
Figure 2. Distinguishing major and minor-isoform introns and measuring the rate of alternative splicing.
(A) Definition of the variables used to compute the relative abundance of a spliced isoform compared to other transcripts with alternative splice boundaries (RAS) or compared to unspliced transcripts (RANS): : number of spliced reads corresponding to the precise excision of the focal intron; : number of reads corresponding to alternative splice variants relative to this intron (i.e. sharing only one of the two intron boundaries); : number of unspliced reads, co-linear with the genomic sequence. (B,C) Histograms representing the distribution of RAS and RANS values (divided into 5% bins), for protein-coding gene introns. Each line represents one species. Two representative species are colored: Drosophila melanogaster (red), Homo sapiens (brown). (D) Description of the variables used to compute the AS rate of a given a major-isoform intron, and the ’minor-isoform intron relative abundance’ (MIRA) of each of its splice variants (SVs): : number of spliced reads corresponding to the excision of the major-isoform intron; : number of spliced reads corresponding to the excision of a minor-isoform intron (i); : total number of spliced reads corresponding to the excision of minor-isoform introns. (E) Definitions of the main variables used in this study.

Figure 2—figure supplement 1. Transcriptome sequencing depth affects intron detection power and AS rate estimates.

Figure 2—figure supplement 2. The power to detect AS events is positively correlated with transcriptome sequencing depth.
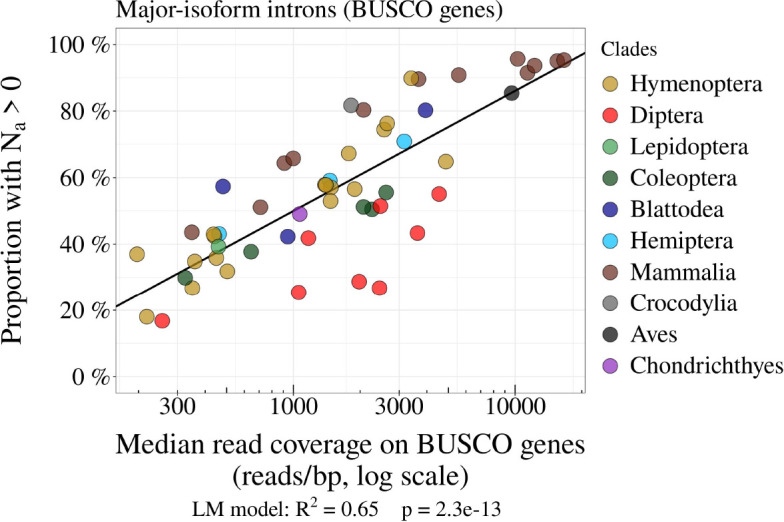
Figure 2—figure supplement 3. Description of the bioinformatic analyses pipeline.
