

Abstract
Inhaled delivery of mRNA has the potential to treat a wide variety of diseases. However, nebulized mRNA lipid nanoparticles (LNPs) face several unique challenges including stability during nebulization and penetration through both cellular and extracellular barriers. Here we develop a combinatorial approach addressing these barriers. First, we observe that LNP formulations can be stabilized to resist nebulization-induced aggregation by altering the nebulization buffer to increase the LNP charge during nebulization, and by the addition of a branched polymeric excipient. Next, we synthesize a combinatorial library of ionizable, degradable lipids using reductive amination, and evaluate their delivery potential using fully differentiated air–liquid interface cultured primary lung epithelial cells. The final combination of ionizable lipid, charge-stabilized formulation and stability-enhancing excipient yields a significant improvement in lung mRNA delivery over current state-of-the-art LNPs and polymeric nanoparticles.
In vivo delivery of mRNA has the promise to treat and prevent a wide variety of genetic, infectious and other diseases1–17. Most notably, intramuscularly delivered lipid nanoparticle (LNP)-based mRNA vaccines have showed excellent efficacy against the SARS-CoV-2 virus driving the COVID-19 pandemic13,14. Other therapies, including vaccines6,18–27, protein replacement therapy28 and gene editing15, are in various stages of investigation.
Targeting the respiratory epithelium for lung-targeted mRNA-based protein replacement therapy and/or gene editing could treat lung diseases such as cystic fibrosis (CF)29,30, primary ciliary dyskinesia31,32 α−1 antitrypsin deficiency33 and asthma34,35. Additionally, mucosal vaccination targeting the deep lung may provide enhanced protection against respiratory infections compared with intramuscular36–46 or even intranasal vaccines46,47. Lung epithelial delivery has been reported with both nebulized hyperbranched poly(β amino ester) (hPBAE)-based nanoparticles48 and LNPs49. However, a recent clinical trial for CF gene therapy using nebulized mRNA LNPs did not show notable efficacy, suggesting that further improvements are necessary for this delivery modality50.
The difficulty of LNP-based nebulized mRNA delivery is related to several nebulization- and lung-specific barriers. First, nebulization induces powerful shear forces that can disrupt nanoparticle structure and induce aggregation49. Second, transfecting the bronchial epithelium requires penetration of a mucus layer that is a steric and chemical barrier to diffusion51–54. Furthermore, while NP uptake is most efficient on the basolateral side of polarized lung epithelial cells, the basolateral side is rendered inaccessible to apically delivered LNPs by tight junctions55,56.
To address the nebulization challenge, we first optimized LNP component ratios for stability and further refined LNP stability through rational design of buffer conditions and excipients. Next, we screened a library of lipids for mRNA delivery to the lung epithelium using in vitro air–liquid interface (ALI) cultures. ALI cultures proved to have good predictive capability and yielded two candidates with excellent in vivo performance, IR-117-17 and IR-19-Py.
When nebulized under optimized formulation conditions, these two lipids substantially outperform state-of-the-art mRNA delivery formulations to the lung and nose, respectively. Nebulized IR-117-17 LNPs produced a 300-fold improvement in lung mRNA delivery over the top previously described LNP49, and a twofold improvement over the previously reported PBAE48, including up to a 45-fold improvement in delivery to the large airways48.
Results
Formulation optimization of LNPs for nebulized delivery
Given the vast chemical space available for LNP formulations and the material-intensive nature of nebulization experiments, we used a design-of-experiment (DOE) approach57,58 to optimize LNPs for stability during nebulization. Initial experiments with a liver-optimized C12–200 based LNP formulation57 showed that LNPs collected after nebulization with an Aeroneb vibrating mesh nebulizer exhibited reduced in vitro transfection ability in A549 cells, loss of mRNA encapsulation and increased size (Supplementary Fig. 1a–c). Using cryogenic transmission electron microscopy (cryo-TEM), we observed randomly stacked lamellar structures on LNP surfaces suggestive of surface damage following nebulization in contrast to ordinary sphere and electron-dense appearances displayed by pre-nebulized LNPs (Supplementary Fig. 1d)59. We thus screened for LNP formulations which demonstrated maximal post-nebulization stability as defined by (1) minimal loss in in vitro transfection efficiency or mRNA encapsulation efficiency and (2) minimal increase in LNP size following nebulization.
To identify a nebulized LNP delivery baseline, we performed a pilot experiment comparing two LNP formulations57,60 previously developed for intravenous delivery of mRNA. As previously described48, we delivered 0.5 mg of firefly luciferase (FFL) mRNA to mice via nebulization to a whole-body exposure chamber and measured luminescence in excised lungs 6 h after delivery48. The C12–200-based liver-targeting formulation57 resulted in higher lung luminescence than the cKK-E12-based lung-targeting formulation60 (Supplementary Fig. 2) and therefore we selected the C12–200-based formulation as our central formulation for screening.
For our mixture-constrained DOE screen, we varied the molar ratios of the ionizable lipid, helper phospholipid and cholesterol, and the identity of the ionizable lipid and helper phospholipid (Fig. 1a). In total, we prepared 19 formulations, referred to as T1–1 to T1–19, including our initial liver-targeting C12–200 formulation (T1–1), encapsulating FFL mRNA via microfluidic mixing for evaluation (Fig. 1b and Supplementary Table 1).
Fig. 1 |. Formulation optimization of LNPs for nebulized delivery using DOE.

a, Molar ratios of the ionizable lipid, helper phospholipid and cholesterol were varied along with the identity of the ionizable lipid and phospholipid.
b, Molar ratios of the resultant 19 LNP formulations with T1–1 representing the original formulation previously developed in our laboratory around which the formulation design is centred. c, In vitro transfection efficiencies of formulations before (blue) and after (red) nebulization in A549 cells. FFL mRNA (50 ng) was delivered for pre-nebulized formulations, normalized by total mRNA in solution as measured by Ribogreen, and volume-matched doses of post-nebulized formulations. Mean ± s.d., n = 3 wells per group. RLU, relative light unit.
d, Encapsulation efficiency of FFL mRNA before (blue) and after (red) nebulization. Mean ± s.d., n = 3 technical replicates. e, Lung luminescence 6 h after nebulized delivery of 0.5 mg FFL mRNA to mice demonstrating that T1–5 significantly increasing luminescent signal compared with the original T1–1 formulation. Representative images of lungs imaged by IVIS are shown. One-way analysis of variance (ANOVA) with post hoc Tukey test, mean± s.d., n = 3 mice for PBS, n = 5 mice for others.
The formulations were evaluated for in vitro transfection ability, encapsulation efficiency and size before and after nebulization (Fig. 1c,d, and Supplementary Fig. 3). Of the 19, we identified one formulation (T1–5), which is a 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP)-containing formulation with high transfection efficiency in A549 cells both before and after nebulization, and showing no loss in encapsulation efficiency due to nebulization (Fig. 1c). As expected, the DOTAP-containing T1–5 formulation had a more positive zeta potential than the original, 1,2-dioleoyl-sn-glycero-3-p hosphoethanolamine (DOPE)-containing T1–1 (Supplementary Fig. 4a). The addition of DOTAP also improved the preservation of the pre-nebulization morphology of LNPs following nebulization because minimal surface damage was observed (Supplementary Fig. 4b) compared with T1–1 (Supplementary Fig. 1d). This may contribute to the observed ability for T1–5 LNPs to preserve mRNA encapsulation following nebulization (Fig. 1b).
After confirming the reproducibility of our promising T1–5 results (Supplementary Fig. 5), we next evaluated its in vivo performance in nebulized FFL mRNA delivery. Luminescence was significantly higher in lungs transfected with T1–5 compared with T1–1 (Fig. 1e). While these results were promising, the potential of the T1–5 formulation was hindered by its substantial size increase following nebulization which in contrast was not observed for a nebulizable hPBAE polymer nanoparticle (Supplementary Fig. 3)48. This nebulization-induced LNP aggregation suggested further opportunities to improve LNP stability and potentially improve in vivo efficacy.
Excipients and buffer modifications to improve LNP stability
We therefore performed a rationally designed screen of stabilizing excipients and buffer alternatives to PBS (Fig. 2a). We first hypothesized that the disaccharides trehalose and sucrose, which stabilize liposomes and LNPs to freeze–thaw cycles61–64, could also stabilize LNPs to nebulization. We also hypothesized that a hydrophilic polymer may inhibit LNP aggregation through reducing steric hindrance, and therefore we evaluated polysaccharides (Dextran and Ficoll), and linear (PEG6K and PEG20K) and branched (bPEG20K) polyethylene glycols (PEGs). Although neither disaccharide improved the nebulized performance of T1–5 in mice when compared with T1–5 alone, all of the polymers improved performance. Most notably, bPEG20K led to a ~13-fold increase in lung luminescence (Fig. 2b). To evaluate whether bPEG20K improved the in vivo performance of T1–5 by impacting lung physiology, we nebulized a 2% bPEG20K solution without LNPs to mice immediately followed by T1–5 LNPs alone. Pretreatment with bPEG20K solution did not improve T1–5 performance compared with T1–5 delivered without pretreatment (Supplementary Fig. 6a). Also, inhalation of a bPEG20K solution did not result in notable morphological changes to the lung (Supplementary Fig. 6b). This indicates that bPEG20K’s effect on nebulized performance is tied to its presence in the formulation. A 2% bPEG20K solution was used for all subsequent nebulizations unless specified because concentrations beyond 2% were not observed to improve the stability of LNPs during nebulization (Supplementary Fig. 6c).
Fig. 2 |. Excipients and buffer modifications for improved LNP stability and in vivo delivery.
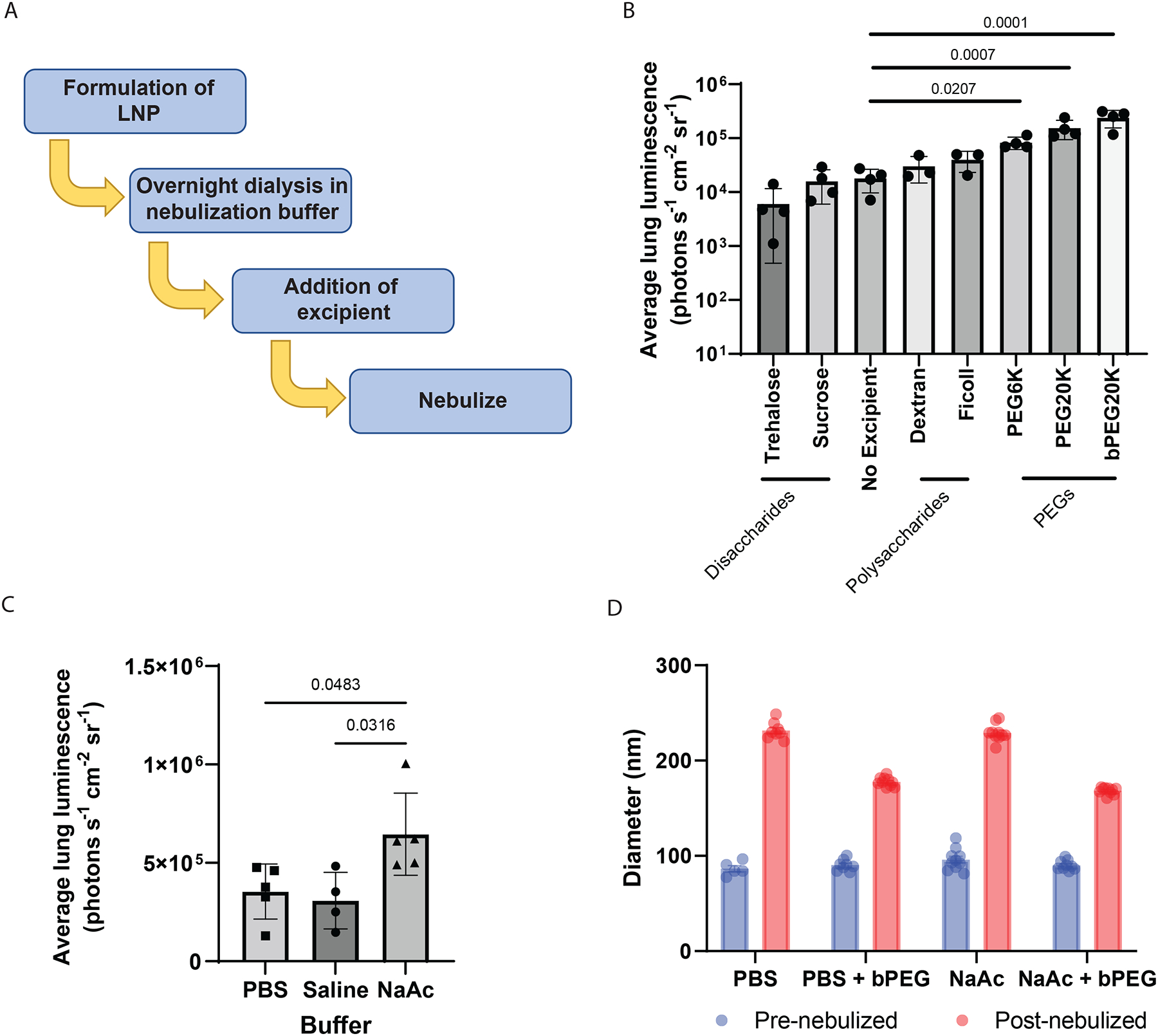
a, Schematic of the workflow for evaluating addition of excipients and buffer modifications to improve LNP nebulization. b,c, Bioluminescence in lung 6 h after nebulized administration of 0.5 mg FFL mRNA with T1–5 and PEG excipients showing significant increase over no excipients (b) (n = 3 mice for Ficoll and Dextran, n = 4 mice for all others; final excipient concentration 20% w/v for the disaccharides and 2% w/v for all other excipients), and overnight dialysis of T1–5 into 100 mM NaAc buffer, pH 5.2, improving luminescent signal over dialysis into 0.9% saline or PBS (c) (n = 4 mice for saline, n = 5 mice for all others). Mean ± s.d., one-way ANOVA with post hoc Tukey test. d, DLS measurements of LNP size before and after nebulization demonstrating reduction in size increase following nebulization for T1–5 formulation when bPEG20K is added to PBS and further reduction when PBS is replaced with NaAc buffer in the presence of bPEG20K. Mean ± s.e.m., n = 6–10 repeated measurements.
During nebulization, we noticed mice often huddled in the chamber, potentially causing inconsistent aerosol exposure. To address this, we used individual restrainers for mice, connecting them to a nose-only exposure system where their noses were exposed to a central chamber through which the aerosol flowed. This system resulted in increased lung luminescence compared with the whole-body chamber (Supplementary Fig. 7). Therefore, we adopted the nose-only system for further experiments.
We next hypothesized that modifications to the LNP buffer may further improve nebulized delivery efficacy of the T1–5 formulation with bPEG20K excipient. We compared two previously tested nebulization buffers to PBS: 0.9% saline, pH 7.0 (ref. 65), and 100 mM sodium acetate (NaAc), pH 5.2 (ref. 48). We reasoned that the slightly acidic NaAc buffer would protonate the ionizable lipids present in LNPs, leading to reduced LNP aggregation via electrostatic repulsion. To evaluate the buffers, T1–5 LNPs were microfluidically mixed57, initially dialysed into PBS before secondary, overnight dialysis in the selected buffer, and 2% bPEG20K excipient was added before nebulization. We found that NaAc buffer, but not 0.9% saline, significantly improved nebulized delivery efficacy compared with PBS (Fig. 2c). As predicted, NaAc buffer dialysis increased the zeta potential of LNPs compared with those dialysed only in PBS (Supplementary Fig. 8a). Additionally, dialysis into NaAc buffer did not result in an appreciable change in the morphology of the LNPs as observed by cryo-TEM (Supplementary Fig. 8b).
To test whether the bPEG20K stabilizing excipient and NaAc buffer reduced LNP aggregation during nebulization, we measured the size of LNPs before and after nebulization in the presence or absence of the excipient and buffer modification. Incorporating bPEG20K into either buffer attenuated the increase in LNP size following nebulization, with the smallest post-nebulization size achieved by combining 2% bPEG20K with NaAc buffer (Fig. 2d). These conditions also mostly preserved LNP morphology following nebulization, although we noticed formation of small (<10 nm) liposomal structures after nebulization, presumably as a result of nebulization-induced shearing (Supplementary Fig. 8b).
We also hypothesized that the combination of excipient, NaAc buffer and T1–5 formulation might stabilize the mRNA from fragmentation during nebulization. Indeed, nebulization drove fragmentation of naked mRNA and T1–1-formulated mRNA, but had a minimal to non-existent effect on the stability of T1–5-formulated mRNA with excipients (Supplementary Fig. 9). Because fragmented mRNA is unlikely to be functional, this is a potential mechanism by which our approach improves nebulized delivery. Given the improvements observed for nebulizer stability and in vivo transfection efficiency, secondary dialysis into NaAc buffer and addition of 2% w/v bPEG20K excipient were used for all subsequent in vivo experiments unless otherwise noted.
Synthesis and screening of a biodegradable lipid library
We next sought to identify lipids that facilitated transfection of the lung epithelium. First, we designed a biodegradable lipid synthesis strategy using reductive amination chemistry (Fig. 3a), in which ester-containing lipid aldehydes were reacted with amines. Typical conversion rates as estimated by thin-layer chromatography were ~50–80% before purification. To test whether we could screen the lipids without individual purification as has been done with other libraries6,66,67, we prepared 22 lipids, both purified and crude, formulated into LNPs using the T1–5 formulation, and tested their FFL mRNA delivery in A549 cells. We observed no statistically significant positive correlation between purified and unpurified lipids with this chemistry (Supplementary Fig. 10), suggesting that a screen of crude lipids would not be informative.
Fig. 3 |. Synthesis and screening of a biodegradable lipid library.

a, Four-step synthetic sequence, comprising esterification, oxidation and aldol reaction, for making our ionizable lipid library. Tails are made in the aldol reaction (step 3). DIPEA, N,N-diisopropylethylamine; DCM, dichloromethane; STAB, sodium triacetoxyborohydride. b, Individual tails and amine components for combinatorial reaction. c, Schematic of difference between (1) classic submerged culture (undifferentiated cells, no tight junctions, non-physiological environment) and (2) ALI cultures. d, Representative H&E-stained histology images of 3-week-old large (top) and small (bottom) airway ALI cultures. Scale bars, 50 μm. e, Delivery of 1 μg FFL mRNA using T1–5-formulated, microfluidically mixed, non-nebulized C12–200 LNPs, 2–13 days after airlift. n = 3 ALI culture wells per day and airway cell line type, two-way ANOVA with Dunnett’s multiple-comparisons test, mean ± s.e.m. f, Screen of the top 22 A549 hits for delivery to small-airway ALI cultures sorted by sum of log luminescence in small and large airways. n = 4 ALI culture wells per airway cell line type, that is, eight wells total per lipid, 500 ng mRNA per well, mean ± s.e.m. g, Structures of two top hits, IR-117-17 and IR-19-Py.
We therefore restricted ourselves to a smaller library that combined a broad array of headgroups with a restricted set of tails, reasoning that we could optimize tail structure in subsequent steps. We predicted that unsaturated tails would have optimal activity, and therefore we primarily synthesized lipids with oleic and linoleic acid tails (Fig. 3b), although we also synthesized four lipids with C16 tails. We synthesized and screened a total of 46 lipids in A549 cells, and found that several of them had efficiency comparable to that of C12–200 (Supplementary Fig. 11). Of the C16 tail lipids, three-quarters had worse activity than headgroup-matched unsaturated tail variants, while the fourth was not in the top 20 lipids by activity, thus preliminarily validating our focus on unsaturated tails.
Ionizable lipid screening in ALI cultures
Because nebulization is low throughput, and in vitro mRNA delivery assays with classic cell-culture techniques are unreliable, we generated ALI cultures derived from primary human bronchial epithelial cells as an intermediate, medium-throughput technique. ALI cultures are grown on porous Transwell inserts such that media can be removed from the top (apical) side of the insert and the cells can be fed from the bottom (basolateral) side (Fig. 3c). ALI cultures recapitulate key characteristics of the in vivo bronchial epithelium including differentiation, mucus secretion, periciliary layer formation and tight junctions68–71. ALI cultures are difficult to transfect71, probably due to these in vivo-like characteristics, which suggested that these cultures could be a good model for screening LNPs.
We therefore generated both primary large-airway and small-airway ALI cultures because both airways are important for respiratory disease72–75. As expected, both airway cultures formed tight junctions (Supplementary Fig. 12a,b)76 and large airway cultures grew more thickly, with dense visible ciliation (Fig. 3d).
To validate ALI cultures as a model for LNP delivery, we tested T1–5-formulated C12–200 LNPs for FFL delivery to ALI cultures for up to 2 weeks after airlift. Transfection efficiency dropped over that period by 2.5–3 orders of magnitude (Fig. 3e), consistent with the previous observation that siRNA delivery by traditional in vitro transfection reagents decreases sharply over the first week of ALI culture71 due to cell differentiation, polarization and/or tight junction formation, leading to poor apical uptake and reduced access to the more endocytic basolateral side of cells56,71.
Next, we screened the 22 top-performing LNPs from the A549 assay for ALI culture mRNA delivery. Nebulization is resource intensive and while microfluidic mixing of LNPs is a standard procedure in the production of RNA LNPs77, it can be time intensive, and replacing microfluidic mixing with pipette mixing during screening can yield useful results6,67. Therefore, to accelerate initial screening, we performed our screening using handmixed, non-nebulized LNPs. The top two performers were A10-Lin/IR-117-17 and A20-Ol/IR-19-Py (Fig. 3f,g).
To identify the structure–activity relationships of our top-performing structures, we screened variants of IR-117-17 and IR-19-Py. First, we screened saturated tails of lengths C9–C18 for each lipid, and found that intermediate tail lengths in the range of C12–C14 were optimal, although the unsaturated tails were still superior (Supplementary Fig. 13). Increasing the level of unsaturation for IR-117-17 using an α-linolenic acid (ALA)-tailed variant did not affect activity.
We also tested headgroup variants of IR-117-17 with oleic, linoleic and/or ALA tails. Shortening the alkyl chains branching off from the tertiary amine impaired activity for the linoleic and ALA tails, while activity for the oleic tails was unaffected or improved by tail shortening, (Supplementary Fig. 14a,b). IR-19-Py activity was insensitive to replacement of the pyrazole group with an imidazole, and to demethylation of one of the headgroup nitrogens (Supplementary Fig. 14c,d).
In vivo evaluation of lead-ionizable lipid LNPs
Next, we nebulized T1–5-formulated IR-117-17 and IR-19-Py to mice, along with C12, C15 and ALA tail variants of IR-117-17 to test the predictive capacity of ALI cultures. As controls, we also nebulized our previously reported lung-delivery-optimized hPBAE48 and NLD1, an LNP that has been reported to offer good nebulized mRNA delivery49. We measured whole-lung luminescence at 6 h for the LNPs, 24 h for the PBAE and 48 h for NLD1, according to the reported peak expression for the latter two particles. As observed in ALI cultures, IR-117-17 (1.1 × 106 photons s−1 cm−2 sr−1) significantly outperformed C12–200 (6.1 × 105 photons s−1 cm−2 sr−1) (Fig. 4a). IR-117-17 also outperformed the polymeric PBAE nanoparticle (5.2 × 105 photons s−1 cm−2 sr−1) by twofold and NLD1 (2.8 × 103 photons s−1 cm−2 sr−1) by 300-fold (Fig. 4a). Nebulized delivery of 1 mg of FFL mRNA via IR-117-17 LNPs resulted in uniform distribution of luminescence across all five lobes of the lung (Supplementary Fig. 15) and in significantly higher levels of FFL protein expression in the lung compared with delivery via hPBAE at respective peak expression time points (Supplementary Fig. 16), consistent with the results observed by IVIS imaging.
Fig. 4 |. In vivo testing and in vitro–in vivo comparison of top lipids.

a, Nebulized mRNA delivery to mouse lung after 1 mg dose delivered via nose cone, imaged at peak expression (6 h for LNPs, 24 h for hPBAE48, 48 h for NLD149). Mean ± s.d., n = 3 for PBS, NLD1, IR-117-17-ALA, n = 4 for all others, one-way ANOVA with post hoc Tukey test. Left: all nebulized NPs, right: zoomed-in image showing significant NLD1 delivery compared with PBS. b–e, Correlation of in vivo nebulized lung delivery data from a to FFL delivery to ALI cultures (500 ng per well) using handmixed non-nebulized LNPs (b) (no significant correlation, P = 0.37; ALI culture measurements given in Supplementary Fig. 12), FFL delivery to A549 cells (20 ng per well) using handmixed non-nebulized LNPs (c) (no significant correlation, P = 0.85), FFL delivery to ALI cultures (500 ng per well) using microfluidically mixed post-nebulized LNPs (d) (ρ = 0.64, P = 0.0022), and FFL delivery to A549 cells (20 ng per well) using microfluidically mixed post-nebulized LNPs (e) (ρ = 0.658, P = 0.0076). In b–e, correlation P values were calculated using an F-test, mean ± s.e.m., n ≥ 3 ALI cultures wells for each measurement.
The ALI culture experiments correctly predicted that IR-117-17-C15 would perform poorly relative to IR-117-17-C12 and the original IR-117-17. However, our ALI cultures incorrectly predicted good IR-117-17-ALA activity: it performed the worst of all tested lipids. Because of this, there was no statistically significant correlation between the ALI culture and the in vivo data (Fig. 4b) when only taking LNPs into account. Meanwhile, the A549 data were substantially worse, predicting nearly everything incorrectly (Fig. 4c).
We reasoned that one possible explanation for this lack of correlation was that our nebulizer stabilization techniques were not universally applicable to all LNPs. We therefore generated LNPs from C12–200 and the IR-117-17 variants via microfluidic synthesis, nebulized the LNPs using bPEG20K and NaAc excipients, and treated A549 cells and ALI cultures with the post-nebulized LNPs. For post-nebulized LNPs, both A549 cells (P = 0.0076) and ALI cultures (P = 0.0022) showed significant correlations between in vitro and in vivo activity (Fig. 4d,e), primarily because both predicted IR-117-17 to be effective and IR-117-17-ALA was adversely affected by nebulization. However, C12–200 continued to be the most effective lipid for A549 cell transfection, whereas ALI cultures correctly identified IR-117-17 as the top lipid.
Based on these results, we further characterized IR-117-17 and IR-19-Py, which had statistically comparable delivery to C12–200 and a more chemically distinct headgroup than IR-117-17-C12. As with C12–200, optimized buffer and excipient conditions improved delivery of LNPs formulated with each biodegradable lipid (Supplementary Fig. 17). Likewise, bPEG20K and NaAc buffer reduced post-nebulization size when compared with PBS alone for both biodegradable lipids (Supplementary Fig. 18a), and attenuated a slight loss in encapsulation efficiency observed with nebulization in PBS buffer only (Supplementary Fig. 18b). Also like C12–200, the zeta potentials of IR-117-17 and IR-19-Py LNPs dialysed in NaAc buffer were more positive than those dialysed in PBS (Supplementary Fig. 18c).
IR-117-17 LNPs in PBS contained ‘bleb’ structures on top of the amorphous electron-dense core on around two-thirds of the LNPs, as observed under cryo-TEM (Supplementary Fig. 18d). Such morphology has been previously reported78,79. When the bPEG20K and NaAc buffer were used, two populations, one with liposome morphology and one with the conventional, concrete electron-dense morphology, emerged for the IR-117-17 LNP formulation. Such formation of the same dual populations under a pH 4 NaAc buffer was previously reported for both siRNA LNPs80 and mRNA LNPs81, and is believed to be associated with charge repulsion between protonated ionizable lipids. Overall, the bPEG20K and NaAc buffer resulted in better preservation of the pre-nebulized LNP structures compared with PBS buffer (Supplementary Fig. 18d). Notably, IR-117-17 LNPs nebulized in the presence of bPEG20K and NaAc buffer had an average diameter of only 116 nm (Supplementary Fig. 18a), the smallest size we have observed for any LNP following nebulization, and the mRNA remained stable following nebulization (Supplementary Fig. 9).
Pharmacokinetics and pharmacodynamics of top LNPs
To determine whether lung protein expression could be controlled by nebulized dose, we performed a dose–response experiment with C12–200, IR-117-17 and IR-19-Py. For all three LNPs, increased lung luminescence correlated with dose (Fig. 5a). We next performed a time-course study following nebulized delivery, and found that protein expression in both the lungs (Fig. 5b,d) and nose (Fig. 5c,d) lasted in some cases at least 48 h after nebulized dosing. IR-19-Py exhibited prolonged nasal expression, suggesting potential for use in nasal vaccines. For mRNA protein-replacement therapies to treat pulmonary diseases such as CF, repeat dosing of the therapeutic will be necessary. Therefore, we characterized the ability to repeat-dose nebulized LNPs by administering 1 mg of FFL mRNA every 72 h and measured lung luminescence 6 h post-nebulization. Using 72 h intervals ensures that previous luciferase signals would minimally impact new ones. For all LNPs, no loss in lung luminescence was observed over the course of the three administrations (Fig. 5e). To confirm that our top candidate IR-117-17 could be degraded via esterase cleavage and thus be unlikely to accumulate upon repeated dosing, we incubated it with pooled human liver and lung S9 fractions containing important metabolic enzymes82. We detected an increase in the predicted esterase-cleaved metabolites in the presence of S9 but not in a PBS control (Supplementary Fig. 19). Inhaled delivery of 1 mg of FFL mRNA with IR-117-17 LNPs or hPBAE resulted in normal tissue histology (Supplementary Fig. 20a) and no elevation in levels of proinflammatory cytokines in bronchoalveolar lavage fluid (BALF) 24 h after inhalation (Supplementary Fig. 20b). In contrast, intranasal administration of 5 μg of LPS resulted in severe lung injury and elevated levels of several proinflammatory cytokines in BALF (Supplementary Fig. 20).
Fig. 5 |. Pharmacokinetics and pharmacodynamics of top-performing LNPs.

a, Dose–response study of T1–5 LNPs comprising the two biodegradable ionizable lipids, IR-117-17 and IR-19-Py, compared with the non-biodegradable lipid C12–200. Mean ± s.d., n = 3 mice per dose. b,c, Time course of protein expression in the lung (b) and nose (c) of mice following nebulized delivery of 1 mg mRNA. Mean ± s.d., n = 5 mice per experimental group. d, Representative whole-body images of mice 6 h after nebulized delivery of 1 mg mRNA. e, Repeat-dosing of 1 mg of mRNA every 3 days. Mean ± s.d., n = 5 mice per experimental group. f, Luminescence in the lungs of Scnn1b-Tg mice, a model of the CF phenotype, following nebulized delivery of 1 mg of FFL mRNA with either IR-117-17 LNPs or hPBAE. Mean ± s.d., n = 3 for PBS, n = 8 for all others, one-way ANOVA with post hoc Tukey test.
To evaluate nebulized delivery of mRNA in a relevant disease model, we examined delivery performance in the Scnn1b-Tg mouse model. This strain overexpresses the ENaC sodium channel, leading to sodium hyperabsorption in the lung and CF-like inflammatory and muco-obstructive lung disease. This model is reported to mimic CF lung disease better than actual knockout of CFTR, which mostly causes intestinal disease in mice83–85. Given the physiological differences between Scnn1b-Tg and wild-type mice, we performed a time-course study in Scnn1b-Tg mice to identify peak luciferase expression following nebulized mRNA delivery with either IR-117-17 or hPBAE. Peak luminescence was observed at 12 and 24 h for IR-117-17 and hPBAE, respectively (Supplementary Fig. 21). As with wild-type mice, inhaled delivery of 1 mg of FFL mRNA with IR-117-17 resulted in significantly higher peak luminescence compared with hPBAE (Fig. 5f), indicating the potential of IR-117-17 LNPs to deliver mRNA even in the presence of CF-like lung disease.
Quantifying cell transfection with IR-117-17 LNPs
Finally, to quantify transfection of specific cell types, we tested nebulized delivery of Cre recombinase mRNA to the Ai14 mouse model. Cells in these mice express tdTomato only upon Cre activity (Fig. 6a)60. We nebulized 9 mg of Cre mRNA in IR-117-17 LNPs or hPBAEs to mice over the course of three doses and measured lung transfection in large and small airways using histology. A 9 mg dose was selected because nebulization of aerosols to mice results in <1% of the dose depositing in the lungs86,87. Additionally, it is estimated that in a previous study of nebulized primary ciliary dyskinesia therapy, >10 mg of mRNA needed to be nebulized to mice to achieve therapeutic results88. Supplementary Fig. 22a shows representative whole-lung images and Fig. 6b shows representative images of the large and small airways in lungs of mice that received nebulized IR-117-17.
Fig. 6 |. Evaluating functional mRNA delivery to lung epithelium with IR-117-17 LNPs in the Ai14 mouse model.

a, Schematic of Cre delivery experiment to quantify transfected cells in Ai14 mice. A total of 9 mg of Cre mRNA evenly divided over the course of three doses was nebulized to mice using IR-117-17 LNPs or hPBAE. PBS was nebulized to mice as a control. Created with BioRender.com b, Representative images of large and small airways with or without nebulized IR-117-17 LNP treatment. Left column: co-visualization of transfected cells (tdTomato+) with DAPI. Right column: co-visualization of transfected cells (tdTomato+), club cells (CCSP+) and ciliated cells (acetylated tubulin (AcTub)+, AcTub is membrane-localized, hence colour is not present throughout the cell). Green arrows indicate transfected club cells (tdTomato+ CCSP+), purple arrows indicate transfected ciliated cells (tdTomato+ AcTub+). c, Quantification of transfection of large and small airways by IR-117-17 LNPs and hPBAEs. n = 3 mice; individual points are averages over ≥5 large airways per mouse and ≥25 small airways per mouse; mean ± s.e.m., two-way ANOVA. d, Some epithelial regions in the lung had near 100% transfection.
By quantifying the images, we observed 10.3% and 8.8% transfection of large- and small-airway cells, respectively, for IR-117-17 versus 0.23% and 1.92% with hPBAE (Fig. 6c and Supplementary Fig. 22a). This represents 45-fold and 4.6-fold better transfection for IR-117-17 in the large and small airways, respectively. We next quantified transfection of club cells, which are the largest source of CFTR in the lung89,90. Additionally, they are promising targets for both gene therapy and gene editing for CF because they can act as progenitor cells and are easier to target via nebulization than basal cells91. Slightly smaller percentages of club cells (8.4% and 7.6%) were transfected using IR-117-17 LNPs (Supplementary Fig. 22b), although the difference between club cell transfection and all cell transfection was not significant (P = 0.35 for large airways, P = 0.61 for small airways, t-test). hPBAE transfected 0.15% and 1.2% of large- and small-airway club cells, respectively.
IR-117-17 LNPs also transfected other cell types including alveolar macrophages and alveolar epithelium (Supplementary Fig. 23). hPBAE also transfected alveoli, consistent with other reports of majority alveolar-directed expression of modified PBAE-like materials (Supplementary Fig. 23)92. This probably explains the comparable overall lung transfection efficiency observed by bulk luminescence for inhaled hPBAE delivery (Fig. 5a) despite poor conducting airway delivery (Fig. 6c).
Additionally, for LNP-treated mice, all 21 large airways we quantified and 140 out of 151 (93%) of small airways had at least one transfected cell (Supplementary Fig. 22c,d), although some individual stretches of airway showed near-complete transfection (Fig. 6d). Heterogeneity in transfection may be due to differences in transfectability and/or particle deposition and is an interesting area for future investigation.
Conclusions
We describe three advances toward safe and effective mRNA delivery. First, we identified a combination of formulation, excipient and buffer that improved LNP stability and activity following nebulization. Second, we showed that in vitro screens based on primary ALI cultures can effectively evaluate mRNA delivery LNPs for the lung. These screens have higher throughput than in vivo testing and greater accuracy than submerged cell-culture assays. In comparison with barcoded in vivo screening, ALI culture screening avoids nonlinear effects inherent to nanoparticle pooling93. Lastly, we introduce IR-117-17 and IR-19-Py, which under our optimized conditions yield state-of-the-art nebulized delivery to the lung and nose, respectively. IR-117-17 targets the conducting airways far better than the control PBAE, making it a more promising candidate to treat diseases that manifest outside alveolar spaces. Although additional work is needed to advance inhaled nanotherapy, the technology described here is one of the most potent mRNA delivery formulations reported for nebulized, in vivo mRNA delivery.
Methods
Nanoparticle formulation
LNPs were synthesized by mixing an aqueous phase containing the mRNA with an ethanol phase containing the lipids either by pipetting or in a microfluidic chip device94. The aqueous phase was prepared in a 10 mM citrate buffer with corresponding mRNA (FFL and Cre recombinase mRNA provided by Translate Bio). The ethanol phase was prepared by solubilizing a mixture of ionizable lipid, helper phospholipid (DOTAP, DOPE or 1,2-distearoyl-sn-glycero-3-p hosphocholine (DSPC) (Avanti)), cholesterol (Chol, Sigma-Aldrich) and C14-PEG2000 (Avanti) at predetermined molar ratios with an ionizable lipid/mRNA weight ratio of 10:1. For microfluidically prepared LNPs, the aqueous and ethanol phases were mixed in a microfluidic device at a 3:1 ratio by syringe pumps to a final mRNA concentration of 0.1 mg ml‒1 for in vitro studies or 0.2 mg ml−1 for in vivo studies. The resultant formulation was dialysed against PBS, unless otherwise specified, overnight in a 20 kDa molecular weight cut-off dialysis cassette (ThermoFisher) at 4 °C. For formulations in other buffers, LNPs were first dialysed against PBS for 4 h followed by overnight dialysis against 0.9% saline, pH 7.0 or 100 mM NaAc buffer prepared by diluting 3 M, pH 5.2 NaAc with deionized H2O. Following dialysis, LNPs were concentrated using Amicon 100 kDa molecular weight cut-off centrifugation filters (Sigma) at 4 °C. For pipette-mixed LNPs, the two phases were mixed by repeated pipetting and immediately diluted in PBS to the specified concentrations.
For screening of ionizable lipids generated by reductive amination, screening took place holding mass ratios from the T1–5 formulation (with C12–200 as the ionizable lipid) constant. This corresponded to mass ratios of 50.1:24.6:16.8:8.5 ionizable lipid:DOTAP:cholesterol: C14-PEG2000. This constant mass ratio formulation was used in all subsequent experiments.
NLD1 LNPs and hPBAE polymer nanoparticles were prepared as previously described48,49.
LNP characterization
mRNA encapsulation efficiencies were measured by a modified Quanti-iT Ribogreen RNA assay (Invitrogen) as previously described95. The diameter of the LNPs was measured by dynamic light scattering (DLS; Dyna Pro Plate Reader, Wyatt or Zetasizer Nano, Malvern). LNPs were diluted to 0.5 ng mRNA μl−1 in PBS for DLS measurements. LNP diameters are reported as the largest intensity mean peak average, constituting >90% of the nanoparticles present in the sample. For DLS measurements on the Dyna Pro, only acquisitions meeting the instrument’s data quality criteria were included. Zeta potential was measured using a Malvern Zetasizer Nano. LNPs were diluted to 1 ng mRNA μl−1 in either 0.1× PBS or 10 mM NaAc to match the overnight dialysis buffer for zeta potential measurements.
Cryo-TEM
LNPs were diluted to 3 mg ml−1 lipid concentration in deionized water and 3 μl was dropped on a copper grid coated with carbon film and blotted using a Gatan Cryo Plunge III. The grid was mounted on a Gatan 626 single-tilt cryoholder and cooled by liquid nitrogen. Imaging was done on a JEOL 2100 FEG microscope at 200 kV with magnification of between 10,000× and 60,000×, recorded on a Gatan 2kx2k UltraScan charge-coupled device camera.
Nebulization of nanoparticles
To characterize the effects of nebulization on LNP size, encapsulation efficiency and transfection efficiency, 100 μl of LNPs at 150 ng μl−1 were loaded into an Aeroneb vibrating mesh nebulizer (Aerogen) and nebulized into 1.5 ml microcentrifuge tubes. Where specified, 2% w/v of excipients were added to the LNP formulation following dialysis and immediately prior to nebulization.
Cell culture
A549 lung epithelial cells (ATCC) were grown in DMEM high glucose + sodium pyruvate + GlutaMax (Gibco number 10569-010) + 10% FBS.
Primary large airway cells (University of North Caroline at Chapel Hill MLI Tissue Procurement and Cell Culture Core) and primary small airway cells (ATCC) were expanded using PneumaCult-ExPlus (Stem-Cell) according to the instructions for the media and plated for ALI cultures at P3 and P4, respectively.
ALI cultures were plated either onto 0.4 μm pore, 6.5 mm diameter PET Transwell inserts for 24-well plates (Corning) or 0.4 μm pore polycarbonate inserts for 96-well plates (Corning). The 24-well plates were used for histology, the 2 week transfection time course, and transepithelial electrical resistance measurements; 96-well plates were used for all other experiments. Cells were seeded at levels of 33,000 cells per well in the 24-well inserts and 15,000 cells per well in the 96-well plate inserts.
Per the manufacturer’s instructions, cultures were grown to confluence with both apical and basolateral (ExPlus) media for 2–4 days before airlift. Because polycarbonate inserts are opaque, rendering confluence not observable, each time cells were thawed and plated on a 96-well insert plate, the same cells were plated on 6.5 mm PET inserts. The 96-well insert plates were airlifted 1 day after the concurrently plated PET inserts because initial experiments showed that the extra day helped tight junctions form more consistently. Upon airlift, apical media was removed and basolateral media was changed to PneumaCult-ALI (for large-airway cells) or ALI-S (for small-airway cells) media (StemCell). Qualitative tight junction formation, as measured by a lack of leakage of media overnight into the apical space, was observed for both 24-well and 96-well ALI cultures within 1 week of airlift. Media was changed three times per week and cells were not transfected until at least 20 days of ALI culture.
In vitro transfection experiments
For DOE screening and optimization of the IR-117-17 and IR-19-Py leads, 10,000 A549 cells per well were plated in white-sided, clear-bottom 96-well plates. After 24 h of incubation, FFL mRNA encapsulated in either LNPs diluted in PBS or RNAiMAX (ThermoFisher) was added at 50 ng per well except for post-nebulized LNP samples which were added at the same volume as the respective pre-nebulized sample. For the initial reductive amination lipid screen, 2,000 A549 cells per well were plated in a white 384-well plate and allowed to grow overnight. The next day, LNPs were generated by pipette mixing (except where otherwise noted) to a final concentration of 100 ng μl−1 mRNA, diluted in PBS to 10 ng μl−1 mRNA, and wells were treated with 20 ng mRNA per well. For ALI cultures, LNPs were likewise generated by pipette mixing or microfluidic mixing as noted in the main text, and diluted to 50 ng μl−1 total mRNA with PBS. Then, 10 μl of this 50 ng solution was added apically to 96-well inserts, or 20 μl to 24-well inserts.
Bioluminescence was measured 24 h after transfection using Bright-Glo (Promega) according to the manufacturer’s instructions. Bioluminescence was quantified using a Tecan Infinite M200 Pro plate reader (Tecan).
Biodegradability analysis
IR-117-17 (5 ng μl−1) was incubated with 2 mg ml−1 pooled mixed-gender human liver S9 fraction, lung S9 fraction or PBS only. After 0, 1 or 7 days, samples were quantified via liquid chromatography–mass spectrometry on an Agilent 6410 MSQQQ using an Acquity C8 column (Waters) and peak areas for the relevant ions were quantified.
Animal studies
All procedures were performed under an animal protocol approved by the Massachusetts Institute of Technology Committee on Animal Care (#2303000482) and the guidelines for animal care in an MIT animal facility. C57BL/6J (female, 6–8 weeks old) and B6.Cg-Gt(ROSA)26Sortm14( CAG-tdTomato)Hze/J (female, 3–4 months old) were purchased from Jackson Laboratories. C57BL/6N Scnn1b-Tg (purchased from Jackson Laboratories) were bred in house, genotyped by ear punch (Transnetyx), and male mice were used for experiments (10–14 weeks). Mice were housed in an MIT animal facility.
In vivo nebulized delivery
In vivo nebulized delivery of mRNA to mice was performed either in a whole-body exposure chamber or a nose-only exposure chamber (CH Technologies) where specified. LNPs at 0.3 mg ml−1 of mRNA were loaded into the nebulizer at required volumes with 2% w/v excipients where specified. For the whole-body exposure chamber, the nebulizer was connected to the chamber via a tee and an oxygen flow rate of 15 standard cubic feet per hour was used to direct aerosol into the chamber. For the nose-only exposure chamber, mice were immobilized in restrainers and the restrainers connected to the chamber. A flow rate of 2 standard liters per minute of oxygen was used and pressure within the chamber was maintained at −0.1 deci-inches H2O. Nebulizations were performed until no more aerosol was observed in the chamber.
For luciferase protein quantification, mice were killed at specified time points following nebulization, and their lungs removed and flash frozen in liquid nitrogen. Tissues were homogenized in a pestle and mortar on dry ice. Homogenates were resuspended in PBS and centrifuged at 20,000g for 5 min. Supernatant was assayed for luciferase content (Bright-Glo, Promega) using a luciferin standard curve (Quanti-Lum, Promega) and for total protein content (Pierce BCA kit, Invitrogen).
In vivo bioluminescence
Six hours after nebulization, unless otherwise noted, mice were injected intraperitoneally with 0.2 ml XenoLight d-luciferin (10 mg ml−1 in DPBS; PerkinElmer). For whole-body imaging, mice were anaesthetized in a ventilated anaesthesia chamber with 2.5% isofluorane in oxygen and imaged 10 min after luciferin injection. Otherwise, mice were killed 10 min after luciferin injection and organs were collected for imaging. Luminescence was measured with an in vivo imaging system (IVIS, PerkinElmer) and quantified using Living Image software (PerkinElmer).
Nebulization of bPEG20K to mice
For pretreatment of mice with bPEG20K prior to nebulized delivery of mRNA, 1.5 ml of 2% w/v bPEG20K in PBS was nebulized to C57BL/6J mice immediately followed by nebulized delivery of FFL mRNA. To evaluate the impact of bPEG20K on lung morphology, 1.5 ml of 2% w/v bPEG20K in PBS was nebulized to C57BL/6 J mice, and mice were killed 6 h after nebulization. Lungs were harvested and placed overnight in 4% paraformaldehyde (PFA) prior to transfer to 70% ethanol and embedding in a paraffin block. Sections of embedded tissue were mounted on slides and stained with haematoxylin and eosin (H&E). Slides were scanned by an Aperio Digital Pathology slide scanner.
Toxicity study of LNPs and hPBAE
Twenty-four hours after nebulization of 1 mg of FFL mRNA or intranasal administration of 5 μg LPS, mice were killed for either BALF collection to measure proinflammatory cytokine levels or for tissue (lung, liver) harvest to determine tissue injury. PBS was nebulized as a control. Lungs and livers were placed overnight in 4% PFA prior to being transferred to 70% ethanol and embedded in a paraffin block. Sections of embedded tissue were mounted on slides and stained with H&E. Slides were scanned by an Aperio Digital Pathology slide scanner.
For BALF collection and analysis, mouse lungs were lavaged with 1 ml PBS via an incision in the trachea, and lavage fluid was collected. The lavage fluid was centrifuged for 5 min, 13,000g, at 4 °C. Supernatant was harvested and analysed by Eve Technologies using the Mouse Cytokine Proinflammatory Focused 10-Plex Discovery Assay Array (MilliporeSigma) on a Luminex 200 System according to the manufacturer’s protocol.
mRNA stability study
LNPs or naked mRNA were collected either with or without nebulization and mixed in a 1:1 ratio with 10% Triton X-100 (Sigma) to access encapsulated mRNA. RNA was then extracted using a Monarch RNA cleanup kit (New England Biolabs) and run in an Advanced Analytical Fragment Analyzer.
Histology study with Ai14 mice
B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J mice (Jackson Laboratory) were administered three 3 mg doses of Cre mRNA via nebulization in the nose-only exposure chamber. Control Ai14 mice were administered PBS. Nebulizations were separated by 48 h and mice were killed 5 days after the final dose. Upon CO2 euthanasia, the mouse lungs were first perfused with PBS via the right ventricle. The left lobe was inflated through the trachea with 0.25 ml of fresh 4% PFA and then placed in 4% PFA overnight. The following day, the fixed tissue was submitted for paraffin processing. The lobe was embedded with the ventral side down. Serial sections (5 μm each) were obtained up to the middle of the lung. For each mouse, two slides were used for immunofluorescent imaging. One section was taken from the middle of the lung where more large airways are present and the other was taken from a quarter of the way into the lung where more small airways are present. The slides were deparaffinized, permeabilized, blocked and then stained with rabbit anti-RFP antibody (abcam, ab62341, 1:200) for 1 h. The slides were washed and labelled with Dylight 594 goat anti-rabbit antibody (Vector Labs, DI-1594, 1:200), Alexa Fluor 647 anti-acetylated tubulin antibody (Santa Cruz, SC-23950, 1:200) and Alexa Fluor 488 anti-Uteroglobin/SCGB1A1/CC10 antibody (Santa Cruz, SC-390313, 1:200) for 1 h. Autofluorescence was blocked with Vector Labs autofluorescence quencher and slides were then mounted with 4,6-diamidino-2-phenylindole (DAPI) mounting media. Slides were imaged on a Nikon spinning-disk confocal microscope. The number of tdTomato-positive cells in all airways on these sections were counted manually using FIJI.
Statistical analysis
DOE was performed using JMP 13 software (JMP, SAS Institute). Standard deviations for encapsulation efficiencies were calculated in Microsoft Excel by error propagation based on the error in total mRNA and non-encapsulated mRNA as measured by Ribgoreen. All statistical tests were two-sided where applicable.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
This work was supported by the NIH (grant numbers UG3HL147367 and R01 HL162564-02 to A.Y.J., F.E., C.M. and D.G.A.) and Sanofi (formerly Translate Bio, to I.O.R., Y.H., R.S.M. and D.G.A.). J.W. was supported by the Cystic Fibrosis Foundation under award WITTEN19XX0. S.M. and F.A.O. were supported by the MIT Undergraduate Research Opportunities Program. We thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the Animal Imaging & Preclinical Testing, Histology, Nanotechnology Materials, BioMicro Center and Microscopy core facilities. This work was also supported in part by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute.
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41565-023-01548-3.
Competing interests
A.Y.J., J.W., I.O.R. and D.G.A. have filed a patent for the biodegradable lipid library described herein (US Patent Application No. 18080299). D.G.A. receives research funding from Sanofi/Translate Bio, and is a founder of oRNA Tx. R.L. is co-founder and a director of Moderna. He also serves on the board and has equity in Particles For Humanity. For a list of entities with which R.L. is, or has been recently involved, compensated or uncompensated, see https://www.dropbox.com/s/yc3xqb5s8s94v7x/Rev%20Langer%20COI.pdf?dl=0. The other authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41565-023-01548-3.
Data availability
All data that support the findings of this study are provided within the paper and its Supplementary Information. Source data are provided with this paper.
References
- 1.Kowalski PS, Rudra A, Miao L & Anderson DG Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol. Ther 27, 710–728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajj KA & Whitehead KA Tools for translation: non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater 2, 1–17 (2017). [Google Scholar]
- 3.Han X et al. An ionizable lipid toolbox for RNA delivery. Nat. Commun 12, 7233 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiu M et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl Acad. Sci. USA 118, e2020401118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swingle KL, Hamilton AG & Mitchell MJ Lipid nanoparticle-mediated delivery of mRNA therapeutics and vaccines. Trends Mol. Med 27, 616–617 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Miao L et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol 37, 1174–1185 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Zhang X et al. Functionalized lipid-like nanoparticles for in vivo mRNA delivery and base editing. Sci. Adv 6, eabc2315 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Billingsley MM et al. Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. 20, 1578–1589 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riley RS et al. Ionizable lipid nanoparticles for in utero mRNA delivery. Sci. Adv 7, eaba1028 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sabnis S et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther 26, 1509–1519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fenton OS et al. Synthesis and biological evaluation of ionizable lipid materials for the in vivo delivery of messenger RNA to B lymphocytes. Adv. Mater 29, 1606944 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Liu J et al. Fast and efficient CRISPR/Cas9 genome editing in vivo enabled by bioreducible lipid and messenger RNA nanoparticles. Adv. Mater 31, 1902575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Polack FP et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med 383, 2603–2615 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baden LR et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med 384, 403–416 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gillmore JD et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med 385, 493–502 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Cornebise M et al. Discovery of a novel amino lipid that improves lipid nanoparticle performance through specific interactions with mRNA. Adv. Funct. Mater 10.1002/adfm.202106727 (2021). [DOI] [Google Scholar]
- 17.Barbier AJ, Jiang AY, Zhang P, Wooster R & Anderson DG The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol 40, 840–854 (2022). [DOI] [PubMed] [Google Scholar]
- 18.Chakraborty C, Sharma AR, Bhattacharya M & Lee S-S From COVID-19 to cancer mRNA vaccines: moving from bench to clinic in the vaccine landscape. Front. Immunol 12, 2648 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cafri G et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Invest 130, 5976–5988 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oberli MA et al. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 17, 1326–1335 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Espeseth AS et al. Modified mRNA/lipid nanoparticle-based vaccines expressing respiratory syncytial virus F protein variants are immunogenic and protective in rodent models of RSV infection. NPJ Vaccines 5, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aliprantis AO et al. A phase 1, randomized, placebo-controlled study to evaluate the safety and immunogenicity of an mRNA-based RSV prefusion F protein vaccine in healthy younger and older adults. Hum. Vaccines Immunother 17, 1248–1261 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bahl K et al. Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol. Ther 25, 1316–1327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feldman RA et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37, 3326–3334 (2019). [DOI] [PubMed] [Google Scholar]
- 25.John S et al. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 36, 1689–1699 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Medina-Magües LG et al. mRNA vaccine protects against zika virus. Vaccines 9, 1464 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mu Z, Haynes BF & Cain DW HIV mRNA vaccines—progress and future paths. Vaccines 9, 134 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zabaleta N, Torella L, Weber ND & Gonzalez-Aseguinolaza G mRNA and gene editing: late breaking therapies in liver diseases. Hepatology 10.1002/hep.32441 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson E et al. Lipid nanoparticle-delivered chemically modified mRNA restores chloride secretion in cystic fibrosis. Mol. Ther 26, 2034–2046 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Da Silva Sanchez A, Paunovska K, Cristian A & Dahlman JE Treating cystic fibrosis with mRNA and CRISPR. Hum. Gene Ther 31, 940–955 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai M et al. Gene editing of DNAH11 restores normal cilia motility in primary ciliary dyskinesia. J. Med. Genet 53, 242–249 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Paff T, Omran H, Nielsen KG & Haarman EG Current and future treatments in primary ciliary dyskinesia. Int. J. Mol. Sci 22, 9834 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guan S, Darmstädter M, Xu C & Rosenecker J In vitro investigations on optimizing and nebulization of IVT-mRNA formulations for potential pulmonary-based α−1-antitrypsin deficiency treatment. Pharmaceutics 13, 1281 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeyer F et al. mRNA-mediated gene supplementation of Toll-like receptors as treatment strategy for asthma in vivo. PLoS ONE 11, e0154001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mays LE et al. Modified Foxp3 mRNA protects against asthma through an IL-10–dependent mechanism. J. Clin. Invest 123, 1216–1228 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rakhra K et al. Exploiting albumin as a mucosal vaccine chaperone for robust generation of lung-resident memory T cells. Sci. Immunol 6, eabd8003 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bivas-Benita M et al. Pulmonary delivery of chitosan-DNA nanoparticles enhances the immunogenicity of a DNA vaccine encoding HLA-A*0201-restricted T-cell epitopes of Mycobacterium tuberculosis. Vaccine 22, 1609–1615 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Rajapaksa AE et al. Effective pulmonary delivery of an aerosolized plasmid DNA vaccine via surface acoustic wave nebulization. Respir. Res 15, 60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu M et al. Intranasal vaccination with mannosylated chitosan formulated DNA vaccine enables robust IgA and cellular response induction in the lungs of mice and improves protection against pulmonary mycobacterial challenge. Front. Cell. Infect. Microbiol 7, 445 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.King RG et al. Single-dose intranasal administration of AdCOVID elicits systemic and mucosal immunity against SARS-CoV-2 and fully protects mice from lethal challenge. Vaccines 9, 881 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An X et al. Single-dose intranasal vaccination elicits systemic and mucosal immunity against SARS-CoV-2. iScience 24, 103037 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim YC et al. Strategy to enhance dendritic cell-mediated DNA vaccination in the lung. Adv. Ther 3, 2000013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu D & Hickey AJ Pulmonary vaccine delivery. Expert Rev. Vaccines 6, 213–226 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Sou T et al. New developments in dry powder pulmonary vaccine delivery. Trends Biotechnol. 29, 191–198 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Huang J et al. A novel dry powder influenza vaccine and intranasal delivery technology: induction of systemic and mucosal immune responses in rats. Vaccine 23, 794–801 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Minne A et al. The delivery site of a monovalent influenza vaccine within the respiratory tract impacts on the immune response. Immunology 122, 316–325 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z et al. Exosomes decorated with a recombinant SARS-CoV-2 receptor-binding domain as an inhalable COVID-19 vaccine. Nat. Biomed. Eng 6, 791–805 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel AK et al. Inhaled nanoformulated mRNA polyplexes for protein production in lung epithelium. Adv. Mater 31, 1805116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lokugamage MP et al. Optimization of lipid nanoparticles for the delivery of nebulized therapeutic mRNA to the lungs. Nat. Biomed. Eng 5, 1059–1068 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson C Future therapies for cystic fibrosis. Lancet Respir. Med 10, e75–e76 (2022). [DOI] [PubMed] [Google Scholar]
- 51.Witten J, Samad T & Ribbeck K Selective permeability of mucus barriers. Curr. Opin. Biotechnol 52, 124–133 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Witten J & Ribbeck K The particle in the spider’s web: transport through biological hydrogels. Nanoscale 9, 8080–8095 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cone RA Barrier properties of mucus. Adv. Drug Deliv. Rev 61, 75–85 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Lieleg O & Ribbeck K Biological hydrogels as selective diffusion barriers. Trends Cell Biol. 21, 543–551 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim N, Duncan GA, Hanes J & Suk JS Barriers to inhaled gene therapy of obstructive lung diseases: a review. J. Controlled Release 240, 465–488 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coyne CB, Kelly MM, Boucher RC & Johnson LG Enhanced epithelial gene transfer by modulation of tight junctions with sodium caprate. Am. J. Respir. Cell Mol. Biol 23, 602–609 (2000). [DOI] [PubMed] [Google Scholar]
- 57.Kauffman KJ et al. Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. 15, 7300–7306 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Billingsley MM et al. Orthogonal design of experiments for optimization of lipid nanoparticles for mRNA engineering of CAR T cells. Nano Lett. 22, 533–542 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li S et al. Payload distribution and capacity of mRNA lipid nanoparticles. Nat. Commun 13, 5561 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kauffman KJ et al. Rapid, single-cell analysis and discovery of vectored mRNA transfection in vivo with a loxP-flanked tdTomato reporter mouse. Mol. Ther. Nucleic Acids 10, 55–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ball RL, Bajaj P & Whitehead KA Achieving long-term stability of lipid nanoparticles: examining the effect of pH, temperature, and lyophilization. Int. J. Nanomed 12, 305–315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao P et al. Long-term storage of lipid-like nanoparticles for mRNA delivery. Bioact. Mater 5, 358–363 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crowe JH, Oliver AE, Hoekstra FA & Crowe LM Stabilization of dry membranes by mixtures of hydroxyethyl starch and glucose: the role of vitrification. Cryobiology 35, 20–30 (1997). [DOI] [PubMed] [Google Scholar]
- 64.Ohtake S, Schebor C, Palecek SP & de Pablo JJ Phase behavior of freeze-dried phospholipid–cholesterol mixtures stabilized with trehalose. Biochim. Biophys. Acta Biomembr 1713, 57–64 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Eastman SJ et al. Optimization of formulations and conditions for the aerosol delivery of functional cationic lipid:DNA complexes. Hum. Gene Ther 8, 313–322 (1997). [DOI] [PubMed] [Google Scholar]
- 66.Whitehead KA et al. Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nat. Commun 5, 4277 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu S et al. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR–Cas gene editing. Nat. Mater 20, 701–710 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pezzulo AA et al. The air–liquid interface and use of primary cell cultures are important to recapitulate the transcriptional profile of in vivo airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol 300, L25–L31 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hill DB & Button B in Mucins: Methods and Protocols (eds McGuckin MA & Thornton DJ) 245–258 (Humana Press, 2012); 10.1007/978-1-61779-513-8_15 [DOI] [Google Scholar]
- 70.Ramachandran S et al. Efficient delivery of RNA interference oligonucleotides to polarized airway epithelia in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol 305, L23–L32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krishnamurthy S et al. Manipulation of cell physiology enables gene silencing in well-differentiated airway epithelia. Mol. Ther. Nucleic Acids 1, e41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burgel P-R, Montani D, Danel C, Dusser DJ & Nadel JA A morphometric study of mucins and small airway plugging in cystic fibrosis. Thorax 62, 153–161 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ratjen F Cystic fibrosis: the role of the small airways. J. Aerosol Med. Pulm. Drug Deliv 25, 261–264 (2012). [DOI] [PubMed] [Google Scholar]
- 74.van den Berge M, ten Hacken NHT, Cohen J, Douma WR & Postma DS Small airway disease in asthma and COPD: clinical implications. Chest 139, 412–423 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Tiddens HAWM, Donaldson SH, Rosenfeld M & Paré PD Cystic fibrosis lung disease starts in the small airways: can we treat it more effectively? Pediatr. Pulmonol 45, 107–117 (2010). [DOI] [PubMed] [Google Scholar]
- 76.Tatsuta M et al. Effects of cigarette smoke on barrier function and tight junction proteins in the bronchial epithelium: protective role of cathelicidin LL-37. Respir. Res 20, 251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maeki M, Uno S, Niwa A, Okada Y & Tokeshi M Microfluidic technologies and devices for lipid nanoparticle-based RNA delivery. J. Control. Release 344, 80–96 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheng MHY et al. Induction of bleb structures in lipid nanoparticle formulations of mRNA leads to improved transfection potency. Adv. Mater 10.1002/adma.202303370 (2023). [DOI] [PubMed] [Google Scholar]
- 79.Brader ML et al. Encapsulation state of messenger RNA inside lipid nanoparticles. Biophys. J 120, 2766–2770 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kulkarni JA et al. On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and siRNA. ACS Nano 12, 4787–4795 (2018). [DOI] [PubMed] [Google Scholar]
- 81.Kulkarni JA et al. Fusion-dependent formation of lipid nanoparticles containing macromolecular payloads. Nanoscale 11, 9023–9031 (2019). [DOI] [PubMed] [Google Scholar]
- 82.Richardson SJ, Bai A, Kulkarni AA & Moghaddam MF Efficiency in drug discovery: liver S9 fraction assay as a screen for metabolic stability. Drug Metab. Lett 10, 83–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scholte BJ, Davidson DJ, Wilke M & de Jonge HR Animal models of cystic fibrosis. J. Cyst. Fibros 3, 183–190 (2004). [DOI] [PubMed] [Google Scholar]
- 84.McCarron A, Donnelley M & Parsons D Airway disease phenotypes in animal models of cystic fibrosis. Respir. Res 19, 54 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim N et al. Inhaled gene therapy of preclinical muco-obstructive lung diseases by nanoparticles capable of breaching the airway mucus barrier. Thorax 77, 812–820 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Phillips JE, Zhang X & Johnston JA Dry powder and nebulized aerosol inhalation of pharmaceuticals delivered to mice using a nose-only exposure system. J. Vis. Exp 10.3791/55454 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Beck SE et al. Deposition and expression of aerosolized rAAV vectors in the lungs of rhesus macaques. Mol. Ther 6, 546–554 (2002). [DOI] [PubMed] [Google Scholar]
- 88.Woo CJ et al. Inhaled delivery of a lipid nanoparticle encapsulated messenger RNA encoding a ciliary protein for the treatment of primary ciliary dyskinesia. Pulm. Pharmacol. Ther 75, 102134 (2022). [DOI] [PubMed] [Google Scholar]
- 89.Okuda K et al. Secretory cells dominate airway CFTR expression and function in human airway superficial epithelia. Am. J. Respir. Crit. Care Med 203, 1275–1289 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carraro G et al. Transcriptional analysis of cystic fibrosis airways at single-cell resolution reveals altered epithelial cell states and composition. Nat. Med 27, 806–814 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hodges CA & Conlon RA Delivering on the promise of gene editing for cystic fibrosis. Genes Dis. 6, 97–108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vanover D et al. Nebulized mRNA-encoded antibodies protect hamsters from SARS-CoV-2 infection. Adv. Sci 9, 2202771 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rhym LH, Manan RS, Koller A, Stephanie G & Anderson DG Peptide-encoding mRNA barcodes for the high-throughput in vivo screening of libraries of lipid nanoparticles for mRNA delivery. Nat. Biomed. Eng 7, 901–910 (2023). [DOI] [PubMed] [Google Scholar]
- 94.Chen D et al. Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation. J. Am. Chem. Soc 134, 6948–6951 (2012). [DOI] [PubMed] [Google Scholar]
- 95.Heyes J, Palmer L, Bremner K & MacLachlan I Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J. Control. Release 107, 276–287 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that support the findings of this study are provided within the paper and its Supplementary Information. Source data are provided with this paper.