

Abstract
Viral mutations within patients nurture the adaptive potential of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) during chronic infections, which are a potential source of variants of concern. However, there is no integrated framework for the evolutionary analysis of intra-patient SARS-CoV-2 serial samples. Herein, we describe Viral Intra-Patient Evolution Reporting and Analysis (VIPERA), a new software that integrates the evaluation of the intra-patient ancestry of SARS-CoV-2 sequences with the analysis of evolutionary trajectories of serial sequences from the same viral infection. We have validated it using positive and negative control datasets and have successfully applied it to a new case, which revealed population dynamics and evidence of adaptive evolution. VIPERA is available under a free software license at https://github.com/PathoGenOmics-Lab/VIPERA.
Keywords: SARS-CoV-2, within-host evolution, serially sampled infection, intra-patient diversity, snakemake workflow, bioinformatics
Background
During the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, more than seven million deaths have been reported by the World Health Organization (WHO) due to Coronavirus disease 2019 (COVID-19) (World Health Organization 2024). The pandemic has been driven by SARS-CoV-2 variants of concern (VOC), which are variants with an increased pathogenicity (Centers for Disease Control and Prevention 2020). These VOCs have appeared several times in the COVID-19 pandemic, and it has been observed that the clades containing the VOCs are preceded by a stem branch that shows, on average, a four-fold increase in the substitution rate (Tay et al. 2022), which was usually around 10–3 substitutions per site and year in 2020 (Duchene et al. 2020; van Dorp et al. 2020).
Different hypotheses—such as undetected acute infections (E. Wilkinson et al. 2021) or secondary hosts—have been proposed to explain the increase in the substitution rate and thus, the appearance of VOCs. Nowadays, several pieces of evidence support the hypothesis that VOCs originated in chronic infections. First, the immune system of immunocompromised patients can fail to clear acute SARS-CoV-2 infections leading to long-term infections (Clark et al. 2021). The high number of viral mutations from long-term infections, most of them in the spike protein-coding region (Harari et al. 2022), would suggest an increased evolutionary rate, as observed in branches that give rise to VOC clades (Msomi et al. 2021). Second, defining mutations of several VOCs have been detected in sequences from chronic infections (S. A. J. Wilkinson et al. 2022). Following these findings, there has been an effort not only to study SARS-CoV-2 chronic infections, trying to enhance the surveillance of VOCs, but also to better understand the mechanisms behind their emergence (Harari et al. 2022; Nussenblatt et al. 2022; Chaguza et al. 2023; Gonzalez-Reiche et al. 2023). While there are pipelines that integrate reproducible workflows to analyze genomic diversity between patients (Hadfield et al. 2018; Bukur et al. 2023), there is a lack of easily deployable, accessible, and integrated workflows for analyzing and reporting the evolutionary trajectories of SARS-CoV-2 chronic infections. Current pipelines for processing serially sampled sequencing data that consider the particularities of intra-host samples are restricted to certain analyses, such as detecting mixed viral populations, or identifying chronic infections but using only consensus sequences (Valieris et al. 2022; Gonzalez-Reiche et al. 2023; Goya et al. 2023; Harari et al. 2024; Pipek et al. 2024). For this reason, carrying out this type of studies through public databases is a difficult task especially without further clinical information.
Here, we present Viral Intra-Patient Evolution Reporting and Analysis (VIPERA), a user-friendly workflow to easily identify and study within-host evolution in SARS-CoV-2 serially sampled infections. First, it provides an aggregate of population genomics and phylogenetic analyses that allows researchers to determine if a collection of SARS-CoV-2 samples originates from a single virus serially sampled infection. Furthermore, VIPERA provides insights into intra-host evolutionary dynamics, tracking variant trajectories and selective pressure over time. The generated report serves as a valuable guide for prioritizing and refining subsequent analyses using the newly generated data for tailored in-depth intra-host studies. Overall, this streamlined approach provides a comprehensive overview of evolutionary trends in intra-host evolution.
Results
A comprehensive report of a serially sampled SARS-CoV-2 infection
VIPERA offers an integrated framework for detecting and studying serially sampled SARS-CoV-2 infections. The necessary data inputs are the read mappings (in BAM format) and the consensus genomes (in FASTA format) for each sequence of the target dataset, as well as the associated sample metadata. The main output from VIPERA is a report file in HTML format summarizing all the analyses in three main sections: ‘1. Summary of the target dataset’, ‘2. Evidence for single, serially-sampled infection’, and ‘3. Evolutionary trajectory of the serially-sampled SARS-CoV-2 infection’. In addition, the intermediate files which are instrumental in the creation of the final report—such as the lineage demixing summary, the maximum-likelihood phylogeny of the target dataset within its spatiotemporal context, the pairwise weighted-distance matrix for the target dataset, or the variant calling results with the dataset ancestor as reference—are also made available to the user (see Supplementary Table 1 for a full list). This offers a great degree of flexibility and control over the data, allowing for further in-depth analysis if required. The three sections of the report are described hereafter.
Summary of the target samples dataset
First, the report displays a summary of the target sample dataset that includes the date and location of sampling. This summary also reports the lineage assignment and a time-sorted index of each sample that is used to identify the samples in the downstream analyses.
Evidence for single, serially sampled infection
The first aim of VIPERA is to streamline the process of confirming that samples originate from a single, serially sampled infection collected from the same patient at different time points—as opposed to multiple successive infections, co-infections, or instances of sample contamination. For this, the following analyses are conducted.
Lineage admixture
A lineage composition profile of each sample based on read mappings is reported to detect if different viral lineages are present in the sample (e.g. in co-infections or contaminations).
Phylogenetic reconstruction
A maximum-likelihood tree including target and context samples is displayed in the VIPERA output. Although onward transmission from a chronic host could result in samples that their monophyly is not evident due to sampling bias, a group of SARS-CoV-2 sequences originating from a serially sampled infection tend to be monophyletic. The phylogeny enables users to assess whether the target samples are monophyletic based on ultrafast bootstrap (UFBoot) and Shimodaira–Hasegawa-like approximate likelihood ratio test (SH-aLRT) support values.
Nucleotide diversity comparison
The nucleotide diversity (π) for the target samples is compared with the distribution of π obtained for random subgroups extracted from a patient-independent context dataset. If the target dataset has a significantly lower π than the distribution of π-values for sequences from different patients, then we can assume that they come from the same viral infection. The report includes the estimated significance of π being lower in the target samples.
Evolutionary trajectory of the serially sampled SARS-CoV-2 infection
The next step is to characterize within-host evolution. To this end, VIPERA reports a set of analyses focused on describing the intra-host evolutionary trajectory of the target samples. This serves as a comprehensive overview of temporal trends for guiding researchers towards informed research decisions.
Number of polymorphic sites
To investigate the within-host viral diversity, we use the number of polymorphic sites as a measure of diversity. The default lower threshold for minor allele frequency is 0.05. The report displays the number of polymorphic sites of each sample and the correlation of this parameter with time, which allows for the observation of fluctuations in diversity throughout the course of the infection.
Description of within-host nucleotide variants
The report includes a summary of within-host nucleotide variants with respect to its predicted ancestral sequence. The summary includes a genome-wide depiction of the proportion of sites in which we find a polymorphism. This allows for the identification of mutation hotspots. The summary also depicts each individual mutation throughout the genome for each sample. Mutations are represented according to their classification in single-nucleotide variants (SNVs) or insertions and deletions (indels) and colored depending on whether they are synonymous or non-synonymous SNVs, in-frame or frameshift indels, or intergenic nucleotide changes. Due to the relevance of the spike protein for SARS-CoV-2 adaptation, a zoom-in of the summary is also generated for the S gene.
Temporal signal of the intra-host mutations
The temporal signal of the target samples is also assessed. First, a neighbor-joining tree of the target samples is constructed using weighted pairwise distances based on allele frequencies. Then, root-to-tip distances measured on this tree are correlated with time. The estimated evolutionary rate is reported as the number of changes per year. We also evaluate the correlation of allele frequencies at each polymorphic site with time, calculating the Pearson’s correlation coefficients and the adjusted significance of the linear fit. Then, allele frequencies with a significant, positive correlation—which are assumed to be affected by selective pressures or hitchhiking—are displayed on a time series of allele frequencies along the viral genome. All sites with more than one alternative allele are also displayed.
Correlation between alternative alleles
To evaluate if there are interactions between mutations, the report includes an interactive heatmap of pairwise allele frequency correlation coefficients, which includes the relationships between alleles. The interactive heatmap enables the user to easily obtain correlation values and restrict the region for visualization.
Non-synonymous and synonymous substitution rates over time
The report includes a time series of the synonymous mutations per synonymous site (dS) and non-synonymous mutations per non-synonymous site (dN) of each sample with respect to the ancestor sequence, as well as their ratio ω (dN/dS).
Validating the detection of serially sampled infections
To validate the evidence of serially sampled infection, we tested the pipeline with two control sets of samples. The positive control dataset includes thirty sequences from a chronic infection collected in Yale between 8 February 2021 and 7 March 2022 (Chaguza et al. 2023). All the sequences from the positive control were designated as the B.1.517 lineage. Its context dataset (n = 170) was automatically fetched from the Global Initiative on Sharing All Influenza Data (GISAID), searching for samples assigned to the same lineage, and collected in the same location, from 1 February 2021 to 12 March 2022.
The negative control dataset combines fifteen sequences from two different patients. The Patient A dataset, which includes twelve samples, represented a serially sampled infection. Subsequently, it was selected as the target dataset for our novel case study, elaborated in the ensuing ‘Results’ section. The Patient A dataset was combined with three samples from a different patient (Patient B) to compose an artificial but robust negative control, which finally contained an unbalanced number of sequences (4:1 ratio) from two potentially serially sampled infections, as opposed to a single infection from a single patient. All samples were collected in Barcelona between 24 March 2020 and 16 November 2020, and designated as lineage B.1 (see ‘Methods’ section). Its context dataset (n = 84) was also automatically fetched from GISAID by searching for the same lineage, and collected in the same location, from 11 March 2020 to 28 November 2020.
Lineage composition analysis to confirm homogeneity within patient
After estimating the lineage admixture of each sample, two different landscapes appeared in the positive and negative control datasets. All thirty samples from the positive control had a 100 per cent estimated abundance of the B.1.517 lineage (Fig. 1A). Conversely, for the negative control, five samples were mostly B.1 or B.1.399, while in the remaining ten samples, B.1 and B.1 sublineages had an estimated abundance of up to 88 per cent (Fig. 1B). The similarity between lineage B.1 and its sublineages (one or two SNPs) was not high enough to conclude that the variety of sublineages was a consequence of different evolutionary origins.
Figure 1.
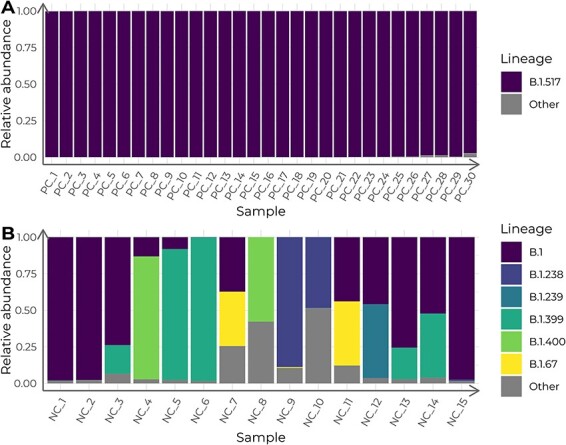
Lineage admixture of the control datasets, calculated with Freyja. Columns depict the estimated relative lineage abundance in each sample in (A) the positive control (PC) dataset and in (B) the negative control (NC) dataset. Samples in the x-axis are ordered chronologically, from more ancient to newer.
Phylogenetic reconstruction to assess monophyly
A maximum-likelihood tree was constructed with both the target and the context datasets for the two validation cases. In the positive control, all thirty samples fell into a robust clade together with the other eight sequences from the context dataset (UFboot: 97 per cent; SH-aLRT: 87 per cent; Fig. 2A). Those eight samples were later confirmed to have been sampled from the same patient (personal communication with Dr Anne Hahn and Dr Nathan Grubaugh). This highlights the capacity of VIPERA for effectively identifying sequences related to the dataset of interest without any prior knowledge. Thus, considering the eight additional sequences as part of our study dataset, and not part of the context, we can conclude that the positive control sequences were monophyletic.
Figure 2.
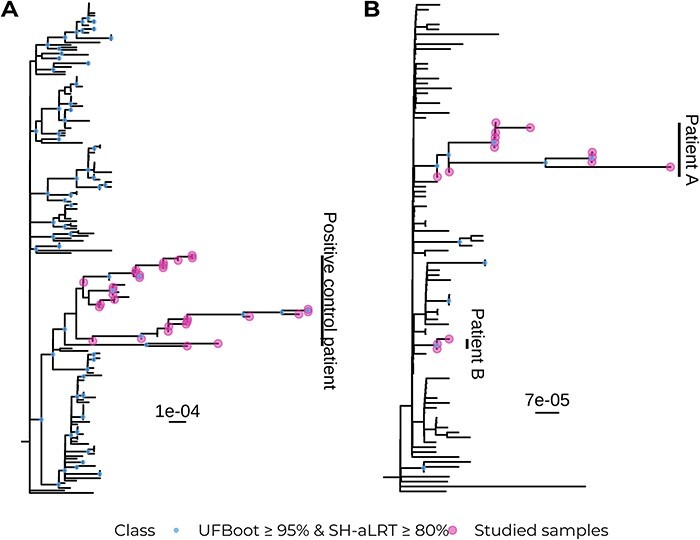
Maximum-likelihood phylogenies of the control datasets and their context samples with 1000 support replicates. (A) Positive control dataset. (B) Negative control dataset.
As for the negative control, all fifteen sequences were paraphyletic and fell into a clade with weak support (UFBoot: 7.0 per cent; SH-aLRT: 0.00 per cent) together with another unrelated sixty-one context sequences. However, sequences were divided into two strongly supported monophyletic clades that correspond with the two groups of samples coming from two different patients that we had artificially mixed. One clade contained the three sequences from the patient B of the negative control (UFBoot: 96 per cent; SH-aLRT: 92 per cent) and the other clade contained the twelve sequences from patient A of the negative control (UFBoot: 97 per cent; SH-aLRT: 87 per cent; Fig. 2B).
Nucleotide diversity to confirm the lower diversity of virus samples with a common origin
For each validation dataset, we calculated the nucleotide diversity of the target samples and compared it with the nucleotide diversity of 1,000 subsets of samples of the same size as the target dataset, extracted from each corresponding context dataset. The nucleotide diversity of the positive control (π = 1.85 · 10−4) was significantly lower than that of its corresponding context dataset (average = 5.30 · 10−4, SD = 2.87 · 10−5; t-test t = 376.27, P < 0.001; Fig. 3A) assuming a normal distribution of the context π-values (Shapiro–Wilk test W = 0.997, P = 0.076). Conversely, the negative control dataset did not show a significantly lower nucleotide diversity (π = 1.03 · 10−4) compared to its context dataset π distribution (average = 1.34 · 10−4, SD = 3.55 · 10−5; empirical P = 0.137; Fig. 3B) without assuming normality (Shapiro–Wilk test W = 0.98, P < 0.001).
Figure 3.
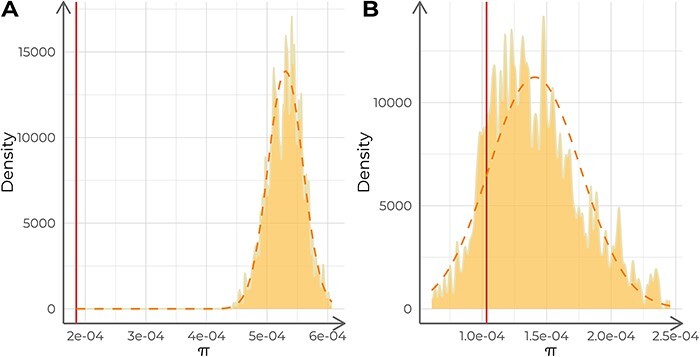
Analysis of the nucleotide diversity (π) of each control dataset. The dashed lines describe a normal distribution with the same mean and standard deviation as the distribution of π-values. The solid vertical lines indicate the π-value for the target samples. (A) Analysis of the positive control against 1,000 replicates (n = 15 each) of its context dataset. (B) Analysis of the negative control against 1,000 replicates (n = 30 each) of its context dataset.
Furthermore, we repeated the analysis of the positive control, this time including in the target dataset the eight additional samples that were extracted from the same patient, as we later discovered. Nucleotide diversity was lower compared with the original analysis (π = 1.3 · 10−4) and with the nucleotide diversity distribution of its corresponding context (average = 5.20 · 10−4, SD = 2.45 · 10−5; t-test t = 514.19, P < 0.001).
Using VIPERA to analyze a novel case
We applied the pipeline to study the within-host evolution in a set of twelve SARS-CoV-2 samples collected from the same host (Patient A) and designated to lineage B.1. The patient was an 82-year-old woman diagnosed with diffuse large B-cell lymphoma (DLBCL) in palliative treatment, who tested positive in SARS-CoV-2 after contact tracing (day zero) a year after DLBCL diagnosis. The patient tested positive again on day thirty-five. She was included in a clinical trial with remdesivir, starting on day forty-five and finalizing on day fifty-five with good tolerance and remission of respiratory symptoms. After three more positive tests on days fifty-five, seventy, and ninety, two doses of hyperimmune plasma were administered on days 128 and 129. The patient tested positive again on days 132, 136, 148, 227, 233, and 237. A general clinical worsening occurred afterwards and the patient passed away sometime after day 281. Further details about the medical history of the patient are available in Supplementary Table 2. Genome sequencing was performed on the fourteen samples collected on each day of positive SARS-CoV-2 testing, but only twelve passed the quality requirements and were analyzed here. These samples were also incorporated in the negative control dataset, as previously described.
The context dataset for the case study was automatically constructed searching for B.1 sequences collected in Barcelona between 24 March 2020 and 16 November 2020, in the GISAID database, and included eighty-five sequences. Additionally, another custom context dataset was also constructed with 110 samples manually selected from the SEQCOVID Consortium. These were collected in Barcelona from independent patients between 11 March 2020 and 28 November 2020 and classified as B.1. The results obtained using both context datasets were consistent, so we report those with the automatically constructed context dataset because it is the default VIPERA option.
Evidence for single, serially sampled infection
First, we investigated the most probable lineage admixture for all twelve samples. We observed two pairs of samples with an estimated lineage abundance of nearly 100 per cent for lineages B.1 and B.1.399, respectively. The remaining samples were further classified as B.1 sublineages with their estimated abundances ranging from 0.07 per cent to 88 per cent (Fig. 4A). The small number of mutations between B.1 and B.1 sublineages (one or two SNPs) might reflect variations during the evolution of the virus over time rather than the mixture of different viruses. Second, the maximum-likelihood phylogeny revealed that the case study dataset formed a monophyletic cluster. The clade that contained all the target samples was supported by UFBoot score of 97 per cent and an SH-aLRT score of 92 per cent (Fig. 5A and B). Third, the nucleotide diversity (π = 4.11 · 10−5) was significantly lower than that of its corresponding context dataset (average = 1.44 · 10−4, SD = 4.04 · 10−5; empirical P < 0.001; Fig. 4B) without assuming a normal distribution of the context π values (Shapiro–Wilk test W = 0.967, P < 0.001). This finding supports the hypothesis of these sequences coming from a serially sampled single-virus infection.
Figure 4.
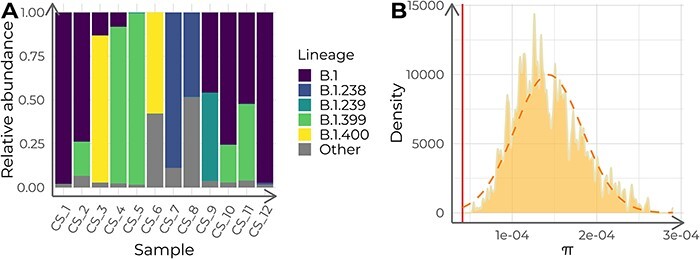
Lineage admixture and nucleotide diversity (π) analysis of the twelve case study samples. (A) Estimated relative lineage abundance in each of the twelve target samples from the case study, calculated with Freyja. Samples in the x-axis are ordered chronologically, from more ancient to newer. (B) Nucleotide diversity (π) distribution for 1,000 samples (n = 12) of context sequences for the case study. The orange dashed curve depicts a normal distribution with the same mean and standard deviation as the π-value distribution. The red vertical line indicates the π of the case study dataset.
Figure 5.
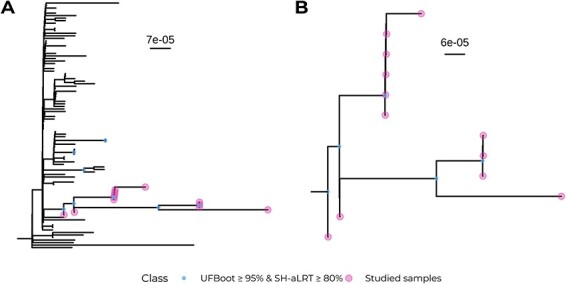
Phylogenetic analysis of the case study dataset. (A) Maximum-likelihood phylogeny with 1,000 supporting replicates for both target samples and samples composing the case study context dataset. (B) Zoom of the clade containing all target samples in (A).
All the evidence that included lineage assignment, monophyly, and nucleotide diversity indicated a proximal common origin. Therefore, we proceeded to examine intra-host evolution, which is described in the third section of the report.
Evolutionary trajectory of the serially sampled SARS-CoV-2 infection
Nucleotide variants associated with within-host evolution.
Genomic variation was not evenly distributed along the SARS-CoV-2 genome. The non-structural protein (NSP) 3 coding region in ORF1ab, the S gene and the N gene, reached peaks of 1 percent of polymorphic sites (Fig. 6). We found ten indels, six of which led to frameshifts: two in the ORF1ab, two in the ORF7b, one in the ORF3a and N gene. Additionally, ninety-nine different SNPs were found, sixty-seven of which were non-synonymous (see the automatically generated variant calling results in Supplementary Table 3).
Figure 6.
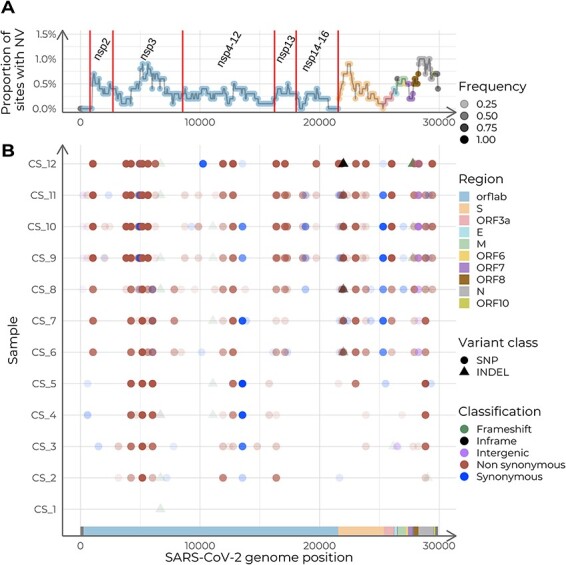
Summary of the intra-host accumulation of nucleotide variants (NV), using the dataset ancestor as reference. (A) Nucleotide variants per site along the SARS-CoV-2 genome. Relative abundance of NVs is calculated with a sliding window of width 1,000 nucleotides and a step of fifty. Labels indicate the coding regions of the non-structural proteins (NSP) within ORF1ab. (B) Genome variation along the genome for each sample. The y-axis displays samples in chronological order, with the earliest collection date at the bottom and the latest at the top.
We evaluated the correlation of allele frequencies with time for all detected variants. Eight out of 109 showed a significant correlation with time, being positive for all of them (Pearson’s coefficients ranging from 0.873 to 0.957; Fig. 7A and B). Additionally, we found two positions with more than one alternative allele (Fig. 7B). Interestingly, all variants that showed a significant correlation with time, showed a change in allele frequencies after the administration of hyperimmune plasma (samples labeled CS_5 and CS_6; Fig. 7B). Therefore, we evaluated all changes in viral allele frequencies associated with the administration of therapeutic agents that were reported in the medical history. We based this analysis on the aforementioned variant calling output (Supplementary Table 2), which demonstrates the usefulness of the VIPERA output to perform further downstream analyses. We found thirty-nine genetic variants that changed in frequency before and after the administration of hyperimmune plasma. Twenty-three of them were found in ORF1ab, ten in gene S, two in gene N, two in ORF3a, one in ORF8, and one in an intergenic region (Supplementary Fig. 1A). We also evaluated viral variation before and after the remdesivir clinical trial (samples labeled CS_2 and CS_3). We found twenty-seven genetic variants that changed frequency, including seventeen in ORF1ab, two in gene S, two in gene N, one in each ORF3a, ORF7, and ORF8, and three intergenic (Supplementary Fig. 1B). Among these alleles, eleven changed frequency after both treatments, with eight of them having opposite trajectories (ORF1ab:T1322I and ORF1ab:T1322K in NSP3, ORF1ab:A1923V in NSP3, ORF1ab:K3886N in NSP7, ORF1ab:I4429I in NSP12, ORF1ab:P5371S in NSP13, S:G35G, and N:S194L), and three having the same trajectory (ORF1ab:T1638I in NSP3, ORF1ab:D4166N in NSP9, and S:I770V).
Figure 7.
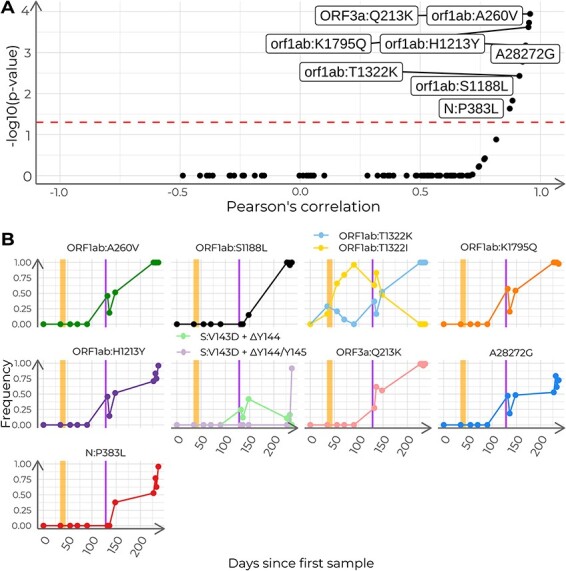
Analysis of the frequency of polymorphisms with time in the case study. (A) Pearson’s correlation coefficients and adjusted P-values for all 110 detected nucleotide variants. Dashed line indicates adjusted P = 0.05. Labeled dots represent nucleotide variants correlated with time (adjusted P < 0.05). B) Time series of relative allele frequencies. The shown positions include nucleotide variants with a significant correlation with time and sites with more than two possible states. Each subplot depicts the progression of the allele frequencies in time for a given genome position. The vertical stripes in orange indicate the span of the remdesivir clinical trial. The vertical stripes in purple indicate the days of administration of hyperimmune plasma.
Finally, we evaluated the correlation in time-dependent trajectories between different alleles. We found pairwise correlation coefficients above 0.85 between the trajectories of ORF1ab:A260V (NSP2), ORF1ab:S1188L (NSP3), ORF1ab:T1322K (NSP3), ORF1ab:K1795Q (NSP3), A28272G, ORF1ab:H1213Y (NSP13), N:P383L, and ORF3a:Q213K (Figs 7 and 8). In addition, these variants correlated with ORF8:I121L and ORF1ab:P970S (NSP13) as well (Fig. 8B).
Figure 8.
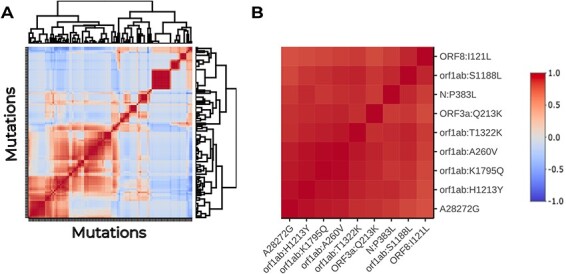
Heatmap of the association between polymorphism trajectories in the case study. (A) Hierarchically clustered heatmap of the pairwise Pearson’s correlation coefficients between the time series of allele frequencies in the case study. The cluster containing the previously found mutations is squared in black. (B) Subset of the correlation heatmap, restricted to the cluster marked in (A).
Evolutionary dynamics.
Using the number of polymorphic sites as an estimate of genetic diversity, we observed that diversity was positively correlated with time in days since the first sample and, time since the initial sampling significantly predicted the number of polymorphic sites (R2 = 0.70, F(1, 10) = 22.69, P < 0.001). The estimated substitution rate was 32.02 substitutions per year, 95 per cent CI [26.62, 37.41]. This was not significantly higher than the estimate for the positive control (24.94 substitutions per year, 95 per cent CI [19.59, 30.28]; F(1, 38) = 1.72, P = 0.194).
We calculated the number of non-synonymous substitutions per non-synonymous site (dN) and the number of synonymous substitutions per synonymous site (dS) for each sample. The initial phases of the infection showed very low diversity, but both dN and dS increased over time, reaching values of 0.0007 and 0.0001, respectively. The dN/dS ratio (ω) ranged between 1.11 and 5.98, with an average value of 2.36 (Fig. 9A). Interestingly, ω was higher than in another immunocompromised patient (Chaguza et al. 2023), where dN/dS was generally below one (Fig. 9B). Moreover, we observed distinct ω trajectories after the administration of each therapeutic agent. In the case study, two distinct treatments were used. During the remdesivir clinical trial, ω-value decreased with a fold change of 0.53. However, during hyperimmune plasma administration ω increased with a fold change of 2.40 due to an increase in dN and a slight decrease in dS. The patient from the positive control started a palliative radiation treatment on day 278 and immediately after that day, ω drastically increased due to a decrease in dS rather than an increase in dN.
Figure 9.
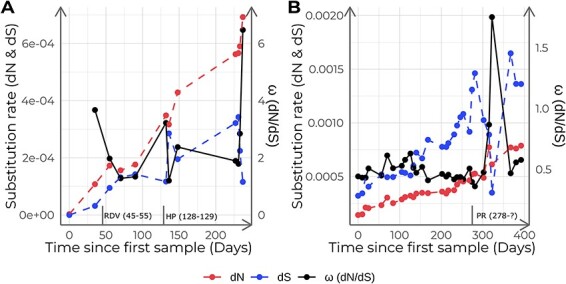
Non-synonymous (dN), synonymous (dS) substitution rates, and ω (dN/dS) for this study samples (A) and the positive control dataset (B). Each point corresponds to a different sample calculated with respect to the ancestor and sorted in chronological order. Vertical lines in the x-axis indicate the administered treatments and their duration: Remdesivir (RDV), hyperimmune plasma (HP), and palliative radiation (PR).
Discussion
Chronic infections are becoming an important issue in SARS-CoV-2 evolutionary studies due to the relationship between the prolonged within-host viral evolution and the emergence of VOCs (Markov et al. 2023). However, the study of serially sampled SARS-CoV-2 samples lacks integrated workflows that facilitate the analyses. To close this gap, we have developed VIPERA, a tool that automatizes the analysis of serially sampled SARS-CoV-2 infections, serving as a valuable baseline for researchers who want to assess the intra-host evolutionary trajectories of SARS-CoV-2 samples. The generated report provides a summary of evolutionary parameters, allowing researchers to make evidence-driven decisions about their research direction using the processed data and prioritize areas for further investigation.
A key strength of VIPERA is the combined use of phylogenetic and population genomics approaches to analyze SARS-CoV-2 samples and provide information to ascertain whether there is a serially sampled infection or not. To do so, mapped reads are used in different ways to consider the entire intra-host viral population. First, the lineage assignment of the samples is calculated using allele frequencies. This analysis enables the user to detect co-infections or viral lineage replacement events, which can go unnoticed in a consensus genome analysis. Second, VIPERA also reports a maximum-likelihood phylogeny including the target and the context dataset. The tree allows the user to assess whether the target samples are monophyletic, which is a good indicator for serially sampled infections. Third, because nucleotide diversity is expected to be reduced for SARS-CoV-2 sequences from the same infection compared to independent samples, we use this metric to evaluate serially sampled infections. Comparison of within and between-host diversity has been previously used for viral outbreak analysis to detect transmission chains (Caro-Pérez et al. 2017), and it has proven to be a strong indicator of serially sampled infection in this work. Even when the context dataset includes some samples from the same patient as the target sequences, we found that nucleotide diversity still contains enough signal to differentiate intra-patient variation. This could be partly due to the robustness of the context dataset. Although VIPERA cannot assess in a systematic manner whether all samples in the context dataset are independent, we found identical results when we compared a customized context dataset with truly independent sequences and the automatic one. Thus, these results support the robustness of our approach to select a context dataset automatically.
Once assessed if all sequences derive from the same infection, VIPERA’s results can be used to study the evolutionary process. Phylodynamic processes of inter-host and intra-host evolutionary dynamics can produce distinctive phylogenetic patterns (Grenfell et al. 2004). In our work, monitoring the evolution of the virus during 8 months allowed for the observation of both intra and between-host phylodynamic patterns within the same phylogeny. We achieved this by including a well-designed context dataset, as described earlier. We observed a balanced topology in the phylogeny at an inter-host population level, but a heavily unbalanced one for within-patient samples, reflecting the different intra-host versus inter-host processes. VIPERA also reports dN/dS estimates through time which can potentially reveal selective or population dynamics. In our case study, dN/dS increased over time because dN increased more than dS during the course of the infection. This result contrasts sharply with the study of another immunocompromised patient that we used as a positive control for VIPERA (Chaguza et al. 2023) in which the dN/dS ratio was much lower. In new viral populations, elevated genome-wide dN/dS ratios might reflect a smaller effective population size (Ne) (Lin et al. 2019). Therefore, the lower dN/dS seen in the positive control might indicate that the lack of immune pressure could result in a higher viral Ne and more efficient selection pressure. This could be linked to the patient receiving chimeric antigen receptor T (CAR-T) cell therapy for relapsed DLBCL post-stem-cell transplantation. CAR-T therapy can trigger a substantial, long-term depletion of antibody-producing B cells and increased susceptibility to infections (as reviewed in Cappell and Kochenderfer 2023), which could significantly modulate the long-term immune pressure over SARS-CoV-2. Throughout the most of the infection in the positive control (Chaguza et al. 2023), dN/dS remained below one, with the exception of day 315, when dS experienced a significant drop. This decrease in substitution rate happened just 37 days after the patient started palliative radiation therapy and was admitted in a hospital several times after that. As the treatment and the immune status of the patient might pose a significant pressure on the virus, the diminished evolutionary potential of the virus at that time could also be influenced by this pressure. In any case, by incorporating patient medical histories, our tool empowers researchers to detect evolutionary trajectory alterations that may be associated with or respond to clinical events.
VIPERA also reports a description of the intra-host nucleotide variants and their relationship with other variables such as collection date or other intra-host nucleotide variants. In our case study, we detected several mutations that are concerning because of their relationship with immune system evasion, such as ORF1ab:T1638I (NSP3), ORF1ab:S1188L (NSP3), and ORF3a:Q213K (de Silva et al. 2021; Zekri et al. 2021). We also found mutations previously found in within-host evolution analyses such as N:P383L, ORF1ab:H1213Y (NSP13), and S:V143D + ΔY144 (Chiara et al. 2021; Sahin et al. 2021; Halfmann et al. 2023). This deletion is within a recurrent deletion region (RDR) of the spike protein that has appeared recurrently in long-term infections and has been fixed independently in different variants of concern (McCarthy et al. 2021). Thanks to the availability of a medical history of the case study, we were also able to compare the allele frequencies before and after the administration of therapeutic agents. After treatment with hyperimmune plasma, we detected the emergence of recurrent mutations such as ORF1ab:K1795Q (NSP3), which was commonly found in variant Gamma, and S:E484Q, which later appeared on variant Delta-related lineages associated with a loss of sensitivity to neutralization (Ferreira et al. 2021; Verghese et al. 2021; Brandolini et al. 2022). We also detected the emergence of ORF1ab:A2529V (NSP3), a marker of the Delta lineage AY.4, and S:T95I, which was later commonly found in Omicron lineage BA.1. Both mutations have also been identified in another immunocompromised patient (Zannoli et al. 2023). ORF1ab:H5614Y (NSP13) and S:L141F, which have been predicted to interfere with antiviral drug binding (Ameen et al. 2021; Ghorbani et al. 2022), also emerged after the plasma administration. ORF1ab:D1323G (NSP3) and ORF1ab:T4065I (NSP8) emerged after the clinical trial with remdesivir. They have also been detected in immunocompromised patients (Weigang et al. 2021; Spinicci et al. 2022). ORF1ab:A4841V (NSP12, the RNA-dependent RNA-polymerase) also emerged after the clinical trial and has been observed as a marker of the basal lineage B.1.13 (Thorne et al. 2022). We also noticed the emergence of S:I770V after both treatments, becoming nearly fixed by the end of the study. This mutation was later found in the Omicron lineage CP.4 (see cov-lineages/pango-designation v1.16 on GitHub). Overall, the phylogenetic patterns of our case study, the appearance of recurrent mutations indicative of adaptation, along with the dN/dS dynamics, might be interpreted as positive selection acting on a few sites despite the immune pressure limiting the viral Ne within a chronic host.
In summary, VIPERA facilitates the analysis of SARS-CoV-2 chronic infections by providing evidence for serially sampled infection, describing the viral within-host evolution, and setting up an environment equipped with the files needed for further customized within-host viral evolution analyses. The generated environment includes consensus alignments, phylogenies, sample compositional data, and results concerning variant calling and allele frequency progression. This wealth of data empowers researchers to perform further comparative analysis measuring genetic diversity, population structure, detecting genetic markers associated with resistance or virulence, among others. For instance, we demonstrate that the availability of a medical history associated with the serial sample opens up the possibility of a deeper understanding of the effect of treatments and immune status in viral evolution. For these reasons, we foresee VIPERA as an enhancer for SARS-CoV-2 serially sampled infection studies, contributing to surveillance of VOCs and to understand the mechanisms behind their emergence. Although VIPERA is designed for reporting on SARS-CoV-2 sequence data, the framework could be extended to other viruses in further iterations of the software.
Conclusions
VIPERA is a new bioinformatic tool for studying and analyzing serially sampled SARS-CoV-2 infections. VIPERA provides an aggregate of analysis for detecting whether there is a serially sampled infection or not, including novel approaches such as genetic diversity and genetic distance at the population levels. It also provides a description of the within-host evolution observed in the target samples. Having undergone rigorous validation through two stringent control cases, our tool has proven its efficacy in a real-world case study. Being on the cusp of a new era in understanding the intra-host evolution of SARS-CoV-2, VIPERA paves the way for a more efficient analysis of serially sampled SARS-CoV-2 infections.
Methods
Pipeline implementation
To facilitate the study of SARS-CoV-2 within-host evolution using data from single-virus serially sampled infections, we have implemented VIPERA, a user-friendly, customizable, and reproducible workflow using Snakemake (Mölder et al. 2021), R v4.1.3 (R Core Team 2021) and Python v3.10 (Van Rossum and Drake 2009) in addition to other software listed in Supplementary Table 4. VIPERA enables the automated analysis of an arbitrary number of samples collected from a single patient at different time points after infection. VIPERA takes as input sorted BAM files, consensus sequences in FASTA format and also a metadata file with collection dates, locations and GISAID IDs. The execution configuration parameters are fully documented in the code repository and explicitly exposed in YAML files that can be easily interpreted and modified by the user. While our tool is suited for the computational capabilities of an average laptop, we leveraged Snakemake profiles to ensure seamless deployment in a high-performance computing (HPC) environment. In our cluster, we achieve a consistent run time of less than 15 min, using one Intel(R) Xeon(R) Gold 6230 CPU @ 2.10 GHz and less than 1 GB of RAM. The run time decreases by up to a factor of five on sixteen cores, using around six GB of RAM. The main output of VIPERA is a report file in HTML format that includes different analytical results and data visualization for detecting single-virus sustained infections and studying within-host evolution.
Dataset retrieval and preprocessing
Three sets of SARS-CoV-2 samples were used in order to test and use VIPERA: a positive control, a negative control, and a novel case.
For the positive control, we used thirty SARS-CoV-2 samples collected in Connecticut between 1 June 2021 and 7 March 2022 described as a chronic infection (Chaguza et al. 2023). FASTQ files were fetched from the SRA using fastq-dump, implemented in the SRA toolkit v3.0.0 (Leinonen, Sugawara, and Shumway 2011). Reads were mapped against the Wuhan-Hu-1 reference genome (NCBI RefSeq accession no. NC_045512.2) (Wu et al. 2020) using BWA-MEM v0.7.17 (Li 2013). ARTIC v4.1 primer schemes (ARTICnetwork 2023) were trimmed from the generated BAM files using iVar v1.4.2 (Grubaugh et al. 2019). Using samtools v1.17 (Danecek et al. 2021) and iVar v1.4.2 (Grubaugh et al. 2019), trimmed BAM files were sorted and indexed to obtain the consensus sequence with a minimum frequency threshold of 0.6 and a minimum depth of twenty reads.
The negative control and the novel case datasets were selected from samples for which we had access to read mappings in BAM format, consensus sequences, and metadata via the SeqCOVID Consortium. Viral samples were collected in the Hospital Clínic de Barcelona and sequenced in the Institute of Biomedicine of Valencia using the ARTIC v3 primer scheme (ARTICnetwork 2023). Libraries were prepared using the Nextera Flex DNA Library Preparation Kit and sequenced on the Illumina MiSeq platform. Reads were processed through the SeqCOVID pipeline for SARS-CoV-2 bioinformatic analysis (SeqCOVID Consortium 2021). The case study comprised twelve samples collected from the same patient (Patient A) in Barcelona, Spain between 24 March 2020 and 16 November 2020, and previously designated as lineage B.1 (see Supplementary Table 5). The medical history of the case study was obtained from the Hospital Clínic de Barcelona after approval by the ethical committee. For the negative control, the previous twelve samples were mixed with three samples from a different patient (Patient B), also collected in Barcelona, Spain between 1 April 2020 and 28 August 2020, and previously designated as B.1 (see also Supplementary Table 5).
Characterizing serially sampled infections from a single virus
Longitudinal analysis of viral lineage assignment and admixture
The descriptive analysis of the target dataset of intra-patient samples includes the assignment of a Pango lineage according to sample consensus sequences, as well as the evaluation of possible lineage admixture within each sample. A lineage is assigned to the genome sequences of each sample using Pangolin v4.3 (O’Toole et al. 2021) in accurate (UShER) mode. A demixing step is performed using Freyja v1.4.2 (Andersen Laboratory 2023), which utilizes read mappings to estimate the lineage admixture of each sample based on lineage-defining mutational barcodes by solving a convex optimization problem.
Construction of a context dataset
The analyses require a collection of independent samples—ideally, samples that originate from different hosts and separate infection events. This set of samples is referred to as the ‘context dataset’ in our study. Automated construction of the context dataset is enabled by default, contingent upon the provision of user credentials for the GISAID SARS-CoV-2 database (Khare et al. 2021), using GISAIDR v0.9.9 (Wirth and Duchene 2022). This facilitates the retrieval of a dataset comprising samples that fulfill the spatial, temporal, and phylogenetic criteria, including a sampling location that corresponds to that of the target samples, a collection date that falls within a time window encompassing 95 per cent of the date distribution of the target samples (with 2.5 per cent trimmed at each end to account for extreme values) plus and minus 2 weeks, and a lineage assignment that is shared by at least one of the target samples.
During the process, a series of tweakable checkpoints are established in the configuration file to ensure a robust downstream analysis. First, samples whose GISAID accession number matches any of the target samples are removed. Second, we enforce a minimum number of context samples to provide a sufficient number of random subsample replicates for the diversity assessment. The workflow determines the minimum number of context samples by applying the formula for calculating the number of C(n, k) possible combinations of n items extracted k at a time (n ≥ k ≥ 0), and solving n to obtain C(n, k) context samples with k samples in each subset. Here, k equals the number of samples in the target dataset. The uniroot function of the stats R library is employed for this calculation. If any of these checkpoints fails, the context dataset cannot be automatically built. Alternatively, a manually constructed context dataset may be provided.
For all the analyses shown in this article, an automatically constructed context dataset has been used. Additionally, a manually constructed context dataset was also used for the case study to compare the results with the ones obtained using an automatically constructed context dataset.
Nucleotide diversity comparison
Nucleotide diversity (π) of the target dataset is compared with that of the context dataset, composed of independent samples. By default, a nucleotide diversity distribution for the context dataset is calculated for 1,000 random sample subsets of size equal to the number of target samples extracted without replacement. The number of replicates can be easily modified by the user. Then, the obtained distribution is compared with the nucleotide diversity obtained for the target dataset; empirically, if the π distribution is not normal, or via parametric tests, if it is. Calculations are performed in R and nucleotide diversities are calculated with pegas v1.2 (Paradis 2010).
Assessing phylogenetic relationships
Consensus sequences of the target and context datasets are aligned to the Wuhan-Hu-1 reference genome (NCBI RefSeq accession number: NC_045512.2) (Wu et al. 2020) using Nextalign v2.13 (Hadfield et al. 2018). Positions classified as problematic (Weilguny 2023) are masked in the alignments. Then, a maximum-likelihood phylogeny is constructed using IQTREE v2.2.2.3 (Minh et al. 2020). By default, inference is performed under a general time-reversible (GTR) substitution model with empirical base frequencies, a heterogeneity model with a proportion of invariable sites and a discrete Gamma distribution with four rate categories, ultrafast bootstrap (UFBoot) (Minh, Nguyen, and von Haeseler 2013; Hoang et al. 2018) with 1,000 replicates, and the Shimodaira–Hasegawa-like approximate likelihood ratio test (SH-aLRT) (Guindon et al. 2010) with 1,000 replicates. This inference enables the study of the taxonomic grouping of the target dataset within the relevant epidemic context.
Describing within-host variability
Variant calling and nucleotide variant description
Variants are called using samtools v1.17 (Danecek et al. 2021) and iVar v1.4.2 (Grubaugh et al. 2019) using a reconstructed ancestral genome as reference to restrict the analysis to sequence variation related to the within-host evolution. Variants are re-annotated using snpEff v5.1d (Cingolani et al. 2012). To reconstruct the ancestral sequence, the target samples are aligned to the Wuhan-Hu-1 reference genome (NCBI RefSeq accession no. NC_045512.2) (Wu et al. 2020) using Nextalign v2.13 (Hadfield et al. 2018). Then, the ancestral genome is obtained with IQTREE v2.2.2.3 (Minh et al. 2020). By default, maximum-likelihood trees are inferred under a GTR substitution model with empirical base frequencies and a heterogeneity model with a proportion of invariable sites and a discrete Gamma distribution with four rate categories. The quality criteria for variant calling were a minimum base quality of twenty, a minimum depth of thirty and a minimum frequency cutoff of 5 percent. Nucleotide variants supported by less than twenty reads or less than two reads in one strand were filtered out.
The distribution for the polymorphisms found along the SARS-CoV-2 genome is calculated using a sliding window (default width: 1,000 nucleotides; step: fifty nucleotides). The number of mutations per site for each window is represented on its right side. Positions are annotated using the Python library gb2seq v0.0.20 (Charité Institute of Virology 2023).
To select the most interesting polymorphisms to plot, we perform a linear regression of the allele frequencies of each polymorphism on the time (in days) elapsed since the first within-patient sample collection. Correlation is measured with the Pearson’s correlation coefficient, and the P-value of the linear regression is adjusted for multiple testing using the Benjamini–Hochberg method (Benjamini and Hochberg 1995). This analysis is performed using the stats R library. Then, polymorphisms that have a significant correlation with time progression are selected for further characterization. Additionally, sites with more than one alternative allele are also selected to monitor potential associations or interactions between the alternative alleles.
Moreover, we calculate pairwise correlations between allele frequencies for all pairs of polymorphisms Mutations are hierarchically clustered based on correlation values. Pairwise correlations are measured with the Pearson’s correlation coefficient using the stats R library. Display of the hierarchical clustering and correlation values is carried out through the heatmaply R library (Galili et al. 2018) with hclust (from the stats R library) as the clustering function.
Temporal signal
To take the within-host variability in the viral population into account, we propose a pairwise distance metric between samples that integrate the differences in allele frequencies across the whole genome. Relying solely on consensus changes under the assumption of low relative entropy (Guang et al. 2016) results in the loss of valuable information about the diversity within the sampled population, which can aid in phylogenetic analyses (Guang et al. 2022; Torres Ortiz et al. 2023). By incorporating allele frequencies into our metric, we gain access to a more comprehensive representation of the evolutionary processes, including drift, mutation, and selection, shaping the data. We define the difference between two vectors of J allele frequencies, based on the FST measure (Wright 1949), such that the distance between two samples (M and N) is the sum for all I polymorphic sites of the differences between allele frequencies at each position (see Equation 1). Then, with this distance matrix, a neighbor-joining tree is constructed in R using ape v5.7 (Paradis, Schliep, and Schwartz 2019). Patristic distances to the root are calculated with adephylo v1.1–13 (Jombart, Balloux, and Dray 2010).
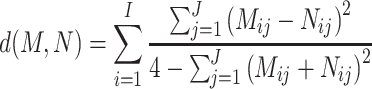 |
(1) |
Finally, the within-host evolutionary rate is estimated by linear regression of the patristic distances to the root in each phylogeny on the days passed since the first within-patient sample collection, using the lm implementation in the stats R library. We performed an analysis of covariance (ANCOVA) to assess the differences between substitution rates of different datasets using the same R library.
Investigating traces of selection
To track selection footprints, the rates of substitutions per synonymous site (dS) and substitutions per non-synonymous site (dN) are calculated for each sample. Synonymous and non-synonymous sites are calculated with respect to the reconstructed ancestral sequence. Then, dN and dS are calculated considering allele frequencies. Calculations are performed in Python using the Nei–Gojobori method (Nei and Gojobori 1986) with support of gb2seq v0.0.20 (Charité Institute of Virology 2023) for codon annotation.
Supplementary Material
Acknowledgements
The computations were performed on the HPC cluster Garnatxa at the Institute for Integrative Systems Biology (I2SysBio), a joint collaborative research institute involving the University of Valencia (UV) and the Spanish National Research Council (CSIC). We thank Dr Anne Hahn and Dr Nathan Grubaugh (Laboratory of Epidemiology of Public Health, Yale School of Public Health, USA) for sharing the information about the sequences we identified as belonging to a previous study.
Contributor Information
Miguel Álvarez-Herrera, Institute for Integrative Systems Biology (I2SysBio, University of Valencia—CSIC), FISABIO Joint Research Unit ‘Infection and Public Health’, C/Agustín Escardino, 9, Paterna 46980, Spain.
Jordi Sevilla, Institute for Integrative Systems Biology (I2SysBio, University of Valencia—CSIC), FISABIO Joint Research Unit ‘Infection and Public Health’, C/Agustín Escardino, 9, Paterna 46980, Spain.
Paula Ruiz-Rodriguez, Institute for Integrative Systems Biology (I2SysBio, University of Valencia—CSIC), FISABIO Joint Research Unit ‘Infection and Public Health’, C/Agustín Escardino, 9, Paterna 46980, Spain.
Andrea Vergara, Department of Clinical Microbiology, CDB, Hospital Clínic of Barcelona; University of Barcelona; ISGlobal, C. de Villarroel, 170, Barcelona 08007, Spain; CIBER of Infectious Diseases (CIBERINFEC), Av. Monforte de Lemos, 3-5, Madrid 28029, Spain.
Jordi Vila, Department of Clinical Microbiology, CDB, Hospital Clínic of Barcelona; University of Barcelona; ISGlobal, C. de Villarroel, 170, Barcelona 08007, Spain; CIBER of Infectious Diseases (CIBERINFEC), Av. Monforte de Lemos, 3-5, Madrid 28029, Spain.
Pablo Cano-Jiménez, Institute of Biomedicine of Valencia (IBV-CSIC), C/ Jaime Roig, 11, Valencia 46010, Spain.
Fernando González-Candelas, Institute for Integrative Systems Biology (I2SysBio, University of Valencia—CSIC), FISABIO Joint Research Unit ‘Infection and Public Health’, C/Agustín Escardino, 9, Paterna 46980, Spain; CIBER of Epidemiology and Public Health (CIBERESP), Av. Monforte de Lemos, 3-5, Madrid 28029, Spain.
Iñaki Comas, Institute of Biomedicine of Valencia (IBV-CSIC), C/ Jaime Roig, 11, Valencia 46010, Spain; CIBER of Epidemiology and Public Health (CIBERESP), Av. Monforte de Lemos, 3-5, Madrid 28029, Spain.
Mireia Coscollá, Institute for Integrative Systems Biology (I2SysBio, University of Valencia—CSIC), FISABIO Joint Research Unit ‘Infection and Public Health’, C/Agustín Escardino, 9, Paterna 46980, Spain.
Data availability
VIPERA is a cross-platform Snakemake (≥7.19) workflow written in Python and R, released as free software under the GNU GPLv3 license. The source code and the report of our case study are available in GitHub (https://github.com/PathoGenOmics-Lab/VIPERA, release v1.2.0). The latest version of VIPERA is also available in the ‘standardized usage’ area of the Snakemake workflow catalog (https://snakemake.github.io/snakemake-workflow-catalog).
Sequencing data from the positive control is available through its source publication by Chaguza et al. (Chaguza et al. 2023). Raw sequencing data from the negative control and the novel case study are available at the ENA. Accession numbers are provided in Supplementary Table 5. Read mappings and consensus genomes can be accessed via DOI: 10.20350/digitalCSIC/15648.
Supplementary data
Supplementary data is available at VEVOLU Journal online.
Funding
This work is a part of project CNS2022-135116 funded by MCIN/AEI/10.13039/501100011033, by the European Union NextGenerationEU/PRTR, and grant PID2021-123443OB-I00 funded by MCIN/AEI/10.13039/501100011033 and ERDF A way of making Europe. M.A.H. and P.R.R. are supported by the PTI+ Salud Global European Commission – NextGenerationEU (Regulation EU 2020/2094). J.S. is supported by the CSIC’s JAE intro program. F.G.C. was funded by project PID2021-127010OB-I00 from the Spanish Ministry of Science and CIPROM2021-053 from the Generalitat Valenciana. I.C. is funded by the European Research Council (101001038-TB-RECONNECT) and the Spanish Ministry of Economy, Industry and Competitiveness (PID2019-104477RB-I00).
Conflict of interest:
The authors declare that they have no competing interests.
References
- Ameen F. et al. (2021) ‘Rilpivirine Inhibits SARS-CoV-2 Protein Targets: A Potential Multi-target Drug’, Journal of Infection and Public Health, Special Issue on COVID-19 – Vaccine, Variants and New Waves 14: 1454–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen Laboratory . (2023) Freyja: Depth-weighted De-Mixing. <https://github.com/andersen-lab/Freyja> accessed 16 Jun 2023.
- ARTICnetwork . (2023) ARTIC-ncov2019: ARTIC Nanopore Protocol for nCoV2019 Novel Coronavirus. <https://github.com/artic-network/artic-ncov2019> accessed 7 Jun 2023.
- Benjamini Y., and Hochberg Y. (1995) ‘Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing’, Journal of the Royal Statistical Society: Series B (Methodological), 57: 289–300. [Google Scholar]
- Brandolini M. et al. (2022) ‘Mutational Induction in SARS-CoV-2 Major Lineages by Experimental Exposure to Neutralising Sera’, Scientific Reports, 12: 12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukur T. et al. (2023) ‘CoVigator—A Knowledge Base for Navigating SARS-CoV-2 Genomic Variants’, Viruses, 15: 1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappell K. M., and Kochenderfer J. N. (2023) ‘Long-term Outcomes following CAR T Cell Therapy: What We Know so Far’, Nature Reviews Clinical Oncology, 20: 359–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Pérez N. et al. (2017) ‘Phylogenetic Analysis of an Epidemic Outbreak of Acute Hepatitis C in HIV-infected Patients by Ultra-deep Pyrosequencing’, Journal of Clinical Virology, 92: 42–7. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention . (2020) Sars-Cov-2 Variant Classifications and Definitions. SARS-CoV-2 Variant Classifications and Definitions. <https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html> accessed 12 Sept 2023.
- Chaguza C. et al. (2023) ‘Accelerated SARS-CoV-2 Intrahost Evolution Leading to Distinct Genotypes during Chronic Infection’, Cell Reports Medicine, 4: 100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charité Institute of Virology . (2023) gb2seq: Use a GenBank File to Extract Sequences for Features and Other Information from Another Genome. <https://github.com/VirologyCharite/gb2seq> accessed 25 May 2023.
- Chiara M. et al. (2021) ‘Comparative Genomics Reveals Early Emergence and Biased Spatiotemporal Distribution of SARS-CoV-2’, Molecular Biology and Evolution, 38: 2547–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P. et al. (2012) ‘A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff’, Fly, 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S. A. et al. (2021) ‘SARS-CoV-2 Evolution in an Immunocompromised Host Reveals Shared Neutralization Escape Mechanisms’, Cell, 184: 2605–2617.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P. et al. (2021) ‘Twelve Years of SAMtools and BCFtools’, GigaScience, 10: giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva T. I. et al. (2021) ‘The Impact of Viral Mutations on Recognition by SARS-CoV-2 Specific T Cells’, iScience, 24: 103353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchene S. et al. (2020) ‘Temporal Signal and the Phylodynamic Threshold of SARS-CoV-2’, Virus Evolution, 6: veaa061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira I. A. T. M. et al. (2021) ‘SARS-CoV-2 B.1.617 Mutations L452R and E484Q are Not Synergistic for Antibody Evasion’, The Journal of Infectious Diseases, 224: 989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili T. et al. (2018) ‘Heatmaply: An R Package for Creating Interactive Cluster Heatmaps for Online Publishing’, Bioinformatics, 34: 1600–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorbani A. et al. (2022) ‘Highlight of Potential Impact of New Viral Genotypes of SARS-CoV-2 on Vaccines and Anti-viral Therapeutics’, Gene Reports, 26: 101537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Reiche A. S. et al. (2023) ‘Sequential Intrahost Evolution and Onward Transmission of SARS-CoV-2 Variants’, Nature Communications, 14: 3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goya S. et al. (2023) ‘Assessing the Hidden Diversity Underlying Consensus Sequences of SARS-CoV-2 Using VICOS, a Novel Bioinformatic Pipeline for Identification of Mixed Viral Populations’, Virus Research, 325: 199035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenfell B. T. et al. (2004) ‘Unifying the Epidemiological and Evolutionary Dynamics of Pathogens’, Science, 303: 327–32. [DOI] [PubMed] [Google Scholar]
- Grubaugh N. D. et al. (2019) ‘An Amplicon-based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and iVar’, Genome Biology, 20: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang A. et al. (2016) ‘An Integrated Perspective on Phylogenetic Workflows’, Trends in Ecology and Evolution, 31: 116–26. [DOI] [PubMed] [Google Scholar]
- Guang A. et al. (2022) ‘Incorporating Within-Host Diversity in Phylogenetic Analyses for Detecting Clusters of New HIV Diagnoses’, Frontiers in Microbiology, 12: 803190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S. et al. (2010) ‘New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0’, Systematic Biology, 59: 307–21. [DOI] [PubMed] [Google Scholar]
- Hadfield J. et al. (2018) ‘Nextstrain: Real-time Tracking of Pathogen Evolution’, Bioinformatics, 34: 4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfmann P. J. et al. (2023) ‘Evolution of a Globally Unique SARS-CoV-2 Spike E484T Monoclonal Antibody Escape Mutation in a Persistently Infected, Immunocompromised Individual’, Virus Evolution, 9: veac104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari S. et al. (2022) ‘Drivers of Adaptive Evolution during Chronic SARS-CoV-2 Infections’, Nature Medicine, 28: 1501–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari S. et al. (2024) ‘Using Big Sequencing Data to Identify Chronic SARS-Coronavirus-2 Infections’, Nat Commun 15: 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang D. T. et al. (2018) ‘UFBoot2: Improving the Ultrafast Bootstrap Approximation’, Molecular Biology and Evolution, 35: 518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T., Balloux F., and Dray S. (2010) ‘adephylo: New Tools for Investigating the Phylogenetic Signal in Biological Traits’, Bioinformatics, 26: 1907–9. [DOI] [PubMed] [Google Scholar]
- Khare S. et al. (2021) ‘GISAID’s Role in Pandemic Response’, China CDC Weekly, 3: 1049–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R., Sugawara H., and Shumway M., on behalf of the International Nucleotide Sequence Database Collaboration (2011) ‘The Sequence Read Archive’, Nucleic Acids Research, 39: D19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2013) ‘Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM’. [Google Scholar]
- Lin -J.-J. et al. (2019) ‘Many Human RNA Viruses Show Extraordinarily Stringent Selective Constraints on Protein Evolution’, Proceedings of the National Academy of Sciences, 116: 19009–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markov P. V. et al. (2023) ‘The Evolution of SARS-CoV-2’, Nature Reviews, Microbiology, 21: 361–79. [DOI] [PubMed] [Google Scholar]
- McCarthy K. R. et al. (2021) ‘Recurrent Deletions in the SARS-CoV-2 Spike Glycoprotein Drive Antibody Escape’, Science, 371: 1139–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B. Q. et al. (2020) ‘IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era’, Molecular Biology and Evolution, 37: 1530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B. Q., Nguyen M. A. T., and von Haeseler A. (2013) ‘Ultrafast Approximation for Phylogenetic Bootstrap’, Molecular Biology and Evolution, 30: 1188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölder F. et al. (2021) ‘Sustainable Data Analysis with Snakemake’, F1000Research, 10: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msomi N. et al. (2021) ‘Africa: Tackle HIV and COVID-19 Together’, Nature, 600: 33–6. [DOI] [PubMed] [Google Scholar]
- Nei M., and Gojobori T. (1986) ‘Simple Methods for Estimating the Numbers of Synonymous and Nonsynonymous Nucleotide Substitutions’, Molecular Biology and Evolution, 3: 418–26. [DOI] [PubMed] [Google Scholar]
- Nussenblatt V. et al. (2022) ‘Yearlong COVID-19 Infection Reveals Within-Host Evolution of SARS-CoV-2 in a Patient with B-Cell Depletion’, The Journal of Infectious Diseases, 225: 1118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole Á. et al. (2021) ‘Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool’, Virus Evolution, 7: veab064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E. (2010) ‘pegas: An R Package for Population Genetics with an Integrated–modular Approach’, Bioinformatics, 26: 419–20. [DOI] [PubMed] [Google Scholar]
- Paradis E., Schliep K., and Schwartz R. (2019) ‘ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R’, Bioinformatics, 35: 526–8. [DOI] [PubMed] [Google Scholar]
- Pipek O. A. et al. (2024) ‘Systematic Detection of Co-infection and Intra-host Recombination in More than 2 Million Global SARS-CoV-2 Samples’, Nature Communications, 15: 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2021) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing.
- Sahin E. et al. (2021) ‘Genomic Characterization of SARS-CoV-2 Isolates from Patients in Turkey Reveals the Presence of Novel Mutations in Spike and Nsp12 Proteins’, Journal of Medical Virology, 93: 6016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SeqCOVID Consortium . (2021) Sars-cov2-mapping. <https://gitlab.com/fisabio-ngs/sars-cov2-mapping> accesed 28 Jul 2023.
- Spinicci M. et al. (2022) ‘Long-term SARS-CoV-2 Asymptomatic Carriage in an Immunocompromised Host: Clinical, Immunological, and Virological Implications’, Journal of Clinical Immunology, 42: 1371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay J. H. et al. (2022) ‘The Emergence of SARS-CoV-2 Variants of Concern is Driven by Acceleration of the Substitution Rate’, Molecular Biology and Evolution, 39: msac013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne L. G. et al. (2022) ‘Evolution of Enhanced Innate Immune Evasion by SARS-CoV-2’, Nature, 602: 487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres Ortiz A. et al. (2023) ‘Within-host Diversity Improves Phylogenetic and Transmission Reconstruction of SARS-CoV-2 Outbreaks’, eLife, 12: e84384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valieris R. et al. (2022) ‘A Mixture Model for Determining SARS-Cov-2 Variant Composition in Pooled Samples’, Bioinformatics, 38: 1809–15. [DOI] [PubMed] [Google Scholar]
- van Dorp L. et al. (2020) ‘Emergence of Genomic Diversity and Recurrent Mutations in SARS-CoV-2’, Infection Genetics & Evolution, 83: 104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rossum G. and Drake F. L. (2009) Python 3 Reference Manual, Scotts Valley, CA: CreateSpace. [Google Scholar]
- Verghese M. et al. (2021) ‘A SARS-CoV-2 Variant with L452R and E484Q Neutralization Resistance Mutations’, Journal of Clinical Microbiology, 59: 10–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigang S. et al. (2021) ‘Within-host Evolution of SARS-CoV-2 in an Immunosuppressed COVID-19 Patient as a Source of Immune Escape Variants’, Nature Communications, 12: 6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilguny L. (2023). ProblematicSites_SARS-CoV2. <https://github.com/W-L/ProblematicSites_SARS-CoV2> accessed 7 May 2023.
- Wilkinson E. et al. (2021) ‘A Year of Genomic Surveillance Reveals How the SARS-CoV-2 Pandemic Unfolded in Africa’, Science, 374: 423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S. A. J. et al. (2022) ‘Recurrent SARS-CoV-2 Mutations in Immunodeficient Patients’, Virus Evolution, 8: veac050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth W. and Duchene S. (2022) GISAIDR. <https://zenodo.org/records/6474693> accessed 21 Jun 2023.
- World Health Organization . (2024) WHO Coronavirus (COVID-19) Dashboard. COVID-19 deaths. <https://covid19.who.int> accessed 9 Feb 2024.
- Wright S. (1949) ‘The Genetical Structure of Populations’, Annals of Eugenics, 15: 323–54. [DOI] [PubMed] [Google Scholar]
- Wu F. et al. (2020) ‘A New Coronavirus Associated with Human Respiratory Disease in China’, Nature, 579: 265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannoli S. et al. (2023) ‘SARS-CoV-2 Coinfection in Immunocompromised Host Leads to the Generation of Recombinant Strain’, International Journal of Infectious Diseases, 131: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zekri A.-R. N. et al. (2021) ‘Characterization of the SARS-CoV-2 Genomes in Egypt in First and Second Waves of Infection’, Scientific Reports, 11: 21632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
VIPERA is a cross-platform Snakemake (≥7.19) workflow written in Python and R, released as free software under the GNU GPLv3 license. The source code and the report of our case study are available in GitHub (https://github.com/PathoGenOmics-Lab/VIPERA, release v1.2.0). The latest version of VIPERA is also available in the ‘standardized usage’ area of the Snakemake workflow catalog (https://snakemake.github.io/snakemake-workflow-catalog).
Sequencing data from the positive control is available through its source publication by Chaguza et al. (Chaguza et al. 2023). Raw sequencing data from the negative control and the novel case study are available at the ENA. Accession numbers are provided in Supplementary Table 5. Read mappings and consensus genomes can be accessed via DOI: 10.20350/digitalCSIC/15648.