

Abstract
Plasmodium falciparum, the human malaria parasite, infects two hosts and various cell types, inducing distinct morphological and physiological changes in the parasite in response to different environmental conditions. These variations required the parasite to adapt and develop elaborated molecular mechanisms to ensure its spread and transmission. Recent findings have significantly improved our understanding of the regulation of gene expression in P. falciparum. Here, we provide an up-to-date overview of technologies used to highlight the transcriptomic adjustments occurring in the parasite throughout its life cycle. We also emphasize the complementary and complex epigenetic mechanisms regulating gene expression in malaria parasites. This review concludes with an outlook on the chromatin architecture, the remodeling systems, and how this 3D genome organization is critical in various biological processes.
Keywords: Plasmodium, single-cell, epigenetics, lncRNA, long noncoding RNA, chromatin architecture
1. INTRODUCTION
Global malaria deaths have gradually declined from 896,000 in 2000 to 558,000 in 2019 (134). This has been possible through an increase in malaria control efforts, including distribution of insecticide-treated mosquito bed nets and systemic insecticide spraying. Recently, RTS,S became the first malaria vaccine to be recommended by the WHO. In a pilot study, 2.3 million doses of the vaccine were administered in three African countries, and an approximately 30% reduction in severe malaria was observed (134). Even if this vaccine does not provide complete protection, it may serve as a significant contributor to a malaria control program.
Malaria is caused by the protozoan Plasmodium, which belongs to the Apicomplexa phylum, a large group of parasites including Toxoplasma, Cryptosporidium, and Babesia. Over 200 species of Plasmodium are reported, but only 5 of them can infect humans: P. falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi. While P. vivax is the most widespread, with high prevalence in Southeast Asia and Latin America, P. falciparum is most common in Africa and causes the greatest morbidity and mortality worldwide.
Despite the progress made over the last decades, the WHO’s World Malaria Report 2021 (134) documented a 12% increase in malaria-related fatalities occurring in 2020. The majority (68%) of this augmentation seemed to be attributable to service disruptions during the COVID-19 pandemic. This crisis highlights the importance of maintaining a steady and continuous regiment to control malaria. In addition to the COVID-19 challenges, the emergence and spread of resistance to antimalarials are proving to be tremendous threats. The mutations that have been observed in the pfkelch13 gene are strongly associated with reduced sensitivity to artemisinin, our most effective and last line of defense against P. falciparum infection, and they are spreading dangerously around the world, including in Africa. These concerns reinforce the importance of deciphering the biological mechanisms of Plasmodium, including gene regulation, to discover novel potential therapeutic targets.
In this review, we present the transcriptome modulations across the different stages of the parasite life cycle. We also discuss the contribution of diverse regulatory mechanisms in gene expression, from epigenetics to chromatin structure.
Epigenetics: field of study focused on factors controlling gene expression without altering DNA sequence
2. PLASMODIUM STAGE-SPECIFIC EXPRESSION PATTERNS
2.1. The Plasmodium falciparum Life Cycle
The human malaria parasite has a complex life cycle involving an intermediate human host and the definitive host and vector, the female Anopheles mosquito. During a blood meal, a Plasmodium-infected mosquito transmits sporozoites from its salivary glands to the human bloodstream. These sporozoites migrate to the liver, invade hepatocytes, and initiate parasite amplification. This asymptomatic pre-erythrocytic phase takes roughly 7–10 days for P. falciparum. In all Plasmodium species, once the pre-erythrocytic cycle is complete, tens of thousands of infectious merozoites are released in the bloodstream to invade red blood cells. Within an erythrocyte, an immature ring stage develops into a large, transcriptionally and metabolically active trophozoite that can then differentiate into a multinucleated schizont. The schizont can divide into up to 32 new merozoites, which burst out of the erythrocyte to reinfect new red blood cells. This rupture is associated with clinical symptoms, and most current treatments target this phase. The erythrocyte invasions are repeated continuously with the new generations of merozoites that perpetuate this asexual cycle. During the intraerythrocytic developmental cycle (IDC), a proportion of parasites differentiate into male and female gametocytes. In P. falciparum, this gametocytogenesis is exceptionally long, with maturation after 9–12 days, and is composed of five morphologically distinct stages (stages I–V). These gametocytes are ingested during a new blood meal by a female Anopheles. A zygote forms inside the mosquito’s gut, a zygote is formed by fertilization of a male microgamete and a female macrogamete. This zygote becomes a mobile ookinete, which in turn migrates and invades the midgut wall to form an oocyst. The oocyst stage can produce thousands of new sporozoites. To complete and perpetuate the life cycle, these sporozoites migrate to the mosquito’s salivary glands and are injected into a human during a subsequent blood meal.
2.2. An Overview of the Plasmodium falciparum Genome and Its Gene Expression
In 2002, the 22.8-megabase nuclear genome of P. falciparum 3D7 clone was published: ~5,500 annotated genes along 14 chromosomes (42). This genome is still one of the most AT-rich genomes that have been sequenced, with ~80% and ~90% AT content for coding and intergenic regions, respectively. The publication of the P. falciparum genome was the foundation for transcriptomic exploration. Consecutively, DNA microarrays (12, 70) were applied to analyze the transcriptome of P. falciparum at different stages of development, including gametocyte and sporozoite stages. They exposed how most of the predicted genes are expressed during the IDC, with an active burst of transcription during trophozoite and gametocyte stages. The genes exhibit a periodic transcriptional pattern with timely expression; for example, the genes involved in invasion are expressed during late schizont development, and some genes are specifically expressed in gametocytes and sporozoites. In 2005, a similar approach applied following sexual differentiation identified a subset of genes highly transcribed and expressed in gametocytes, confirming high correlation between Plasmodium transcriptome and the distinct stages of development (113).
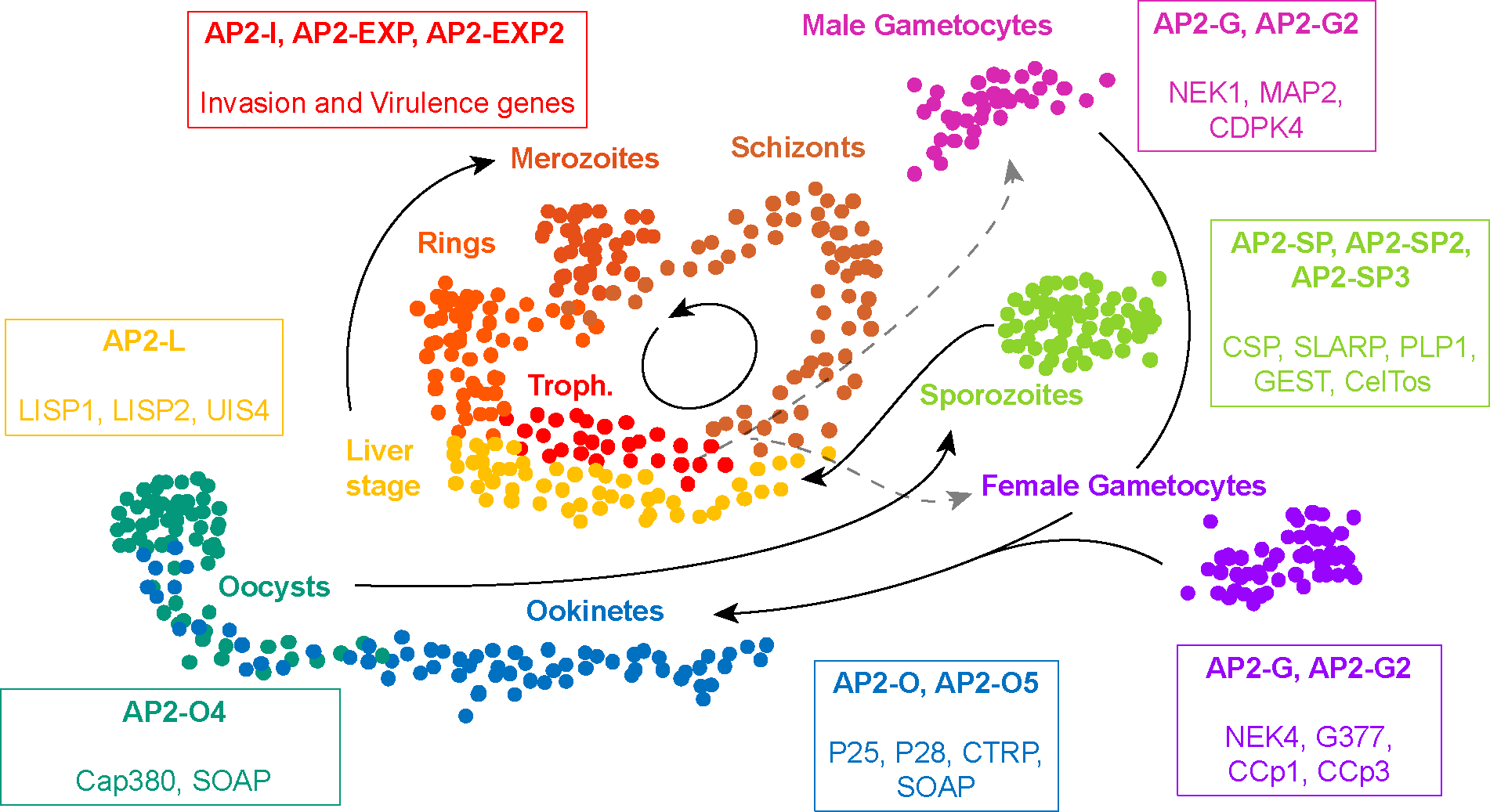
The advent of next-generation sequencing brought about new opportunities to decipher the malaria parasite’s transcriptome more thoroughly. Plasmodium RNA-seq was first performed in 2010, providing novel understanding of overall global gene expression patterns in asexual stages of P. falciparum (87). With this method, previous predictions of gene boundaries and splice sites were corrected and novel transcripts were detected, significantly improving the parasite transcriptome resolution. Additional RNA-seq data sets were subsequently generated for asexual (6, 53, 73, 119, 121, 132), sexual (68, 73), and mosquito (44, 72, 140) stages of P. falciparum, providing access to a previously untapped source of knowledge. These high-throughput sequencing efforts contributed to detection of antisense variants, noncoding RNAs (ncRNAs), and stage-specific transcripts and to characterization of mutant lines. In 2018, nascent mRNAs were explored by 4-thiouracil incorporation, yielding a high-resolution RNA transcriptome data set for the IDC of P. falciparum (88). Sex-specific genes were also identified in P. falciparum and P. berghei, a rodent malaria species, with NIMA-related kinase 4 (NEK4), CCp1, CCp3, and G377 described as markers for female gametocytes while NEK1, CDP4, and MAP2 kinases were found to be enriched in male gametocytes (65, 68) (Figure 1).
Figure 1.
Model of single-cell transcriptomes across the Plasmodium life cycle. Cells are colored according to their distinct stage of development. Master ApiAP2 transcription factors are in bold font along with stage-specific or characteristic markers expressed at the particular stages. The model is based on the single-cell atlas of P. berghei parasites (55). Abbreviation: ApiAP2, apicomplexan Apetala2.
Host–parasite interactions are critical to fully understand malaria infection and adaptation within the human host. Although dual RNA-seq allows simultaneous capture of host and pathogen transcriptomes, this powerful strategy has been used only sparingly in Plasmodium. It was applied in the analysis of infected and uninfected hepatocytes to identify mucin-13 as a robust human marker of Plasmodium-hepatic infection, regardless of the species tested (67). Despite its sporadic application in malaria research, dual RNA-seq delivers an attractive opportunity to gain knowledge on parasite and host cell interactions in parallel that is not yet available through alternative platforms.
Recently, single-cell sequencing technologies have emerged as the state-of-the-art platforms for transcriptomic study. Single-cell RNA-seq (scRNA-seq) provides an unbiased cell-to-cell level of RNA transcript detection at a resolution that is unprecedented to date. This technology has revealed previously uncharacterized and rare phenotypes hidden by classical bulk RNA-seq. scRNA-seq was applied to different morphological stages of P. falciparum, including asexual (85, 102), sexual (85, 93, 127), and mosquito (83, 101) stages (Figure 1). These studies have, among other things, assessed the sex-specific markers of gametocytes (102, 127) and described transcriptional signatures among committed and sexual stages (13, 85, 93). Moreover, additional regulators involved in sexual commitment were identified, such as transcription factors (TFs), SNF2 helicases, and histone-modifying enzymes (93). scRNA-seq data sets and computational methods provide the opportunity to resolve (pseudo)trajectories and deduce the developmental dynamics of the pathogen down to individual cells. Such models were obtained for asexual (102) and mosquito (83, 101) stages of the parasite cell cycle, which provided a more insightful and continuous transcriptomic repertoire instead of the distinct snapshots derived through conventional RNA-seq techniques. Thus far, only the entire life cycle of P. berghei has been fully completed and published (55). The authors of this paper generated single-cell transcriptomes from ten stages and detected 5,156 genes divided into 20 clusters. They also compared the blood stage trajectories of P. falciparum, P. knowlesi, and P. berghei. Despite the vastly distinct hosts and IDC periods, their transcriptional activities had an overall pattern. Single-cell technology is also a powerful tool to analyze clinical samples (55) or study the impact of environment changes. Plasmodium parasite cultures incubated at 40°C presented a global decrease of transcription and greater transcriptional heterogeneity when compared to control parasites, especially for genes involved in gametocytogenesis and stress response (99).
Overall, the plethora of technologies, optimized over time, have made it possible to decipher the transcriptomes of Plasmodium and identify specific transcriptional signatures (Figure 1). The Malaria Cell Atlas, managed by the Wellcome Sanger Institute, has already provided an open access platform for single-cell transcriptomics data sets across different parasite cell cycle stages of P. falciparum, P. knowlesi, and P. berghei (https://www.sanger.ac.uk/tool/mca/mca). This project will facilitate the global understanding of transcriptional regulation and benefit the malaria research community.
2.3. Transcription Factors and Their Role in Plasmodium Gene Regulation
In eukaryotic cells, TFs are key regulators of transcriptional activity. Roughly 1,600 human TFs have been described (66). As described above, Plasmodium presents some specific transcriptional signatures suggesting a fine-tuned and well-controlled regulation.
RNA polymerase II is responsible for the transcription of messenger RNAs along with several small nuclear RNAs (Figure 2). The polymerase requires general TFs to form a multiprotein complex that assembles onto targeted promoters. Although several of these partners have been identified in P. falciparum, the lack of clear orthologs based on homology of critical components found in other eukaryotic lineages suggests that alternative mechanisms may be implicated in the parasite transcriptional machinery (21). For example, recruitment of the preinitiation complex, composed of general TFs and RNA polymerase II, is independent of transcriptional status and histone acetylation in P. falciparum, in contrast to the classical eukaryotic model (45). One chromatin immunoprecipitation (ChIP)-on-chip–type experiment demonstrated how RNA polymerase II is bound to promoters in early and late asexual stages (97). During this early phase, the polymerase is engaged with promoter regions yet prevented from further transcriptional elongation. GRO-seq analysis, capturing nascent RNAs, confirmed this pausing, which could contribute to gene regulation and the timely transcriptional burst found in the trophozoite stages (77).
Figure 2.
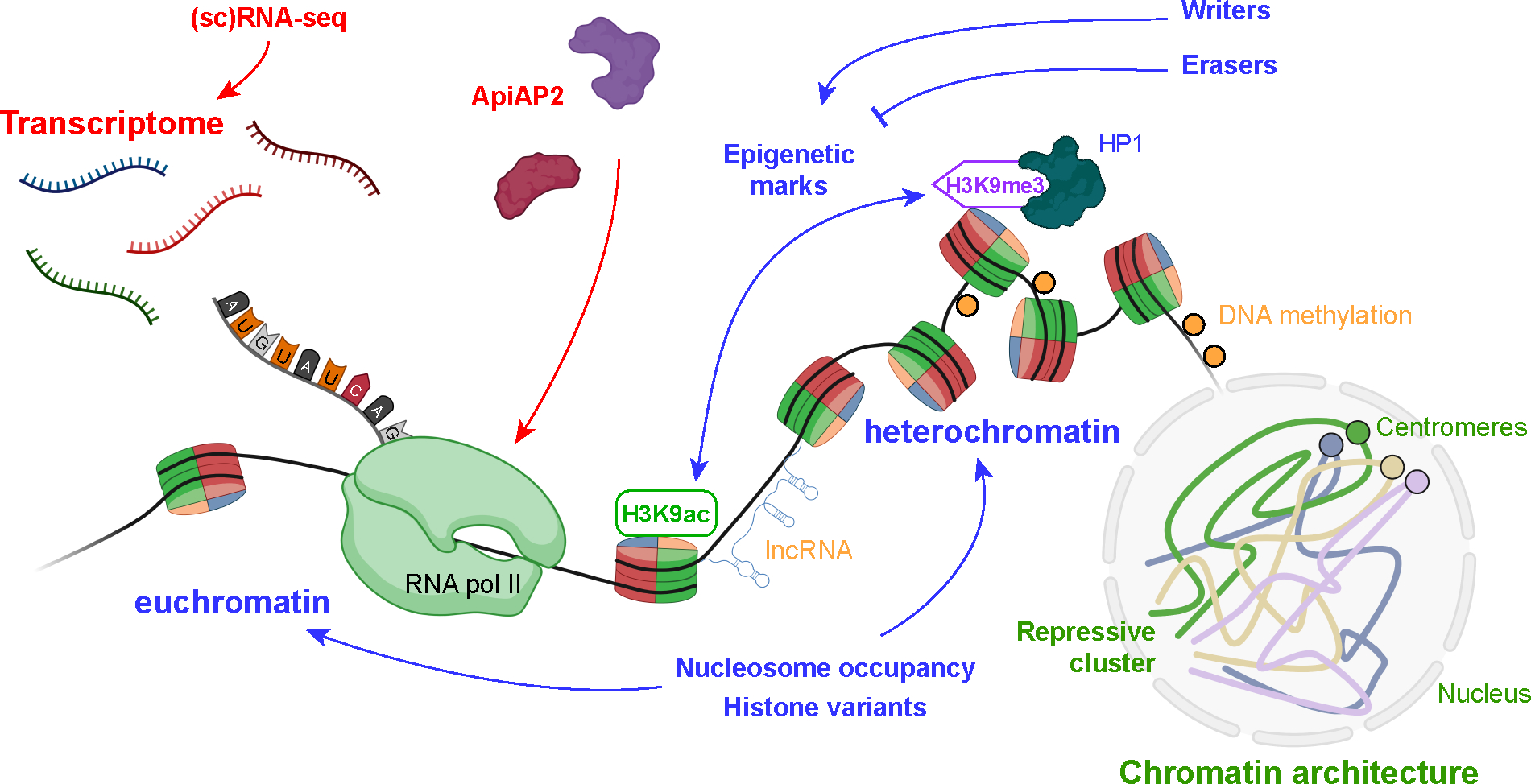
Epigenetic mechanisms in Plasmodium. RNA-sequencing platforms (red) contribute to the identification of distinct transcriptomic signatures across the different life stages of Plasmodium. These gene expression patterns define cell function(s) and are under control of master ApiAP2 transcription factors. Nucleosome occupancy and histone modifications (blue) are major factors regulating chromatin condensation. Other epigenetic mechanisms include lncRNAs and DNA methylation patterns (orange). Finally, the overall chromatin landscape is well organized, forming a repressive cluster including telomeres and var genes positioned at opposing poles of centromeres within the nucleus (green). Abbreviations: ApiAP2, apicomplexan Apetala2; lncRNA, long noncoding RNA; RNAP, RNA polymerase; scRNA-seq, single-cell RNA sequencing; HP1, heterochromatin protein 1.
Besides general TFs, the transcriptional machinery is also dependent on specific TFs, required for the recruitment of chromatin-remodeling and chromatin-modifying enzymes. In 2004, a comparative analysis showed a ratio of one TF per 800 genes in Plasmodium (compare to one TF per 29 genes in Saccharomyces cerevisiae), indicating a relative paucity of TFs (120). Subsequently, the main specific family of TFs in apicomplexan parasites was discovered (3). These proteins contain Apetala2 (AP2) domains, originally described in plants as DNA-binding domains, and about 20–27 members of this apicomplexan AP2 (ApiAP2) family are highly conserved across all Plasmodium species as well as in other apicomplexans. Each protein contains one to three AP2 domains and recognizes multiple distinct DNA motifs (22).
Different studies have reported several of these AP2 TFs as master regulators (activators or repressors) involved in developmental-stage transition (Figures 1 and 2). The most characterized of these ApiAP2 is AP2-G, for which gene deletion resulted in abolishment of gametocytogenesis in P. falciparum (60) and in P. berghei (41). Conversely, AP2-G2 acts as a global repressor of transmission-specific genes, promoting gametocyte maturation (82, 115, 116, 135, 137). Although gametocytogenesis implies a cascade of multiple ApiAP2 TFs (recently reviewed in Reference 94), certain ApiAP2 TFs such as AP2-G5 (107), AP2-G, and AP2-G2 function as main contributors by regulating hundreds of genes and operating as a transcriptional switch from asexual to sexual programs. Similarly, additional master regulators were described in other stages of P. berghei, with AP2-O and AP2-Z shown to be essential for ookinetes (61, 86, 139), AP2-SP for sporozoites (138), and AP2-L for liver stages (56). In 2017, two systematic knockout screens of AP2 TFs in P. berghei (82) and P. yoelii (141) malaria models infecting rodents confirmed the importance of these previous TFs but also identified new ApiAP2 members fundamental for mosquito stages, such as AP2-O2/O3/O4 for ookinetes and AP2-SP2/SP3 for sporogony.
Although the majority of the ApiAP2 family TFs are associated with developmental-stage transitions, some of them seem to be required for supplementary mechanisms whose biological functions remain elusive. AP2-exp and AP2-exp2 mutants showed upregulation of antigenic variant genes (rifin, stevor, and Pfmc-2TM) and reduction of genes involved in cell remodeling, respectively (79, 109). Different AP2 TFs are also associated with heterochromatin (108), such as AP2-HC (23), while AP2Tel and PfSIP2 were found to be enriched in subtelomeric regions (38, 112).
Antigenic variant genes: multicopy gene families associated with virulence and immune evasion in Plasmodium
Heterochromatin: highly condensed chromatin found mostly within telomeres and repressed genes, as opposed to euchromatin, which corresponds to transcriptionally active regions
Recently, machine learning algorithms, used to build predictive models of gene expression, suggested that upstream regulatory AP2-binding sites may play a more limited role in transcriptional regulation during the IDC than previously thought (100). Several hierarchical players are required to establish and maintain the distinct transcriptional signatures across developmental stages in Plasmodium.
3. EPIGENETIC REGULATION IN MALARIA PARASITES: THE ROLE OF NUCLEOSOMES AND HISTONES
3.1. Chromatin Organization, Nucleosome Positioning, and Remodeling
In all eukaryotes, nuclear DNA is compacted and organized into chromatin through the binding of nucleosomes. The core unit of the nucleosome is composed of a histone octamer (two copies each of H2A, H2B, H3, and H4) that wraps ~147 DNA base pairs. Nucleosome occupancy and positioning are widely conserved in eukaryotes, including Plasmodium. Transcriptionally active gene promoters are characterized by a nucleosome-depleted region located just upstream of the transcription start site. Comparative studies have found that P. falciparum presents relatively weak nucleosome positioning at the +1 and +2 sites compared to model organisms (64); however, the parasite also exhibited significantly higher nucleosome occupancy at the start and end of the gene body (19, 92, 131). Regardless of the methods used to elucidate nucleosome positioning and occupancy—spanning from micrococcal nuclease digestion with deep sequencing (MNase-seq) to formaldehyde-assisted isolation of regulatory elements with deep sequencing (FAIRE-seq) to assay for transposase-accessible chromatin with deep sequencing (ATAC-seq)—it is remarkably clear that the chromatin accessibility correlates with mRNA abundance in Plasmodium (19, 64, 92, 100, 103, 121, 131). This association confirms that the depletion of these nucleosomes is fundamental to allow access and binding of TFs and diverse DNA-binding (co)factors/proteins. De novo motif searches using the accessible regions of the chromatin identified motifs already associated with specific AP2 TFs (103, 121), such as AP2-I, which plays an essential role for expression of invasion genes (105).
In Plasmodium, nucleosome occupancy is dynamic and varies considerably throughout the cell cycle. During the IDC, the overall histone coverage is minimal at trophozoite stages, whereas maximal coverage is detected during late schizont stages. These patterns clearly correlate to the pathogen’s transcription activity and chromatin condensation states (19, 92). Additional studies on sexual and mosquito stages could potentially highlight important dynamic nucleosome changes on regulation of stage-specific genes, as already observed for asexual-specific genes (103, 121).
The four core histones can be substituted by histone variants conferring distinct physical properties, dynamics, and alternative functions. In Plasmodium, H2A.Z, H2B.Z, H2Bv, CenH3, and H3.3 were identified as histone variants (52, 80, 118). Interestingly, some of them have specific and unique features, such as H2A.Z/H2B.Z, which is associated with intergenic regions of P. falciparum (54, 90), and PfCENH3, shown to be linked with centromeres (52). H2A.Z also seems to be enriched at the transcription start site of the single active var gene, suggesting a contribution of this particular histone variant in regulation of this virulence gene family (89, 90).
var genes: multicopy gene family contributing to antigenic variation whose expression is mutually exclusive to express a single gene at a time
Although DNA compaction associated with nucleosomes is understood to hinder accessibility of the transcription machinery, the occupancy and positioning of these nucleosomes provide a complementary and dynamic regulatory mechanism crucial to controlling gene expression in malaria parasites.
3.2. Chromatin Organization and Histone Modifications
In addition to the fundamental roles of nucleosome organization, the histone core itself can be subject to a versatile set of reversible epigenetic marks. These reversible posttranslational modifications (PTMs), which include acetylation, methylation, and phosphorylation, occur on histone tails and are distributed along the genome, mainly on promoter regions. In Plasmodium, several dozen PTMs have been identified through quantitative mass spectrometry for each histone, including variants, revealing some PTMs unique to the parasite (27, 80, 104, 106, 123). Interestingly, acetylation of H4 seems to occur from N terminus to C terminus, whereas deacetylation occurs in the opposite direction (106, 123). This model contrasts with other organisms and seems to be a feature unique to Plasmodium.
Although the impact on transcriptional activity of these epigenetic modifications depends on global chromatin environments, some of them are considered to be hallmarks of euchromatin or heterochromatin. H3K9me3 and H3K36me3 are associated with inactive promoters and heterochromatin and are found to be mutually exclusive of H3K9ac and H3K4me3, signatures of active transcription (Figure 2). H3K9me3 is recognized and bound by heterochromatin protein 1 (HP1), an interaction conserved across eukaryotes, and their coupled role(s) in silencing var genes has been well studied in Plasmodium. A clear enrichment of H3K9me3 was observed for inactive var genes located in the nuclear periphery of the parasite (26, 75, 98, 104). Conversely, the single active var gene was shown to be marked by H3K9ac and H3K4 di- and trimethylation (40, 74).
The majority of these studies were performed on asexual stages, with some specific subsets of these modifications observed during distinct stages of the IDC. In ring stages, histones are enriched in H4K20me1 or H3K56me1, whereas trophozoites are associated with H3K9me2 and H3K18me1 (27). Although there is little information about other stages of P. falciparum, recent works have unveiled epigenetic modifications throughout different gametocyte stages (27, 110). Various histone marks were identified as stage specific, such as H3K36me2/me3 for early gametocytes or H3K27me1 and H3K36me1 for late gametocytes (27, 29). However, a recent study has revealed some discrepancies in the PTM patterns (110), substantiating the need to validate histone marks identified by mass spectrometry using alternative tools such as ChIP-seq. In addition, the overall layout of histone marks would stand to benefit from comparative studies across Plasmodium species. Complementary studies scrutinizing certain epigenetic modifications in sexual and mosquito stages of P. berghei (133) and P. vivax (84) were published. In this rodent parasite, for example, the H3K9ac mark correlated with the 5′ untranslated region of active genes throughout different stages, except for female gametocytes. This is most likely because mRNAs are stored and translationally repressed at this stage (133).
ChIP-seq (chromatin immunoprecipitation sequencing): technique for identifying the DNA-binding sites of DNA-binding proteins or histone modifications
Most histone PTM studies have been accomplished at single-site resolution, whereas combinational epigenetic modifications co-occurring on histones have long been neglected. Combinatorial associations were clearly identified in asexual and sexual stages of P. falciparum, exposing different forms of cross talk between histone modifications that seem to be fundamental for gene regulation and may be stage specific (27, 106, 110, 125). Similarly, ubiquitination and sumoylation are described in various eukaryotes as histone marks, but they remain poorly characterized in malaria parasites, despite ubiquitinated PfH2B (80, 106, 110, 123) as well as arginine methylation having been reported (110, 125). Although additional studies will be required to decipher the complexity of the histone code, there is no doubt that these modifications are critical regulators of gene expression throughout the life cycle of P. falciparum.
3.3. Chromatin Organization and Histone-Modifying Enzymes
As discussed above, epigenetic modifications are essential for gene regulation in Plasmodium. The histone PTMs are reversible and themselves under the control of a wide range of enzymes. These proteins, identified as histone-modifying enzymes, are categorized as writers and erasers and respectively deposit and remove epigenetic marks (Figure 2).
Methylation of the histone lysines is controlled by the opposing activity of histone lysine methyltransferases (HKMTs) and demethylases (HKDMs) (30). These enzymatic classes are composed of ten SET domain proteins for HKMTs, and three Jumonji C (JmjC) domain–containing proteins and two LSD1s for HKDMs (30, 57). Similarly, histone acetyltransferases, including MYST and GNAT families, along with histone deacetylases (HDACs), composed of HDAC and sirtuin proteins, have antagonistic effects on the acetylation of histone tails (62). The activities of these enzymes were extensively studied in the regulation of var genes in P. falciparum. PfSETvs has been shown to be a key regulator through deposition of H3K36me3 on inactive var genes (57, 124), as well as the sirtuins Sir2A and Sir2B involved in deacetylation and silencing of these virulence genes (33, 122). These complementary enzymatic activities orchestrate the mutually exclusive expression of var genes. Apicidin and curcumin, respectively inhibitors of HDACs and histone acetyltransferase PfGCN5, have been shown to drastically alter the epigenome and transcriptome of P. falciparum during the IDC (24, 31). Thus, inhibitors of histone-modifying enzymes are considered potent antimalarial compounds retaining efficacy against multiple stages of P. falciparum (28).
In addition to histone-modifying enzymes, chromatin-associated factors play a role in the interpretation of epigenomic marks. These reader proteins recognize specific histone modifications and promote the recruitment of chromatin-remodeling complexes and/or transcriptional machinery. Nearly 30 putative readers have been identified and classified in P. falciparum, such as bromodomain and zinc finger proteins, among others (53). Although most of these readers remain uncharacterized, others have been documented, such as PHD1, a PHD finger protein. PHD1 showed high affinity to H3K4me2/me3 and mediated the recruitment of ADA2, GCN5, and PF3D7_1402800, subunits of the transcription coactivator SAGA complex (53). In another instance, disruption of PfGCN5 led to drastic changes of the parasite epigenome and transcriptome, and its interaction with AP2-LT suggested assembly of an atypical SAGA complex in P. falciparum (81). The TF PfAP2-I coordinates the expression of invasion genes and may be recruited by PfBDP1, a bromodomain reader recognizing acetylated H3 (59, 105). Taken together, these histone-modifying enzymes have proven to be crucial in regulation of histone modifications and warrant further investigation and characterization, as are promising therapeutic targets.
4. BEYOND HISTONES: ADDITIONAL REGULATORY FACTORS
4.1. Regulation and Function of DNA Methylation
Besides the fundamental role of nucleosome positioning and histone marks in gene expression, epigenetic traits also encompass additional regulatory mediators. DNA methylation is one example and requires direct chemical modification occurring on the DNA. In mammalian genomes, this reaction is catalyzed by DNA methyltransferases, which transfer a methyl group to the fifth carbon of a cytosine residue to form 5-methylcytosine (5mC) (46). An additional oxidized intermediate, 5-hydroxymethylcytosine (5hmC), can also be generated during the demethylation process. Recruitment of readers to these epigenetic marks affects gene repression or hinders the binding of TFs (Figure 2). In Plasmodium, the contribution of this mechanism remains poorly understood. Although a unique DNA methyltransferase was identified in the parasite, a pioneering study provided a genome-wide map of 5mC distribution (91). Using bisulfite sequencing (BS-seq) technology, the authors detected that 0.58% of the total genomic cytosines were methylated. However, this method does not distinguish between 5mC and 5hmC. More recently, 5mC levels were detected at 0.01–0.02% of the P. falciparum genome, while a new modification, 5hmC-like, was identified as predominant, with 0.19–0.38% of total genomic cytosines (50). While additional data will be needed to confirm the presence of these modifications in the genome, they were found in gene bodies during the IDC; however, correlation with gene expression in asexual stages was found for only a small subset of genes. These findings highlight the need for renewed focus on the processes of DNA methylation in Plasmodium.
N6-Methyladenosine (m6A) has been known to be present mainly in prokaryotic DNA. Its identification in mammalian cells was controversial, and possibly due to bacterial contamination, nonspecific antibodies, and/or technical limitations. In Plasmodium a restriction enzyme–based approach identified the presence of m6A sites at very low levels in the P. falciparum genome, suggesting that this modification may be involved in gene regulation (78). Although these studies confirmed the presence of DNA methylation, the biological mechanisms remain to be investigated.
4.2. The Role of Noncoding RNAs in Gene Regulation
Over the last decades evidence has been mounting on the importance of ncRNAs in cell regulation and gene expression. Although they were initially overlooked and regarded as transcriptional noise, the advent of deep RNA-seq platforms provided an unprecedented advantage to uncover the roles of these previously unacknowledged non-protein-coding molecules. They are now a major focus of study and promising targets for drug therapies, vaccine targets, or diagnostic biomarkers.
In eukaryotes, small ncRNAs such as small interfering RNA (siRNA), microRNA (miRNA), and Piwi-interacting RNA (piRNA) average 18 to 200 nucleotides and function primarily in posttranscriptional modification or silencing and degradation processes. Long ncRNAs (lncRNAs), in contrast, are more than 200 nucleotides and vary in structure, forming complex hairpins and loops that dictate their niche epigenetic and regulatory functions. lncRNAs are classified based on their genomic location, transcriptional arrangements, and/or the transcriptomic mechanisms to which they belong. These ncRNAs can be transcribed as sense lncRNA, antisense lncRNA, intronic lncRNA, bidirectional lncRNA, and long-intergenic lncRNA (lincRNA). Regardless of their categories and features, they have been shown to adopt many mRNA-like traits, such as 5′ 7-methylguanosine capping and RNA polymerase II–mediated transcription (95).
In malaria parasites, there is no evidence for the presence of RNA interference mechanisms (miRNAs, siRNAs, piRNAs); nor is there evidence of conventional regulatory mechanisms based on bioinformatics analyses (9). Instead, these protozoans have adapted a higher-than-average repertoire of RNA-binding proteins and lncRNAs that have been implicated in a wide array of regulatory functions (114) (Figure 2). Yet, despite the categorical evidence on the diverse functional relevance of ncRNAs in higher eukaryotes, the functional and mechanistic repertoire of these ncRNAs in most apicomplexan parasites remains elusive. The malaria research community has begun to tap into this new aspect of Plasmodium biology, and prominent studies have identified hundreds to thousands of P. falciparum ncRNAs (15, 16, 25, 96, 136). Recently, a total of 1,768 intergenic lncRNAs have been identified (7). The subcellular localization and stage-specific expression of several of these putative lncRNAs were validated using RNA fluorescence in situ hybridization (RNA-FISH) and scRNA-seq. Additionally, the genome-wide occupancy of several candidate nuclear lncRNAs was explored using chromatin isolation by RNA purification (ChIRP). ChIRP-seq of candidate lncRNAs revealed that lncRNA occupancy sites within the parasite genome are focal and sequence specific, with a particular enrichment for several parasite-specific gene families, including those involved in pathogenesis, erythrocyte remodeling, and regulation of sexual differentiation. This study also demonstrated that the presence of some of these lncRNAs correlates with changes in gene expression (7). Moreover, disruption of one of these identified lncRNAs, lncRNA-ch14, resulted in a defect during sexual differentiation and development including sexual reproduction, validating the importance of some of these lncRNAs in gene regulation and stage transition. These lncRNAs could be the missing epigenetic regulators in Plasmodium. By interacting with DNA, RNA, and proteins, they can modulate chromatin structure by recruiting histone-modifying enzymes and transcription factors and control transcription.
One of the best-studied features of Plasmodium is found with the regulation of var genes. lncRNAs are implicated in the modulation of antigenic switching of the PfEMP1 variants through transcription of both a sense lncRNA and an antisense lncRNA. These lncRNAs originate from a bidirectional promoter in the var intron and are extended to exon I and exon II for antisense and sense lncRNAs, respectively (36). Expression of antisense lncRNAs from a particular silent var increases the activation of this specific gene and triggers, in trans, this var gene switching (1, 58). Recently, thioredoxin peroxidase I was identified to interact and colocalize with the antisense lncRNA, and its knockdown delayed var switching, suggesting a redox sensor role of this protein allowing the activation of a var gene depending on the nuclear environment (51). However, CRISPR/Cas9 editing of the var2csa intron demonstrated that these lncRNAs are not fundamental for activation and inactivation but may instead play a more subtle role in the switching mechanism than previously believed (17). Additional GC-rich ncRNAs are also described to be involved in the regulation of var expression (4, 48, 129). Although the means by which these regulatory mechanisms occur are still under debate and require further study, the current findings clearly validate the prominent implications of ncRNAs in the regulation of antigenic variation.
The antisense lncRNA transcribed from the gametocyte development protein 1 (GDV1) has also been shown to play a critical role in regulation of AP2-G. Identified as disrupted in a gametocyte-deficient line, its complementation restored gametocyte production whereas its overexpression led to higher gametocyte production, confirming the fundamental role of GDV1 in sexual development in P. falciparum (35). During unfavorable environmental conditions, the nuclear protein GDV1 evicts HP1 from the H3K9me3 on the ap2-g promoter, resulting in destabilization of the silencing mechanisms in place (37). With displacement of HP1, chromatin conformational changes result in euchromatin formation and facilitate transcription of ap2-g, initiating gametocytogenesis. It was revealed that during asexual parasite development, a five-exon antisense lncRNA transcribed from the gdv1 downstream locus is responsible for self-regulation and repression of the gene and indirectly of ap2-g (15). Additionally, knockout of this lncRNA increased expression of several genes associated with sexual differentiation (ap2-g, dblmsp2, pfg14_748, and pf3d7_1477400) (37). Recently, an inducible gametocyte producer line was engineered to overexpress GDV1, leading to a sexual conversion rate of 75%, a striking difference to the 8% found in the control population (11), validating the essentiality of this gene-lncRNA in this mechanism.
Despite the perplexity and debate maintained in the study of ncRNAs, the relative importance of these molecules across parasite development and transmission is undeniable. From pre- and posttranscriptional modifications to chromatin arrangements and gene regulation, ncRNAs have significant therapeutic and diagnostic potential. Advancements in capture technologies and enhanced sequencing platforms have helped dispel much of the mystery surrounding these molecules, yet there remains much to be resolved.
4.3. DNA Secondary Structures and G-Quadruplex
DNA is usually a canonical right-handed DNA helix but also has the ability to form various secondary structures such as hairpins and quadruplexes. G-quadruplex (G4) is one such structure and is formed by self-aggregation of four guanines in DNA and RNA. With the AT-rich genome of P. falciparum, G4 structures have long been confined to GC-rich repeats found in telomere ends and var genes located in subtelomeric regions (117). Recently, prediction algorithms showed enrichment of G4s not only in telomere regions and subtelomeric and internal var genes but also in nucleosome-depleted regions (10, 43). The use of pyridostatin, a G4 ligand, altered the P. falciparum transcriptome, resulting in the downregulation of 56–60% of genes containing G4s in promoters or exons (43). To our knowledge, only G-strand-binding protein 2 (GBP2) has been characterized as a G4-binding protein in P. falciparum. This protein interacted with G4 structures and telomeres in vivo, protected the parasite from pyridostatin treatment, and played a role in var gene regulation (34, 49). Although these pioneering studies support the existence of G4 structures and mediators in P. falciparum, their indirect or direct role(s) in gene regulation warrants further investigation.
5. NUCLEAR AND CHROMATIN ARCHITECTURE: DYNAMIC STRUCTURAL CHANGES ACROSS THE MALARIA PARASITE LIFE CYCLE
1. Nuclear Architecture
As discussed previously, the transcriptional status of malaria parasites is dynamic and controlled by various and interconnected layers of regulatory mechanisms associated with phenotypic and morphological changes. In the ring stage, for example, the genome is compacted and enriched in nucleosomes, with each nucleus encompassing 3 to 7 tightly clustered nuclear pores (2, 19, 130). This condensation hampers the binding of the transcriptional machinery and is consistent with the low transcription activity detected during ring stages. Conversely, the nuclear volume and the number of nuclear pore complexes (NPCs) are significantly increased in trophozoite stages, validating the open chromatin structure and active transcriptional activity (2, 130). These NPCs are roughly 60 in number and seem not to colocalize with HP1 and H3K9me3, two main heterochromatin markers, suggesting they are largely distributed in euchromatin and active regions (32). The number of pores gradually decreases throughout schizogony, with 6–16 NPCs for mid-schizonts and 2–6 for late schizonts, clustering once more to a single location near the euchromatin region (130). At this stage, nucleosomes are repackaged and the chromatin is compacted to facilitate the processes of cytokinesis and reinvasion, leading to a reduced nuclear volume akin to the ring stage.
Cytokinesis: a process of cell division in which cytoplasm is divided between the different nuclei forming the daughter cells
Yet, little information is known about the nuclear architecture of the parasite for the sexual and mosquito stages. Female gametocytes have been shown to develop a smaller and more compacted nucleus, with transcripts stored and translationally repressed, while male gametocytes, needing to prepare for mitosis and gamete formation, show larger nucleus volumes. In P. berghei, nuclear pores are distant from DNA, but P. falciparum has morphologically atypical gametocytes, which has hindered the ability to draw reliable conclusions (63). The remarkable plasticity and patterns of nuclear landscapes between Plasmodium spp. and within transitional stages warrant further analysis to help describe these complex parasites.
5.2. Chromatin Architecture
The pathogen is also reliant on the overall chromosomal architecture to mechanistically control the spatial proximity between essential genome components such as promotors and enhances cross talk between regions otherwise located far apart on the same chromosome or on different chromosomes.
These contact points were first recorded during early studies employing FISH, revealing important loci belonging to var gene families in the subtelomeric regions of heterologous chromosomes at the nuclear periphery (39, 75). Clustering of the chromosomal telomeres as a repressive center enriched in H3K9me3 and HP1 protein enables interaction and communication between the var gene families, crucial for mutual exclusive expression. These perinuclear repressive clusters congregate additional subtelomeric multigene families associated with reinvasion such as stevor and Pfmc-2TM. Like their var gene neighbors, these clonally variant gene families are subject to mutually exclusive expression during the parasite cell cycle (69).
The intra- and interchromosomal organizational patterns were further divulged following the development of 3D chromosome capture tools. Chromosome conformation capture coupled with next-generation sequencing technology (Hi-C) provided the opportunity to record and capture important contact loci in an all-versus-all approach and revealed the complex nature of the chromosomal landscape throughout parasite development (Figure 3). The unique patterns of chromosomal condensation were found to reflect the transcriptomic activity of parasite cell cycle progression. This genomic landscape adopts tight condensation formation throughout the pathogen’s early ring stage and during late schizogony, consistent with the low transcriptional activity associated with these parasite stages (2). Conversely, contact maps and 3D models uncovered a more open chromatin throughout the trophozoite stages, when transcription peaks. Hi-C studies revealed the spatiotemporal nature of the parasite’s genome whereby centromeres are directed to one end of the nuclear periphery along with discrete telomeric heterochromatin clusters allocated to the opposing end (2, 20, 71). Although no well-defined topologically associating domains were identified in Plasmodium, these Hi-C analyses have helped highlight the formation of domain-like structures surrounding the genes involved in antigenic variation bringing the subtelomeric and internal var genes loci closer together (Figure 3). In P. falciparum, a correlation between gene expression and genome organization was observed, with a gradient of expression increasing from the telomere to the centromere (20). A weaker relation was demonstrated for P. vivax, P. knowlesi, and P. berghei, but no correlation was observed for P. yoelii and Babesia microti.
Figure 3.
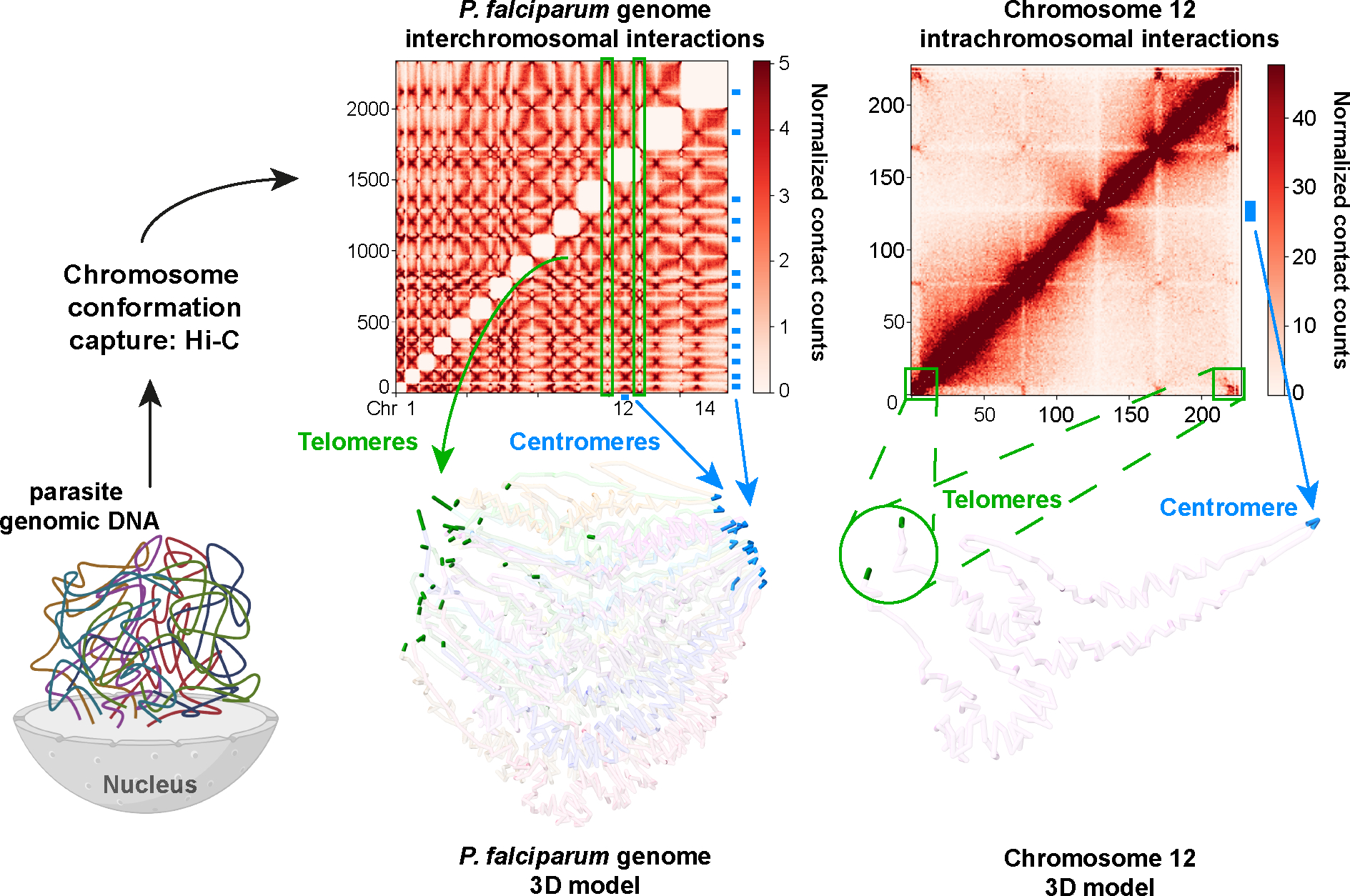
Hi-C data and 3D modeling of the Plasmodium falciparum genome. The chromatin conformation of the parasite genome is captured by Hi-C. Analysis of Hi-C data allows the generation of chromosomal contact maps depicting inter- and intrachromosomal interactions. This technique also enables reliable computer modeling of genome 3D structure. Interchromosomal interactions (left) between telomeres (green) and centromeres (blue) of the parasite genome are highlighted. The intrachromosomal interactions and the 3D model of chromosome 12 have been emphasized (right).
Hi-C: unbiased method to capture the 3D conformation of genomes based on chromatin fixation, enzymatic digestion, proximity ligation, and deep sequencing
Distinct changes were also observed for the transmission stages, such as rearrangement of the ap2-g locus moving it from the repressive heterochromatin cluster during the asexual stage to the active euchromatin at the early gametocyte stage, which led to its expression (18). Large chromosomal rearrangements were also observed for genes encoding proteins involved in invasion and exported proteins associated with gametocytogenesis and erythrocyte remodeling, indicating their relocation with the repressed virulence genes at the gametocyte stage (18). These interactions were even stronger in sporozoites, confirming their tight repression at this stage. Additional intrachromosomal interactions were detected and involved regions containing genes important for sporozoite migration and invasion such as csp, plp1, and gest (18) (Figure 1).
With the advancements in chromatin capture technologies, crucial features have been revealed about the importance of the dynamic nature of the Plasmodium chromosomal landscape driving developmental stages of the parasite. Future studies will most likely allow us to learn more about the molecular components regulating chromatin structure in Plasmodium.
5.3. Chromatin-Associated Modulators
Chromatin structure is essential for maintaining stage-specific gene expression. This structure is highly dynamic and requires fine organizational regulation by chromatin-associated proteins and lncRNAs and others.
In most metazoan organisms, the nucleus is demarcated by two lipid bilayers and reinforced by a lamina system that serves as a site for mechanosignaling, epigenetic regulation, and nuclear stability (47). Despite the fundamental relevance of this scaffolding hub, evidence of lamina components in many organisms, including Plasmodium, has yet to be found. A recent study successfully identified hundreds of chromatin-associated proteins using chromatin enrichment for proteomics (ChEP) (8). The authors also successfully characterized a CRWN-like protein (PF3D7_1325400), a plant-related protein functionally analogous to animal nuclear lamina proteins. Phenotypic analyses of combining mutations demonstrated the essentiality of the CRWN family for the viability of Arabidopsis and for proper nuclear organization (128), suggesting a similar role in Plasmodium. Findings obtained through immunofluorescence assays revealed this CRWN-like protein was colocalized with H3K9me3, insinuating a role in heterochromatin regulation (8).
High-mobility group box (HMGB) proteins have been a subject of interest, serving critical architectural organization and gene expression roles across the sexual and asexual stages of the parasite. In human cells, these proteins can bind to nucleosomes, lncRNAs, and diverse DNA structures, including G4, and regulate chromatin organization, telomere maintenance, and DNA repair (126). In Plasmodium, four HMGB proteins have been characterized through sequence homology of the conserved HMGB domains (14). Recently, PfHMGB1 was reported to be enriched in the centromeres, and its knockdown increased the distances among the centromeres at the nuclear periphery (76). This local chromatin disorganization also altered telomere clustering, which may explain the complete silencing of all var genes upon PfHMGB1 depletion and confirm the critical architectural role of this factor.
lncRNAs also have major roles in chromatin conformational regulation. The most prominent example of lncRNA functional implications lies in the X inactive specific transcript (Xist) expressed in mammalian cells. In eukaryotic cells, Xist regulates gene expression by mediating the inactivation of the X chromosome during zygote development after fertilization (41). In P. falciparum, a unique family of roughly 22 lncRNAs labeled as lncRNA-TAREs (telomere-associated repeat elements) was characterized (16). These TERRA (telomere repeat–containing RNA)-like lncRNAs are transcribed by RNA polymerase II from TAREs on almost all chromosome ends (16, 111). lncRNA TERRA is recruited to yeast and mammalian telomeres and is involved in telomerase recruitment, telomere maintenance, and stabilization of protein–telomere interactions (5). lncRNA-TAREs are long intergenic lncRNAs (>4 kb) and may play roles in DNA replication and telomere maintenance (15, 16, 111). lncRNA-TARE-6 consists of degenerate 21-bp repeats and forms a stable secondary structure composed of 12 predicted hairpin loops (111). This lncRNA was shown to bind nuclear proteins and histone H3, suggesting it may be implicated in the recruitment of nuclear factors to regulate chromosomal conformation. Another prominent example is lncRNA-TARE-4. ChIRP-seq experiments demonstrated lncRNA-TARE-4 interaction with most of the subtelomeric regions in a specific manner and revealed two potential motifs associated with lncRNA-TARE-4 binding (7). Taken together, chromatin-associated proteins and lncRNAs have proven to be fundamental regulators of chromatin architecture and warrant further investigation to help decipher at the mechanistic level the role of chromatin structure in gene regulation.
6. CONCLUSION
Collectively, the recent advances discussed in this review demonstrate the array of complex epigenetic mechanisms required to control transcriptional regulation in Plasmodium. Nucleosome positioning, TFs, histone modifications, and ncRNAs, among others, participate in a systematic and complementary manner in the dynamics of nuclear organization to coordinate gene regulation throughout parasite development and transmission. Although important formerly underappreciated or unknown regulators have been identified in recent years, our understanding is far from complete.
ACKNOWLEDGMENTS
We thank Todd Lenz for the Hi-C contact maps and 3D models. This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (grants R01 AI136511, R01 AI142743-01, and R21 AI142506-01 to K.L.R.) and the University of California, Riverside (NIFA-Hatch-225935 to K.L.R.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Amit-Avraham I, Pozner G, Eshar S, Fastman Y, Kolevzon N, et al. 2015. Antisense long noncoding RNAs regulate var gene activation in the malaria parasite Plasmodium falciparum. PNAS 112(9):E982–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ay F, Bunnik EM, Varoquaux N, Bol SM, Prudhomme J, et al. 2014. Three-dimensional modeling of the P. falciparum genome during the erythrocytic cycle reveals a strong connection between genome architecture and gene expression. Genome Res. 24(6):974–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balaji S, Madan Babu M, Iyer LM, Aravind L. 2005. Discovery of the principal specific transcription factors of Apicomplexa and their implication for the evolution of the AP2-integrase DNA binding domains. Nucleic Acids Res. 33(13):3994–4006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barcons-Simon A, Cordon-Obras C, Guizetti J, Bryant JM, Scherf A. 2020. CRISPR interference of a clonally variant GC-rich noncoding RNA family leads to general repression of var genes in Plasmodium falciparum. mBio. 11(1):e03054–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barral A, Déjardin J. 2020. Telomeric chromatin and TERRA. J. Mol. Biol. 432(15):4244–56 [DOI] [PubMed] [Google Scholar]
- 6.Bártfai R, Hoeijmakers WAM, Salcedo-Amaya AM, Smits AH, Janssen-Megens E, et al. 2010. H2A.Z demarcates intergenic regions of the Plasmodium falciparum epigenome that are dynamically marked by H3K9ac and H3K4me3. PLOS Pathog. 6(12):e1001223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batugedara G, Lu XM, Abel S, Chahine Z, Hristov B, et al. 2022. Deciphering the non-coding code of pathogenicity and sexual differentiation in the human malaria parasite. bioRxiv 2022.10.12.511630, Oct. 14 [Google Scholar]
- 8.Batugedara G Lu XM, Saraf A, Sardiu ME, Cort A, et al. 2020. The chromatin bound proteome of the human malaria parasite. Microb. Genom. 6(2):e000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baum J, Papenfuss AT, Mair GR, Janse CJ, Vlachou D, et al. 2009. Molecular genetics and comparative genomics reveal RNAi is not functional in malaria parasites. Nucleic Acids Res. 37(11):3788–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhartiya D, Chawla V, Ghosh S, Shankar R, Kumar N. 2016. Genome-wide regulatory dynamics of G-quadruplexes in human malaria parasite Plasmodium falciparum. Genomics 108(5–6):224–31 [DOI] [PubMed] [Google Scholar]
- 11.Boltryk SD, Passecker A, Alder A, Carrington E, van de Vegte-Bolmer M, et al. 2021. CRISPR/Cas9-engineered inducible gametocyte producer lines as a valuable tool for Plasmodium falciparum malaria transmission research. Nat. Commun. 12(1):4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bozdech Z, Llinás M, Pulliam BL, Wong ED, Zhu J, DeRisi JL. 2003. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLOS Biol. 1(1):e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brancucci NMB, de Niz M, Straub TJ, Ravel D, Sollelis L, et al. 2018. Probing Plasmodium falciparum sexual commitment at the single-cell level. Wellcome Open Res. 3:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briquet S, Boschet C, Gissot M, Tissandié E, Sevilla E, et al. 2006. High-mobility-group box nuclear factors of Plasmodium falciparum. Eukaryot. Cell 5(4):672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broadbent KM, Broadbent JC, Ribacke U, Wirth D, Rinn JL, Sabeti PC. 2015. Strand-specific RNA sequencing in Plasmodium falciparum malaria identifies developmentally regulated long non-coding RNA and circular RNA. BMC Genom. 16(1):454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Broadbent KM, Park D, Wolf AR, van Tyne D, Sims JS, et al. 2011. A global transcriptional analysis of Plasmodium falciparum malaria reveals a novel family of telomere-associated lncRNAs. Genome Biol. 12(6):R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryant JM, Regnault C, Benatar CS, Baumgarten S, Guizetti J, Scherf A. 2017. CRISPR/Cas9 genome editing reveals that the intron is not essential for var2csa gene activation or silencing in Plasmodium falciparum. mBio 8(4):729–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bunnik EM, Cook KB, Varoquaux N, Batugedara G, Prudhomme J, et al. 2018. Changes in genome organization of parasite-specific gene families during the Plasmodium transmission stages. Nat. Commun. 9(1):1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bunnik EM Polishko A, Prudhomme J, Ponts N, Gill SS, et al. 2014. DNA-encoded nucleosome occupancy is associated with transcription levels in the human malaria parasite Plasmodium falciparum. BMC Genom. 15(1):347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bunnik EM, Venkat A, Shao J, McGovern KE, Batugedara G, et al. 2019. Comparative 3D genome organization in apicomplexan parasites. PNAS 116(8):3183–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Callebaut I, Prat K, Meurice E, Mornon JP, Tomavo S. 2005. Prediction of the general transcription factors associated with RNA polymerase II in Plasmodium falciparum: conserved features and differences relative to other eukaryotes. BMC Genom. 6:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell TL, de Silva EK, Olszewski KL, Elemento O, Llinás M. 2010. Identification and genome-wide prediction of DNA binding specificities for the ApiAP2 family of regulators from the malaria parasite. PLOS Pathog. 6(10):e1001165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrington E, Cooijmans RHM, Keller D, Toenhake CG, Bártfai R, Voss TS. 2021. The ApiAP2 factor PfAP2-HC is an integral component of heterochromatin in the malaria parasite Plasmodium falciparum. iScience 24(5):102444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaal BK, Gupta AP, Wastuwidyaningtyas BD, Luah Y-H, Bozdech Z. 2010. Histone deacetylases play a major role in the transcriptional regulation of the Plasmodium falciparum life cycle. PLOS Pathog. 6(1):e1000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chappell L, Ross P, Orchard L, Russell TJ, Otto TD, et al. 2020. Refining the transcriptome of the human malaria parasite Plasmodium falciparum using amplification-free RNA-seq. BMC Genom. 21(1):395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chookajorn T, Dzikowski R, Frank M, Li F, Jiwani AZ, et al. 2007. Epigenetic memory at malaria virulence genes. PNAS 104(3):899–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coetzee N, Sidoli S, van Biljon R, Painter H, Llinás M, et al. 2017. Quantitative chromatin proteomics reveals a dynamic histone post-translational modification landscape that defines asexual and sexual Plasmodium falciparum parasites. Sci. Rep. 7(1):607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coetzee N, von Grüning H, Opperman D, van der Watt M, Reader J, Birkholtz LM. 2020. Epigenetic inhibitors target multiple stages of Plasmodium falciparum parasites. Sci. Rep. 10(1):2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connacher J, Josling GA, Orchard LM, Reader J, Llinás M, Birkholtz LM. 2021. H3K36 methylation reprograms gene expression to drive early gametocyte development in Plasmodium falciparum. Epigenet. Chromatin 14(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui L, Fan Q Cui L, Miao J. 2008. Histone lysine methyltransferases and demethylases in Plasmodium falciparum. Int. J. Parasitol. 38(10):1083–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui L, Miao J, Furuya T, Li X, Su XZ, Cui L. 2007. PfGCN5-mediated histone H3 acetylation plays a key role in gene expression in Plasmodium falciparum. Eukaryot. Cell 6(7):1219–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dahan-Pasternak N, Nasereddin A, Kolevzon N, Pe’er M, Wong W, et al. 2013. Pfsec13 is an unusual chromatin-associated nucleoporin of Plasmodium falciparum that is essential for parasite proliferation in human erythrocytes. J. Cell Sci. 126(14):3055–69 [DOI] [PubMed] [Google Scholar]
- 33.Duraisingh MT, Voss TS, Marty AJ, Duffy MF, Good RT, et al. 2005. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell 121(1):13–24 [DOI] [PubMed] [Google Scholar]
- 34.Edwards-Smallbone J, Jensen AL, Roberts LE, Totañes FIG, Hart SR, Merrick CJ. 2022. Plasmodium falciparum gbp2 is a telomere-associated protein that binds to G-quadruplex DNA and RNA. Front. Cell Infect. Microbiol. 12:782537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eksi S, Morahan BJ, Haile Y, Furuya T, Jiang H, et al. 2012. Plasmodium falciparum gametocyte development 1 (Pfgdv1) and gametocytogenesis early gene identification and commitment to sexual development. PLOS Pathog. 8(10):e1002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Epp C, Li F, Howitt CA, Chookajorn T, Deitsch KW. 2009. Chromatin associated sense and antisense noncoding RNAs are transcribed from the var gene family of virulence genes of the malaria parasite Plasmodium falciparum. RNA 15(1):116–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Filarsky M, Fraschka SA, Niederwieser I, Brancucci NMB, Carrington E, et al. 2018. GDV1 induces sexual commitment of malaria parasites by antagonizing HP1-dependent gene silencing. Science 359(6381):1259–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flueck C, Bartfai R, Niederwieser I, Witmer K, Alako BTF, et al. 2010. A major role for the Plasmodium falciparum ApiAP2 protein PfSIP2 in chromosome end biology. PLOS Pathog. 6(2):e1000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freitas-Junior LH, Bottius E, Pirrit LA, Deitsch KW, Scheidig C, et al. 2000. Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature 407(6807):1018–22 [DOI] [PubMed] [Google Scholar]
- 40.Freitas-Junior LH, Hernandez-Rivas R, Ralph SA, Montiel-Condado D, Ruvalcaba-Salazar OK, et al. 2005. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 121(1):25–36 [DOI] [PubMed] [Google Scholar]
- 41.Furlan G, Galupa R. 2022. Mechanisms of choice in X-chromosome inactivation. Cells 11(3):535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gardner MJ, Hall N, Fung E, White O, Berriman M, et al. 2002. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419(6906):498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gazanion E, Lacroix L, Alberti P, Gurung P, Wein S, et al. 2020. Genome wide distribution of G-quadruplexes and their impact on gene expression in malaria parasites. PLOS Genet. 16(7):e1008917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gómez-Díaz E, Yerbanga RS, Lefèvre T, Cohuet A, Rowley MJ, et al. 2017. Epigenetic regulation of Plasmodium falciparum clonally variant gene expression during development in Anopheles gambiae. Sci. Rep. 7(1):40655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gopalakrishnan AM, Nyindodo LA, Ross Fergus M, López-Estraño C. 2009. Plasmodium falciparum: preinitiation complex occupancy of active and inactive promoters during erythrocytic stage. Exp. Parasitol. 121(1):46–54 [DOI] [PubMed] [Google Scholar]
- 46.Greenberg MVC, Bourc’his D. 2019. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20(10):590–607 [DOI] [PubMed] [Google Scholar]
- 47.Gruenbaum Y, Foisner R. 2015. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 84:131–64 [DOI] [PubMed] [Google Scholar]
- 48.Guizetti J, Barcons-Simon A, Scherf A. 2016. Trans-acting GC-rich non-coding RNA at var expression site modulates gene counting in malaria parasite. Nucleic Acids Res. 44(20):9710–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gurung P, Gomes AR, Martins RM, Juranek SA, Alberti P, et al. 2021. PfGBP2 is a novel G-quadruplex binding protein in Plasmodium falciparum. Cell Microbiol. 23(4):e13303. [DOI] [PubMed] [Google Scholar]
- 50.Hammam E, Ananda G, Sinha A, Scheidig-Benatar C, Bohec M, et al. 2020. Discovery of a new predominant cytosine DNA modification that is linked to gene expression in malaria parasites. Nucleic Acids Res. 48(1):184–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heinberg A, Amit-Avraham I, Mitesser V, Simantov K, Goyal M, et al. 2022. A nuclear redox sensor modulates gene activation and var switching in Plasmodium falciparum. PNAS 119(33):e2201247119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoeijmakers WAM, Flueck C, Françoijs KJ, Smits AH, Wetzel J, et al. 2012. Plasmodium falciparum centromeres display a unique epigenetic makeup and cluster prior to and during schizogony. Cell Microbiol. 14(9):1391–401 [DOI] [PubMed] [Google Scholar]
- 53.Hoeijmakers WAM, Miao J, Schmidt S, Toenhake CG, Shrestha S, et al. 2019. Epigenetic reader complexes of the human malaria parasite, Plasmodium falciparum. Nucleic Acids Res. 47(22):11574–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoeijmakers WAM, Salcedo-Amaya AM, Smits AH, Françoijs KJ, Treeck M, et al. 2013. H2A.Z/H2B.Z double-variant nucleosomes inhabit the AT-rich promoter regions of the Plasmodium falciparum genome. Mol. Microbiol. 87(5):1061–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Howick VM, Russell AJC, Andrews T, Heaton H, Reid AJ, et al. 2019. The malaria cell atlas: single parasite transcriptomes across the complete Plasmodium life cycle. Science 365(6455):eaaw2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwanaga S, Kaneko I, Kato T, Yuda M. 2012. Identification of an AP2-family protein that is critical for malaria liver stage development. PLOS ONE 7(11):e47557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang L, Mu J, Zhang Q, Ni T, Srinivasan P, et al. 2013. PfSETvs methylation of histone H3K36 represses virulence genes in Plasmodium falciparum. Nature 499(7457):223–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jing Q, Cao L, Zhang L, Cheng X, Gilbert N, et al. 2018. Plasmodium falciparum var gene is activated by its antisense long noncoding RNA. Front. Microbiol. 9:3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Josling GA, Petter M, Oehring SC, Gupta AP, Dietz O, et al. 2015. A Plasmodium falciparum bromodomain protein regulates invasion gene expression. Cell Host Microbe 17(6):741–51 [DOI] [PubMed] [Google Scholar]
- 60.Kafsack BFC, Rovira-Graells N, Clark TG, Bancells C, Crowley VM, et al. 2014. A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature 507(7491):248–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaneko I, Iwanaga S, Kato T, Kobayashi I, Yuda M. 2015. Genome-wide identification of the target genes of AP2-O, a Plasmodium AP2-family transcription factor. PLOS Pathog. 11(5):e1004905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanyal A, Rawat M, Gurung P, Choubey D, Anamika K, Karmodiya K. 2018. Genome-wide survey and phylogenetic analysis of histone acetyltransferases and histone deacetylases of Plasmodium falciparum. FEBS J. 285(10):1767–82 [DOI] [PubMed] [Google Scholar]
- 63.Kehrer J, Kuss C, Andres-Pons A, Reustle A, Dahan N, et al. 2018. Nuclear pore complex components in the malaria parasite Plasmodium berghei. Sci. Rep. 8(1):11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kensche PR, Hoeijmakers WAM, Toenhake CG, Bras M, Chappell L, et al. 2015. The nucleosome landscape of Plasmodium falciparum reveals chromatin architecture and dynamics of regulatory sequences. Nucleic Acids Res. 44(5):2110–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khan SM, Franke-Fayard B, Mair GR, Lasonder E, Janse CJ, et al. 2005. Proteome analysis of separated male and female gametocytes reveals novel sex-specific Plasmodium biology. Cell 121(5):675–87 [DOI] [PubMed] [Google Scholar]
- 66.Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, et al. 2018. The human transcription factors. Cell 172(4):650–65 [DOI] [PubMed] [Google Scholar]
- 67.LaMonte GM, Orjuela-Sanchez P, Calla J, Wang LT, Li S, et al. 2019. Dual RNA-seq identifies human mucosal immunity protein Mucin-13 as a hallmark of Plasmodium exoerythrocytic infection. Nat. Commun. 10(1):488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lasonder E, Rijpma SR, van Schaijk BCL, Hoeijmakers WAM, Kensche PR, et al. 2016. Integrated transcriptomic and proteomic analyses of P. falciparum gametocytes: molecular insight into sex-specific processes and translational repression. Nucleic Acids Res. 44(13):6087–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lavazec C, Sanyal S, Templeton TJ. 2007. Expression switching in the stevor and Pfmc-2TM superfamilies in Plasmodium falciparum. Mol. Microbiol. 64(6):1621–34 [DOI] [PubMed] [Google Scholar]
- 70.Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, et al. 2003. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301(5639):1503–8 [DOI] [PubMed] [Google Scholar]
- 71.Lemieux JE, Kyes SA, Otto TD, Feller AI, Eastman RT, et al. 2013. Genome-wide profiling of chromosome interactions in Plasmodium falciparum characterizes nuclear architecture and reconfigurations associated with antigenic variation. Mol. Microbiol. 90(3):519–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lindner SE, Swearingen KE, Shears MJ, Walker MP, Vrana EN, et al. 2019. Transcriptomics and proteomics reveal two waves of translational repression during the maturation of malaria parasite sporozoites. Nat. Commun. 10(1):4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.López-Barragán MJ, Lemieux J, Quiñones M, Williamson KC, Molina-Cruz A, et al. 2011. Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genom. 12:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lopez-Rubio JJ, Gontijo AM, Nunes MC, Issar N, Hernandez Rivas R, Scherf A. 2007. 5′ flanking region of var genes nucleate histone modification patterns linked to phenotypic inheritance of virulence traits in malaria parasites. Mol. Microbiol. 66(6):1296–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lopez-Rubio JJ, Mancio-Silva L, Scherf A. 2009. Genome-wide analysis of heterochromatin associates clonally variant gene regulation with perinuclear repressive centers in malaria parasites. Cell Host Microbe 5(2):179–90 [DOI] [PubMed] [Google Scholar]
- 76.Lu B, Liu M, Gu L, Li Y, Shen S, et al. 2021. The architectural factor HMGB1 is involved in genome organization in the human malaria parasite Plasmodium falciparum. mBio 12(2):e00148–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu XM, Batugedara G, Lee M, Prudhomme J, Bunnik EM, Le Roch KG. 2017. Nascent RNA sequencing reveals mechanisms of gene regulation in the human malaria parasite Plasmodium falciparum. Nucleic Acids Res. 45(13):7825–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luo GZ, Wang F, Weng X, Chen K, Hao Z, et al. 2016. Characterization of eukaryotic DNA N6-methyladenine by a highly sensitive restriction enzyme-assisted sequencing. Nat. Commun. 7:11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martins RM, Macpherson CR, Claes A, Scheidig-Benatar C, Sakamoto H, et al. 2017. An ApiAP2 member regulates expression of clonally variant genes of the human malaria parasite Plasmodium falciparum. Sci. Rep. 7(1):14042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miao J, Fan Q, Cui L, Li J, Li J, Cui L. 2006. The malaria parasite Plasmodium falciparum histones: organization, expression, and acetylation. Gene 369(1–2):53–65 [DOI] [PubMed] [Google Scholar]
- 81.Miao J, Wang C, Lucky AB, Liang X, Min H, et al. 2021. A unique GCN5 histone acetyltransferase complex controls erythrocyte invasion and virulence in the malaria parasite Plasmodium falciparum. PLOS Pathog. 17(8):e1009351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Modrzynska K, Pfander C, Chappell L, Yu L, Suarez C, et al. 2017. A knockout screen of ApiAP2 genes reveals networks of interacting transcriptional regulators controlling the Plasmodium life cycle. Cell Host Microbe 21(1):11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mohammed M, Dziedziech A, Sekar V, Ernest M, Silva TLAE, et al. 2023. Single-cell transcriptomics to define Plasmodium falciparum stage transition in the mosquito midgut. Microbiol. Spectr. 11(2):e03671–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Muller I, Jex AR, Kappe SHI, Mikolajczak SA, Sattabongkot J, et al. 2019. Transcriptome and histone epigenome of Plasmodium vivax salivary-gland sporozoites point to tight regulatory control and mechanisms for liver-stage differentiation in relapsing malaria. Int. J. Parasitol. 49(7):501–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ngara M, Palmkvist M, Sagasser S, Hjelmqvist D, Björklund ÅK, et al. 2018. Exploring parasite heterogeneity using single-cell RNA-seq reveals a gene signature among sexual stage Plasmodium falciparum parasites. Exp. Cell Res. 371(1):130–38 [DOI] [PubMed] [Google Scholar]
- 86.Nishi T, Kaneko I, Iwanaga S, Yuda M. 2022. Identification of a novel AP2 transcription factor in zygotes with an essential role in Plasmodium ookinete development. PLOS Pathog. 18(8):e1010510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Otto TD, Wilinski D, Assefa S, Keane TM, Sarry LR, et al. 2010. New insights into the blood-stage transcriptome of Plasmodium falciparum using RNA-seq. Mol. Microbiol. 76(1):12–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Painter HJ, Chung NC, Sebastian A, Albert I, Storey JD, Llinás M. 2018. Genome-wide real-time in vivo transcriptional dynamics during Plasmodium falciparum blood-stage development. Nat. Commun. 9(1):2656. Erratum. 2022. Nat. Commun. 13:1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Petter M, Lee CC, Byrne TJ, Boysen KE, Volz J, et al. 2011. Expression of P. falciparum var genes involves exchange of the histone variant H2A.Z at the promoter. PLOS Pathog. 7(2):e1001292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Petter M, Selvarajah SA, Lee CC, Chin WH, Gupta AP, et al. 2013. H2A.Z and H2B.Z double-variant nucleosomes define intergenic regions and dynamically occupy var gene promoters in the malaria parasite Plasmodium falciparum. Mol. Microbiol. 87(6):1167–82 [DOI] [PubMed] [Google Scholar]
- 91.Ponts N, Fu L, Harris EY, Zhang J, Chung DWD, et al. 2013. Genome-wide mapping of DNA methylation in the human malaria parasite Plasmodium falciparum. Cell Host Microbe 14(6):696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ponts N, Harris EY, Lonardi S, Le Roch KG. 2011. Nucleosome occupancy at transcription start sites in the human malaria parasite: a hard-wired evolution of virulence? Infect. Genet. Evol. 11(4):716–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Poran A, Nötzel C, Aly O, Mencia-Trinchant N, Harris CT, et al. 2017. Single-cell RNA sequencing reveals a signature of sexual commitment in malaria parasites. Nature 551(7678):95–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Quansah E, Pappoe F, Shen J, Liu M, Yang S, et al. 2022. ApiAP2 gene-network regulates gametocytogenesis in Plasmodium parasites. Cell Microbiol. 2022:5796578 [Google Scholar]
- 95.Quinn JJ Chang HY. 2015. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 17(1):47–62 [DOI] [PubMed] [Google Scholar]
- 96.Raabe CA, Sanchez CP, Randau G, Robeck T, Skryabin BV, et al. 2009. A global view of the nonprotein-coding transcriptome in Plasmodium falciparum. Nucleic Acids Res. 38(2):608–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rai R, Zhu L, Chen H, Gupta AP, Sze SK, et al. 2014. Genome-wide analysis in Plasmodium falciparum reveals early and late phases of RNA polymerase II occupancy during the infectious cycle. BMC Genom. 15(1):959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ralph SA, Scheidig-Benatar C, Scherf A. 2005. Antigenic variation in Plasmodium falciparum is associated with movement of var loci between subnuclear locations. PNAS 102(15):5414–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rawat M, Srivastava A, Johri S, Gupta I, Karmodiya K. 2021. Single-cell RNA sequencing reveals cellular heterogeneity and stage transition under temperature stress in synchronized Plasmodium falciparum cells. Microbiol. Spectr. 9(1):e0000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Read DF, Cook K, Lu YY, Le Roch KG, Noble WS. 2019. Predicting gene expression in the human malaria parasite Plasmodium falciparum using histone modification, nucleosome positioning, and 3D localization features. PLOS Comput. Biol. 15(9):e1007329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Real E, Howick VM, Dahalan FA, Witmer K, Cudini J, et al. 2021. A single-cell atlas of Plasmodium falciparum transmission through the mosquito. Nat. Commun. 12(1):3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reid AJ, Talman AM, Bennett HM, Gomes AR, Sanders MJ, et al. 2018. Single-cell RNA-seq reveals hidden transcriptional variation in malaria parasites. eLife 7:e33105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ruiz JL, Tena JJ, Bancells C, Cortés A, Gómez-Skarmeta JL, Gomez-Díaz E. 2018. Characterization of the accessible genome in the human malaria parasite Plasmodium falciparum. Nucleic Acids Res. 46(18):9414–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Salcedo-Amaya AM, van Driel MA, Alako BT, Trelle MB, van den Elzen AMG, et al. 2009. Dynamic histone H3 epigenome marking during the intraerythrocytic cycle of Plasmodium falciparum. PNAS 106(24):9655–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Santos JM, Josling G, Ross P, Joshi P, Orchard L, et al. 2017. Red blood cell invasion by the malaria parasite is coordinated by the PfAP2-I transcription factor. Cell Host Microbe 21(6):731–41.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Saraf A Cervantes S, Bunnik EM, Ponts N, Sardiu ME, et al. 2016. Dynamic and combinatorial landscape of histone modifications during the intraerythrocytic developmental cycle of the malaria parasite. J. Proteome Res. 15(8):2787–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shang X, Shen S, Tang J, He X, Zhao Y, et al. 2021. A cascade of transcriptional repression determines sexual commitment and development in Plasmodium falciparum. Nucleic Acids Res. 49(16):9264–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shang X, Wang C, Fan Y, Guo G, Wang F, et al. 2022. Genome-wide landscape of ApiAP2 transcription factors reveals a heterochromatin-associated regulatory network during Plasmodium falciparum blood-stage development. Nucleic Acids Res. 50(6):3413–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shang X, Wang C, Shen L, Sheng F, He X, et al. 2022. PfAP2-EXP2, an essential transcription factor for the intraerythrocytic development of Plasmodium falciparum. Front. Cell Dev. Biol. 9:3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shrestha S, Lucky AB, Brashear AM, Li X, Cui L, Miao J. 2022. Distinct histone post-translational modifications during Plasmodium falciparum gametocyte development. J. Proteome Res. 21(8):1857–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sierra-Miranda M, Delgadillo DM, Mancio-Silva L, Vargas M, Villegas-Sepulveda N, et al. 2012. Two long non-coding RNAs generated from subtelomeric regions accumulate in a novel perinuclear compartment in Plasmodium falciparum. Mol. Biochem. Parasitol. 185(1):36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sierra-Miranda M, Vembar S-S, Delgadillo DM, Ávila-López PA, Herrera-Solorio A-M, et al. 2017. PfAP2Tel, harbouring a non-canonical DNA-binding AP2 domain, binds to Plasmodium falciparum telomeres. Cell Microbiol. 19(9):e12742. [DOI] [PubMed] [Google Scholar]
- 113.Silvestrini F, Bozdech Z, Lanfrancotti A, di Giulio E, Bultrini E, et al. 2005. Genome-wide identification of genes upregulated at the onset of gametocytogenesis in Plasmodium falciparum. Mol. Biochem. Parasitol. 143(1):100–10 [DOI] [PubMed] [Google Scholar]
- 114.Simantov K, Goyal M, Dzikowskiid R. 2022. Emerging biology of noncoding RNAs in malaria parasites. PLOS Pathog. 18(7):e1010600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Singh S, Santos JM, Orchard LM, Yamada N, van Biljon R, et al. 2021. The PfAP2-G2 transcription factor is a critical regulator of gametocyte maturation. Mol. Microbiol. 115(5):1005–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sinha A Hughes KR, Modrzynska KK, Otto TD, Pfander C, et al. 2014. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature 507(7491):253–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Smargiasso N, Gabelica V, Damblon C, Rosu F, de Pauw E, et al. 2009. Putative DNA G-quadruplex formation within the promoters of Plasmodium falciparum var genes. BMC Genom. 10:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sullivan WJ. 2003. Histone H3 and H3.3 variants in the protozoan pathogens Plasmodium falciparum and Toxoplasma gondii. DNA Seq. 14(3):227–31 [DOI] [PubMed] [Google Scholar]
- 119.Tang J, Chisholm SA, Yeoh LM, Gilson PR, Papenfuss AT, et al. 2020. Histone modifications associated with gene expression and genome accessibility are dynamically enriched at Plasmodium falciparum regulatory sequences. Epigenet. Chromatin 13(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Templeton TJ, Iyer LM, Anantharaman V, Enomoto S, Abrahante JE, et al. 2004. Comparative analysis of Apicomplexa and genomic diversity in eukaryotes. Genome Res. 14(9):1686–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Toenhake CG, Fraschka SAK, Vijayabaskar MS, Westhead DR, van Heeringen SJ, Bártfai R. 2018. Chromatin accessibility-based characterization of the gene regulatory network underlying Plasmodium falciparum blood-stage development. Cell Host Microbe 23(4):557–69.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tonkin CJ, Carret CK, Duraisingh MT, Voss TS, Ralph SA, et al. 2009. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLOS Biol. 7(4):e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Trelle MB, Salcedo-Amaya AM, Cohen AM, Stunnenberg HG, Jensen ON. 2009. Global histone analysis by mass spectrometry reveals a high content of acetylated lysine residues in the malaria parasite Plasmodium falciparum. J. Proteome Res. 8(7):3439–50 [DOI] [PubMed] [Google Scholar]
- 124.Ukaegbu UE, Kishore SP, Kwiatkowski DL, Pandarinath C, Dahan-Pasternak N, et al. 2014. Recruitment of PfSET2 by RNA polymerase II to variant antigen encoding loci contributes to antigenic variation in P. falciparum. PLOS Pathog. 10(1):e1003854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.von Grüning H, Coradin M, Mendoza MR, Reader J, Sidoli S, et al. 2022. A dynamic and combinatorial histone code drives malaria parasite asexual and sexual development. Mol. Cell Proteom. 21(3):100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Voong CK, Goodrich JA, Kugel JF. 2021. Interactions of HMGB proteins with the genome and the impact on disease. Biomolecules 11(10):1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Walzer KA, Kubicki DM, Tang X, Chi J-TA. 2018. Single-cell analysis reveals distinct gene expression and heterogeneity in male and female Plasmodium falciparum gametocytes. mSphere 3(2):e00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang H, Dittmer TA, Richards EJ. 2013. Arabidopsis CROWDED NUCLEI (CRWN) proteins are required for nuclear size control and heterochromatin organization. BMC Plant Biol. 13(1):200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wei G, Zhao Y, Zhang Q, Pan W. 2015. Dual regulatory effects of non-coding GC-rich elements on the expression of virulence genes in malaria parasites. Infect. Genet. Evol. 36:490–99 [DOI] [PubMed] [Google Scholar]
- 130.Weiner A, Dahan-Pasternak N, Shimoni E, Shinder V, von Huth P, et al. 2011. 3D nuclear architecture reveals coupled cell cycle dynamics of chromatin and nuclear pores in the malaria parasite Plasmodium falciparum. Cell Microbiol. 13(7):967–77 [DOI] [PubMed] [Google Scholar]
- 131.Westenberger SJ, Cui L, Dharia N, Winzeler E, Cui L. 2009. Genome-wide nucleosome mapping of Plasmodium falciparum reveals histone-rich coding and histone-poor intergenic regions and chromatin remodeling of core and subtelomeric genes. BMC Genom. 10:610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wichers JS, Scholz JAM, Strauss J, Witt S, Lill A, et al. 2019. Dissecting the gene expression, localization, membrane topology, and function of the Plasmodium falciparum STEVOR protein family. mBio 10(4):e01500–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Witmer K, Fraschka SA, Vlachou D, Bártfai R, Christophides GK. 2020. An epigenetic map of malaria parasite development from host to vector. Sci. Rep. 10(1):6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.World Health Organ. 2021. World Malaria Report 2021. Geneva: World Health Organ. [Google Scholar]
- 135.Xu Y, Qiao D, Wen Y, Bi Y, Chen Y, et al. 2021. PfAP2-g2 is associated to production and maturation of gametocytes in Plasmodium falciparum via regulating the expression of PfMDV-1. Front. Microbiol. 11:3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang M, Shang X, Zhou Y, Wang C, Wei G, et al. 2021. Full-length transcriptome analysis of Plasmodium falciparum by single-molecule long-read sequencing. Front. Cell Infect. Microbiol. 11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yuda M, Iwanaga S, Kaneko I, Kato T. 2015. Global transcriptional repression: ainitial and essential step for Plasmodium sexual development. PNAS 112(41):12824–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yuda M, Iwanaga S, Shigenobu S, Kato T, Kaneko I. 2010. Transcription factor AP2-Sp and its target genes in malarial sporozoites. Mol. Microbiol. 75(4):854–63 [DOI] [PubMed] [Google Scholar]
- 139.Yuda M, Iwanaga S, Shigenobu S, Mair GR, Janse CJ, et al. 2009. Identification of a transcription factor in the mosquito-invasive stage of malaria parasites. Mol. Microbiol. 71(6):1402–14 [DOI] [PubMed] [Google Scholar]
- 140.Zanghì G, Vembar SS, Baumgarten S, Ding S, Guizetti J, et al. 2018. A Specific PfEMP1 is expressed in P. falciparum sporozoites and plays a role in hepatocyte infection. Cell Rep. 22(11):2951–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang C, Li Z, Cui H, Jiang Y, Yang Z, et al. 2017. Systematic CRISPR-Cas9-mediated modifications of Plasmodium yoelii ApiAP2 genes reveal functional insights into parasite development. mBio. 8(6):e01986–17 [DOI] [PMC free article] [PubMed] [Google Scholar]