

Abstract
Androgen binding to the androgen receptor (AR) in the cytoplasm induces the AR to translocate to the nucleus, where it regulates the expression of target genes. Here, we found that androgens rapidly activated a plasma membrane–associated signaling node that enhanced nuclear AR functions. In murine primary osteoblasts, dihydrotestosterone (DHT) binding to a membrane-associated form of AR stimulated plasma membrane–associated protein kinase G type 2 (PKG2), leading to the activation of multiple kinases, including ERK. Phosphorylation of AR at Ser515 by ERK increased the nuclear accumulation and binding of AR to the promoter of Ctnnb1, which encodes the transcription factor β-catenin. In male mouse osteoblasts and human prostate cancer cells, DHT induced the expression of Ctnnb1 and CTNN1B, respectively, and β-catenin target genes, stimulating the proliferation, survival, and differentiation of osteoblasts and the proliferation of prostate cancer cells in a PKG2-dependent fashion. Because β-catenin is a master regulator of skeletal homeostasis, these results explain the reported male-specific osteoporotic phenotype of mice lacking PKG2 in osteoblasts and imply that PKG2-dependent AR signaling is essential for maintaining bone mass in vivo. Our results suggest that widely used pharmacological PKG activators such as sildenafil could be beneficial for male and estrogen-deficient female patients with osteoporosis but detrimental in patients with prostate cancer.
INTRODUCTION
Signaling by androgens and estrogens was initially shown to occur by binding to their corresponding receptors in the cytoplasm (the AR and ER, respectively), thereby inducing the receptors to translocate to the nucleus and associate with specific DNA response elements and transcriptional coactivators or corepressors (1, 2). In addition, AR and ER can regulate transcription without direct DNA binding through interactions with other transcription factor complexes, such as the AP-1 complex comprising Fos and Jun family proteins (1, 2).
Androgens and estrogens also signal through rapid, non-genomic events, such as inducing a rise in intracellular Ca2+ concentration within seconds, and activating the tyrosine kinase Src and extracellular signal–regulated kinases 1 and 2 (ERK1/2) within minutes (1, 3). For estrogens, this rapid, non-genomic signaling occurs through well-characterized plasma membrane-associated ER-α and ER-β, and the membrane-associated and nuclear ERs appear to cooperate during adipocyte differentiation, where activation of the kinase Akt by ER-mediated signaling at the membrane is required for optimal nuclear translocation and DNA binding of ER-α (4). Signaling through plasma membrane–associated androgen receptors has been less well studied (3, 5). Extranuclear androgen signaling has been shown in several systems including prostate cancer, ovarian granulosa cells, and pancreatic β cells, but the precise protein complexes involved remain incompletely defined (3, 6–9).
The skeletal differences between males and females are primarily the result of the actions of androgens and estrogens, with males attaining higher peak bone mass, greater bone size, and stronger bones compared to females (1). Testosterone increases bone mass mainly by activating the AR in cells of the osteoblast lineage (1, 10, 11). In addition, testosterone acts by way of conversion to estrogens, because bone loss occurs in males with loss-of-function mutations affecting the enzyme that aromatizes testosterone to estradiol, and in males lacking the main estrogen receptor, ER-α (1, 12, 13). In females, estrogens signal largely through ER-α to increase bone mass, with ER-β restraining ER-α functions in trabecular bone; the AR plays only a minor role in female skeletal homeostasis (10, 11).
The nitric oxide–cyclic guanosine monophosphate–protein kinase G (NO-cGMP-PKG) signaling pathway mediates anabolic responses of bone to hormonal and mechanical stimuli (14). In osteoblasts, NO synthase (NOS) is activated in response to estrogens and fluid shear stress (15–18). The resulting NO stimulates guanylyl cyclase-1 (GC-1) to generate cGMP, which can activate cytosolic protein kinase G type 1 (PKG1) and plasma membrane–associated protein kinase G type 2 (PKG2), leading to enhanced osteoblast proliferation, differentiation, and survival (14). Osteoblast-specific knockout of PKG1 in mice does not produce obvious skeletal abnormalities, although the mice show delayed fracture healing (19). However, male mice with osteoblast-specific PKG2 knockout (OB Prkg2-KO) have low trabecular and cortical bone mass due to reduced bone formation by osteoblasts (20). Correspondingly, male mice over-expressing a partially activated form of PKG2 (OB PKG2R242Q-TG) in osteoblasts have high bone mass and increased bone formation compared to wild-type littermates (21). In contrast, female OB Prkg2-KO and OB PKG2R242Q-TG mice have normal bone formation and microarchitecture (20, 21). In both mouse lines, males, but not females, show altered expression of genes related to Wnt-β-catenin signaling in bones and primary osteoblasts (POBs) compared to bones and cells from wild-type littermates (20, 21). Wnt-β-catenin signaling positively regulates osteoblast proliferation, survival, and differentiation and is central to skeletal homeostasis, with mutations affecting Wnt pathway components causing severe skeletal disorders (22).
Based on the sexual dimorphism in the skeletal phenotype caused by osteoblast-specific PKG2 knockout or overexpression, we hypothesized that PKG2 may play an important role in AR signaling. Here, we show that androgens activate a plasma membrane–associated AR-NOS3-PKG2-Src signaling node that promotes AR nuclear accumulation and binding to the promoter of the gene encoding β-catenin (Ctnnb1), increased Ctnnb1 transcription, and induction of β-catenin target genes.
RESULTS
Androgens rapidly induce NO production and activate Src, ERK, and Akt through membrane-associated PKG2
We used dihydrotestosterone (DHT) to examine androgen signaling because, unlike testosterone, DHT is not converted to estrogens (23). We found that DHT rapidly increased the intracellular Ca2+ concentration in POBs from male mice (Fig. 1A), as reported previously for testosterone in rat and chicken osteoblasts in studies that showed testosterone induces Ca2+ influx through L-type Ca2+ channels (24, 25). We did not observe Ca2+ oscillations in DHT-treated osteoblasts, which is in contrast to the effects of DHT in skeletal muscle (26).
Fig. 1. Androgens rapidly induce NO production and activate Src, ERK, and Akt through membrane-associated PKG2.
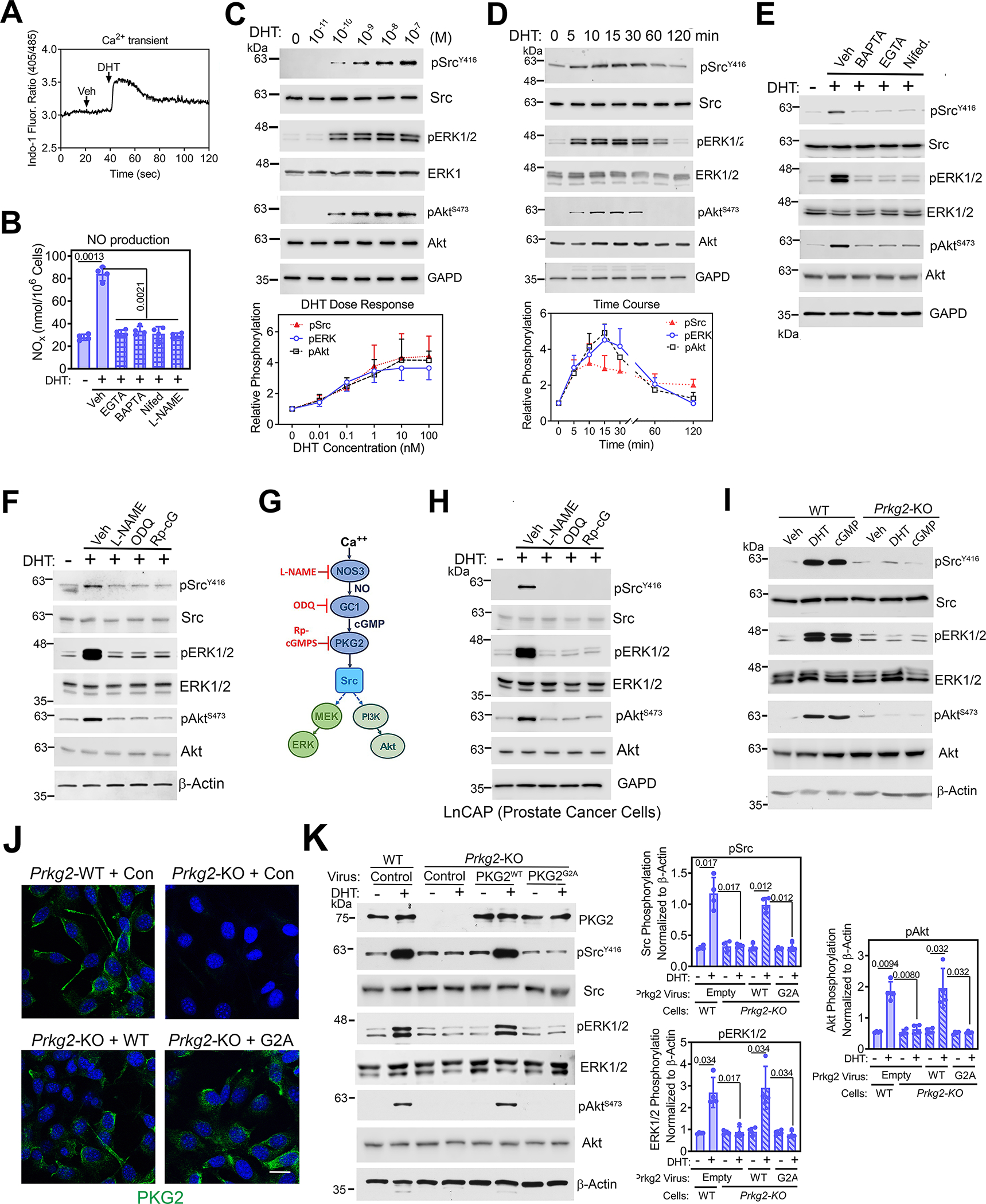
(A) Quantification of the 405 nm/485 nm fluorescence ratio in male WT POBs loaded with the intracellular Ca2+ indicator Indo1-AM and treated with vehicle (Veh) followed by 1 nM DHT. (B) Quantification of NO derivatives in the media of male WT POBs pretreated with vehicle, EGTA, BAPTA-AM, nifedipine (Nifed), or L-NAME before treatment with DHT for 30 min. (C and D) Immunoblotting for total and phosphorylated (p) forms of Src, ERK1/2, and Akt in male WT POBs treated with the indicated DHT concentrations for 10 min (C), or with 1nM DHT for the indicated amounts of time (D). GAPD is a loading control. Amounts of pSrcY416, pERKY204, and pAktS473 were quantified by densitometry. (E and F) Immunoblotting for the indicated proteins in male WT POBs pretreated with Ca2+ modulators (E) or pretreated with L-NAME, ODQ, or Rp-8-CPT-PET-cGMPS (Rp-cG) (F) and treated with 1 nM DHT for 10 min. GAPD and β-actin are loading controls. (G) Schematic showing the Ca2+-induced activation of NOS3 and the NO-cGMP-PKG2 pathway resulting in Src, ERK, and Akt activation, with pathway inhibitors shown in red. (H) Immunoblotting of LnCAP cells pretreated with L-NAME, ODQ, or Rp-8-CPT-PET-cGMPS and treated with 1 nM DHT for 10 min. (I) Immunoblotting of POBs from male Prkg2-WT and OB Prkg2-KO mice after treatment with DHT or 8-CPT-cGMP (cGMP) for 10 min. (J) Immunofluorescence showing PKG2 localization in POBs from male Prkg2-WT and OB Prkg2-KO mice that were infected with virus expressing LacZ (control) or WT, membrane-associated PKG2 (PKG2WT) or mutant, cytosolic PKG2 (PKG2G2A) before DHT stimulation. Scale bar, 25 μm. (K) Immunoblotting for and quantification of the indicated proteins in cells treated as in (J). All data are representative of at least three independent experiments; n=4 for (B) and (J); graphs show means ± S.D. P values for the indicated comparisons by ANOVA. For (E) to (I), quantification of three independent experiments is shown in fig. S1B, D, and E and S2A. Separate, identical gels loaded with equal amounts of cell lysates were run for analysis of total and phospohorylated forms of Src, ERK, and Akt.
DHT rapidly increased NO production about 4-fold in the POBs (Fig. 1B). This was likely due to the stimulation of NOS by the increase in intracellular Ca2+, because this effect was blocked by the NOS inhibitor L-NAME, extra- and intracellular Ca2+ chelation with EGTA or BAPTA, respectively, and or by the L-type Ca2+ channel blocker nifedipine (Fig. 1B). These data are consistent with a Ca2+-induced increase in NOS activity in the osteoblasts, similar to 17β-estradiol activating NOS in endothelial cells in a Ca2+-dependent fashion (27). The increased NO activated guanyl cyclase-1 (GC-1), because DHT increased intracellular cGMP in a NOS-dependent manner (fig. S1A).
At concentrations as low as 0.1–1 nM, DHT induced phosphorylation of Src, ERK1/2, and Akt in POBs on sites known to stimulate activity of the kinases, with maximal phosphorylation occurring at 15 min and declining to baseline by 120 min (Fig. 1, C and D). Total amounts of Src, ERK, and Akt were unchanged over the concentration range and time course studied (Fig. 1, C and D). The DHT-induced phosphorylation of all three proteins was completely blocked by BAPTA, EGTA, or nifedipine (Fig. 1E; fig. S1B). In contrast, epidermal growth factor (EGF)-induced phosphorylation of Src, ERK1/2, and Akt was only partially reduced by BAPTA, EGTA, or nifedipine (fig. S1C), indicating that partial activation of these kinases can occur independently of a rise in intracellular Ca2+. DHT-induced Src, ERK, and Akt phosphorylation was also blocked by L-NAME, the GC-1 inhibitor ODQ, and the PKG inhibitor Rp-8-CPT-PET-cGMPS (Fig. 1, F and G; fig. S1D). Thus, kinase activation by DHT was dependent on Ca2+ and NO-cGMP-PKG signaling.
The AR is an important treatment target in prostate cancer, and rapid, non-genomic androgen signaling through Src leads to increased proliferation of prostate cancer cells (6, 28, 29). In the androgen-dependent human prostate cancer cell line LnCAP, we found that DHT activated Src, ERK1/2, and Akt, and that activation of these three signaling kinases required an intact NO-cGMP-PKG signaling pathway because their activation was inhibited by L-NAME, ODQ, or Rp-8-CPT-PET-cGMPS (Fig. 1H and fig. S1E).
The DHT-induced Src, ERK1/2, and Akt phosphorylation was mimicked by activating PKG with the membrane-permeable cGMP analog 8-CPT-cGMP (Fig. 1I and fig. S2A). DHT and cGMP effects were abolished in PKG2-deficient POBs (derived from OB Prkg2-KO mice) but could be restored when the cells were reconstituted with wild-type PKG2 (Fig. 1, I to K and fig. S2A). These results are consistent with our previous finding that PKG2 activation in osteoblasts leads to the activation of Src in a manner that depends on the phosphatases Shp1 and Shp2 (Shp1/2), which in turn activates ERK1/2 and Akt by way of mitogen-activated protein kinase kinase (MEK) and phosphoinositide 3-kinase (PI3K), respectively (Fig. 1G) (15, 30). Reconstitution of the PKG2-deficient cells with PKG2G2A, a mutant form of PKG2 that cannot localize to the plasma membrane due to lack of an N-terminal myristoylation site (Fig. 1J), failed to restore signaling (Fig. 1K) (15, 31). In wild-type osteoblasts, endogenous PKG2 colocalized with Src at the plasma membrane (fig. S2B). Thus, PKG2 must be plasma membrane–associated to mediate androgen-induced activation of Src, ERK1/2, and Akt.
Rapid androgen signaling in osteoblasts occurs through a membrane-associated AR
We found that the DHT-induced increase in NO production in male POBs was prevented when cells were pre-treated with enzalutamide, a selective AR antagonist (32, 33), or when the AR was depleted by siRNA transfection (Fig. 2, A and B; fig. S3A). Enzalutamide also blocked DHT-induced activation of Src, ERK1/2, and Akt (fig. S3B) and reduced DHT-induced phosphorylation of NOS3 on Ser1177, a site targeted by Akt and associated with NOS activation (34, 35) (fig. S3C). The PI3K inhibitor LY29004 reduced DHT-induced Akt activation and NOS3 phosphorylation (fig. S3C).
Fig. 2. Rapid androgen signaling occurs through a membrane-associated AR.
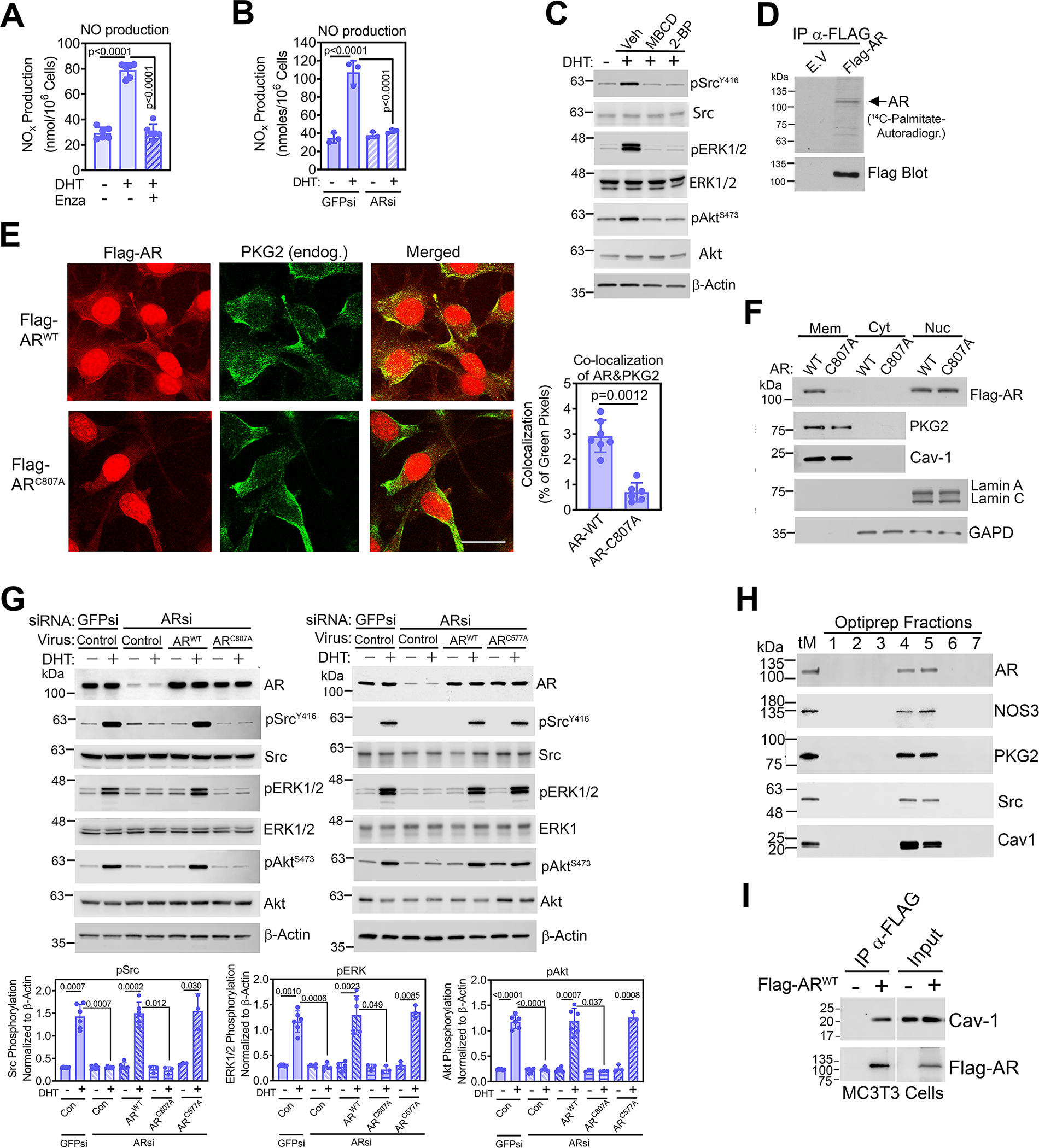
(A and B) Quantification of NO derivatives in the media of male WT POBs pre-treated with enzalutamide (A), or transfected with siRNAs targeting AR (ARsi) or green fluorescent protein (GFPsi, control) (B); some cells received DHT for 30 min. (C) Immunoblotting for total and phosphorylated (p) forms of Src, ERK1/2, and Akt in male WT POBs pretreated with methyl-β-cyclodextrin (MBCD) or 2-bromopalmitate (2-BP) and treated with DHT for 10 min. Actin is a loading control. (D) Autoradiograph of and Western blotting for Flag in Flag immunoprecipitates from MC3T3 cells transfected with empty vector (E.V.) or Flag-tagged AR labelled with 14C-palmitate for 4 h. (E) Immunofluorescence staining for Flag (red) and endogenous PKG2 (green) in POBs transfected with Flag-tagged WT AR (ARWT) or AR with a mutation in a conserved membrane localization motif (ARC807A) and treated with DHT for 18 h. Scale bar, 25 μm. Quantification of AR and PKG2 colocalization, indicated by yellow pixels in the merged image, is shown in the graph; 7 images from 3 independent experiments were analyzed, representing ~30 cells per condition. (F) Immunoblotting for the indicated proteins in membrane (Mem), cytosol (Cyt), and nuclear (Nuc) fractions of POBs infected with virus encoding Flag-tagged ARWT or ARC807A and treated with DHT for 18 h. Caveolin-1 (Cav-1) and lamin A/C are markers for plasma membranes and nuclei, respectively. GAPD is a loading control for cytosol. (G) Immunoblotting for the indicated proteins in POBs transfected with siRNAs targeting GFP or AR and then infected with LacZ virus (control) or virus encoding Flag-tagged WT AR (ARWT), ARC807A with a mutation in a conserved membrane localization motif (left), or ARC577A with a mutation in the first DNA-binding zinc finger motif (right). Cells received DHT for 10 min; three independent experiments with each AR mutant are summarized in the bar graphs below. (H) Immunoblotting for the indicated proteins in POB membranes from a Percoll™ gradient (total membranes, tM) that were further fractionated over a density gradient of Optiprep™. Caveolin-1 is a marker of caveolar fractions. (I) Flag immunoprecipitates from MC3T3 cells transfected with empty vector or Flag-tagged ARWT were blotted with antibodies specific for caveolin-1 or Flag. 10% input lysate was analyzed in parallel. All data are representative of three independent experiments, except n=6 for (A), n=7 for (E), and n=2 for (D). Graphs show means ± S.D.; p values for the indicated comparisons by one-way ANAOVA (A), two-tailed Mann-Whitney test (B), or two-way ANOVA (B and G). For (C), quantification of three independent experiments is shown in fig. S3D. Separate, identical gels loaded with equal amounts of cell lysates were run for analysis of total and phospohorylated forms of Src, ERK, and Akt.
Rapid, non-genomic estrogen signaling through ER-α and ER-β requires targeting of the receptors to cholesterol-rich plasma membrane subdomains by palmitoylation of a specific cysteine residue within a nine amino acid motif that is highly conserved among multiple steroid receptors, including the AR (36). To test if an analogous mechanism exists for the AR, we treated POBs with the cholesterol-depleting agent methyl-β-cyclodextrin (MBCD) and the palmitoylation inhibitor 2-bromopalmitate (2-BP) to examine the drugs’ effect on androgen signaling. Both agents prevented DHT-induced activation of Src, ERK1/2, and Akt, suggesting that a posttranslational lipid modification of AR and association with cholesterol-rich membrane fractions are important for rapid androgen signaling (Fig. 2C and fig. S3D). We then showed that the AR undergoes palmitoylation by following 14C-palmitate incorporation into Flag-tagged wild-type AR (Flag-AR) immunoprecipitated from transfected MC3T3 osteoblast-like cells (Fig. 2D). Some low-abundance bands apparent in the Flag immunoprecipitates may represent AR breakdown products and/or other palmitoylated proteins that co-immunoprecipitate with Flag-AR (Fig. 2D). Flag-AR colocalized with PKG2 at the plasma membrane in osteoblasts treated with DHT to minimize cytoplasmic AR concentrations by inducing AR nuclear translocation (Fig. 2E upper row).
To distinguish between the functions of membrane-associated AR and cytoplasmic AR that can translocate to the nucleus and bind DNA, we employed two mutant constructs: ARC807A, which has a mutation in the conserved palmitoylation site necessary for membrane-binding of steroid receptors (36), and ARC577A, which has a mutation in the receptor’s first zinc finger motif necessary for DNA binding (37). Like wild-type Flag-AR (Flag-ARWT), Flag-ARC807A localized to the nucleus in DHT-treated POBs, but unlike the wild-type protein, it did not colocalize with PKG2 at the plasma membrane (Fig. 2E lower row). Whereas Flag-ARWT localized to the plasma membrane in MC3T3 osteoblast-like cells, which produce little endogenous AR, Flag- ARC807A did not (fig. S3E). Lack of membrane association of ARC807A in DHT-treated POBs was also apparent on Western blots of fractionated cell extracts, with caveolin-1 and lamin-A/C serving as markers for plasma membranes and nuclei, respectively (Fig. 2F). In male POBs, siRNA-mediated depletion of AR completely prevented DHT-induced Src, ERK1/2, and Akt phosphorylation (Fig. 2G). Reconstitution of AR-depleted osteoblasts with wild-type AR (ARWT) or DNA-binding mutant AR (ARC577A) fully restored DHT-induced Src, ERK1/2, and Akt phosphorylation, whereas reconstitution with the membrane-association mutant AR (ARC807A) failed to restore signaling (Fig. 2G). Thus, membrane association of the AR, but not its DNA-binding activity, was necessary for the rapid response of POBs to DHT.
We used density gradient centrifugation to assess if the membrane-associated AR resided in membrane lipid rafts, which are cholesterol-rich plasma membrane microdomains that contain caveolin-1 and function as signal transduction platforms (38). Endogenous AR from male POBs (Fig. 2H) and Flag-ARWT expressed in MC3T3 cells (fig. S3F) was present in the same fractions as caveolin-1, NOS3, PKG2, and Src. Moreover, endogenous caveolin-1 coimmunoprecipitated with Flag-ARWT in MC3T3 cells (Fig. 2I). Thus, DHT-induced rapid signaling through Src, ERK1/2, and Akt required membrane-associated AR, which colocalized with PKG2 and Src in caveolin-1-enriched membrane fractions.
Rapid androgen signaling through PKG2 and ERK enhances AR nuclear accumulation in osteoblasts
Androgen binding to cytoplasmic AR induces its dissociation from chaperones and subsequent translocation into the nucleus, and androgens inhibit nuclear AR degradation, thereby enhancing AR accumulation in the nucleus (39, 40). Phosphorylation of AR on Ser515 within an ERK1/2 consensus sequence may increase AR stability and transcriptional activity in transfected cells (41, 42).
To determine if PKG2-dependent ERK activation regulates AR nuclear accumulation, we performed immunofluorescence staining with antibodies recognizing total AR or AR phosphorylated on Ser515 (Fig. 3, A and B). In the absence of androgen, only ~6% of POBs from wild-type mice showed more AR in the nucleus than in the cytoplasm, but after just 4 h of DHT treatment, ~20% of the cells showed more AR in the nucleus than in the cytoplasm (Fig. 3A). This is to be contrasted with PKG2-deficient osteoblasts from OB Prkg2-KO mice, in which DHT had a very modest effect and <10% of cells showed more nuclear than cytoplasmic AR after 4 h of stimulation (Fig. 3A). When the AR was evaluated using an antibody recognizing the form that is phosphorylated on Ser515, ~45% of DHT-treated wild-type cells showed more nuclear than cytoplasmic staining, whereas essentially no phosphorylated AR was observed in PKG2-deficient cells (Fig. 3B). In unstimulated wild-type cells, the PKG-activating cGMP analog 8-CPT-cGMP induced nuclear accumulation of total and Ser515-phosphorylated AR, but no nuclear accumulation or phosphorylation of the AR occurred in PKG2-deficient cells (Fig. 3, A and B). The MEK inhibitor U0126, which blocks ERK activation, completely prevented AR Ser515 phosphorylation in wild-type cells and reduced or eliminated AR nuclear accumulation in response to DHT or 8-CPT-cGMP, respectively (Fig. 3, A and B). Thus, DHT-induced nuclear accumulation of the AR partly depends on PKG2 and ERK, and cGMP can induce some AR nuclear accumulation in the absence of androgen, likely through activation of PKG2 and ERK.
Fig. 3. Rapid androgen signaling through PKG2-dependent activation of ERK enhances AR nuclear accumulation.
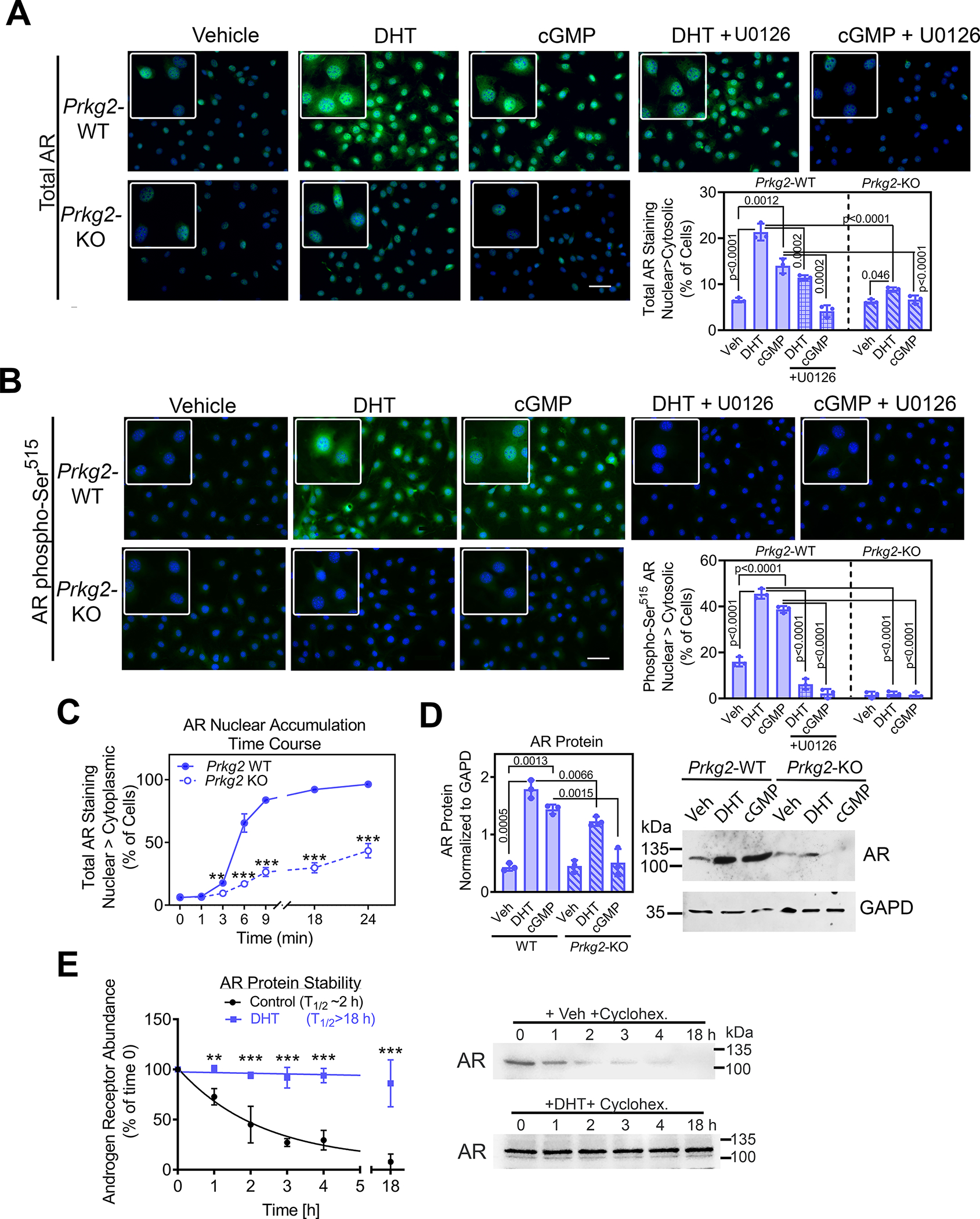
(A and B) Immunofluorescence staining for total AR (A) or AR phosphorylated on Ser515 (B) in POBs from male Prkg2-WT mice or OB Prkg2-KO mice. Cells were treated for 4 h with vehicle, DHT, or the cGMP analog 8-CPT-cGMP, and some cells were pre-treated with the MEK inhibitor U1026. Graphs show the percentage of cells in which nuclear fluorescence was greater than cytosolic fluorescence; at least 200 cells from 3 independent experiments were analyzed per condition, and the threshold for cytosolic fluorescence intensity was defined in untreated WT cells. Effects of U1026 in WT cells were analyzed separately by one-way ANOVA; comparison between WT and KO cells treated with DHT or cGMP was by two-way ANOVA. (C) WT and KO POBs were treated with DHT for the indicated times, with nuclear and cytosolic fluorescence staining for total AR assessed as in (A). (D) Immunoblotting for and quantification of total AR protein in whole cell lysates of WT and KO POBs treated for 24 h with vehicle, DHT or 8-CPT-cGMP. GAPD is a loading control. (E) AR protein abundance assessed by immunoblotting at the indicated times after addition of cycloheximide at time 0 to male WT POBs; cells were treated with vehicle or DHT for 4 h prior to and during incubation with cycloheximide. The graphs in (C) to (E) show means ± S.D of three independent experiments; p values for the indicated comparisons by two-way ANOVA, with **p<0.01 and ***p<0.001 for the comparison between WT and KO cells (C) or between vehicle and DHT (E) at each timepoint.
Nuclear AR accumulation was detectable as early as 3 h after adding DHT to wild-type cells and by 18 h was observed in almost all cells; in contrast, nuclear accumulation of AR was delayed and reduced in PKG2-deficient cells, reaching only 30% at 18 h (Fig. 3C and fig. S4A). Total AR protein was increased ~2-fold after 4 h of DHT treatment and was increased almost 4-fold after 24 h of DHT or 8-CPT-cGMP treatment (Fig.3D, fig. S4D). The DHT effect was blunted and the cGMP effect was absent in PKG2-deficient cells (Fig. 3D). The amount of AR mRNA paralleled the changes in total AR protein, with DHT treatment increasing Ar mRNA and AR protein stability in wild-type osteoblasts (Fig. 3E and fig. S4, B and C). These results are consistent with androgens increasing AR abundance through both transcriptional and posttranscriptional mechanisms in prostate cancer (43). Thus, DHT-induced nuclear AR accumulation in wild-type osteoblasts likely results from a combination of increased synthesis, decreased degradation, and increased nuclear import (39). Although DHT-induced nuclear AR accumulation was defective in PKG2-deficient osteoblasts, basal amounts of Ar mRNA and protein were similar in wild-type and PKG2-deficient cells (Fig. 3D and fig. S4B).
Androgen stimulation increases β-catenin abundance and target gene expression in osteoblasts through membrane-associated PKG2, Src, Erk, and Akt
We previously found that male, but not female, OB Prkg2-KO mice express less β-catenin mRNA and protein in bone compared to wild-type littermates, and POBs from male OB Prkg2-KO mice show defects in differentiation and Wnt-β-catenin–related gene expression that are not seen in female cells (20). To better understand PKG2 regulation of Wnt-β-catenin–related genes in males, we examined the effects of DHT and cGMP on the expression of Ctnnb1, which encodes β-catenin, and the β-catenin target genes Axin2 (axin-2), Ccnd1 (cyclin D1), Ccn1 (cellular communication network factor-1), and Tnfrsf11b (osteoprotegerin) (44–47).
DHT or 8-CPT-cGMP treatment of POBs from male Prkg2-WT mice increased β-catenin protein and Ctnnb1 mRNA ~4-fold, but neither agent affected cells from OB Prkg2-KO mice (Fig. 4, A and B). DHT did not affect Ctnnb1 mRNA stability (fig, S5A), but it increased the half-life of β-catenin protein from about 3 h to >18 h (fig. S5C). This stabilization of β-catenin was accompanied by an increase in the phosphorylation of glycogen synthase kinase–3β (GSK-3β) on Ser9 (fig. S5B). We and others have previously reported on β-catenin stabilization through the inhibitory phosphorylation of GSK-3β on Ser9, mediated by PKG2 and Akt (21, 22, 30, 48).
Fig. 4. Androgen stimulation of β-catenin and its target genes requires signaling by PKG2, Src, Erk, and Akt.
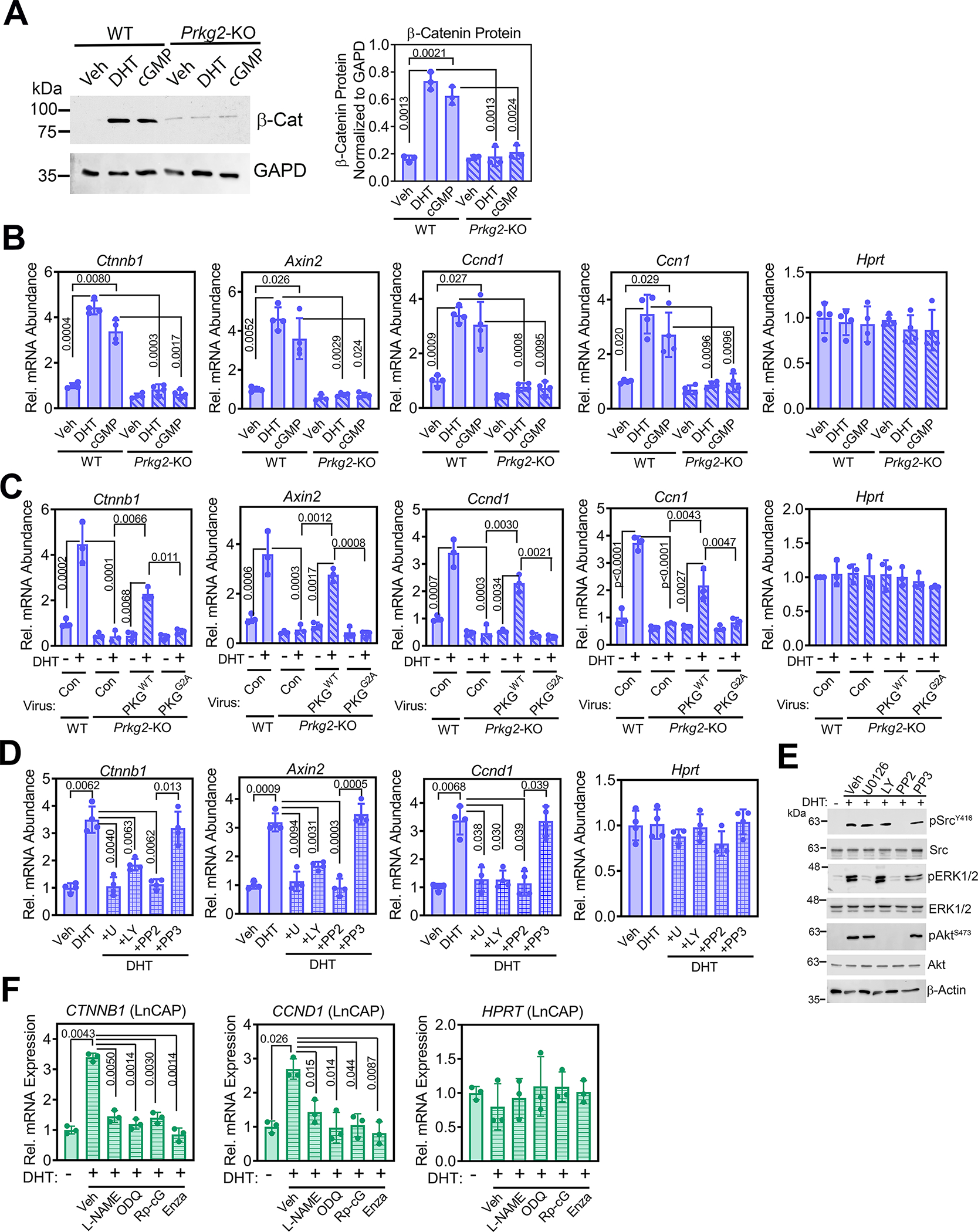
(A) Immunoblotting for and quantification of total β-catenin protein in POBs from male Prkg2-WT and OB Prkg2-KO mice treated with DHT or 8-CPT-cGMP for 24 h. GAPD is a loading control. (B) Relative mRNA abundance of Ctnnb1 and β-catenin target genes (Axin2, Ccnd1, and Ccn1) normalized to 18S RNA in POBs from male Prkg2-WT and OB Prkg2-KO mice treated with DHT or 8-CPT-cGMP for 24 h. Hprt served as a control. Mean ΔCt values obtained in vehicle-treated Prkg2-WT cells were assigned a value of one. (C) Quantification of the indicated mRNAs in Prkg2-KO osteoblasts reconstituted with virus encoding LacZ (con), membrane-associated WT PKG2 (PKGWT), or cytosolic mutant PKG2 (PKGG2A); cells were treated as in (B). (D) Quantification of the indicated mRNAs in WT POBs pre-treated with U0126, LY29004, PP2, or PP3 (inactive PP2 analog) prior to treatment with DHT for 24 h. (E) Immunoblotting for total and phosphorylated (p) forms of Src, ERK1/2, and Akt in male WT POBs pretreated with U0126, LY29004, PP2, or PP3 and treated with DHT for 10 min; quantification of 3 independent experiments is in fig. S5E. Actin is a loading control. (F) Quantification of Ctnnb1 and Ccnd1 mRNAs in LnCAP prostate cancer cells pre-treated with L-NAME, ODQ, Rp-8-CPT-PET-cGMPS (Rp-cG), or enzalutamide (enza), prior to receiving DHT for 24 h. All graphs show means ± S.D. of three (A, C, E, and F) or four (B and D) independent experiments; p values for the indicated comparisons by two-way (A to C) or one-way (D and F) ANOVA. Separate, identical gels loaded with equal amounts of cell lysates were run for analysis of total and phospohorylated forms of Src, ERK, and Akt.
Consistent with their effects on β-catenin abundance, DHT and 8-CPT-cGMP each induced all four β-catenin target genes (Axin2, Ccnd1, Ccn1, and Tnfrsf11b) in wild-type osteoblasts, but DHT only partly induced Tnfrsf11b and neither agent affected Axin2, Ccnd1, or Ccn1 expression in PKG2-deficient cells (Fig. 4B and fig. S5D). Adenoviral expression of wild-type PKG2 restored DHT-induced expression of β-catenin and its targets in PKG2-deficient osteoblasts, whereas expression of the membrane-binding–incompetent PKG2G2A was ineffective (Fig. 4C). Inhibition of Src (PP2) or MEK and ERK (U0126) completely blocked, and inhibition of PI3K and Akt (LY294002) reduced DHT-induced expression of Ctnnb1 and β-catenin target genes in wild-type osteoblasts (Fig. 4, D and E; fig. S5E). Thus, androgen signaling, through membrane-associated PKG2 activating Src, ERK, and Akt, is required for DHT-induced increases in Ctnnb and β-catenin target gene expression in male osteoblasts.
Androgen stimulation of β-catenin and CCND1 expression in prostate cancer cells depends on NO-cGMP-PKG signaling
Genome-wide analyses of prostate cancers and their metastases have shown that the Wnt-β-catenin pathway is important for the development and progression of this cancer, but most studies have concentrated on direct interactions between AR and β-catenin proteins in this context (49). We found that DHT increased CTNNB1 and CCND1 expression in LnCAP cells in an NO-cGMP-PKG–dependent manner, and enzalutamide prevented these effects (Fig. 4F). These results are consistent with PKG-dependent Src and ERK1/2 activation in DHT-treated LnCAP cells (Fig. 1H).
DHT increases Ctnnb1 transcription in osteoblasts by promoting AR binding near the transcription start site
DHT and cGMP increased Ctnnb1 expression in male POBs largely by stimulating transcription from the β-catenin promoter, because both agents increased luciferase expression from a human CTNNB1 promoter reporter in POBs (Fig. 5A), similarly to their effects on endogenous Ctnnb1 expression in POBs (Fig. 4B). Consistent with Src and MEK inhibitors blocking DHT-induced endogenous Ctnnb1 expression in POBs (Fig. 4D), the inhibitors blocked DHT-induced activation of the CTNNB1 promoter reporter in POBs (Fig. 5B).
Fig. 5. DHT increases Ctnnb1 transcription by inducing AR binding near the transcription start site.
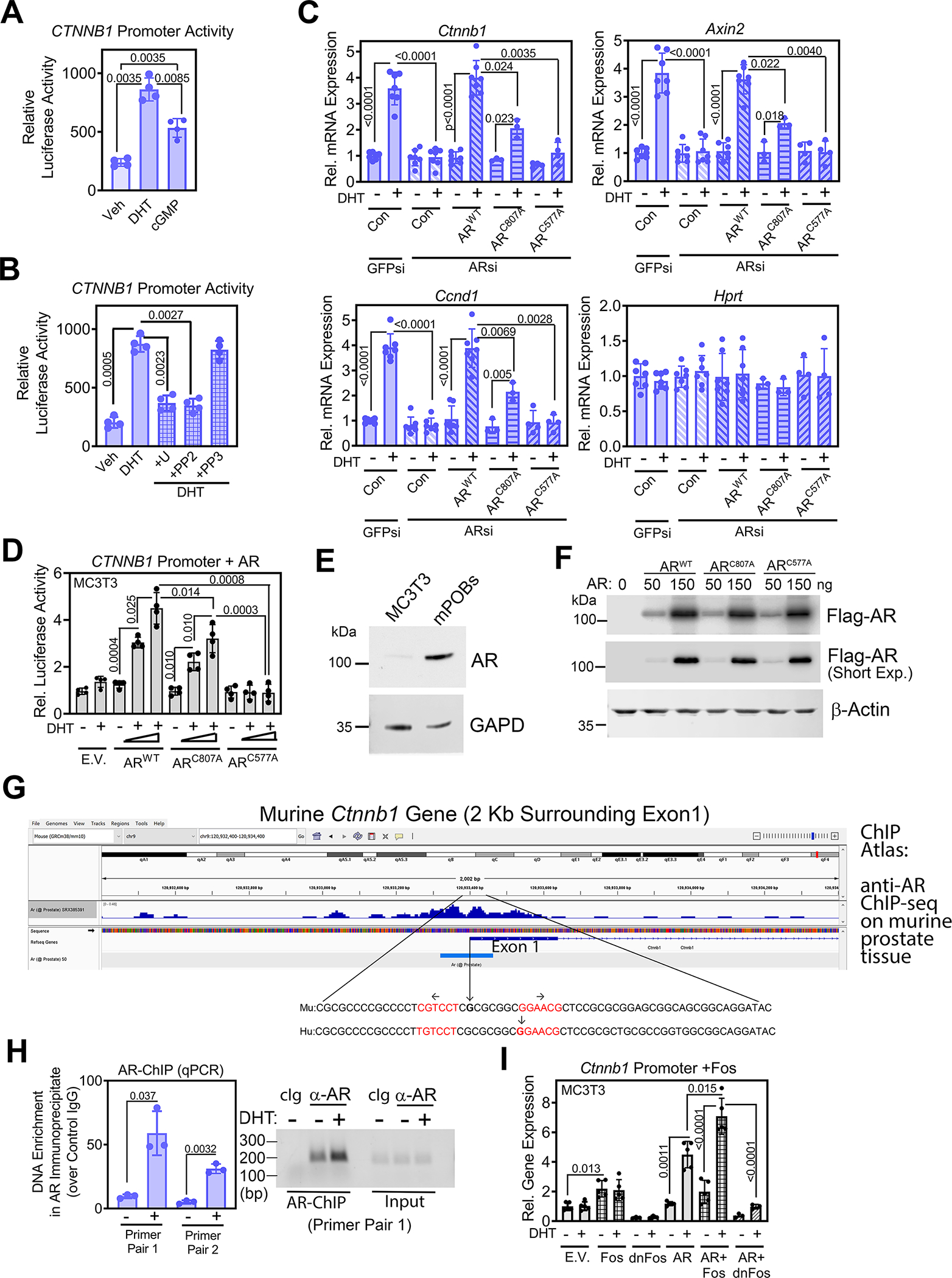
(A) Luciferase activity from a human CTNNB1 promoter–driven luciferase reporter in male WT POBs after 36 h treatment with vehicle, DHT, or 8-CPT-cGMP. (B) Luciferase activity from the CTNNB1 reporter in POBs pre-treated with U0126, PP2, or PP3 prior to treatment with DHT. (C) Relative abundance of Ctnnb1 mRNA and β-catenin target genes (Axin2, Ccnd1), normalized to 18S RNA, in male WT POBs. Cells were transfected with siRNAs targeting GFP or AR and received LacZ virus (Con) or virus encoding Flag-tagged ARWT, membrane binding-deficient ARC807A, or DNA binding-deficient ARC577A prior to treatment with vehicle or DHT for 24 h. Hprt served as a control. Mean ΔCt values obtained in vehicle-treated cells were assigned a value of one. (D) Luciferase activity in MC3T3 cells co-transfected with the CTNNB1 reporter and either empty vector (E.V.) or increasing amounts of ARWT, ARC807A, or ARC577A; cells were treated with vehicle or DHT for 36 h. (E) Immunoblotting for endogenous AR in untransfected MC3T3 cells and male WT POBs. GAPD is a loading control. (F) Immunoblotting for the Flag-tagged AR constructs in MC3T3 cells transfected as in (D). Quantification in shown fig. S6D. Actin is a loading control. (G) Published ChIP-seq results for AR binding to sequences surrounding exon 1 of the Ctnnb1 gene in murine prostate tissue (50). The enlargement shows conserved murine and human sequences with the transcription start sites marked by vertical arrows, and an inverted repeat of an ARE-like motif highlighted in red. (H) Quantification of Ctnnb1 enrichment in chromatin immunoprecipitation using an AR-specific antibody from POBs treated with vehicle or DHT for 6 h. Sequences near the Ctnnb1 transcription start site were amplified by two independent primer pairs. QPCR results are expressed as a fold increase over the signal obtained with control IgG; PCR products were also analyzed by agarose gel electrophoresis (right). (I) Luciferase activity in MC3T3 cells co-transfected with the CTNNB1 reporter and empty vector (E.V.) or vectors encoding c-Fos, a dominant-negative Fos (dnFos), and/or ARWT as indicated and treated with vehicle or DHT for 36 h. Bar graphs show means ± S.D. of 3 (H), 4 (A, B, C, D) or 5 (I) independent experiments; in (C), 3 independent experiments comparing ARWT to ARC807 and 4 experiments comparing ARWT to ARC577A were combined. All blots are representative of 3 independent experiments. P values for the indicated comparisons by one-way ANOVA (A and B) or two-way ANOVA (C, D, and I); for H, comparison by two-tailed Welch’s t test.
Enzalutamide prevented the DHT-induced expression of Ctnnb1, Axin2, and Ccnd1 mRNAs in male POBs (fig. S6A). Enzalutamide also blocked DHT-induced increases in Ar mRNA and AR protein (fig. S6, A and B), consistent with AR autoregulation observed in other cell types (43). Correspondingly, siRNA-mediated AR depletion prevented the DHT-induced increases in Ctnnb1, Axin2, and Ccnd1 mRNAs (Fig. 5C). Reconstitution of AR-depleted cells with ARWT completely restored DHT-induced Ctnnb1, Axin2, and Ccnd1 mRNA expression, whereas reconstitution with the membrane binding–deficient ARC807A only partly restored DHT-induced gene expression (Fig. 5C). In contrast, reconstitution of AR-depleted cells with the DNA binding–deficient ARC577A failed to restore any DHT-induced Ctnnb1, Axin2, or Ccnd1 mRNA expression (Fig. 5C). Taken together, these results indicate that DHT induction of Ctnnb1 and β-catenin target genes requires DNA binding of the AR and is partly dependent on rapid signaling from membrane-associated AR through Src and ERK1/2.
In MC3T3 pre-osteoblast–like cells, DHT had very little effect on expression of the luciferase CTNNB1 promoter reporter (Fig. 5D), consistent with the low AR protein and Ar mRNA abundance in these cells (Fig. 5E, and fig. S6C). Although MC3T3 cells were originally derived from murine embryos of both sexes, it is possible that the subclone used here, MC3T3-E1 subclone 4, represents a female subclone, potentially explaining the lack of AR in these cells. Transfection of Flag-ARWT into MC3T3 cells dose-dependently increased CTNNB1 promoter activity in the presence of DHT (Fig. 5D). Flag-ARC807A was partly active, and Flag-ARC577A was inactive in this system (Fig, 5D), despite production of similar amounts of the tagged proteins in the cells (Fig. 5F, and fig. S6D). These results are consistent with the reconstitution experiments in AR-depleted male POBs, showing partial or no activity of the membrane binding-deficient or DNA binding-deficient AR mutants, respectively (Fig. 5C).
Published ChIP-seq data from normal murine prostate tissue, human fetal prostate, and human breast and prostate cancer cell lines show AR binding near the Ctnnb1 and CTNNB1 transcription start sites (Fig. 5G and fig. S7A) (50). The highly conserved sequences surrounding the start of Ctnnb1 and CTNNB1 exon 1 include an inverted repeat of an androgen response element (ARE)-like motif with an eight-nucleotide spacer (Fig. 5G), resembling an established ARE found in the TMPRSS2 gene (51). We performed ChIP assays using an AR-specific antibody in unstimulated murine POBs treated with vehicle or DHT for 6 h, and found DHT-inducible AR binding to these sequences near the β-catenin transcription start site (Fig. 5H). Thus, AR binds to the Ctnnb1 promoter in murine POBs, confirming published ChIP-Seq data from other cell types and tissues.
Because the Ctnnb1 promoter contains several functional AP-1 binding sites (52), we sought to determine whether c-Fos contributes to DHT regulation of the Ctnnb1 promoter. DHT and 8-CPT-cGMP increased c-Fos protein 4–5-fold in wild-type murine osteoblasts, but DHT had less effect and cGMP had no effect in PKG2-deficient cells (fig. S7, B and C). These results are consistent with our previous report that PKG2 induces c-Fos mRNA expression through ERK in osteoblasts (53). Transfection of c-Fos into MC3T3 cells doubled luciferase expression from the CTNNB1 promoter reporter, and cotransfection of c-Fos with ARWT had an additive effect (Fig. 5I). In contrast, expression of a dominant-negative Fos protein (54) severely reduced DHT-induced AR effects on the promoter (Fig. 5I). Thus, DHT increases Ctnnb1 transcription by inducing AR binding to the Ctnnb1 promoter, and PKG2-dependent induction of c-Fos likely contributes to androgen regulation of the promoter.
Androgen stimulation of cell proliferation, mitochondrial activity, survival, and osteoblastic differentiation requires NO-cGMP-PKG2 signaling
To determine if stimulation of the NO-cGMP-PKG2 pathway mediated physiological functions of androgens, we measured cell proliferation and mitochondrial activity in POBs and LnCAP cells and apoptosis and osteoblastic differentiation in POBs. We found that DHT increased cell proliferation, as measured by bromo-deoxyuridine (BrdU) incorporation into S-phase nuclei, about 2-fold in male wild-type POBs but had no effect on Prkg2-KO cells (Fig. 6A). DHT also increased proliferation in LnCAP cells, with the increase in proliferation completely abolished by NO-cGMP-PKG2 pathway inhibitors (Fig. 6B), This increase in proliferation is consistent with our observation that DHT treatment increased CCND1 expression in LnCAP cells (Fig. 4F). In both murine osteoblasts and LnCAP cells, DHT increased mitochondrial activity, as measured by MTT reduction to formazan, and again this effect was blocked by NO-cGMP-PKG2 pathway inhibitors (Fig. 6, C and D).
Fig. 6. Androgen stimulation of cell proliferation, mitochondrial activity, and survival requires NO-cGMP-PKG2 signaling.
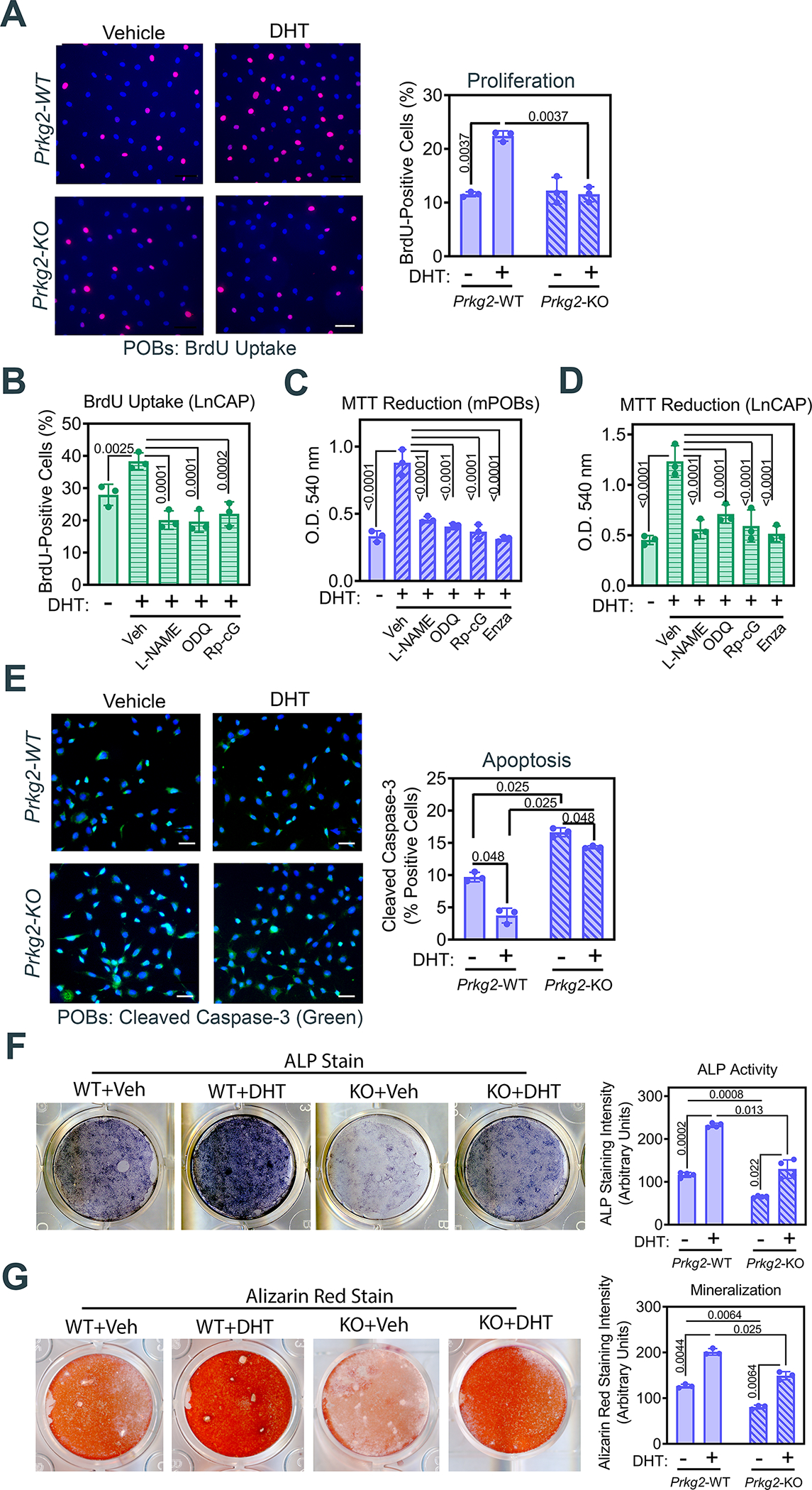
(A) Immunofluorescence staining for and quantification of bromo-deoxyuridine (BrdU) incorporation into S-phase nuclei in POBs from male Prkg2-WT or OB Prkg2-KO mice treated with DHT for 24 h. Scale bar, 50 μm. (B) Quantification of immunofluorescence staining for BrdU uptake in LnCAP prostate cancer cells pretreated with L-NAME, ODQ, or Rp-8-CPT-PET-cGMPS (Rp-cG), prior to receiving BrdU and DHT or vehicle for 24 h. (C and D) Mitochondrial MTT reduction to formazan measured spectrophotometrically in WT POBs (C) and LnCAP cells (D) pretreated with L-NAME, ODQ, Rp-8-CPT-PET-cGMPS, or enzalutamide, and treated with DHT for 24 h. (E) Immunofluorescence staining for and quantification of cleaved caspase-3 in serum-starved POBs from male Prkg2-WT or OB Prkg2-KO mice treated with DHT for 24 h. Scale bar, 50 μm. (F and G) Quantification of alkaline phosphatase (ALP) activity (F) and alizarin red staining (G) in Prkg2-WT or OB Prkg2-KO POBs cultured in differentiation medium and treated with vehicle or DHT for 14 d (F) or 21 d (G), respectively. All graphs show means ± S.D. of 3 (A to E, G) or 4 (F) independent experiments. P values for the indicated comparisons by one-way ANOVA (B to D) or two-way ANOVA (A, E, F and G).
DHT markedly reduced apoptosis caused by serum-starvation in wild-type POBs, as measured by cleaved caspase-3 staining, but had a more modest effect in the Prkg2-KO cells, which showed a higher amount of basal cleaved caspase-3 (Fig. 6E). To test the involvement of PKG2-mediated AR signaling in osteoblastic differentiation, we induced differentiation in post-confluent cultures to ensure that the results were not influenced by cell proliferation. DHT promoted osteoblastic differentiation, as assessed by alkaline phosphatase activity and Alizarin Red staining of mineralized matrix, when added to post-confluent POBs in medium containing ascorbate and β-glycerolphosphate (Fig. 6, F and G). The effects of DHT on osteoblastic differentiation were more pronounced in wild-type compared to Prkg2-KO cells (Fig. 6, F and G). Thus, DHT’s positive effects on osteoblast proliferation, survival, and differentiation were at least partly mediated by PKG2.
Estrogen regulation of β-catenin and its target genes is PKG2-independent
To better understand why female OB Prkg2-KO mice do not have reduced bone mass as occurs in male OB Prkg2-KO mice, we compared the effects of estrogen and 8-CPT-cGMP on Ctnnb1, Axin2, and Ccnd1 mRNA expression in male and female POBs from wild-type and OB Prkg2-KO mice. 17β-estradiol (E2, the main circulating estrogen) increased expression of the three genes ~ 2-fold in male and 4–6-fold in female wild-type cells, whereas 8-CPT-cGMP stimulated gene expression 4–5-fold in wild-type cells of both sexes (Fig. 7, A and B). The weaker estrogen effect in male osteoblasts was likely due to lower ER-α mRNA (Esr1) and protein expression in male compared to female cells (Fig. 7C and fig. S8, A and B). Esr1 mRNA and ER-α protein abundance did not vary between Prkg2-WT and Prkg2-KO osteoblasts, and ER-β mRNA (Esr2) expression did not differ between sexes or genotypes (Fig. 7C and fig. S8, A and B). As might be expected, Ar mRNA and AR protein were considerably higher in male than female cells (Fig. 7C, and fig. S8, A and B).
Fig. 7. Estrogen induction of β-catenin and its target genes is PKG2-independent.
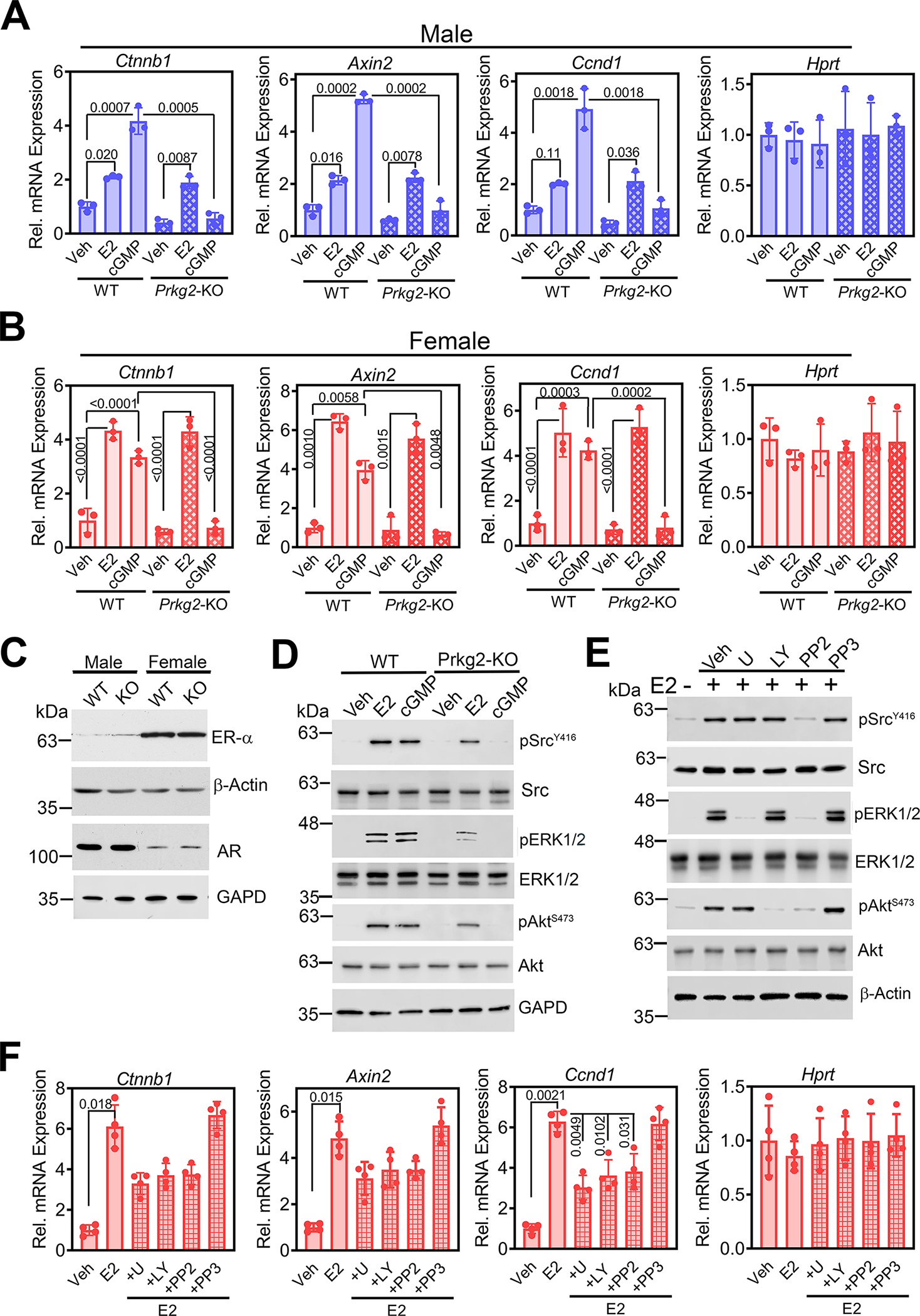
(A and B) Relative abundance of Ctnnb1 mRNA and the β-catenin target genes Axin2 and Ccnd1 normalized to 18S RNA in POBs from male (A) or female (B) Prkg2-WT or OB Prkg2-KO mice treated with 17β-estradiol (E2) or 8-CPT-cGMP (cGMP) for 24 h. Hprt served as a control. Mean ΔCt values in vehicle-treated WT cells were assigned a value of one. (C) Immunoblotting for ER-α and AR in whole-cell extracts of male and female WT and KO POBs. β-actin and GAPD are loading controls. Quantification of three independent experiments is shown in fig. S8B. (D) Immunoblotting for total and phosphorylated (p) forms of Src, ERK1/2, and Akt in female Prkg2-WT and Prkg2-KO POBs treated with E2 or 8-CPT-cGMP for 10 min. Quantification of 5 independent experiments is shown in fig. S8D. (E) Immunoblots of the indicated proteins in female WT POBs pre-treated with U1026, LY294002, PP2, or PP3 and treated with E2 for 10 min. Quantification of 2 independent experiments is shown in fig. S8E. (F) Relative abundance of Ctnnb1, Axin2, and Ccnd1 transcripts in female WT POBs pretreated with the indicated drugs and treated with E2 for 24 h. Graphs show means ± S.D. of 3 (A and B) or 4 (F) independent experiments; P values for the indicated comparisons by one-way (F) or two-way ANOVA (A and B). Separate, identical gels loaded with equal amounts of cell lysates were run for analysis of total and phospohorylated forms of Src, ERK, and Akt.
In contrast to the effects of DHT in male POBs (Fig. 4B and fig. S5D), effects of estradiol on Ctnnb1, Axin2, Ccnd1, and Tnfrsf11b mRNA expression were PKG2-independent in cells from both sexes (Fig. 7, A and B, and fig. S8C). However, cGMP effects were abolished in both male and female PKG-deficient cells (Fig. 7, A and B). Similar to DHT, estradiol rapidly induced Src, ERK1/2, and Akt activation in female wild-type cells, but, in contrast to DHT, estradiol still increased Src, ERK1/2, and Akt phosphorylation in Prkg2-KO osteoblasts, albeit at reduced amounts (Fig. 7D and fig. S8D) compared to male cells (Fig. 1I and fig. S2A). Furthermore, MEK/ERK, PI3K/Akt, or Src inhibitors only modestly reduced estradiol-induced Ctnnb1, Axin2, and Ccnd1 mRNA expression in female wild-type cells, with the inhibitors reducing the three mRNAs by <50%, and their effects did not reach statistical significance in most cases (Fig. 7, E and F, and fig. S8E);. This is in contrast to near-complete inhibition of DHT-induced Ctnnb1, Axin2, and Ccnd1 expression in male cells (Fig. 4D and fig. S5D).
We conclude that androgens are fully dependent on PKG2 for rapid activation of Src, Erk, and Akt and consequent induction of β-catenin and its target genes in osteoblasts and prostate cancer cells. In contrast, estrogens act largely in a PKG-independent fashion.
DISCUSSION
We showed that full-length plasma membrane–associated AR was part of an AR-NOS3-PKG2-Src signaling node that mediated rapid, non-genomic androgen signaling that enhanced nuclear AR accumulation and AR-induced transcription of the gene encoding β-catenin and increased expression of β-catenin target genes in mouse osteoblasts and prostate cancer cells (Fig. 8). β-catenin is important for skeletal homeostasis because it stimulates the expression of genes that promote osteoblast proliferation, survival, and differentiation. By establishing a requirement for PKG2 in androgen signaling and androgen regulation of β-catenin, we explain the sexually dimorphic phenotype of OB Prkg2-KO and OB PKG2R424Q-TG mice.
Fig. 8. Transcriptional regulation of β-catenin through PKG2-mediated cooperation between membrane-associated and nuclear androgen receptors.
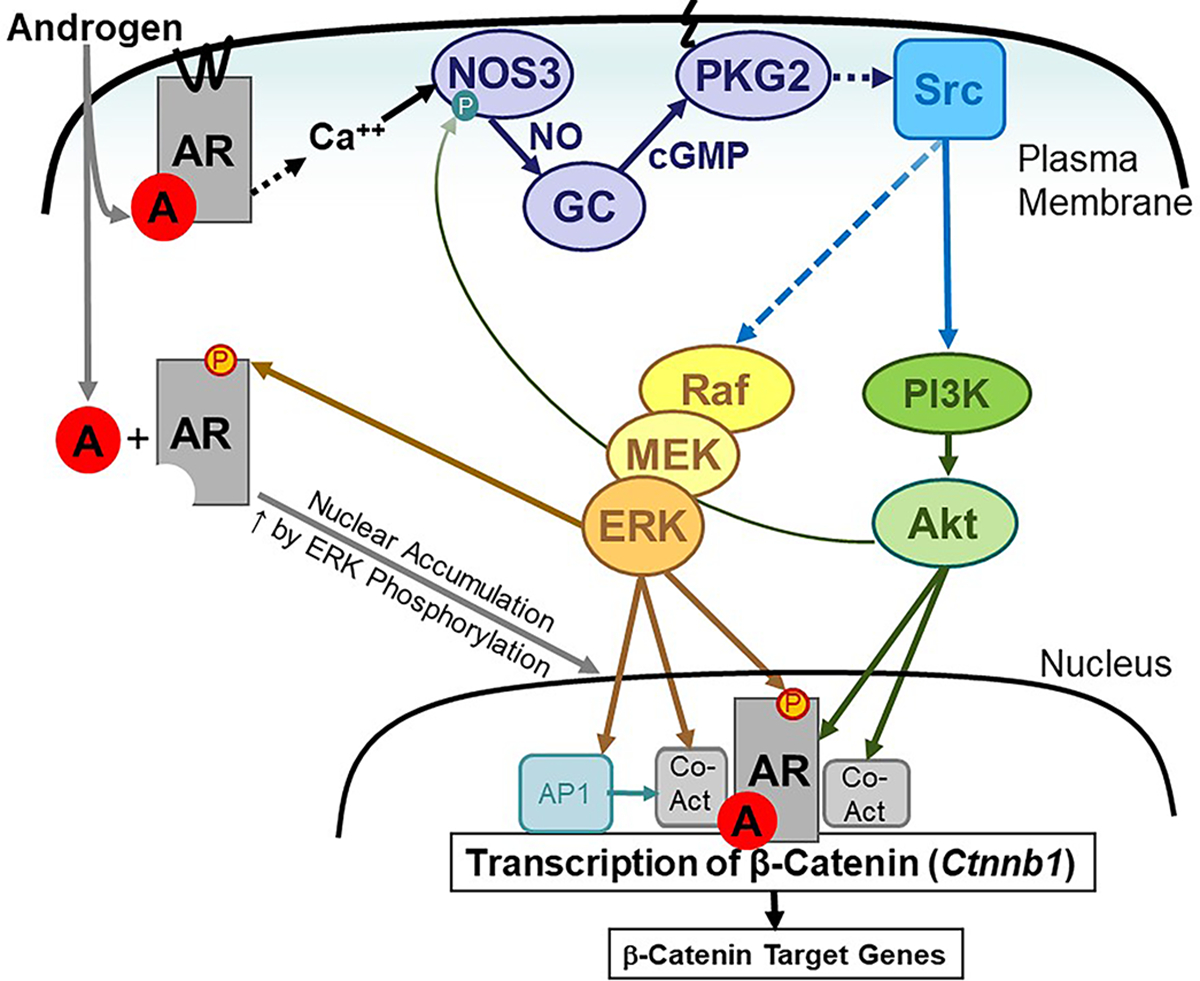
Androgen (A) binding to the membrane-associated AR in osteoblasts increases the intracellular Ca2+ concentration, which activates NOS3 to produce NO. NO activates guanylyl cyclase-1 (GC) to produce cGMP, which activates membrane-bound PKG2. As we have previously shown, PKG2 activates Src in a Shp1/2–dependent manner to increase ERK1/2 activity through Raf and MEK (15) and Akt activity through phosphoinositol-3-kinase (PI3K) (30). ERK-mediated phosphorylation of AR on Ser515 enhances AR nuclear accumulation. Akt can phosphorylate AR, and ERK and Akt can phosphorylate transcriptional co-activators to increase AR transcriptional activity (40). A positive feedback mechanisms enhances NOS activity through Akt-mediated phosphorylation of NOS3. AR binds near the transcription start site of the Ctnnb1 gene to increase transcription, thereby promoting the expression of β-catenin target genes In addition, PKG2-dependent ERK activation increases c-Fos, which can bind to the Ctnnb1 promoter as part of the AP-1 complex (52) and cooperate with AR to enhance transcription.
Rapid signaling by membrane-associated steroid hormone receptors
Androgens bind to plasma membrane fractions (24), and several candidate membrane receptor proteins have been proposed, including G-protein–coupled receptors, cation channels, and the full-length as well as truncated forms of the AR (3, 5, 33). We now demonstrate that the full-length AR colocalized with NOS3, PKG2, and Src in caveolar fractions of osteoblast plasma membranes, and that on ligand binding, the membrane receptor initiated NO-cGMP-PKG2 signaling, thereby regulating cell growth, survival, and differentiation (Fig. 8). Extra-nuclear AR signaling through Src, ERK, and Akt has been previously noted to increase proliferation and survival of prostate cancer cells, stimulate growth and development of ovarian granulosa cells, and enhance glucose-stimulated insulin secretion in pancreatic β-cells (3, 6–8).
Membrane-initiated estrogen, androgen, and progesterone signaling pathways appear to share many similarities (3). Membrane-proximal events for all three steroid receptors include direct interactions between AR, ER-α, and the progesterone receptor with Src and the p85 subunit of PI3K to activate the Src-Ras-ERK and PI3K-Akt signaling pathways, respectively (2, 6, 28, 33, 34). We now demonstrate that DHT-induced Src, ERK, and Akt activation occurred downstream of NO-cGMP-PKG2 signaling in both osteoblasts and prostate cancer cells (Fig. 8). We previously showed that PKG2 phosphorylates and thereby activates the phosphatases Shp1 and Shp2 during NO-mediated mechanotransduction in osteoblasts (15). Shp1 and Shp2 dephosphorylate an inhibitory site on Src (Tyr529), and the activated Src stimulates the Ras-Raf-MEK-ERK and PI3K-PDK1-Akt pathways (15, 30). Src is likely activated by a similar mechanism in DHT-treated osteoblasts.
ER-α and ER-β are targeted to the plasma membrane by the palmitoylation of a cysteine residue within a nine amino acid motif in the ligand-binding domain; this membrane-targeting motif is highly conserved among steroid receptors (2, 36). We showed that mutation of the conserved cysteine produced a mutant AR that did not localize to the plasma membrane and could not reconstitute rapid androgen signaling through Src, ERK, and Akt.
Additionally, we found that estrogens and androgens differed in their requirements for PKG2 in activating Src, ERK, and Akt in osteoblasts: 17β-estradiol signaling was only partly diminished, whereas DHT signaling was completely abolished in PKG2-deficient cells. In endothelial cells, estrogens stimulate NOS3 activity through Src and PI3K/Akt activation in a calcium-dependent fashion (27, 55, 56). Because estrogens also stimulate NO and cGMP production in osteoblasts (14, 57), one would expect a role for PKG2 signalling downstream of ER-α, and we found a partial PKG2-dependence of estrogen-induced Src, ERK, and Akt activation. The PKG2-independent effects of estrogens may involve direct ER-α interactions with growth factor receptors; for example in breast cancer cells, estrogens activate Src, ERK, and Akt by ER-α interaction with insulin-like growth factor-1 receptor (2, 58).
Cooperation between membrane and nuclear AR involving PKG2 and ERK
Rapid androgen, estrogen, and progesterone signaling through membrane-localized steroid receptors can potentiate transcriptional activities of the cognate nuclear receptors though phosphorylation-dependent stimulation of nuclear import, DNA binding, and coactivator recruitment (3, 4). In prostate cancer cells, epidermal growth factor activation of ERK induces AR phosphorylation on Ser515, and mutation of this site (ARS515A) reduces AR transcriptional activity (42). We found that androgens, through a signaling node consisting of membrane AR, NOS3, PKG2, and Src, induced ERK-dependent AR Ser515 phosphorylation to enhance AR nuclear accumulation in osteoblasts. PKG2-dependent ERK activation was necessary for DHT to stimulate Ctnnb1 transcription by inducing AR binding to the β-catenin promoter. Thus, rapid androgen signaling by membrane AR enhanced nuclear AR functions. In contrast, 17β-estradiol regulation of Ctnnb1 was largely PKG2-, Src- and ERK-independent. Estrogen stimulation of β-catenin expression has been attributed to direct interactions between ER-α and β-catenin, with β-catenin autoregulating its own promoter (59). To our knowledge, androgen regulation of Ctnnb1 and CTNNB1 transcription has not been previously reported, although AR crosstalk with the Wnt-β-catenin pathway has been described in prostate cancer, where β-catenin acts as a coactivator of the AR (60, 61).
DHT-induced expression of several β-catenin target genes (Axin2, Ccnd1, and Ccn1) was completely dependent on the presence of membrane-associated AR and PKG2, in agreement with our previous report of reduced expression of these genes in bones of male OB Prkg2-KO mice compared to their wild-type littermates (20). However, DHT could still induce Tnfrsf11b (osteoprotegerin) mRNA in PKG2-deficient osteoblasts, albeit less than in wild-type cells; this corresponds to near-normal Tnfrsf11b mRNA expression in the bones of male OB Prkg2-KO mice, which do not show alterations in osteoclast numbers (20).
Transcriptional versus posttranslational regulation of β-catenin by PKG2
β-catenin is regulated posttranslationally by proteosomal degradation, initiated by its sequential phosphorylation by casein kinase-Iα and GSK-3β (62). We showed previously that PKG2 can inhibit GSK-3β in osteoblasts, thereby positively regulating β-catenin stability and nuclear translocation (30, 48, 63). We found that DHT treatment increased the stability of β-catenin protein, consistent with PKG2 either directly or indirectly, through activation of Akt, phosphorylating and inhibiting GSK-3β (30, 48, 63). Because we and others have previously studied β-catenin stabilization through GSK-3β inhibition (21, 22, 30, 48), we focused in this work on the previously-unrecognized transcriptional regulation of β-catenin by DHT. DHT stimulated transcription from a CTNNB1-dependent reporter, increased Ctnnb1 mRNA expression without affecting Ctnnb1 mRNA stability, and induced AR binding to a conserved ARE-like sequence near the transcription start site of the Ctnnb1 gene. Thus, the increase in β-catenin protein observed in DHT-treated osteoblasts is likely from a combination of AR-dependent transcriptional activation of the Ctnnb1 gene and posttranslational regulation of β-catenin by PKG2 and GSK-3β.
PKG2 requirement for AR signaling in vivo
Using osteoblast-specific PKG2 knockout and transgenic mice (OB Prkg2-KO and OB PKG2R242Q-TG, respectively), we previously showed that PKG2 increases β-catenin protein and the expression of its target genes in the bones of male, but not female, mice (20, 21). Male OB Prkg2-KO mice have low bone mass with reduced bone formation and decreased Wnt-β-catenin–related gene expression in bones and primary osteoblasts, whereas male OB PKG2R242Q-TG mice have the opposite skeletal phenotype with increased Wnt-β-catenin–related gene expression compared to wild-type littermates (20, 21). Females in both of these mouse lines have normal bone formation and microarchitecture, with no alterations in Wnt-β-catenin–related gene expression compared to wild-type littermates (20, 21). We propose that this sexual dimorphism in the skeletal phenotype of OB Prkg2-KO and OB PKG2R242Q-TG mice is explained by androgens requiring membrane-associated PKG2 to stimulate Src, ERK, and Akt, induce transcription of β-catenin and its target genes, and enhance osteoblast proliferation, survival, and differentiation. Similar to male OB Prkg2-KO mice, male osteoblast-specific Ar knockout mice have reduced trabecular bone volumes, whereas females have no change in bone volume or a much milder phenotype, respectively (11, 20, 64). A question to be addressed in future work is whether DHT administration to gonadectomized male and female OB Prkg2-KO will increase bone mass to a lesser degree than it does in wild-type mice.
Although we observed that estradiol, like DHT, rapidly activated Src, ERK, and Akt in osteoblasts, these events were partly PKG2-independent and dispensable for estrogen induction of β-catenin and its target genes. However, transcriptional ER-α effects and direct interactions between ER-α and β-catenin are required to maintain Wnt-β-catenin signaling in female mice and osteoblasts (65).
Bone-protective effects of PKG-activating agents
We and others have shown bone-protective effects of agents that increase cGMP in rodents: NO donors, direct GC-1 stimulators, and phosphodiesterase-5 inhibitors prevent osteoporosis in diabetic male mice and ovariectomized female rodents by stimulating bone formation with modest effects on bone resorption (18, 48, 57, 66, 67). Compounds that increase cGMP increase bone formation and trabecular bone volumes to a lesser extent in intact female mice, suggesting that PKG activation can have bone-anabolic effects even in the presence of estrogens (48, 57, 68). These latter data are consistent with our current and previous results that PKG2 plays some, albeit a minor, role in estrogen signaling in osteoblasts and osteocytes (16).
Epidemiological studies indicate that taking NO-producing nitrates (for example to treat coronary artery disease) is associated with lower fracture risk in older humans (69–71), and some clinical trials suggest that nitrates increase bone formation in postmenopausal, estrogen-deficient females (72, 73). However, a large trial of nitroglycerin for postmenopausal osteoporosis yielded negative results (74). Together with the undesirable effects of long-term nitrate therapy, these results indicate the need to test newer drugs that increase cGMP with more favorable safety profiles. Our results suggest that PKG-activating agents, such as the phosphodiesterase-5 inhibitor sildenafil, might be most effective as a treatment for osteoporosis in male patients and in estrogen-deficient females. However, our data also suggest that PKG-activating drugs may be detrimental in patients with prostate cancer.
MATERIALS AND METHODS
Materials.
Primary antibodies are described in table S1; FITC- or TRITC-labeled secondary antibodies were from Jackson Laboratory. Bromodeoxyuridine (BrdU), 2-bromopalmitate, deoxyribonuclease-1, dihydrotesterone (DHT), calcein, methyl-β-cyclodextrin, L-NG-nitroarginine methylester (L-NAME), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), nifedipine, and thapsigargine were from Millipore Sigma. The Src inhibitor PP2 and its inactive analog PP3, the PI3K inhibitor LY294002, and the MEK inhibitor U1026 were from Calbiochem/EMD. Indo-1-AM was from InVitrogen and enzalutamide was from Selleck Chemicals. 8-(4-chlorophenylthio)-cGMP (8-CPT-cGMP) and the Rp isomer of 8-(4-chlorophenylthio)-β-phenyl-1,N2-ethenoguanosine-3’,5’-cyclic monophosphorothioate (Rp-8-CPT-PET-cGMPS) were from BioLog. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) was from Tocris Bioscience.
Vector constructs.
Adenoviral vectors encoding wild type and G2A-mutant rat PKG2 were described previously (75). A pcDNA3-based expression vector for human AR with an N-terminal Flag epitope tag (Flag-AR) was generated by PCR using the following primers: 5’-GTTGGATCCGCCGCTATGGAAGTGCAGTTAGGG-3’ and 5’-TGTGAAGGTTGCTGTTCCTC-3’ with pCMV-hAR (Addgene #89078) as a template. The BamHI/SmaI-digested PCR product was ligated together with a SmaI/XbaI fragment from pCMV hAR into pFlag-D. pFlag-ARC577A was constructed by inserting a HindIII/Xba fragment of pCMV-hAR-CA577-Z (Addgene #89108) into HindIII/XbaI-digested Flag-AR. Flag-ARC807A was constructed by PCR using the following primers: 5’-CAGGAATTCCTGGCCATGAAAGCAC-TGCTACTC-3’ and 5’-GTCTCTAGAGTCACTGGGTGTGGAAATAG-3’ with pCMV-hAR as a template. The PCR product was digested with EcoRI/XbaI and used to replace the EcoRI/XbaI fragment in Flag-AR. All PCR-generated products were sequenced. To produce adenovirus, the AR coding sequences were ligated into a modified pENTR vector placing a Flag-tag at the N-terminus of each construct. The coding sequences were then recombined into pAD/CMV/V5-DEST with LR Clonase II, and virus was produced in 293A cells using the ViraPower Adenoviral Expression System (Invitrogen). Adenoviral control vector encoding β-galactosidase (LacZ) was produced in parallel.
A luciferase reporter under control of the human β-catenin promoter was constructed in pGL2 (Promega) and contained sequences −3000 bp to +93 bp relative to the transcription start site (obtained by PCR using the following primers: 5’-TTGGAGAGTTGGGATTGTTTG-3’ and 5’-CGA GAGGCTTAAAATGGCG-3’) with genomic DNA from HEK293 cells serving as a template.
Osteoblast-specific PKG2 knockout mice (OB Prkg2-KO).
Prkg2f/f mice (referred to as Prkg2-WT mice) were generated in a C57BL/6NHsd background, and osteoblast-specific Prkg2-KO mice were obtained by crossing Prkg2f/f mice with transgenic mice expressing CRE recombinase under control of the 2.3-kb collagen type-1α1 promoter [B6.Cg-TG(col1a1-cre)-Haak mice from RIKEN BioResource Research Center (RBRC05524), also in a C57BL/6 background], as described previously (20, 76). Maintenance of a breeding colony and generation of POBs from Prkg2-WT and OB Prkg2-KO mice were approved by the Institutional Animal Care and Use Committee of the University of California, San Diego (protocol # S10121). All mice were housed 3–4 animals per cage in a temperature-controlled environment with a 12 h light/dark cycle and ad libitum access to water and food (Teklad Rodent Diet #8604).
Osteoblast isolation, culture, and drug treatment.
POBs were isolated from femurs and tibiae of 10–12 week-old male and female Prkg2-WT and Prkg2-KO mice as explant cultures (77). Epiphyses and bone marrow were removed, and diaphyses were cut into small fragments with a scalpel; the bone fragments were incubated with collagenase II (Worthington, 2 mg/ml) and Trypsin/EDTA (Gibco, 0.25 mg/ml and 0.5 mM) for 2 h at 37 °C. The supernatant was discarded, and the bone chips were washed prior to plating in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/ml), streptomycin (100 μg/ml), and amphotericin B (0.25 μg/ml). Every 3 d, media was exchanged and bone chips rearranged; after 11–15 d, cells were nearly confluent and were passaged by trypsinization. Each POB preparation was characterized for osteoblastic differentiation by alkaline phosphatase (ALP) staining (>85% ALP+ cells) and mineralization capacity (Alizarin Red staining). To induce osteoblastic differentiation, cells grown to confluency were changed to differentiation medium supplemented with 0.3 mM ascorbate and 10 mM β-glycerolphosphate; the medium was exchanged every two days for 14 d (ALP stain) or 21 d (Alizarin Red stain) (48). Cells were used at passages 1–4. MC3T3-E1 cells (subclone 4) were obtained from the ATCC (#CRL-2593) and cultured in α-Minimum Essential Medium (α-MEM) with ribo- and deoxyribonucleosides, glutamine, pyruvate, and 10% FBS; they were characterized by their ability to express Bglap mRNA and produce mineralized colonies in differentiation medium (30). LnCAP cells were from the ATCC (CRL-1740) and were cultured in RPMI1640 media with 10% FBS.
Prior to treatment with dihydrotestosterone (DHT, 0.1 nM unless otherwise indicated), 17β-estradiol (E2, 100 nM), or 8-CPT-cGMP (cGMP, 100 μM), cells were cultured for 18 h in phenol red-free DMEM supplemented with charcoal-stripped FBS: 0.1% (for rapid signaling < 2 h) or 1% (for gene expression changes measured after 24–48 h). As indicated, cells were pre-treated with the following inhibitors prior to receiving DHT: EGTA (2 mM), BAPTA-AM (10 μM), nifedipine (10 μM) for 10 min; L-NAME (4 mM), ODQ, (10 μM) or Rp-8-CPT-PET-cGMPS (50 μM) for 30 min; U0126 (10 μM), LY29004 (10 μM), PP2 (10 μM) or PP3 (10 μM) for 1 h; enzalutamide (10 μM), methyl-β-cyclodextrin (10 μM) or 2-bromopalmitate (10 μM) for 18 h.
To determine AR and β-catenin mRNA and protein stability, cells were cultured for 18 h in phenol red-free DMEM supplemented with 1% FBS and pre-treated with 1 nM DHT for 4 h prior to the addition of actinomycin D (5 μM) or cycloheximide (30 μg/ml), respectively.
Indo-1 Fluorescence.
Ca++ transients were measured in male POBs cultured on glass coverslips and loaded with Indo-1-AM as described (78). Briefly, cells were loaded in α-MEM containing 8% FBS, 0.02 mg/ml Pluronic-F127, and 3 μM Indo-1-AM for 30 min at 37 °C. After washing in α-MEM, cells were incubated for 30 min in α-MEM (1.8 mM Ca++) at 37 °C, and then mounted in a Nikon Diaphot fluorescence microscope equipped with a dual-emission lamp (excitation 365 nm) and Felix GX software (version 4.0.1). Simultaneous fluorescence recordings at 405 and 485 nm were carried out at room temperature from the cytoplasmic region of individual cells; care was taken to avoid fluid shear stress when adding vehicle or DHT (at 6x concentration). Relative cytoplasmic Ca++ concentrations were expressed as Indo-1 fluorescence ratio (405/485 nm).
Quantitation of NOx and cGMP.
NO production was measured as the sum of nitrite and nitrate accumulation in the cell culture medium, using a two-step colorimetric assay (17). The intracellular cGMP concentration was measured by ELISA according to the manufacturer’s instructions (Cayman Chemicals).
Quantitative RT-PCR.
POBs grown in 6-well dishes were harvested in Trizol Reagent (Molecular Research Center) for RNA purification, and quantitative RT-PCR was performed with 1 μg of RNA as described (18). Primers are summarized in table S2; they were tested with serial cDNA dilutions. Genes of interest were normalized to 18S rRNA, and the mean ΔCT obtained for untreated Prkg2-WT osteoblasts was used to calculate relative changes in mRNA expression using the ΔΔCT method.
Cell fractionation.
Cells were extracted by Dounce homogenization, nuclei were collected by centrifugation at 1,000 X g, and membranes were collected on a 30% Percoll™ step gradient by centrifugation at 84,000 X g, as previously described (17). Cytosolic fractions (overlying the membranes) were concentrated 10-fold using Amicon-Ultra (10k) centrifugal filters (Millipore). Membranes were further fractionated over a discontinuous gradient of 5–45% Optiprep™ (17).
14C-Palmitate incorporation into AR.
MC3T3 cells were transfected with empty plasmid or with plasmid encoding FLAG-tagged ARWT and were incubated for 4 hours at 37°C in medium containing 0.1% dialyzed FBS and 50 μCi/ml 14C-palmitic acid (50 mCi/mmol, Perk Elmer). Cell lysates were subjected to immunoprecipitation with anti-FLAG antibody, and immunoprecipitates were analyzed by SDS-PAGE. Gels were soaked in Fluoro-Hance (Research Products International) and exposed for autoradiography for three months at −80°C.
Western blotting and immunofluorescence staining.
Western blots were generated using horseradish peroxidase-conjugated secondary antibodies detected by enhanced chemiluminescence; blots were scanned with a LI-COR Odyssey Imager or using ImageJ for film exposures in the linear range (15). For loading control, blots were reprobed with GAPD or β-actin antibodies. Duplicate gels were run for total Src, ERK, and Akt, because reprobing blots for total Src, ERK, or Akt after they were first developed with phospho-specific antibodies caused interference.
POBs plated on glass coverslips were fixed in 4% paraformaldehyde, permeabilized with 1% Triton-X-100, and incubated with cleaved caspase-3–specific antibody (1:100 dilution), followed by a secondary FITC-conjugated antibody. For BrdU incorporation, cells were incubated with 200 μM BrdU in 0.1% FBS for 24 h; then cells were fixed, permeabilized, and incubated with DNAse I prior to staining with anti-BrdU antibody (1:100 dilution) and secondary TRITC-conjugated antibody. For PKG2, Src, and Flag-AR immunofluorescence staining, the fixed and permeabilized (0.5% Triton) cells were incubated with mouse anti-PKG2 and rabbit anti-Src or rabbit anti-Flag antibodies (1:50 dilutions) followed by TRITC- or FITC-conjugated secondary antibodies (1:100 dilutions). Nuclei were counter-stained with Hoechst 33342, and images were analyzed with a Keyence BZ-X700 fluorescence microscope (for cleaved caspase-3 and BrdU staining) or a Leica SP8 confocal microscope with lightning deconvolution (for PKG2, Src, and Flag-AR).
Liposomal transfection and adenoviral infection of cells.
POBs were transfected in 6-well dishes, when they were 60 to 80% confluent (for DNA) or 40 to 50% confluent (for siRNA), using 6 μl of Lipofectamine-3000™ (Thermo Fischer Scientific) per ml of medium containing 10% FBS; cells received a total of 2 μg/ml DNA or 100 pmol/ml siRNA. SiRNAs targeting green fluorescent protein (GFP, serving as a control) or the AR were purchased from Sigma (AR sense: CCCUAUCCCAGUCCCAAUUG, AR antisense: CAAUUGGGACUGGGAUAGGG). After 6 h, cells were transferred to medium containing 10% charcoal-stripped FBS. At 24 h after siRNA transfection, cells were infected with adenovirus at a multiplicity of infection of ~10. At 24 h post adenovirus infection, cells were serum-starved in 0.1 % or 1 % charcoal-stripped FBS for 18 h, as indicated, followed by the experimental treatment for the indicated time.
MC3T3-E1 cells were transfected in 24-well dishes using Lipofectamine-2000™ and 600 ng of DNA according to the manufacturer’s protocol (Thermo Fischer Scientific).
ChIP Assay.
POBs were grown to sub-confluence in 15 cm dishes, transferred to phenol red-free DMEM supplemented with 1% charcoal-stripped FBS for 18 h, and treated with vehicle or 1 nM DHT for 6 h. Cells were fixed in 1% formaldehyde for 8 min at R.T., neutralized with glycine and pellets were flash-frozen. ChIP assays were performed using the Simple ChIP™ enzymatic chromatin immunoprecipitation kit (Cell Signalling). Chromatin was digested with micrococcal nuclease (0.4 μl/100 μl Simple ChIP™ enzymatic cell lysis buffer B, 15 min at 37°C) to produce fragments of ~150–900 bp length. The nuclear pellet was resuspended in 300 μl ChIP buffer with protease inhibitor cocktail for 10 min on ice, and sonicated 3 × 30 sec to break nuclear membranes. The clarified lysate (9000 × g, 10 min) was subjected to immunoprecipitation overnight at 4°C with anti-AR antibody (2 μg/300 μl) or matched control IgG, using Protein G coupled to magnetic beads. After extensive washing, DNA was eluted from the beads, digested with proteinase K, and purified using spin columns. DNA was amplified using the following primers: F1 5’-CGCTCTCGATTCCTTTAGTTTC-3’ and R1 5’-AAAGTAGTCCCCGCCAGTC-3’ (−144 to −34 relative to the transcription start site of the CtnnB1 gene); F2 5’-GCCGCGCCGCTTATAAATC-3’ and R2 5’-GGCTTCACAGGACACGAGC-3’ (+48 to +136 relative to the transcription start).
Statistics.
Data are presented as means ± SD; all data sets were tested for normality using the Shapiro-Wilk test and for equality of variances by F test. Only data that passed both tests were analyzed by standard repeated measures one-way or two-way ANOVA, and paired comparisons were made using the Holm-Sidak’s multiple comparisons test. For data that exhibited normality but not equality of variances, we used a repeated measures ANOVA with Geisser-Greenhouse correction. If there were missing values, we used a mixed effects model and corrected for multiple comparisons using the two-stage step-up method of Benjamini, Krieger, and Yekutieli. Data that did not exhibit normality were analyzed using the Friedman test for multiple groups and corrected for multiple comparisons by the method of Benjamini, Krieger, and Yekutieli. For comparison of two groups, p values refer to an unpaired, two-tailed Welch’s t test, which does not assume equal variances, or a Whitney-Mann test for data that were not normally distributed. All tests were conducted in GraphPad Prism 9.5.1, except the F test for variances was conducted using an on-line calculator (https://www.statskingdom.com/220VarF2.html). A two-sided p value < 0.05 was considered significant.
Supplementary Material
Acknowledgments:
We are grateful to the staff of the UC San Diego Microscopy Core Facility for help with imaging. We thank Dr. Beatrice A. Golomb of UCSD for her independent review of statistical methods used to analyze the data.
Funding:
This work was supported by the National Institutes of Health grants R01-AR-068601 and R01-AG070778 (to RBP) and P30-NS047101 (UCSD Microscopy Shared Facility),
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The ChIP-seq data were previously published and can be downloaded from https://chip-atlas.org using the following IDs: SRX385391; SRX2388755; SRX5286305; SRX734411; and SRX250092. Constucts and mice generated for this manuscript are available with a UCSD Material Transfer Agreement.
REFERENCES AND NOTES
- 1.Vanderschueren D, Laurent MR, Claessens F, Gielen E, Lagerquist MK, Vandenput L, Borjesson AE, Ohlsson C, Sex steroid actions in male bone. Endocr Rev 35, 906–960 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levin ER, Hammes SR, Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat Rev Mol Cell Biol 17, 783–797 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mauvais-Jarvis F, Lange CA, Levin ER, Membrane-Initiated Estrogen, Androgen, and Progesterone Receptor Signaling in Health and Disease. Endocr Rev 43, 720–742 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahluwalia A, Hoa N, Ge L, Blumberg B, Levin ER, Mechanisms by Which Membrane and Nuclear ER Alpha Inhibit Adipogenesis in Cells Isolated From Female Mice. Endocrinology 161, (2020). [DOI] [PubMed] [Google Scholar]
- 5.Thomas P, Membrane Androgen Receptors Unrelated to Nuclear Steroid Receptors. Endocrinology 160, 772–781 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Unni E, Sun S, Nan B, McPhaul MJ, Cheskis B, Mancini MA, Marcelli M, Changes in androgen receptor nongenotropic signaling correlate with transition of LNCaP cells to androgen independence. Cancer Res 64, 7156–7168 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Sen A, De Castro I, Defranco DB, Deng FM, Melamed J, Kapur P, Raj GV, Rossi R, Hammes SR, Paxillin mediates extranuclear and intranuclear signaling in prostate cancer proliferation. J Clin Invest 122, 2469–2481 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sen A, Prizant H, Light A, Biswas A, Hayes E, Lee HJ, Barad D, Gleicher N, Hammes SR, Androgens regulate ovarian follicular development by increasing follicle stimulating hormone receptor and microRNA-125b expression. Proc Natl Acad Sci U S A 111, 3008–3013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu W, Qadir MMF, Nasteska D, Mota de Sa P, Gorvin CM, Blandino-Rosano M, Evans CR, Ho T, Potapenko E, Veluthakal R, Ashford FB, Bitsi S, Fan J, Bhondeley M, Song K, Sure VN, Sakamuri S, Schiffer L, Beatty W, Wyatt R, Frigo DE, Liu X, Katakam PV, Arlt W, Buck J, Levin LR, Hu T, Kolls J, Burant CF, Tomas A, Merrins MJ, Thurmond DC, Bernal-Mizrachi E, Hodson DJ, Mauvais-Jarvis F, Architecture of androgen receptor pathways amplifying glucagon-like peptide-1 insulinotropic action in male pancreatic beta cells. Cell Rep 42, 112529 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Almeida M, Laurent MR, Dubois V, Claessens F, O’Brien CA, Bouillon R, Vanderschueren D, Manolagas SC, Estrogens and Androgens in Skeletal Physiology and Pathophysiology. Physiol Rev 97, 135–187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maatta JA, Buki KG, Ivaska KK, Nieminen-Pihala V, Elo TD, Kahkonen T, Poutanen M, Harkonen P, Vaananen K, Inactivation of the androgen receptor in bone-forming cells leads to trabecular bone loss in adult female mice. Bonekey Rep 2, 440 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.S. R. Hammes, E. R. Levin, Impact of estrogens in males and androgens in females. J Clin Invest 129, 1818–1826 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Venken K, De Gendt K, Boonen S, Ophoff J, Bouillon R, Swinnen JV, Verhoeven G, Vanderschueren D, Relative impact of androgen and estrogen receptor activation in the effects of androgens on trabecular and cortical bone in growing male mice: a study in the androgen receptor knockout mouse model. J Bone Miner Res 21, 576–585 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Kalyanaraman H, Schall N, Pilz RB, Nitric oxide and cyclic GMP functions in bone. Nitric Oxide 76, 62–70 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rangaswami H, Schwappacher R, Marathe N, Zhuang S, Casteel DE, Haas B, Chen Y, Pfeifer A, Kato H, Shattil S, Boss GR, Pilz RB, Cyclic GMP and protein kinase G control a Src-containing mechanosome in osteoblasts. Sci. Signal 3, ra91 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marathe N, Rangaswami H, Zhuang S, Boss GR, Pilz RB, Pro-survival Effects of 17beta-Estradiol on Osteocytes Are Mediated by Nitric Oxide/cGMP via Differential Actions of cGMP-dependent Protein Kinases I and II. J. Biol. Chem 287, 978–988 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalyanaraman H, Schwappacher R, Joshua J, Zhuang S, Scott BT, Klos M, Casteel DE, Frangos JA, Dillmann W, Boss GR, Pilz RB, Nongenomic thyroid hormone signaling occurs through a plasma membrane-localized receptor. Sci. Signal 7, ra48 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalyanaraman H, Schwaerzer G, Ramdani G, Castillo F, Scott BT, Dillmann W, Sah RL, Casteel DE, Pilz RB, Protein Kinase G Activation Reverses Oxidative Stress and Restores Osteoblast Function and Bone Formation in Male Mice With Type 1 Diabetes. Diabetes 67, 607–623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schall N, Garcia JJ, Kalyanaraman H, China SP, Lee JJ, Sah RL, Pfeifer A, Pilz RB, Protein kinase G1 regulates bone regeneration and rescues diabetic fracture healing. JCI Insight 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalyanaraman H, Pal China S, Cabriales JA, Moininazeri J, Casteel DE, Garcia JJ, Wong VW, Chen A, Sah RL, Boss GR, Pilz RB, Protein Kinase G2 Is Essential for Skeletal Homeostasis and Adaptation to Mechanical Loading in Male but not Female Mice. J Bone Miner Res 38, 171–185 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramdani G, Schall N, Kalyanaraman H, Wahwah N, Moheize S, Lee JJ, Sah RL, Pfeifer A, Casteel DE, Pilz RB, cGMP-dependent protein kinase-2 regulates bone mass and prevents diabetic bone loss. J Endocrinol 238, 203–219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baron R, Kneissel M, WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med 19, 179–192 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Grino PB, Griffin JE, Wilson JD, Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology 126, 1165–1172 (1990). [DOI] [PubMed] [Google Scholar]
- 24.Armen TA, Gay CV, Simultaneous detection and functional response of testosterone and estradiol receptors in osteoblast plasma membranes. J Cell Biochem 79, 620–627 (2000). [PubMed] [Google Scholar]
- 25.Lieberherr M, Grosse B, Androgens increase intracellular calcium concentration and inositol 1,4,5-trisphosphate and diacylglycerol formation via a pertussis toxin-sensitive G-protein. J Biol Chem 269, 7217–7223 (1994). [PubMed] [Google Scholar]
- 26.Estrada M, Espinosa A, Muller M, Jaimovich E, Testosterone stimulates intracellular calcium release and mitogen-activated protein kinases via a G protein-coupled receptor in skeletal muscle cells. Endocrinology 144, 3586–3597 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Chambliss KL, Yuhanna IS, Mineo C, Liu P, German Z, Sherman TS, Mendelsohn ME, Anderson RG, Shaul PW, Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ. Res 87, E44–E52 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, Barone MV, Ametrano D, Zannini MS, Abbondanza C, Auricchio F, Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J 19, 5406–5417 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leung JK, Sadar MD, Non-Genomic Actions of the Androgen Receptor in Prostate Cancer. Front Endocrinol (Lausanne) 8, 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rangaswami H, Schwappacher R, Tran T, Chan GC, Zhuang S, Boss GR, Pilz RB, Protein Kinase G and Focal Adhesion Kinase Converge on Src/Akt/beta-Catenin Signaling Module in Osteoblast Mechanotransduction. J. Biol. Chem 287, 21509–21519 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaandrager AB, Ehlert EME, Jarchau T, Lohmann SM, De Jonge HR, N-terminal myristoylation is required for membrane localization of cGMP-dependent protein kinase type II. J. Biol. Chem 271, 7025–7029 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL, Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davey RA, Grossmann M, Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin Biochem Rev 37, 3–15 (2016). [PMC free article] [PubMed] [Google Scholar]
- 34.Yu J, Akishita M, Eto M, Ogawa S, Son BK, Kato S, Ouchi Y, Okabe T, Androgen receptor-dependent activation of endothelial nitric oxide synthase in vascular endothelial cells: role of phosphatidylinositol 3-kinase/akt pathway. Endocrinology 151, 1822–1828 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Farah C, Michel LYM, Balligand JL, Nitric oxide signalling in cardiovascular health and disease. Nat Rev Cardiol 15, 292–316 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Pedram A, Razandi M, Sainson RC, Kim JK, Hughes CC, Levin ER, A conserved mechanism for steroid receptor translocation to the plasma membrane. J. Biol. Chem 282, 22278–22288 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Zhou ZX, Sar M, Simental JA, Lane MV, Wilson EM, A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the DNA-binding domain and modulation by NH2-terminal and carboxyl-terminal sequences. J Biol Chem 269, 13115–13123 (1994). [PubMed] [Google Scholar]
- 38.Galbiati F, Razani B, Lisanti MP, Emerging themes in lipid rafts and caveolae. Cell 106, 403–411 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Cole R, Pascal LE, Wang Z, The classical and updated models of androgen receptor nucleocytoplasmic trafficking. Am J Clin Exp Urol 9, 287–291 (2021). [PMC free article] [PubMed] [Google Scholar]
- 40.Koryakina Y, Ta HQ, Gioeli D, Androgen receptor phosphorylation: biological context and functional consequences. Endocr Relat Cancer 21, T131–145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chymkowitch P, Le May N, Charneau P, Compe E, Egly JM, The phosphorylation of the androgen receptor by TFIIH directs the ubiquitin/proteasome process. EMBO J 30, 468–479 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ponguta LA, Gregory CW, French FS, Wilson EM, Site-specific androgen receptor serine phosphorylation linked to epidermal growth factor-dependent growth of castration-recurrent prostate cancer. J Biol Chem 283, 20989–21001 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burnstein KL, Regulation of androgen receptor levels: implications for prostate cancer progression and therapy. J Cell Biochem 95, 657–669 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Tetsu O, McCormick F, Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F, Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 22, 1172–1183 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Si W, Kang Q, Luu HH, Park JK, Luo Q, Song WX, Jiang W, Luo X, Li X, Yin H, Montag AG, Haydon RC, He TC, CCN1/Cyr61 is regulated by the canonical Wnt signal and plays an important role in Wnt3A-induced osteoblast differentiation of mesenchymal stem cells. Mol Cell Biol 26, 2955–2964 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glass DA, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G, Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 8, 751–764 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Kalyanaraman H, Ramdani G, Joshua J, Schall N, Boss GR, Cory E, Sah RL, Casteel DE, Pilz RB, A Novel, Direct NO Donor Regulates Osteoblast and Osteoclast Functions and Increases Bone Mass in Ovariectomized Mice. J. Bone Miner. Res 32, 46–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murillo-Garzon V, Kypta R, WNT signalling in prostate cancer. Nat Rev Urol 14, 683–696 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Zou Z, Ohta T, Miura F, Oki S, ChIP-Atlas 2021 update: a data-mining suite for exploring epigenomic landscapes by fully integrating ChIP-seq, ATAC-seq and Bisulfite-seq data. Nucleic Acids Res, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denayer S, Helsen C, Thorrez L, Haelens A, Claessens F, The rules of DNA recognition by the androgen receptor. Mol Endocrinol 24, 898–913 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Q, Dashwood WM, Zhong X, Al-Fageeh M, Dashwood RH, Cloning of the rat beta-catenin gene (Ctnnb1) promoter and its functional analysis compared with the Catnb and CTNNB1 promoters. Genomics 83, 231–242 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Rangaswami H, Marathe N, Zhuang S, Chen Y, Yeh JC, Frangos JA, Boss GR, Pilz RB, Type II cGMP-dependent protein kinase mediates osteoblast mechanotransduction. J. Biol. Chem 284, 14796–14808 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olive M, Krylov D, Echlin DR, Gardner K, Taparowsky E, Vinson C, A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis. J. Biol. Chem 272, 18586–18594 (1997). [DOI] [PubMed] [Google Scholar]
- 55.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK, Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407, 538–541 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH, Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha. Proc Natl Acad Sci U S A 101, 17126–17131 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joshua J, Schwaerzer GK, Kalyanaraman H, Cory E, Sah RS, Li M, Vaida F, Boss GR, Pilz RB, Soluble guanylate cyclase as a novel treatment target for osteoporosis. Endocrinology 155, 4720–4730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song RX, Barnes CJ, Zhang Z, Bao Y, Kumar R, Santen RJ, The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor alpha to the plasma membrane. Proc Natl Acad Sci U S A 101, 2076–2081 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiong W, Zhang L, Yu L, Xie W, Man Y, Xiong Y, Liu H, Liu Y, Estradiol promotes cells invasion by activating beta-catenin signaling pathway in endometriosis. Reproduction 150, 507–516 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schweizer L, Rizzo CA, Spires TE, Platero JS, Wu Q, Lin TA, Gottardis MM, Attar RM, The androgen receptor can signal through Wnt/beta-Catenin in prostate cancer cells as an adaptation mechanism to castration levels of androgens. BMC Cell Biol 9, 4 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Truica CI, Byers S, Gelmann EP, Beta-catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res 60, 4709–4713 (2000). [PubMed] [Google Scholar]
- 62.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X, Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Zhao X, Zhuang S, Chen Y, Boss GR, Pilz RB, Cyclic GMP-dependent protein kinase regulates CCAAT enhancer-binding protein beta functions through inhibition of glycogen synthase kinase-3. J. Biol. Chem 280, 32683–32692 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Notini AJ, McManus JF, Moore A, Bouxsein M, Jimenez M, Chiu WS, Glatt V, Kream BE, Handelsman DJ, Morris HA, Zajac JD, Davey RA, Osteoblast deletion of exon 3 of the androgen receptor gene results in trabecular bone loss in adult male mice. J Bone Miner Res 22, 347–356 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Modder UI, Rudnik V, Liu G, Khosla S, Monroe DG, A DNA binding mutation in estrogen receptor-alpha leads to suppression of Wnt signaling via beta-catenin destabilization in osteoblasts. J Cell Biochem 113, 2248–2255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hukkanen M, Platts LA, Lawes T, Girgis SI, Konttinen YT, Goodship AE, MacIntyre I, Polak JM, Effect of nitric oxide donor nitroglycerin on bone mineral density in a rat model of estrogen deficiency-induced osteopenia. Bone 32, 142–149 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Pal S, Rashid M, Singh SK, Porwal K, Singh P, Mohamed R, Gayen JR, Wahajuddin M, Chattopadhyay N, Skeletal restoration by phosphodiesterase 5 inhibitors in osteopenic mice: Evidence of osteoanabolic and osteoangiogenic effects of the drugs. Bone 135, 115305 (2020). [DOI] [PubMed] [Google Scholar]
- 68.Kim SM, Taneja C, Perez-Pena H, Ryu V, Gumerova A, Li W, Ahmad N, Zhu LL, Liu P, Mathew M, Korkmaz F, Gera S, Sant D, Hadelia E, Ievleva K, Kuo TC, Miyashita H, Liu L, Tourkova I, Stanley S, Lizneva D, Iqbal J, Sun L, Tamler R, Blair HC, New MI, Haider S, Yuen T, Zaidi M, Repurposing erectile dysfunction drugs tadalafil and vardenafil to increase bone mass. Proc Natl Acad Sci U S A 117, 14386–14394 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rejnmark L, Vestergaard P, Mosekilde L, Decreased fracture risk in users of organic nitrates: a nationwide case-control study. J. Bone Miner. Res 21, 1811–1817 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Pouwels S, Lalmohamed A, van ST, Cooper C, Souverein P, Leufkens HG, Rejnmark L, de BA, Vestergaard P, de VF, Use of organic nitrates and the risk of hip fracture: a population-based case-control study. J. Clin. Endocrinol. Metab 95, 1924–1931 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jamal SA, Browner WS, Bauer DC, Cummings SR, Intermittent use of nitrates increases bone mineral density: the study of osteoporotic fractures. J. Bone Miner. Res 13, 1755–1759 (1998). [DOI] [PubMed] [Google Scholar]
- 72.Jamal SA, Cummings SR, Hawker GA, Isosorbide mononitrate increases bone formation and decreases bone resorption in postmenopausal women: a randomized trial. J. Bone Miner. Res 19, 1512–1517 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Wimalawansa SJ, Nitroglycerin therapy is as efficacious as standard estrogen replacement therapy (Premarin) in prevention of oophorectomy-induced bone loss: a human pilot clinical study. J. Bone Miner. Res 15, 2240–2244 (2000). [DOI] [PubMed] [Google Scholar]
- 74.Wimalawansa SJ, Grimes JP, Wilson AC, Hoover DR, Transdermal nitroglycerin therapy may not prevent early postmenopausal bone loss. J. Clin. Endocrinol. Metab 94, 3356–3364 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gudi T, Hong GK-P, Vaandrager AB, Lohmann SM, Pilz RB, Nitric oxide and cGMP regulate gene expression in neuronal and glial cells by activating type II cGMP-dependent protein kinase. FASEB J 13, 2143–2152 (1999). [PubMed] [Google Scholar]
- 76.Nakanishi R, Akiyama H, Kimura H, Otsuki B, Shimizu M, Tsuboyama T, Nakamura T, Osteoblast-targeted expression of Sfrp4 in mice results in low bone mass. J Bone Miner Res 23, 271–277 (2008). [DOI] [PubMed] [Google Scholar]
- 77.Bakker A, Klein-Nulend J, Osteoblast isolation from murine calvariae and long bones. Methods Mol. Med 80, 19–28 (2003). [DOI] [PubMed] [Google Scholar]
- 78.Suarez J, Belke DD, Gloss B, Dieterle T, McDonough PM, Kim YK, Brunton LL, Dillmann WH, In vivo adenoviral transfer of sorcin reverses cardiac contractile abnormalities of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 286, H68–75 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The ChIP-seq data were previously published and can be downloaded from https://chip-atlas.org using the following IDs: SRX385391; SRX2388755; SRX5286305; SRX734411; and SRX250092. Constucts and mice generated for this manuscript are available with a UCSD Material Transfer Agreement.