
Figure 2.
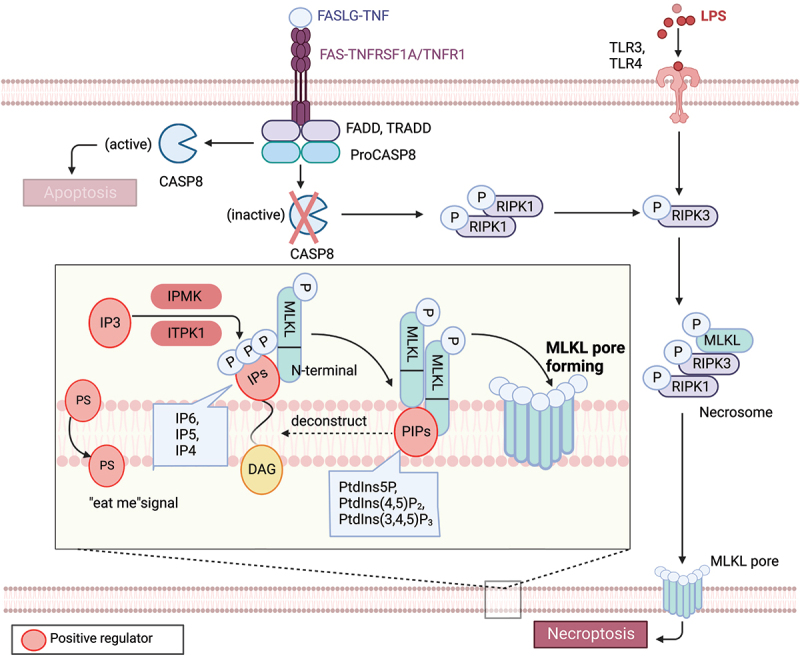
Lipid mechanism in necoroptosis. In the presence of caspase inhibition, necroptosis can be triggered by death receptor stimuli (e.g., TNFRSF1A/TNFR1 and FAS) or pathogen recognition receptor stimuli (e.g., TLR3 and TLR4). When CASP8 is inactive, a complex called the necrosome, comprised of RIPK1, RIPK3, and MLKL, forms. Within this complex, RIPK3-mediated phosphorylation induces a conformational change that activates MLKL, leading to its oligomerization and translocation to the plasma membrane. Subsequently, MLKL forms pores or cation channels at the plasma membrane, resulting in necroptosis. MLKL binds to PtdIns5P, PtdIns(4,5)P2, and PtdIns(3,4,5)P3, but not to unphosphorylated phosphatidylinositol (PtdIns) or other phospholipids. PIPs can be enzymatically deconstructed into diacylglycerol and inositol phosphates. MLKL interacts with highly phosphorylated inositol products (e.g., IP6, IP5, IP4) generated by IP kinases (IPMK and ITPK1). Necroptotic cells also expose phosphatidylserine (PS) on the outer leaflet of the plasma membrane, acting as an “eat-me” signal that can be recognized and phagocytosed.