affymetrix study, phs001681. v1.p1. The eQTL data from GTEx was obtained from https://gtexportal. org/home/datasets (phs000424.GTEx.v7. p2; ref. 16).