

Abstract
Background
Prognosis and disease severity in cystic fibrosis (CF) are linked to declining lung function. To characterise lung function by the number of adults in countries with different levels of Gross National Income (GNI), data from the European Cystic Fibrosis Society Patient Registry were utilised.
Methods
Annual data including age, forced expiratory volume in 1 s (FEV1), anthropometry, genotype, respiratory cultures and CF-related diabetes (CFRD) were retrieved between 2011 and 2021. All countries were stratified into GNI per capita to reflect differences within Europe.
Results
A consistent improvement in FEV1 % pred and survival was observed among the 47 621 people with CF (pwCF), including subjects with chronic Pseudomonas aeruginosa infection, CFRD and/or undernutrition. Mean values of FEV1 % pred changed from 85% to 94.2% for children and from 63.6% to 74.7% for adults. FEV1 % pred further increased among those carrying the F508del mutation in 2021, when elexacaftor/tezacaftor/ivacaftor was available. The number of adult pwCF increased from 13 312 in 2011 to 21 168 in 2021, showing a 60% increase. PwCF living in European lower income countries did not demonstrate a significant annual increase in FEV1 % pred or in the number of adults.
Conclusion
This pan-European analysis demonstrates a consistent improvement in FEV1 % pred, number of adult pwCF and survival over the last decade only in European higher and middle income countries. Urgent action is needed in the lower income countries where such improvement was not observed. The notable improvement observed in pwCF carrying the F508del mutation emphasises the need to develop treatments for all CF mutations.
Tweetable abstract
This analysis demonstrates a consistent improvement in pulmonary function, number of adult pwCF and survival over the past decade, indicating the effectiveness of implementation of the standards of care guidelines in routine CF treatment https://bit.ly/3TX4jyA
Introduction
Despite remarkable improvements in health outcomes for individuals affected with cystic fibrosis (CF), it remains a life-shortening disease with pulmonary insufficiency as the main cause of death. Lung function, which typically decreases over time, is linked to disease severity and predicts prognosis. Forced expiratory volume in 1 s (FEV1) compared with the predicted in a reference population (FEV1 % pred) is considered the best generally available measure for assessing CF lung disease and it is an influential driver for defining disease stage. In 2018, the European Cystic Fibrosis Society Patient Registry (ECFSPR) report on 24 416 patients aged <18 years showed that FEV1 <40% predicted is a risk factor for death [1]. Additionally, FEV1 % pred is a primary outcome measure for clinical studies and comparisons between centres and countries [2–5], and an objective standard utilised for regulatory approval of CF respiratory therapies [6].
The ECFSPR collects demographic and clinical data of over 60 000 consenting people with CF (pwCF) from 40 European and neighbouring countries for a period spanning from 2008 to 2021. Data is collected using a common set of variables and definitions, and is sent to the ECFSPR on an annual basis. The ECFSPR collaborates closely with the CF centres and the national CF registries to ensure that their data is as complete and high quality as possible, in order to accurately reflect the clinical status of CF across Europe [7–10]. The ECFSPR's database provides a unique basis for epidemiological analyses due to its comprehensive and international composition. It offers a cross-national platform facilitating analysis of therapeutic interventions and quality-of-care standards in a diverse population that displays a wide variability of both lung disease and potential risk factors for FEV1 % pred decline [11]. A detailed description of the ECFSPR and its contents (including guidelines, annual reports, etc.) is available at www.ecfs.eu/ecfspr.
Analysis of data reported to the ECFSPR from 2011 to 2016 showed a significant decrease in the overall prevalence and incidence of chronic Pseudomonas aeruginosa and chronic Burkholderia cepacia complex species [12]. In a cross-sectional study, analysis of pwCF enrolled in the ECFSPR during 2007 showed that the age-related decline of FEV1 % pred starts slowly and becomes more rapid at age 12 years. Furthermore, the number of pwCF was relatively stable up to age 18 years, but subsequently decreased, reflecting mortality. Accumulated data from the ECFSPR has also shown that poor nutritional status, chronic P. aeruginosa infection and CF-related diabetes (CFRD) are preventable and/or potentially treatable factors that influence FEV1 % pred deterioration [13].
The effect of socioeconomic status on health is well established; in countries where health expenditure per capita is low, pwCF experience more deprivation-related health disparities than all other individuals in the population [14–16].
The aim of the current study was to investigate the changes in lung function, the number of adult pwCF and survival during the last decade. In addition, we compared theses changes between countries after stratifying into three groups according to their Gross National Income (GNI).
Materials and methods
Variable definitions for the present study
The genotype of pwCF was classified as F508del/F508del for people having the F508del mutation on both alleles, F508del/MF for people having the F508del mutation on one allele and a minimal function mutation on the other allele, MF/MF for people having two minimal function mutations (excluding F508del) and RF/other for people having at least one residual function mutation. The mutation classification according to minimal function and residual function definition was performed consistently with Mei-Zahav et al. [10], with minimal function mutation mainly representing mutations having no or minimal CF transmembrane conductance regulator (CFTR) function (in class I, II or III) and residual function mutation mainly representing mutations having partial CFTR function (in class IV, V or VI).
Most European countries are classified as high income according to the World Bank, having a GNI per capita higher than USD 13 205 in 2021. To take into account economic differences among the European countries within this study we therefore used an ad hoc classification, creating three groups with the same number of countries, according to incremental values of GNI per capita in 2021 computed with the Atlas method (current USD) and extracted from the World Bank database (supplementary table S1). The 27 countries for which longitudinal data were available between 2011 and 2021 were stratified into three groups, Europe lower income countries (LICs), Europe middle income countries (MICs) and Europe higher income countries (HICs), which are different from the World Bank GNI categories of low, middle and high income countries.
Nutritional status, infection with chronic P. aeruginosa and CFRD were collected every year in the annual data of the ECFSPR for each pwCF. Nutritional status was defined using body mass index (BMI) z-score, computed based on the US Centers for Disease Control and Prevention references (www.cdc.gov/growthcharts). BMI z-scores for adults were obtained according to values of pwCF aged ≥20 years. Underweight was defined as BMI z-score < −2. PwCF were defined as chronically infected with P. aeruginosa if >50% of respiratory samples collected during the last 12 months were positive for P. aeruginosa, with at least four samples collected during that period (modified Leeds criteria for chronic infection) and/or significantly raised bacteria-specific antibodies according to local laboratories were present [17]. PwCF can be defined as chronically infected with P. aeruginosa when the aforementioned criteria were fulfilled in recent years and the caring physician had no reason to think the status has changed. Exceptions from this definition for some countries in the ECFSPR are present and details are reported in each ECFSPR annual report. CFRD was defined for the ECFSPR as insulin use from 2011 to 2017 and subsequently as diabetes treated with insulin, hypoglycaemic agents, dietary advice or alternative therapies.
Statistical methods
The aim of the present study was to provide an epidemiological evaluation of changes in lung function over the last decade. To evaluate lung function, values of FEV1 % pred computed with Global Lung Function Initiative equations were considered [18]. The inclusion criteria were all pwCF enrolled in the ECFSPR between 2011 and 2021 who were between 6 and 60 years old and who had not undergone a lung transplant. Countries joining the ECFSPR after 2011 were excluded since they do not have a 10-year follow-up period. Children younger than 6 years were excluded due to their unreliable ability to perform spirometry. People older than 60 years were excluded due to there being scarce data and influential observations in the regression models. PwCF who had had lung transplantation were excluded from the time of transplant, as their FEV1 % pred values do not represent their CF disease stage.
Descriptive statistics were collected for sex, genotype, country group, chronic P. aeruginosa, CFRD, underweight status, neonatal screening and age at diagnosis from 2011 to 2021.
Since the aim of the study was to present an epidemiological overview of the changes in FEV1 % pred mean values in Europe between 2011 and 2021, and not to estimate the changes in FEV1 % pred within the same patient, a marginal model was fitted using generalised estimating equations (GEEs) [11]. The response variable was the value of FEV1 % pred; the explanatory variables were the year of follow-up, included in the model as a dummy variable, and age at FEV1 % pred measurement, included in the model as a continuous variable using a restricted cubic spline with n=5 knots. The number of knots was chosen according to Quasi Information Criterion. The interaction between year of follow-up and age was also included in the model. The correlation among different measurements taken on the same pwCF was taken into account in the GEE model. Predicted values from this model were used to draw descriptive figures of changes of FEV1 % pred over time and age.
Five further GEE models were fitted including separately five prognostic factors for worse lung function in CF. To test if changes of FEV1 % pred over age between 2011 and 2021 were different according to chronic P. aeruginosa, underweight status, CFRD, genotype and country group, the interaction terms with follow-up years were included in the models. Chronic P. aeruginosa, underweight status and CFRD were included in the model as time-varying factors, since they are collected on an annual basis in the ECFSPR.
Since genotype is a variable strongly influencing the changes of FEV1 % pred over the last decade, multiple GEE models were fitted stratified by genotype. Explanatory variables included in the models were year of follow-up (as a dummy variable), age at FEV1 % pred measurement (included as a restricted cubic spline with n=5 knots), chronic P. aeruginosa, CFRD, underweight status and country group. Four forest plots were drawn to summarise the results of these models, in terms of differences in FEV1 % pred values with 95% confidence intervals. The forest plot provides a general overview of the differential contribution of the pwCF characteristics and of the subsequent years of data collection on FEV1 % pred values. Unfortunately, CFTR modulator use cannot be included in the multiple regression models because of missing values between 2011 and 2017, since the ECFSPR only started collecting data on CFTR modulators in 2018. However, the association between elexacaftor/tezacaftor/ivacaftor (ETI) use and time was explored by looking at the percentage of pwCF taking ETI between 2018 and 2021 in the different countries and income groups.
Finally, a Cox regression model was fitted to estimate the differences in survival between 2011 and 2021. A simple model considering only year of follow-up as explanatory variable was considered. In the Cox model, age was used as timescale, accounting for left truncation and right censoring; each pwCF is included in the analysis only for the years he/she is included in the ECFSPR.
Ethical approval
All the participating centres and national registries in the ECFSPR have ethical approval. Informed consent for anonymous data collection and ECFSPR participation, including consent that data may be used for future research, was obtained from all participants. This study was approved by the ECFSPR Scientific Committee and the ECFSPR Steering Committee.
Results
Study population
Data from 65 022 pwCF followed by the participating CF clinics during 2011–2021 were collected by the ECFSPR. Excluded were children younger than 6 years (n=9742), adults older than 60 years (n=486), pwCF with a lung transplant since the beginning of the study period (n=2169) and an additional 1885 (3.6%) pwCF for whom we could not compute FEV1 % pred because of missing values for FEV1 or height. An additional 3564 were excluded because they were residents of countries with <10 years of follow-up in the ECFSPR (Albania, Armenia, Belarus, Bulgaria, Croatia, Cyprus, Finland, Georgia, Iceland, Luxembourg, Norway, Poland and Turkey). The final study population included 47 176 pwCF (52.5% males) (supplementary figure S1).
With regard to the completeness of the dataset, gender is a variable with 0% missing values and underweight has <10% missing values. For chronic P. aeruginosa the percentage of missing values is >10% only from 2011 to 2015 because one large national registry did not collect that variable and another country had a high percentage of missing values. A similar situation occurs for CFRD, having >10% missing values from 2011 to 2014, because of a high percentage of missing values in a large national registry. Overall, the percentage of missing data was similar among the three groups of GNI income countries, with the exception of genotype, being less complete in LICs in 2011. After 2011, there was a global improvement in performing genotyping all over Europe, in particular in the LICs, so that the situation in 2021 is more homogeneous.
The median number of years for which an individual is included in this study is 7 years. This value varies among the countries, being 4 years in LICs, 7 years in MICs and 8 years in HICs.
Table 1 presents the main demographic and clinical characteristics of the included pwCF from 2011 to 2021. In 2021, 9.0% of pwCF had a genotype not classified according to our definition. It has to be noted that the “Unknown” genotype in table 1 includes pwCF with F508del and an unknown mutation. Among the unclassified genotype, in 262 pwCF CFTR mutations analysis was not performed. In 2021, 41.9% of pwCF were F508del/F508del, 26.3% were F508del/MF, 7.4% were MF/MF and 15.4% were RF/other.
TABLE 1.
Clinical and demographic characteristics of people with cystic fibrosis included in this study from 2011 to 2021
| 2011 (n=23 701) | 2012 (n=24 428) | 2013 (n=25 784) | 2014 (n=25 912) | 2015 (n=28 320) | 2016 (n=30 503) | 2017 (n=31 646) | 2018 (n=32 695) | 2019 (n=33 703) | 2020 (n=32 485) | 2021 (n=34 787) | |
| Female | 47.0 | 47.0 | 47.1 | 47.3 | 47.5 | 47.3 | 47.2 | 47.5 | 47.3 | 47.6 | 47.6 |
| Genotype | |||||||||||
| F508del/F508del | 43.3 | 43.3 | 43.0 | 42.2 | 41.4 | 41.6 | 41.6 | 41.7 | 41.6 | 42.3 | 41.9 |
| F508del/MF | 23.5 | 23.5 | 23.7 | 25.3 | 25.2 | 25.5 | 25.5 | 25.9 | 25.9 | 26.2 | 26.3 |
| MF/MF | 5.9 | 6.0 | 6.0 | 6.6 | 6.9 | 7.2 | 7.1 | 7.4 | 7.4 | 7.6 | 7.4 |
| RF/other | 10.8 | 11.1 | 11.3 | 13.0 | 13.7 | 14.2 | 14.8 | 14.9 | 15.4 | 14.8 | 15.4 |
| Unknown | 16.4 | 16.2 | 16.0 | 12.8 | 12.7 | 11.6 | 11.0 | 10.1 | 9.8 | 9.1 | 9.0 |
| Country income group# | |||||||||||
| Lower | 3.4 | 4.9 | 5.7 | 7.4 | 8.0 | 8.1 | 8.3 | 8.3 | 8.0 | 6.3 | 7.1 |
| Middle | 62.7 | 61.1 | 60.3 | 57.9 | 58.0 | 58.8 | 59.3 | 59.0 | 59.7 | 60.0 | 59.6 |
| Higher | 33.9 | 34.0 | 34.0 | 34.7 | 34.0 | 33.1 | 32.5 | 32.7 | 32.3 | 33.7 | 33.4 |
| Chronic P. aeruginosa infection | 39.8 | 39.3 | 37.8 | 36.6 | 35.8 | 35.9 | 35.4 | 34.1 | 33.7 | 32.1 | 26.8 |
| CFRD | 18.2 | 18.2 | 18.9 | 18.8 | 17.8 | 16.4 | 16.7 | 18.7 | 19.8 | 21.2 | 21.3 |
| Underweight | 8.4 | 8.2 | 8.2 | 7.9 | 7.8 | 7.2 | 6.9 | 6.7 | 6.3 | 5.4 | 4.4 |
| Newborn screening performed | 25.6 | 25.6 | 25.9 | 21.3 | 22.7 | 22.6 | 23.7 | 26.8 | 28.4 | 30.0 | 30.6 |
| Median age at diagnosis (months) | 6.5 | 6.5 | 6.5 | 6.0 | 6.0 | 6.0 | 6.0 | 5.2 | 4.9 | 4.8 | 4.3 |
Data are presented as %, unless otherwise stated. MF: minimal function; RF: residual function; P. aeruginosa: Pseudomonas aeruginosa; CFRD: cystic fibrosis-related diabetes. #: the three groups are different from the World Bank Gross National Income categories of low, middle and high income countries.
When comparing the characteristics of people included in the study (table 1) and those living in countries excluded from this study (supplementary table S2) it can be noted that the people living in countries which joined the ECFSPR later are more often from LICs, have a lower percentage of the F508del/F508del genotype and a higher percentage of the MF/MF genotype, and have a lower percentage of CFRD, a higher percentage of underweight and a lower percentage of newborn screening performed. However, these different characteristics do not affect the main results of this study, as we observed in a sensitivity analysis performed at the end of the study.
Main results
As shown in figure 1, there was a gradual and consistent increase during the 10-year study period for FEV1 % pred in all age groups; this was also observed in pwCF with chronic P. aeruginosa (figure 2a), CFRD (figure 2b) and undernutrition (figure 2c). A larger dramatic increase in FEV1 % pred was observed in 2021 when ETI became available for pwCF carrying the F508del mutation. As shown in figure 3, this effect was limited only to pwCF that carry the F508del mutation (p-value for interaction between year and genotype <0.001). Multiple regression analysis according to genotype groups, after adjusting for chronic P. aeruginosa, CFRD, underweight, country group and age, showed that there was a significant annual increase in FEV1 % pred among the F508del homozygous group in 2021 versus 2020 when ETI became available (supplementary figure S2).
FIGURE 1.
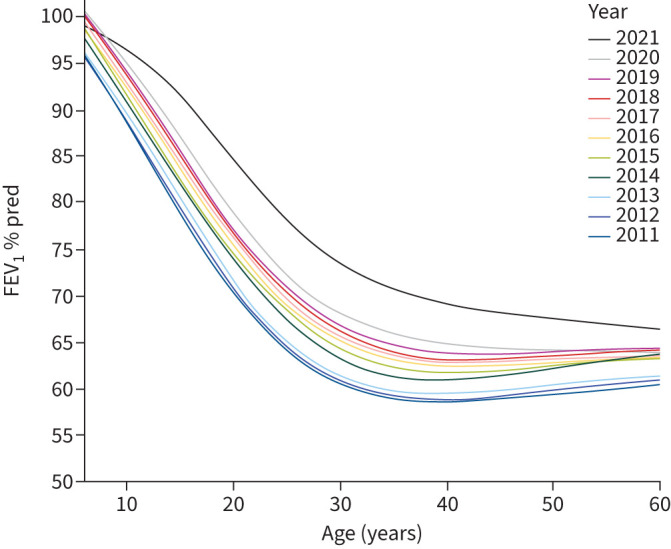
Forced expiratory volume in 1 s (FEV1) percentage predicted values in European people with cystic fibrosis according to age and year.
FIGURE 2.

Forced expiratory volume in 1 s (FEV1) percentage predicted values in European people with cystic fibrosis (pwCF) according to age and year: a) in pwCF with chronic Pseudomonas aeruginosa infection, b) in pwCF with CF-related diabetes (CFRD) and c) in underweight pwCF.
FIGURE 3.

Forced expiratory volume in 1 s (FEV1) percentage predicted values in European people with cystic fibrosis according to age, year and genotype: a) F508del mutation on both alleles (F508del/F508del), b) F508del mutation on one allele and a minimal function mutation on the other allele (F508del/MF), c) two minimal function mutations (excluding F508del) (MF/MF) and d) at least one residual function mutation (RF/other).
The number of pwCF in the adult age group increased along the years as shown in figure 4 and table 2. The number of adult pwCF increased from 13 312 in 2011 to 21 168 in 2021, showing a 60% increase. Furthermore, the distribution of patient age shifted towards a later age. Together with the improved overall survival probability according to age and year of follow-up (figure 5), this is a sign of increased longevity of pwCF starting already before ETI was available. Consistent results were obtained from the Cox regression model showing a significant decrease in hazard ratio of death in 2019, 2020 and 2021 (0.76 (95% CI 0.64–0.90; p=0.001), 0.76 (95% CI 0.63–0.92; p=0.004) and 0.79 (95% CI 0.64–0.98; p=0.032), respectively) (supplementary table S3). However, it has to be noted that the increase in the number and percentage of adults is not similar among the different country groups, as it emerges from figure 4. In MICs and HICs the number of adult pwCF included in this study almost double in the last decade from 4489 to 7498 and from 8539 to 12 850, respectively, and in the meantime the number of children showed only a slight increase; thus the percentage of adults increased. On the other hand, LICs showed a similar increase in children (from 530 to 1634) and adults (from 284 to 820), being mainly due to coverage improvement in these countries. This is reflected in no increase in the percentage of adults from 2011 to 2021.
FIGURE 4.

Median forced expiratory volume in 1 s (FEV1) percentage predicted (graph lines) and number of people with cystic fibrosis (bar charts) by age group and year in Europe: a) lower income countries (LICs), b) middle income countries (MICs) and c) higher income countries (HICs). (The three groups are different from the World Bank Gross National Income categories of low, middle and high income countries.)
TABLE 2.
Number of people with cystic fibrosis according to age group in 2011, 2016 and 2021
| Age group (years) | ||||||
| 6–11 | 12–17 | 18–23 | 24–29 | 30–39 | ≥40 | |
| 2011 | 5004 | 5236 | 4819 | 3443 | 3198 | 1852 |
| 2016 | 6431 | 6154 | 5304 | 4596 | 4625 | 3086 |
| 2021 | 6357 | 6667 | 5612 | 5032 | 5980 | 4544 |
Data are presented as n.
FIGURE 5.
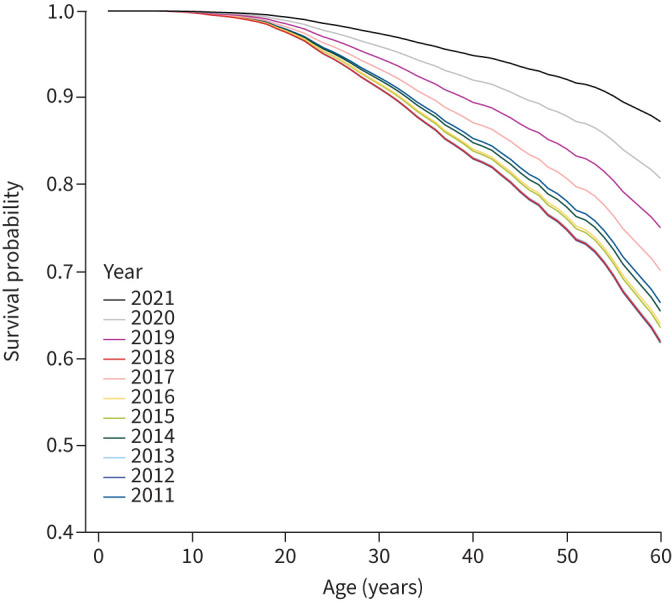
Survival probability in European people with cystic fibrosis according to age and year of follow-up.
When mean FEV1 % pred according to age group, year and GNI group was analysed (figure 6), pwCF living in LICs did not demonstrate the same annual increase in FEV1 % pred, with a significant sharper decline of FEV1 % pred with age. From the GEE model a significant interaction (p<0.001) emerged between country group and year of follow-up, with LICs showing only a small nonsignificant increase in FEV1 % pred values, while MICs and HICs showed a statistically significant increase in FEV1 % pred, in particular from 2020 to 2021. It is interesting to see that among children there is a gradual increase of FEV1 values, without big differences between 2020 and 2021, which is likely due to the lower percentage of paediatric pwCF taking ETI. When FEV1 % pred was analysed according to GNI as a continuous variable and year (figure 7), countries with GNI <USD 20 000 did not show any improvement over the years.
FIGURE 6.

Forced expiratory volume in 1 s (FEV1) percentage predicted values according to age and year in Europe: a) lower income countries (LICs), b) middle income countries (MICs) and c) higher income countries (HICs). (The three groups are different from the World Bank Gross National Income categories of low, middle and high income countries.)
FIGURE 7.
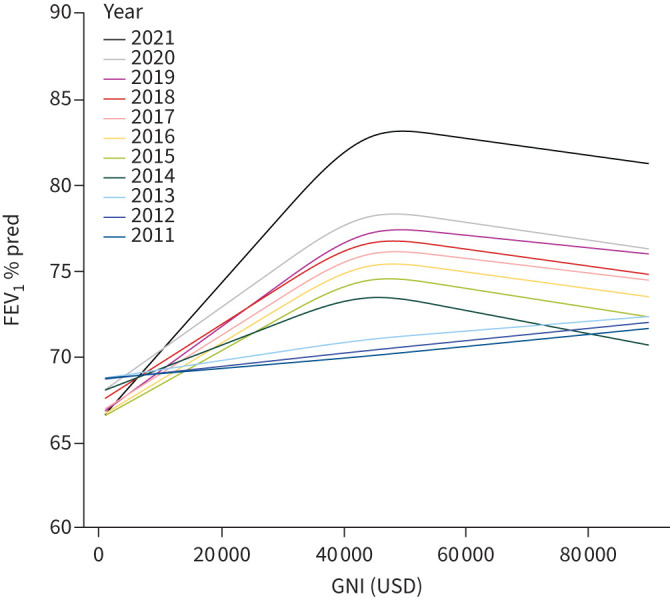
Forced expiratory volume in 1 s (FEV1) percentage predicted values according to Gross National Income (GNI) (as a continuous variable) and year.
The percentage of pwCF taking ETI greatly increased in 2020 and 2021, but only in MICs and HICs, where in 2021 almost half of pwCF used ETI. On the other hand, in LICs the percentage of pwCF using ETI was almost null even in 2021, with the only exception being Greece (supplementary table S4).
Two sensitivity analysis were performed to check the effect of county selection on the results. In the first sensitivity analysis only countries with full coverage (>80%) since 2011 were included. In the second sensitivity analysis all countries were included, without the selection of a minimum follow-up of 10 years. In both cases the results for FEV1 % pred evolution over the last decade and the increase in the number of the adult population are comparable to those reported in the present article.
Discussion
This pan-European analysis of the ECFSPR annual data demonstrates a consistent improvement in pulmonary function, number of adult pwCF and survival over the last decade, which began even before the highly effective CFTR modulators were available. A remarkable increase in FEV1 % pred was observed in 2021 only in pwCF who carry the F508del mutation when ETI became available [19–21]. The improvement in FEV1 % pred was less than the value reported in clinical trials, as not all pwCF who carry F508del started treatment with ETI, and was likely attributable to the highly selected population in clinical trials versus those in the “real world”. Furthermore, we adopted a marginal model providing different estimates as opposed to a conditional model looking at the difference in FEV1 % pred within each patient. In addition to the increase in age-related FEV1 % pred, the current analysis revealed a delay in the downward slope of FEV1 % pred over age among pwCF homozygous for F508del mutations. Future studies should provide more data on how this treatment will affect the survival of pwCF who have irreversible lung disease prior to ETI treatment and those starting ETI prior to the development of structural lung changes. While the findings of this study are impressive, there are still a significant number of pwCF who carry non-ETI-responsive mutations and cannot benefit from ETI, and alternative therapeutic interventions for this group are needed. Until such treatments are available, these individuals should adhere to the standard treatment protocols, which are associated with improved outcome as shown in the present study.
A less optimistic picture was observed in LICs. Despite the availability of guidelines recommending standard treatments, improvement in disease outcomes in LICs was only minimal and lower compared with MICs and HICs. Furthermore, the increase that was observed in MICs and HICs was not observed in LICs. An ECFSPR cohort study by McKone et al. [22] demonstrated that countries in the highest third of healthcare spending had a 46% lower risk of mortality than countries in the lowest third of healthcare spending. The current study strengthens the data from McKone et al. [22] and additionally found that there was no improvement in closing the FEV1 gap despite the discrepancies that have persisted over the last decade.
The effect of socioeconomic status on the health of pwCF is well established [14, 16, 23–27]. Most of the reports on disparities in CF disease outcome came from HICs, demonstrating that children from disadvantaged families have worse outcomes associated with several nongenetic factors, such as material wellbeing, educational attainment, living and working conditions, physical environment and exposures, family environment, social support, health literacy and behaviours, and access to healthcare [16, 24].
The observed improved disease outcomes over the last decades in HICs and MICs are likely due to the result of better insights into the natural course of CF, leading to the development of treatments that target early diagnosis, neonatal screening, respiratory infections, inflammation, mucociliary clearance and nutritional status. A potential contributor to improved outcomes in pwCF is comprehensive care implemented by teams of trained and experienced health professionals that facilitated improved adherence to enhanced modalities of care [28]. Previously described guidelines can assist CF caregivers and health authorities in the evaluation and monitoring of pwCF, detection of complications, and prevention of clinical deterioration [29–31]. PwCF treated at specialised CF centres by a multidisciplinary dedicated team have improved outcomes [31]. Unfortunately, LICs lack the resources to provide this standard of care. In some countries, patients and families need to purchase essential drugs out of pocket. Not all the CF centres have professional nonmedical personnel as part of their teams and patients living far from a CF centre have daily care provided by local physicians. Because of its association with worse outcomes, poor adherence to treatment is considered a potential contributor to disparities in health outcomes observed for various conditions across racial and ethnic groups [32]. Adherence to therapy established by the guidelines is dependent upon a variety of factors. Such factors include individual characteristics of the patient, the patient's family and culture, interactions with healthcare providers, systemic barriers that prevent access to quality healthcare, and financial means to afford expensive therapies. Additional contributing factors associated with ethnicity, including adherence to treatment regimens, self-management, culture, smoke exposure and health literacy, can also contribute to acceleration of the disease process [23, 24, 29, 33].
The results of the current study should inform health authorities of countries with disparities in health outcomes. Increasing awareness and education about CF among healthcare providers, policymakers and the public is essential. National and international advocacy efforts can raise awareness among policymakers and mobilise support for improving CF services in LICs. Additionally, empowering individuals with CF and their families through education and support programmes is vital.
This registry-based study has its limitations, including potential survivor bias, which is a common problem in datasets of this type [34]. Ascertainment bias may also occur due to selection of countries with >10 years of follow-up resulting in the exclusion of very low income countries. Moreover, registries may encounter problems with adherence to the definitions, data quality process, missing data, data entry errors and differences among countries of variable socioeconomic status [12]. Rigorous statistical methods have attempted to partially overcome survivor bias by accounting for left truncation in the survival model and ascertainment through stratified analyses. In recent years, the ECFSPR introduced a data quality control project to check and overcome these limitations. In 2018, the European Medicines Agency qualified the ECFSPR as a resource for collecting CF-specific data for pharmaco-epidemiology studies [35]. A further limitation includes variation in coverage throughout different countries spanning the past 10 years. In 2011, seven countries had <80% coverage. The percentage of pwCF in the ECFSPR has increased over the years and in 2021, only three countries had <80% coverage. However, it cannot be excluded that this increase may also be due to inclusion of less severe pwCF and that most of the increase is due to inclusion of more centres that joined ECFSPR. New centres are similar to those already included since 2011, allowing for the representation of the country to remain the same.
The ECFSPR is an important research tool for tracking the improved care of pwCF. The present study, focusing on the last decade, demonstrates a general improvement in lung function and longevity of European pwCF. The significant impact of ETI for those with F508del mutations was also seen. However, discrepancies in outcomes related to the economic status of different participating countries need to be further addressed to ensure better health outcomes and quality of life for pwCF living in these countries.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary figures and tables ERJ-01241-2023.Supplement (197.2KB, pdf)
Shareable PDF
Acknowledgements
We thank the people with cystic fibrosis, and their families, for consenting to their data being included in the European Cystic Fibrosis Society Patient Registry (ECFSPR). We thank the centres and individual country representatives for providing and allowing the use of the data, and the ECFSPR for providing access to anonymised patient data.
Footnotes
ECFSPR Scientific Committee: Andreas Pfleger, Department of Paediatrics and Adolescent Medicine, Division of Paediatric Pulmonology and Allergology, Medical University of Graz, Graz, Austria; Géraldin Daneau, Sciensano, Epidemiology and Public Health, Health Services Research, Brussels, Belgium; Elise Lammertijn, Cystic Fibrosis Europe and Association Muco ASBL – Mucovereniging VZW, Brussels, Belgium; Milan Macek, Department of Biology and Medical Genetics, Second Faculty of Medicine, Charles University and Motol University Hospital, Prague, Czech Republic; Pavel Drevinek, Department of Medical Microbiology, Second Faculty of Medicine, Charles University and Motol University Hospital, Prague, Czech Republic; Hanne Vebert Olesen, Department of Paediatrics and Adolescent Medicine, Cystic Fibrosis Centre, Aarhus University Hospital, Aarhus, Denmark; Pierre-Régis Burgel, Respiratory Medicine and National Cystic Fibrosis Reference Center, Cochin Hospital, AP-HP, Université de Paris, Institut Cochin, INSERM U1016, Paris, France; Lydie Lemonnier-Videau, Vaincre la Mucoviscidose, Paris, France; Lutz Naehrlich, Universities of Giessen and Marburg Lung Centre, German Centre for Lung Research, Justus Liebig University Giessen, Giessen, Germany; Andrea Párniczky, Heim Pál National Paediatric Institute, Budapest and Institute for Translational Medicine, University of Pécs, Medical School, Pécs, Hungary; Godfrey Fletcher, The Cystic Fibrosis Registry of Ireland, Dublin, Ireland; Meir Mei-Zahav, Pulmonary Institute, Schneider Children's Medical Centre of Israel, Petah Tikva and Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel; Rita Padoan, Cystic Fibrosis Regional Support Centre, Department of Paediatrics, University of Brescia, Brescia and Scientific Board of Italian CF Registry, Rome, Italy; Anna Zolin, Department of Clinical Sciences and Community Health, Laboratory of Medical Statistics, Biometry and Epidemiology “G.A. Maccaccaro”, University of Milan, Milan, Italy; Elina Aleksejeva, Department of Pneumology, Children's Clinical University Hospital, Riga Stradinš University, Riga, Latvia; Kestutis Malakauskas, Adult Cystic Fibrosis Centre, Department of Pulmonology, Lithuanian University of Health Sciences, Kaunas, Lithuania; Oxana Turcu, Department of Paediatrics, Ambulatory Cystic Fibrosis and Other Rare Diseases Centre, Institute for Maternal and Child Healthcare, State University of Medicine and Pharmacy “Nicolae Testemitanu”, Chisinau, Republic of Moldova; Domenique Zomer, Dutch Cystic Fibrosis Foundation (NCFS), Baarn, The Netherlands; Stojka Fustik, Centre for Cystic Fibrosis, University Children's Hospital, Skopje, North Macedonia; Ivana Arnaudova Danevskai, Centre for Cystic Fibrosis, Children and Adults, Institute for Respiratory Diseases in Children, Kozle, North Macedonia; Luísa Pereira, Centre for Cystic Fibrosis, Hospital de Santa Maria, Lisbon, Portugal; Liviu Pop, Victor Babes University of Medicine and Pharmacy Timisoara, National Cystic Fibrosis Centre, Timisoara, Romania; Elen Kondratyeva, Research Centre for Medical Genetics, Moscow, Russian Federation; Milan Rodić, National Centre for Cystic Fibrosis, Mother and Child Health Institute of Serbia “Dr Vukan Čupić”, Belgrade, Serbia; Hana Kayserová, Cystic Fibrosis Centre, University Hospital of Bratislava, Bratislava, Slovakia; Uroš Krivec, Department of Paediatric Pulmonology, University Children's Hospital, Ljubljana University Medical Centre, Ljubljana, Slovenia; Maria Dolores Pastor-Vivero, Paediatric Pneumology and Cystic Fibrosis Unit, Osakidetza, Hospital Universitario Cruces, Bizkaia, Spain; Christina Krantz, Uppsala CF Centre, Department of Women's and Children's Health, Uppsala University, Uppsala, Sweden; Anders Lindblad, Gothenburg CF Centre, Queen Silvia Children's Hospital, The Sahlgrenska Academy at the University of Gothenburg, Gothenburg, Sweden; Andreas Jung, Paediatric Pulmonology, University Children's Hospital Zurich, Zurich, Switzerland; Halyna Makukh, Institute of Hereditary Pathology, Ukrainian National Academy of Medical Sciences, Lviv, Ukraine; Siobhán B. Carr, Department of Respiratory Paediatrics, Royal Brompton Hospital and NHLI, Imperial College, London, UK; Sarah Clarke, Cystic Fibrosis Trust, London, UK.
Ethics statement: All the participating centres and national registries in the ECFSPR have ethical approval. Informed consent for anonymous data collection and ECFSPR participation, including consent that data may be used for future research were obtained from all participants. This study was approved by the ECFSPR Scientific Committee and the ECFSPR Steering Committee.
This article has an editorial commentary: https://doi.org/10.1183/13993003.00328-2024
Conflict of interest: L. Naehrlich reports grants from the German Center for Lung Research, Vertex Pharmaceuticals and Mukoviszidose Institute, participation on a trial steering committee for CF STORM, leadership roles as medical lead of the German CF Registry and pharmacovigilance study manager of the ECFSPR, and medical writing from Articulate Science, outside the submitted work. I. Sermet-Gaudelus reports grants, consulting fees and lecture honoraria from Vertex Pharmaceuticals, and a leadership role as medical lead of the French Pediatric CF Network, outside the submitted work. The remaining authors have no potential conflicts of interest to disclose.
References
- 1.Zolin A, Bossi A, Cirilli N, et al. Cystic fibrosis mortality in childhood. Data from European Cystic Fibrosis Society Patient Registry. Int J Environ Res Public Health 2018; 15: 2020. doi: 10.3390/ijerph15092020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCormick J, Sims EJ, Green MW, et al. Comparative analysis of cystic fibrosis registry data from the UK with USA, France and Australasia. J Cyst Fibros 2005; 4: 115–122. doi: 10.1016/j.jcf.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 3.Rabin HR, Butler SM, Wohl ME, et al. Pulmonary exacerbations in cystic fibrosis. Pediatr Pulmonol 2004; 37: 400–406. doi: 10.1002/ppul.20023 [DOI] [PubMed] [Google Scholar]
- 4.Stern M, Wiedemann B, Wenzlaff P. From registry to quality management: the German Cystic Fibrosis Quality Assessment project 1995–2006. Eur Respir J 2008; 31: 29–35. doi: 10.1183/09031936.00056507 [DOI] [PubMed] [Google Scholar]
- 5.Szczesniak R, Heltshe SL, Stanojevic S, et al. Use of FEV1 in cystic fibrosis epidemiologic studies and clinical trials: a statistical perspective for the clinical researcher. J Cyst Fibros 2017; 16: 318–326. doi: 10.1016/j.jcf.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanders DB, Chmiel JF. Drug development for cystic fibrosis. Pediatr Pulmonol 2021; 56: S10–S22. doi: 10.1002/ppul.25075 [DOI] [PubMed] [Google Scholar]
- 7.Burgel PR, Bellis G, Olesen HV, et al. Future trends in cystic fibrosis demography in 34 European countries. Eur Respir J 2015; 46: 133–141. doi: 10.1183/09031936.00196314 [DOI] [PubMed] [Google Scholar]
- 8.Viviani L, Zolin A, Mehta A, et al. The European Cystic Fibrosis Society Patient Registry: valuable lessons learned on how to sustain a disease registry. Orphanet J Rare Dis 2014; 9: 81. doi: 10.1186/1750-1172-9-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerem E, Orenti A, Zolin A, et al. Clinical outcomes associated with Achromobacter species infection in people with cystic fibrosis. J Cyst Fibros 2022; 22: 334–343. doi: 10.1016/j.jcf.2022.11.001 [DOI] [PubMed] [Google Scholar]
- 10.Mei-Zahav M, Orenti A, Jung A, et al. Disease severity of people with cystic fibrosis carrying residual function mutations: data from the ECFS Patient Registry. J Cyst Fibros 2023; 22: 234–247. doi: 10.1016/j.jcf.2022.07.015 [DOI] [PubMed] [Google Scholar]
- 11.Hatziagorou E, Fieuws S, Orenti A, et al. Risk factors for FEV1 decline in European patients with CF: data from the European Cystic Fibrosis Society Patient Registry (ECFSPR). ERJ Open Res 2023; 9: 00449-2022. doi: 10.1183/23120541.00449-2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hatziagorou E, Orenti A, Drevinek P, et al. Changing epidemiology of the respiratory bacteriology of patients with cystic fibrosis – data from the European Cystic Fibrosis Society Patient Registry. J Cyst Fibros 2020; 19: 376–383. doi: 10.1016/j.jcf.2019.08.006 [DOI] [PubMed] [Google Scholar]
- 13.Kerem E, Viviani L, Zolin A, et al. Factors associated with FEV1 decline in cystic fibrosis: analysis of the ECFS Patient Registry. Eur Respir J 2014; 43: 125–133. doi: 10.1183/09031936.00166412 [DOI] [PubMed] [Google Scholar]
- 14.Kerem E, Cohen-Cymberknoh M. Disparities in cystic fibrosis care and outcome: socioeconomic status and beyond. Chest 2016; 149: 298–300. doi: 10.1016/j.chest.2015.08.021 [DOI] [PubMed] [Google Scholar]
- 15.McGarry ME, Williams WA, McColley SA. The demographics of adverse outcomes in cystic fibrosis. Pediatr Pulmonol 2019; 54: S74–S83. doi: 10.1002/ppul.24434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oates GR, Schechter MS. Socioeconomic status and health outcomes: cystic fibrosis as a model. Expert Rev Respir Med 2016; 10: 967–977. doi: 10.1080/17476348.2016.1196140 [DOI] [PubMed] [Google Scholar]
- 17.Lee TWR, Brownlee KG, Conway SP, et al. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J Cyst Fibros 2003; 2: 29–34. doi: 10.1016/S1569-1993(02)00141-8 [DOI] [PubMed] [Google Scholar]
- 18.Quanjer PH, Stanojevic S, Cole TJ, et al. Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur Respir J 2012; 40: 1324–1343. doi: 10.1183/09031936.00080312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo J, Garratt A, Hill A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J Cyst Fibros 2022; 21: 456–462. doi: 10.1016/j.jcf.2022.01.009 [DOI] [PubMed] [Google Scholar]
- 20.Lee T, Sawicki GS, Altenburg J, et al. Effect of elexacaftor/tezacaftor/ivacaftor on annual rate of lung function decline in people with cystic fibrosis. J Cyst Fibros 2023; 22: 402–406. doi: 10.1016/j.jcf.2022.12.009 [DOI] [PubMed] [Google Scholar]
- 21.Bower JK, Volkova N, Ahluwalia N, et al. Real-world safety and effectiveness of elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis: interim results of a long-term registry-based study. J Cyst Fibros 2023; 22: 730–737. doi: 10.1016/j.jcf.2023.03.002 [DOI] [PubMed] [Google Scholar]
- 22.McKone EF, Ariti C, Jackson A, et al. Survival estimates in European cystic fibrosis patients and the impact of socioeconomic factors: a retrospective registry cohort study. Eur Respir J 2021; 58: 2002288. doi: 10.1183/13993003.02288-2020 [DOI] [PubMed] [Google Scholar]
- 23.Oates GR, Baker E, Rowe SM, et al. Tobacco smoke exposure and socioeconomic factors are independent predictors of pulmonary decline in pediatric cystic fibrosis. J Cyst Fibros 2020; 19: 783–790. doi: 10.1016/j.jcf.2020.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oates GR, Schechter MS. Social inequities and cystic fibrosis outcomes: we can do better. Ann Am Thorac Soc 2021; 18: 215–217. doi: 10.1513/AnnalsATS.202010-1274ED [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schechter MS, Shelton BJ, Margolis PA, et al. The association of socioeconomic status with outcomes in cystic fibrosis patients in the United States. Am J Respir Crit Care Med 2001; 163: 1331–1337. doi: 10.1164/ajrccm.163.6.9912100 [DOI] [PubMed] [Google Scholar]
- 26.O'Connor GT, Quinton HB, Kneeland T, et al. Median household income and mortality rate in cystic fibrosis. Pediatrics 2003; 111: e333–e339. doi: 10.1542/peds.111.4.e333 [DOI] [PubMed] [Google Scholar]
- 27.Taylor-Robinson DC, Smyth RL, Diggle PJ, et al. The effect of social deprivation on clinical outcomes and the use of treatments in the UK cystic fibrosis population: a longitudinal study. Lancet Respir Med 2013; 1: 121–128. doi: 10.1016/S2213-2600(13)70002-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen-Cymberknoh M, Shoseyov D, Kerem E. Managing cystic fibrosis: strategies that increase life expectancy and improve quality of life. Am J Respir Crit Care Med 2011; 183: 1463–1471. doi: 10.1164/rccm.201009-1478CI [DOI] [PubMed] [Google Scholar]
- 29.Kerem E, Conway S, Elborn S, et al. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros 2005; 4: 7–26. doi: 10.1016/j.jcf.2004.12.002 [DOI] [PubMed] [Google Scholar]
- 30.Castellani C, Conway S, Smyth AR, et al. Standards of care for cystic fibrosis ten years later. J Cyst Fibros 2014; 13: S1–S2. doi: 10.1016/j.jcf.2014.03.008 [DOI] [PubMed] [Google Scholar]
- 31.Castellani C, Duff AJA, Bell SC, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros 2018; 17: 153–178. doi: 10.1016/j.jcf.2018.02.006 [DOI] [PubMed] [Google Scholar]
- 32.McQuaid EL, Landier W. Cultural issues in medication adherence: disparities and directions. J Gen Intern Med 2018; 33: 200–206. doi: 10.1007/s11606-017-4199-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farrell MH, Kuruvilla P. Assessment of parental understanding by pediatric residents during counseling after newborn genetic screening. Arch Pediatr Adolesc Med 2008; 162: 199–204. doi: 10.1001/archpediatrics.2007.55 [DOI] [PubMed] [Google Scholar]
- 34.Taylor-Robinson D, Whitehead M, Diderichsen F, et al. Understanding the natural progression in %FEV1 decline in patients with cystic fibrosis: a longitudinal study. Thorax 2012; 67: 860–866. doi: 10.1136/thoraxjnl-2011-200953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.European Medicines Agency . Qualification Opinion 4. The European Cystic Fibrosis Society Patient Registry (ECFSPR). 2018. www.ema.europa.eu/documents/regulatory-procedural-guideline/qualification-opinion-european-cystic-fibrosis-society-patient-registry-ecfspr_en.pdf Date last accessed: 14 January 2024.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary figures and tables ERJ-01241-2023.Supplement (197.2KB, pdf)
This one-page PDF can be shared freely online.
Shareable PDF ERJ-01241-2023.Shareable (437.5KB, pdf)