
Figure 8.
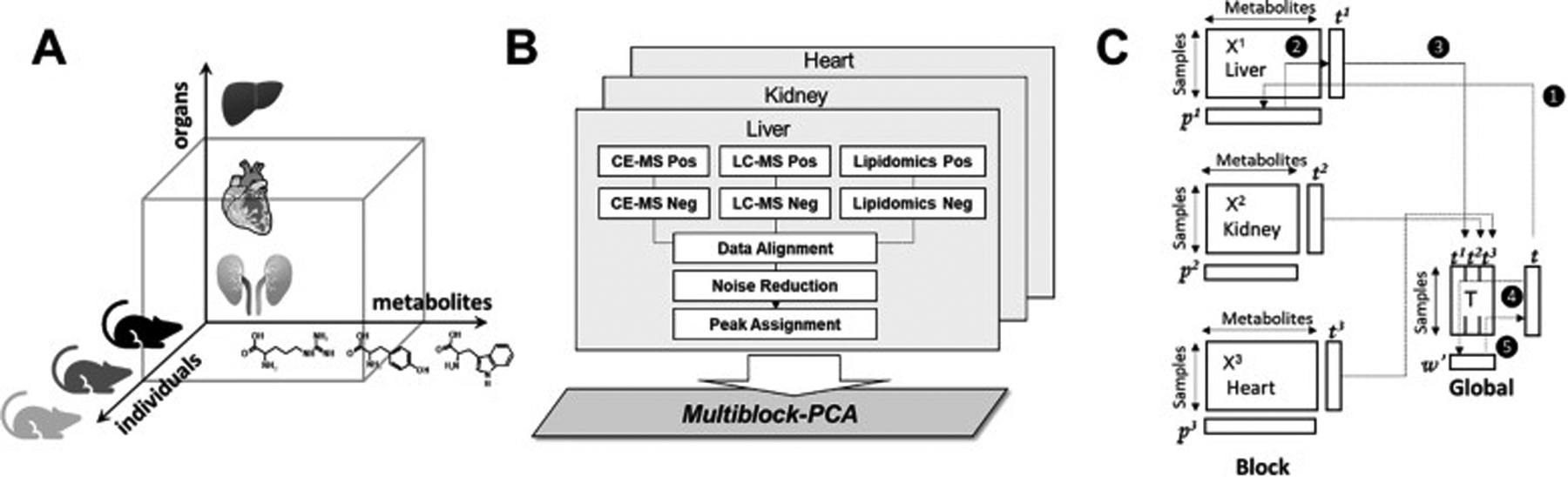
Multiblock metabolomics scheme. (A) Multiblock metabolomics requires a three-dimensional data structure, metabolites, individual samples, and organs. (B) Metabolomics data obtained from CE/MS and LC/MS (hilic and lipid modes) were merged into one data table. After noise reduction, peaks were identified based on the matched m/z values and normalized retention times of the corresponding standard compounds. This process was repeated for the heart, kidney, and liver, and three data matrices were integrated using multiblock PCA. (C) Multiblock PCA architecture: ❶All blocks of X1,2,3 were regressed by an arbitrary global score t to obtain the block loadings p1,2,3. ❷The block scores t1,2,3 were calculated with the normalized block loadings p1,2,3 using the following equation: tb = Xb pb where b = 1, 2, 3. ❸All block scores were combined to a global score matrix T. ❹The global score matrix T was regressed by the global score vector t, resulting in the global weights. ❺ Global weights were normalized to length one and a new global score vector t was then calculated. Reprinted with permission from K. Tanabe, C. Hayashi, T. Katahira, K. Sasaki, K. Igami, Multiblock metabolomics: An approach to elucidate whole-body metabolism with multiblock principal component analysis. Computational and Structural Biotechnology Journal 2021, 19, 1956–1965. Copyright 2021 Elsevier.